
ABSTRACT
In recent years, the development of bispecific antibody (bsAb) has become a major trend in the biopharmaceutical industry. By simultaneously engaging 2 molcular targets, bsAbs show unique mechanisms of action that could lead to clinical benefits unattainable by conventional monoclonal antibodies. Various bsAb generation formats have been described, and several are being investigated in clinical development. However, some bsAb constructs have proven to be problematic due to their unfavorable physicochemical and pharmacokinetic properties, as well as poor manufacturing efficiencies. We describe here a new bispecific design, Fabs-in-tandem immunoglobulin (FIT-Ig), in which 2 antigen-binding fragments are fused directly in a crisscross orientation without any mutations or use of peptide linkers. This unique design provides a symmetric IgG-like bispecific molecule with correct association of 2 sets of VH/VL pairs. We show that FIT-Ig molecules exhibit favorable drug-like properties, in vitro and in vivo functions, as well as manufacturing efficiency for commercial development.
Introduction
The idea of bispecific antibodies (bsAbs) originated at least 3 decades ago. By combining the antigen binding functions of 2 monoclonal antibodies (mAbs) into one molecule, bsAbs have been applied to not only target blockade, but also recruiting effector cells or associating 2 different antigens,Citation1-5 which usually cannot be achieved by the combination of 2 mAbs. The first Food and Drug Administration-approved bsAb drug blinatumomab (Blincyto®), which is a bispecific T-cell engager targeting CD19 and CD3, can harness the cytotoxic potential of T cells and eliminate target cancer cells very efficiently,Citation4,6 and numerous other bsAbs have been developed and are being evaluated in clinical trials.Citation7 Various technologies, including chemical conjugation, quadroma fusion, and co-culture of bacteria,Citation8,9 have been developed for bsAb generation. In recent years, antibody engineering by recombinant methods has emerged as the most prevalent approach for bsAb generation.
Recombinant bsAb can be divided into different categories by their design strategies, i.e., symmetric or asymmetric, with or without a fragment crystallizable (Fc, and bivalent versus tetravalent. Brinkmann and Kontermann recently published a very comprehensive overview of numerous bsAbs formats.Citation8 The common issue associated with immunoglobulin G (IgG)-like bsAb is VH/VL chain mispairing, which is caused by random pairing of 2 light chains with 2 heavy chains of different origin. There are 2 representative design strategies to resolve this problem: 1) asymmetric design, which achieves correct chain pairing by “knobs-into-holes” mutations in the Fc region, such as the CrossMab technology;Citation10 and 2) symmetric design, which adds additional binding domains onto the existing IgG in both arms, such as the DVD-Ig format.Citation11 Many other formats fall within one of these categories of molecular symmetry.Citation12 Despite a rich collection of bispecific design strategies, only a few formats have advanced into clinical stage beyond Phase 1. Many challenges still exist in the areas of drug-like properties, in vivo half-life, steric hindrance in antigen binding, and efficiency in manufacturing process development.
We herein describe a novel symmetric bsAb design, termed Fabs-in-tandem Ig (FIT-Ig), which combines the intact structure of natural antigen-binding fragments (Fabs) from 2 parental mAbs in a unique crisscross orientation without any mutations or peptide linkers, rendering it a generic approach for bispecific generation for a broad applications. Through the engineering and development of several FIT-Ig molecules targeting either soluble proteins or cell surface receptors, we demonstrate that FIT-Ig exhibits favorable drug-like properties, in vitro and in vivo functions, as well as manufacturing feasibility for therapeutic development.
Results
Developing FIT-Ig to neutralize 2 soluble target proteins
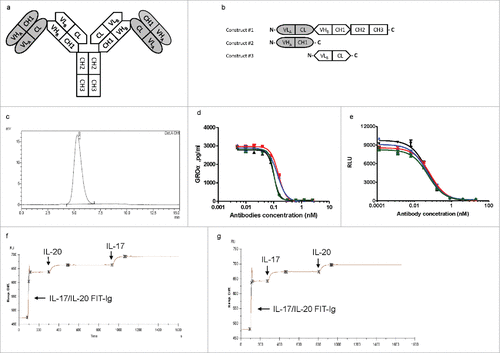
FIT-Ig protein that can bind to 2 soluble proteins of human IL-17 and human IL-20 was generated using anti-IL-17 mAb ixekizumab,Citation13 and anti-IL-20 mAb 15D2.Citation14 Interleukin-17 is linked to the pathogenesis of diverse autoimmune and inflammatory indications, and IL-20 is a proinflammatory cytokine of the IL-10 family. Although both IL-17 and IL-20 are important mediators for inflammatory diseases such as rheumatoid arthritis, they elicit their functions via different cellular and molecular mechanisms. A bsAb targeting both cytokines might enhance efficacy and affect a larger percentage of patients, and might also be faster and less expensive to develop compared with combination therapy.Citation15 To construct the FIT-Ig molecule, the light chain (VL-CL) domains of ixekizumab were directly (IL-17/IL-20 FIT-Ig, ), or through a linker of 3 amino acids “GSG” (IL-17/IL-20 FIT-Ig (SL), SL: Short linker) or 7 amino acids “GGGGSGS” (IL-17/IL-20 FIT-Ig (LL), LL: Long linker) fused in tandem with the heavy chain (VH-CH1-CH2-CH3) of 15D2 at the N terminus (Fig. S1a). The second construct was VH-CH1 of ixekizumab and the third construct was VL-CL of 15D2 ( and Fig. S1b). Different FIT-Ig molecules are named as A/B FIT-Ig protein, where A is the target of the mAb whose Fab domain is positioned on the N terminus of the heavy chain, distal from the Fc region, and B is the target of the mAb whose Fab domain is positioned proximal to the Fc region. FIT-Ig protein have an intact Fc domain, which is critical for the formation of a disulfide-linked full IgG-like molecule, and correct chain pairing allowed 2 sets of VH-CH1 and VL-CL connected structurally in a crisscross orientation. Co-transfection of mammalian cells with expression vectors encoding 3 chains of each FIT-Ig led to the expression and secretion of a single species of an IgG-like molecule with a molecular weight (MW) of ∼240 kDa. The expression titers of FIT-Ig proteins from transiently transfected human embryonic kidney 293 cells (HEK293E) were similar to that of a regular human IgG (as high as 200 mg/L had been observed), which was then easily purified to homogeneity by protein A chromatography. Purified FIT-Ig proteins with or without linker exhibited physical homogeneity as analyzed by size-exclusion chromatography (SEC) (, Fig. S1c, d). In addition, all 26 disulfide bonds of FIT-Ig molecule have been mapped by mass spectrometry (data not shown).
Figure 1. Design, generation and characterization of anti-IL-17/IL-20 Fabs-In-Tandem immunoglobulin (IL-17/IL-20 FIT-Ig) protein. (a) Schematic diagram of IL-17/IL-20 FIT-Ig protein design. (b) DNA construct design of a FIT-Ig. (c) SEC analysis of 293 cell-produced FIT-Ig after one-step Protein A purification. (d,e) IL-17/IL-20 FIT-Ig (
![]()
FIT-Ig protein is bispecific and tetravalent
The IL-17/IL-20 FIT-Ig proteins were evaluated for their neutralization potency and binding affinity for both cytokines. All 3 versions of IL-17/IL-20 FIT-Ig proteins inhibited IL-17-induced growth regulated oncogene-α (GROα) secretion from HS27 cells, with an IC50 similar to that of ixekizumab ( and ). The molecules also inhibited IL-20-induced apoptosis of BAF3 cells overexpressing IL-20 receptors, with an IC50 similar to that of the parental antibody 15D2 ( and ). With Biacore affinity testing, 3 IL-17/IL-20 FIT-Ig proteins exhibited similar antigen binding kinetics compared with both parental mAbs. The slight increase in affinity for IL-17 was likely due to an artifact from the Biacore assay format (). Furthermore, the tetravalent binding property of IL-17/IL-20 FIT-Ig proteins was demonstrated through multiple binding studies by injecting the 2 antigens sequentially over a FIT-Ig protein-immobilized chip on Biacore. When IL-17/IL-20 FIT-Ig protein had been saturated by the first antigen (IL-20), the second antigen (IL-17) was injected and, as expected, a second binding signal was observed (). This observation was reproducible when the antigen injection order was reversed (). Similar multiple binding profiles were observed for IL-17/IL-20 FIT-Ig (SL) and IL-17/IL-20 FIT-Ig (LL) (Fig. S1e-h). All 3 versions of IL-17/IL-20 FIT-Ig proteins exhibited similar binding activity and potency, indicating it was not necessary to introduce linkers between the 2 Fab regions of FIT-Ig. To avoid potential immunogenicity caused by exogenous linkers, FIT-Ig without linkers are preferred and further evaluated.
Table 1. In vitro functional characterization of IL-17/IL-20 FIT-Ig protein.
Developing FIT-Ig to target 2 cell surface receptors
To assess the feasibility of targeting 2 receptors by FIT-Ig, another FIT-Ig molecule targeting hepatocyte growth factor receptor (HGFR or cMet) and epidermal growth factor receptor (EGFR). It was previously shown that a key resistant mechanism for anti-EGFR treatment occurs via cMet activation, and blocking both receptor pathways could result in an enhanced anti-tumor efficacy.Citation16,17 A FIT-Ig was generated using sequences from an anti-cMet mAb and an anti-EGFR mAb (panitumumab) (Fig. S2a).Citation18,19 This cMet/EGFR FIT-Ig was produced with a high titer in transiently transfected 293 cells, and had >99% purity after one-step purification by protein A chromatography (Fig. S2b). Data from SDS-PAGE using materials before and after purification showed that full length FIT-Ig was expressed along with a Fab, which was likely a by-product resulting from association of the 2 short chains and was observed in expressions of all other FIT-Ig proteins. Such mis-paired non-functional Fab was then readily removed from one-step protein-A purification (Fig. S2c). SDS-PAGE analysis also revealed that the FIT-Ig molecule was ∼240 kDa in non-reducing condition, and separated to a ∼75 kDa long chain and 2 ∼25 kDa short chains in reducing condition (Fig. S2d). We also compared the Stokes radii between FIT-Ig and 2 parental mAbs by dynamic light scattering (DLS), and the results revealed Stokes radius of cMet/EGFR FIT-Ig was 6.758 nm, which was obviously larger than that of mAbs (5.864 nm and 5.483 nm, respectively) (Fig. S2e). Furthermore, the precise MWs of full length FIT-Ig (Fig. S3a) and each chain thereof (Fig. S3b, c), measured by mass spectrometry, were consistent with expected values. cMet/EGFR FIT-Ig also retained the binding activity of 2 parental antibodies in protein-based affinity measurement by OctedRed (Table S1). Dual binding testing by Octet Red system indicated cMet/EGFR FIT-Ig was also bispecific and tetravalent (Fig. S4a). Therefore, this indicated cMet/EGFR FIT-Ig had minimal domain steric hindrance in binding to both receptors.
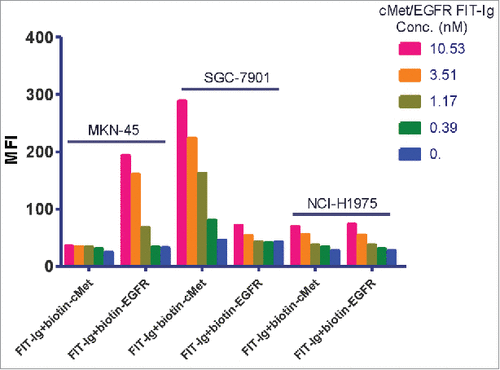
We also explored whether cMet/EGFR FIT-Ig could simultaneously bind to cell surface cMet and EGFR by using 3 cancer cell lines that have distinct EGFR/cMet expression profiles. MKN-45 cells express more cMet than EGFR (mean fluorescence intensities (MFIs) were 6530 and 1578, respectively), SGC-7901 cells express less cMet than EGFR (MFIs were 615 and 3338, respectively), and NCI-H1975 expresses almost equal levels of cMet and EGFR (MFIs were 1500 and 1470, respectively). These cells were first incubated with low to higher concentrations of FIT-Ig, washed, and then stained with either biotinylated soluble EGFR or biotinylated soluble cMet. For binding to MKN-45 cells, at low concentration, cMet/EGFR FIT-Ig saturated its binding domains by binding to both cell surface cMet and EGFR, and therefore could not bind to soluble recombinant biotin-cMet or biotin-EGFR. At higher concentration, cMet/EGFR FIT-Ig fully saturated its cMet binding domains but EGFR binding domains became available, and therefore could bind to soluble recombinant biotin-EGFR (). A similar phenomenon was observed in SGC-7901 cells except the cMet and EGFR expression ratio was reversed, showing only biotin-cMet binding at higher cMet/EGFR FIT-Ig concentrations (). In contrast, for NCI-H1975 cells that express equal amount of EGFR and cMet on the cell surface, no significant binding signal was detected for either biotin-cMet or biotin-EGFR at both low and high concentrations of cMet/EGFR FIT-Ig, indicating that all 4 binding domains of FIT-Ig had been fully occupied (). The same observation was also obtained when the Fab of the FIT-Ig was used in the binding assay instead of full size FIT-Ig (data not shown). It is therefore clearly demonstrated that a cMet/EGFR FIT-Ig molecule could simultaneously bind to both cell surface cMet and EGFR, as a tetravalent molecule.
Figure 2. Dual binding property of cMet/EGFR FIT-Ig on cell surface. FACS analysis of cMet/EGFR FIT-Ig dual binding to cell surface. MKN-45, SGC-7901 and NCI-H1975 cell lines were treated with serial concentration of cMet/EGFR FIT-Ig followed with 1 ug/ml biotinylated cMet or EGFR. Biotinylated cMet or EGFR could bind to cMet/EGFR FIT-Ig free binding cites and be detected by streptavidin Alexa Fluor® 488 conjugate.
FIT-Ig molecules show drug-like properties similar to mAbs
Pharmacokinetic (PK) studies were performed for both IL-17/IL-20 FIT-Ig and cMet/EGFR FIT-Ig. PK properties of the IL-17/IL-20 FIT-Ig protein were assessed upon intravenous (i.v.) and subcutaneous (s.c.) dosing (5 mg/kg) in rats. Serum concentrations were measured by 2 different antigen capture enzyme-linked immunosorbent assay (ELISA) methods, each against one of the 2 target antigens, with anti-human Fc detection. Serum concentrations were very similar when determined by 2 ELISA methods (), indicating that the molecule was intact throughout the 28-day in vivo period and capable of binding both antigens in the presence of serum. Upon i.v. dosing, IL-17/IL-20 FIT-Ig exhibited a biphasic PK profile, consisting of a distribution phase followed by an elimination phase, similar to the PK profile of conventional IgG molecules. The parameters calculated based on the 2 different analytical methods were very similar (). As demonstrated by these results, the in vivo properties of FIT-Ig are very similar to a conventional IgG molecule, with an elimination half-life of approximately 11 d. The PK of cMet/EGFR FIT-Ig was also evaluated with the same strategy as IL-17/IL-20 FIT-Ig, and the PK profile and parameters were also found to be similar to conventional IgG molecules, with a half-life of more than 10 d (Fig. S4b and Table S2).
Figure 3. Pharmacokinetic profile of IL-17/IL-20 FIT-Ig protein in rats Serum concentrations ( ± s.d.) time profile of IL-17/IL-20 FIT-Ig protein after a 5 mg/kg subcutaneous (S.C.) or intravenous (I.V.) administration to male Sprague-Dawley rats. Quantitation was performed using IL-17 capture (a) or IL-20 capture (b) ELISA.
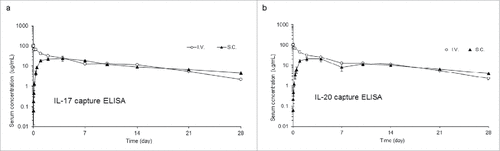
Table 2. Pharmacokinetic parameters of IL-17/IL-20 FIT-Ig proteins in rats.
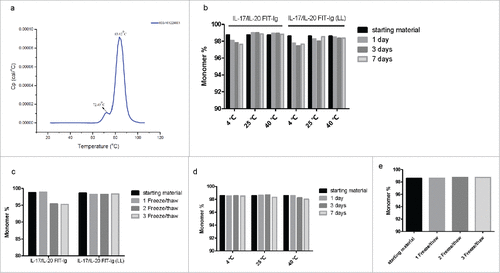
Further developability studies were performed addressing the stability of FIT-Ig molecules. The thermostability of cMet/EGFR FIT-Ig protein was evaluated by differential scanning calorimetry (DSC), and the resulting melting temperatures (Tm) of FIT-Ig protein () were similar to that of 2 parental mAbs (Fig. S4c, d). Additionally, even in a phosphate-buffered saline (PBS) buffer system, no obvious aggregation and degradation were observed by SEC for FIT-Ig proteins at 10 mg/mL after up to one week incubation at various temperature (4 – 40 ℃) or 3 freeze/thaw cycles (). Furthermore, there was no obvious solubility difference between FIT-Ig proteins and mAb in a PEG-induced precipitation assay (data not shown), and several FIT-Ig proteins were formulated to 50 mg/mL while still maintaining stability for an extensive period of time (data not shown).
Figure 4. Stability test of FIT-Ig proteins. (a) Thermostability test of cMet/EGFR FIT-Ig protein by DSC, Tm1 and Tm2 were 72.49 ℃and 83.62 ℃, respectively. (b) SEC analysis of IL-17/IL-20 FIT-Ig and IL-17/IL-20 FIT-Ig (LL) after incubation at different temperature. (c) SEC analysis of IL-17/IL-20 FIT-Ig and IL-17/IL-20 FIT-Ig (LL) after freeze/thaw cycles. (d) SEC analysis of cMet/EGFR FIT-Ig after incubation at different temperature. (e) SEC analysis of cMet/EGFR FIT-Ig after freeze/thaw cycles.
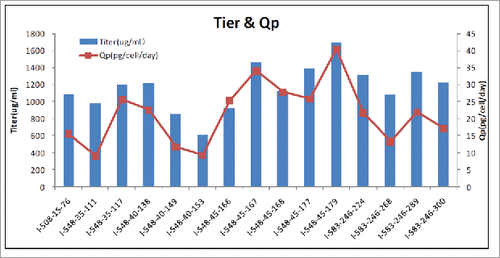
To further determine the manufacturing feasibility of FIT-Ig, Chinese hamster ovary (CHO) cells, the most commonly used mammalian host line for antibody manufacturing, were stably transfected with 2 constructs, one of which contained expression cassettes of long chain (VLA-CL-VHB-CH1-CH2-CH3) and one short chain (VLB-CL), another of which contained expression cassette of one short chain (VHA-CH1). Clone selections and productivity analysis were performed accordingly. A high titer of 1.7 g/L and a cell specific production (Qp) of above 40 pg/cell/day was reached from the second round limited dilution clones (), which also showed a very high level of stability through a 70-passage analysis.
Figure 5. Cell line generation of cMet/EGFR FIT-Ig Titers and Qp of the second round limited dilution clones. Qp, cell specific production.
FIT-Ig triggers synergic effect of two receptor targets on cancer cells
To determine whether our bsAbs can inhibit downstream signaling, the ability of cMet/EGFR FIT-Ig to inhibit hepatocyte growth factor (HGF)-induced AKT phosphorylation in NCI-H292 cell was investigated. The molecule exhibited potent inhibition of phosphor-AKT, with IC50 value in sub-nanomolar level. Moreover, cMet/EGFR FIT-Ig inhibited 80% of AKT phosphorylation while EGFR and cMet antibody combination only inhibited 60% of AKT phosphorylation (). Importantly, the cMet parental antibody and cMet/EGFR FIT-Ig showed no obvious agonistic effect in the absence of ligand (Fig. S5) compared with an agonistic anti-cMet antibody 9.1.2.Citation19
Figure 6. In vitro and in vivo function characterization of cMet/EGFR FIT-Ig (a) cMet/EGFR FIT-Ig inhibited phosphor-AKT in lung carcinoma cell line. NCI-H292 cells were treated with human IgG1 (![]()
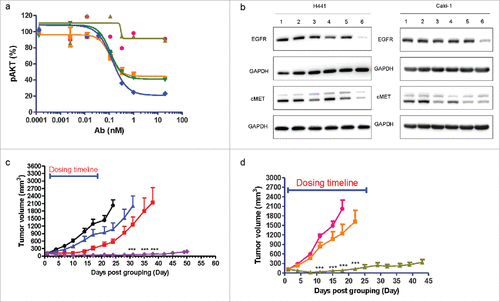
To further understand the function of cMet/EGFR FIT-Ig, we next examined whether it can induce cMet and EGFR depletion in cancer cells. We found that anti-EGFR mAb could hardly induce EGFR depletion, while anti-cMet mAb partially induced cMet depletion. In contrast, both cMet and EGFR was found to be effectively depleted upon cMet/EGFR FIT-Ig treatment in both H441 and Caki-1 cells, while the mAb combination did not show such an effect ().
FIT-Ig showed significantly improved therapeutic efficacy in tumor models
The in vivo anti-tumor activity of cMet/EGFR FIT-Ig was investigated in xenograft models. A recombinant NCI-H1975 cell line expressing human HGF (NCI-H1975-HGF) was generated to enable activation of its cMet pathway. An s.c. NCI-H1975-HGF xenograft tumor model was established to assess in vivo efficacy of cMet/EGFR FIT-Ig. The NCI-H1975 cell line was reported to carry 2 activating EGFR mutations L858R/T790M, and to be resistant to small-molecule EGFR tyrosine-kinase inhibitors, such as erlotinib and gefitinib.Citation20,21 Our data showed that cMet/EGFR FIT-Ig suppressed tumor growth during the treatment period (21 days) and up to approximately 30 d post-treatment. Extensive tumor regression was found (P < 0.001) compared with vehicle-treated animals at day 24, while 76% and 50% tumor growth suppression was found in the anti-EGFR mAb and anti-cMet mAb treatment group (). cMet/EGFR FIT-Ig treatment showed significantly higher tumor inhibition effect compared with the anti-cMet mAb group on day 24 to 31 (P < 0.001); the cMet/EGFR FIT-Ig treatment group also showed a significantly higher tumor inhibition effect compared with the anti-EGFR mAb group on day 31 to 38 (P < 0.001). Tumor distribution of cMet/EGFR FIT-Ig was also evaluated. Mice bearing NCI-H9175-HGF tumors were treated with a single dose of cMet/EGFR FIT-Ig, anti-cMet mAb, or anti-EGFR mAb for 24 hrs, and then tumor lysates were examined by ELISA. cMet/EGFR FIT-Ig showed a tumor distribution profile similar to that of the mAbs, with concentrations of FIT-Ig, anti-EGFR mAb, and anti-cMet mAb at 21.7 μg, 24.1 μg, and 21.9 μg per gram tumor homogenate, respectively.
We also tested cMet/EGFR FIT-Ig in a non-small cell lung cancer patient-derived xenograft (PDX) model LU2503 with wild type EGFR and cMet amplification with Exon 14 deletion. In this study, the cMet/EGFR FIT-Ig group showed significant tumor growth suppression compared with the anti-EGFR mAb group from day 11 to 22 (P < 0.001). ()
Discussion
BsAb development has been an active field in the past decade, demonstrating additive or synergistic effect in neutralizing either soluble antigens or blocking cell surface receptors,Citation22 as well as achieving tumor-specific targeting for engaging check-point inhibitors.Citation23 In addition, bsAbs can work as T cell engagers to direct cytotoxic T cells to malignant cells for efficient elimination,Citation24 or function as a structural framework to bridge 2 proteins together for activation.Citation5 More recently, applications of bsAbs as anti-viral agents has been extensively reported, with encouraging results unachievable by combination of 2 mAbs.Citation25,26 Biological mechanisms and therapeutic areas relevant for bsAbs development continue to increase, with more than 50 bsAbs in clinical development and many more at the pre-clinical stage. Consequently, there has been a constant pursuit of a new generation of bispecific technologies that can produce more stable and active drug candidates with a more efficient manufacturing processes.
Here, we described a generically applicable bsAbs platform technology for therapeutic development. The FIT-Ig format can generate stable IgG-like bispecifics using conventional molecular biology techniques without the need for mutations, linkers, or complex engineering work. We showed that both soluble antigens and cell surface receptors can be targeted using the FIT-Ig format. In the design of FIT-Ig molecules, the 2 Fab domains from parental mAbs were fused directly in tandem in a crisscross orientation. The 3 fragments, when co-expressed in mammalian cells, assembled properly to form a FIT-Ig molecule in the correct structural orientation without VH/VL mismatch pairing in the final molecule. Although a certain level of mis-paired Fab arose from the 2 short chains, such fragment did not seem to have an effect on the expression level of the full-size FIT-Ig, and can be readily removed from a single step purification by protein A chromatography. The FIT-Ig molecules also maintained the complete human Fc region without any mutation, typical for a symmetric design. While the design of a crisscross orientation of the Fabs in tandem is critical for correct association of 2 sets of VH/VL pairs, the lack of requirement for a peptide linker between the 2 Fabs was unexpected. In previous studies, many similar IgG-like bispecific format were described with tetravalent binding patterns. One of the major issues of these format is the heavy chain/light chain mispairing. To solve the issue, paired mutations were introduced into CH1-CL interface to force the correct pairing of the light chain with its cognate heavy chain.Citation27,28 Additionally, in many cases, the described bsAb formats with an appended IgG design also involved the use of peptide linkers between Ig-like domains.Citation11,12,28,29 The design of mutation and linkers was critical to their proper function and in vitro/in vivo stability.Citation30 Although the mutation can be generic and applied to any antibody couple with the same family germline, optimization and new design are still needed for different germline families.Citation31 Structurally, when a variable domain is connected to anther Ig-like domain at its N-terminus, a linker is always required to provide enough space and flexibility for antigen binding, otherwise a strong steric hindrance may occur.Citation11 However, the peptide linkers often create additional complexity into the designs of the bispecific molecules, which likely require optimization of length and amino acid composition for each molecule.Citation32 In addition, both the mutation and linker region may give rise to stability and immunogenicity issues.Citation29 Surprisingly, a peptide linker is not required for the construction of FIT-Ig molecules, even those designed to target large antigens such as receptors. One possible explanation is that the unique design of crisscross connection of the 2 Fabs already provides enough flexibility, allowing independent target engagement by the 2 binding domains, as the first Fab domain may be positioned on the side, rather than on the top of the second Fab domain. Secondly, the 2 Fabs are connected only in one chain, instead of in both chains, which provides tremendous flexibility for the lower domain to engage even a large antigen without any steric hindrance. The structural flexibility of FIT-Ig may also contribute to its capability to bind 2 receptors simultaneously on the cell surface with all of its 4 binding sites occupied, which is the first time this result has been observed with a tetravalent binder.
The elimination of the use of peptide linkers, Fab mutation, and Fc mutations seems to be unique and a key advantage of FIT-Ig technology to enable its efficient engineering from potentially any 2 therapeutic mAbs, as well as broad application for a wide variety of molecular and cellular targets. We have generated and characterized more than 50 FIT-Ig molecules using a large number of mAbs against a variety of target pairs, majority of which exhibited good functional and physicochemical properties without domains optimization. In rare cases, change of domain orientation can give rise to a different expression profile (e.g., production yield, level of aggregation) in HEK 293 cells, which seemed to be a sequence-dependent phenomenon. Therefore, it will be ideal to generate a panel of FIT-Igs using several mAbs for each target, and select the FIT-Ig with the best expression profile for therapeutic development.
Importantly, FIT-Ig molecules exhibited physicochemical and PK properties similar to that of therapeutic mAbs. The intrinsic stability of both Fabs were well maintained in FIT-Ig as indicated in the thermostability assessment by DSC. It was previously reported that alternation or disruption of the Fab structure resulted in reduced thermostability.Citation33 Another critical feature of FIT-Ig molecules is the ease of production and purification, which is a major hurdle for many bispecific or multi-specific antibody-derived molecules.Citation22 We showed that FIT-Ig can be readily generated from transient 293E and stable CHO cells, with productivities within the range of IgGs. Thus, the feasibility to support manufacturing for clinical and commercial development has been established.
A majority of bsAbs in preclinical and clinical development are for oncology indications, and in recent years the field has advanced beyond the original T cell engager mechanism for bispecific application. For example, a bsAb against EGFR and cMet (JNJ-61186372) has been described previously to overcome resistant to anti-EGFR therapy with a synergistic effect in targeting certain tumors in preclinical animal models,Citation3,9,34 and is currently being tested in Phase 1 clinic trials. As a tetravalent bispecific binding protein, cMet/EGFR FIT-Ig induced more extensive co-degradation of cMet and EGFR in H441 and Caki-1 cells compared with mAb alone or in combination. Such differences may due to extensive cell surface receptor clustered by tetravalent antibody binding, which was shown to enhance internalization and trafficking to lysosomes for many receptors, including EGFR.Citation35-38 In addition, cMet/EGFR FIT-Ig showed a more profound inhibition of AKT-phosphorylation compared with the mAb combination. Given that the AKT-phosphorylation was evaluated within 1 h after antibody binding, the inhibition is more related to ligand binding blocking rather than receptor degradation. Furthermore, a more significant and sustainable tumor regression in NCI-H1975-HGF CDX model was achieved in the FIT-Ig treatment group compared with treatment with anti-EGFR mAb or anti-cMet mAb alone. A study with a similar model was also reported previously by another group using a different format of cMet/EGFR bispecific molecule; however, the efficacy was less striking, potentially due to its mono-valent binding mode for each antigen.Citation34 Although one cannot compare 2 complex studies involving animal models from 2 different groups without a common internal control, the impact of avidity effect on the activity of bsAbs have been well documented in the literature.Citation39 In contrast to the anti-EGFR parental mAb, we showed that cMET/EGFR FIT-Ig significantly improved therapeutic efficacy in different tumor models with diverse EGFR and cMet genetic background, and tumor distribution of FIT-Ig was comparable to that of mAbs. Therefore, cMET/EGFR FIT-Ig exerted a unique mechanism to improve therapeutic efficacy and overcome drug resistance through ligand binding blocking and enhanced receptor degradation.
Currently, an increasing number of bsAbs have emerged and are moving into clinical development.Citation40,41 One category of these bsAbs includes antibodies targeting 2 soluble antigens, such as ABT-981 (IL-1-α/IL-1β, Phase 2) and SAR156597 (IL-4/IL-13, Phase 2). Another category includes antibodies targeting 2 cell surface receptors, such as Her2, Her3, IGF-1R, EGFR, cMet, etc. The third class is the conventional T-cell bsAb (TCB), such as blinatumomab (Blincyto®; CD3/CD19) and REGN1979 (CD3/CD20, Phase 1). The FIT-Ig molecules were shown to be capable of targeting both soluble antigens and cell surface receptors, which makes the platform suitable for a broad therapeutic applications. In addition, FIT-Ig format can also support therapeutic development in the design of TCB, which requires monovalent anti-CD3 binding according to studies using other bispecific formats.Citation42-44 A monovalent form of FIT-Ig lacking one side Fab arm (1 + 1 format), but still containing a full Fc region, was competent in inducing T cell-dependent killing of target cells based on our preliminary studies, and this approach is under further exploration.
Materials and methods
Construction, production and characterization of FIT-Ig molecules
FIT-Ig protein was designed as an IgG-like molecule, in which there was an additional Fab domain structurally fused to the N-terminus of an IgG in a crisscross orientation, forming 2 Fabs in tandem with intact Fc region. To validate this technology, we generated 2 FIT-Ig proteins targeting 2 different types of target pairs (cytokines and receptors). For anti-IL-17/IL-20 FIT-Ig construction, the light chain (VL-CL) of ixekizumab was directly (i.e., without a linker), or through a linker of 3 amino acids, or 7 amino acids, fused to the N terminus of the heavy chain (VH-CH1-CH2-CH3) of 15D2 by overlapping polymerase chain reactions, and another 2 chains, VH-CH1 of ixekizumab and VL-CL of 15D2, were also constructed. All 3 chains were subcloned into mammalian expression vectors (pCP), and were transiently co-transfected in HEK293 cells, followed by expression and Protein A purification (GE). The expression titers of FIT-Ig and mAbs were determined by human IgG ELISA. The concentration of purified IL-17/IL-20 FIT-Ig proteins was measured by A280, homogeneity was analyzed by SDS-PAGE and SEC. The construction and production of cMet/EGFR FIT-Ig was completed similarly. The expression of each chain and by-product was analyzed by Western blot with conditioned medium. Molecular weight was measured by mass spectrometry. Twenty-six disulfide bonds of cMet/EGFR FIT-Ig were analyzed by mass spectrometry. Briefly, FIT-Ig protein was digested into peptides by trypsin or chymotrypsin, and then an aliquot was reduced by dithiothreitol (Sigma). The molecular weight of peptides in both type of samples (reducing and non-reducing) was determined and comparably analyzed to identify disulfide bonds formation.
Stability test
Thermostability was analyzed by DSC. The concentration of samples is ∼1 mg/mL. As the temperature increased gradually, different domains of proteins unfolded, and melting temperatures (Tm), which reflected stability, were determined.
An accelerated storage stability testing was also performed. FIT-Ig proteins (adjusted to 10 mg/mL) were incubated in PBS at 4℃, 25℃ and 40℃ for 1, 4 and 7 days, or freeze/thaw once, twice and thrice, respectively. Afterward, all samples were analyzed by SEC to detect levels of aggregation and degradation comparing with starting material.
IL-17 bioassay
One.0 × 104 Hs27 cells (ATCC) with >80% viability were added to each well of 96-well plates (Corning), wherein each well was added with serially diluted FIT-Ig or mAbs proteins (5-fold dilution from 30 nM) and 0.3 nM (final concentration) recombinant human IL-17A (R&D Systems) sequentially. After 17 hours incubation at 37℃, cell culture supernatants were collected, the concentration of secreted GROα were determined by Human CXCL1/GRO α Quantikine PharmPak (R&D Systems) according to manufactured instruction. Data curve was plotted by Graphpad and IC50 was calculated.
IL-20 bioassay
BAF3-IL-20R stable cell line were seeded at 1 × 104 /well to 96-well assay plate and incubated with serially diluted antibodies, followed by 0.2 nM recombinant human IL-20 protein (R&D) at 37℃, 5% CO2 for 3 d. The cytotoxicity was measured by CellTiter-Glo luminescent (Promega). Data curve was plotted by Graphpad and IC50 was calculated.
Stokes radii determination
The samples were centrifuged at 12000 g for 10 minutes. The supernatant were transferred in the 96-well plate and analyzed using DLS instrument of Zetasizer APS (Malvern Instruments Ltd.). The relevant DLS assay parameters were set as follows: the assay temperature was 25℃, the equilibration time was 120 seconds, the measurement duration was chosen as automatic, the number of measurements was 1, the analysis model was protein analysis model, the size-intensity and size-volume profiles were recorded. The antibody samples were analyzed 3 times independently. The average hydrodynamic radiuses were calculated based on the triplicate analysis.
Generation of CHO stable cell line
Cell lines stably expressing cMet/EGFR FIT-Ig were generated. Briefly, glutamine synthetase (GS)-deficient CHO-K1 cells were transfected with FIT-Ig construct DNA in CD CHO medium (Lifetechnologies). After 24 hrs, the cells were split into 96-well plate at a density of 5 × 103 cells/well and grown in medium with 50 uM methionine sulfoximine (MSX) for 2–3 weeks, wherein transfectants were assessed by human IgG ELISA to determine expression concentration of FIT-Ig protein. Clones with high titer were selected for 2 rounds of subcloning by limited dilution to get the pure clone.
Affinity measurements by Biacore
The kinetics of cMet/EGFR FIT-Ig binding to recombinant human cMet and EGFR was determined by surface plasmon resonance-based measurements with a Biacore T200 instrument (GE Healthcare) using standard procedures similar to mAb analysis. Briefly, goat anti-human IgG Fc polyclonal antibody (Cat# GWB-20A705, Genway Biotech) was directly immobilized across a biosensor chip, and FIT-Ig or mAbs samples were injected over reaction matrices at a flow rate of 5 ul/min. The association and dissociation rate constants, kon (M-1s-1) and koff (s-1) respectively, were determined with a continuous flow rate of 30 ul/min. Rate constants were derived by making kinetic binding measurements at 5 different antigen concentrations. The equilibrium dissociation constant KD (M) of the reaction between FIT-Ig and related target proteins was then calculated from the kinetic rate constants using the following formula: KD = koff/kon.
Cell surface dual binding assay by FACS
cMet and EGFR expression cell lines MKN-45, SGC-7901, and NCI-H1975 (ATCC) were incubated with different concentrations of cMet/EGFR FIT-Ig at 4℃ for 1 hr, washed, and then incubated with 1 ug/ml biotinylated human recombinant cMet or EGFR protein (Acrobiosystems) at 4℃ for 1 hr. The specific binding of antibody to antigen was detected by streptavidin Alexa Fluor® 488 conjugate (Invitrogen) using flow cytometry (BD FACSVerse).
AKT phosphorylation assay
Cells were seeded to 96-well assay plate at the density of 2 × 104 /well in complete cell culture medium, and then starved with 0.1% fetal bovine serum (GIBCO) overnight. Tested antibodies were incubated with cells for 30 mins followed by stimulation with 40 ng/ml HGF (R&D) for 5 mins (or without HGF for agonist effect detection assay). The cells were then lysed and phospho-AKT (S473) were measured using Cisbio assay. The signals were not corrected for total levels of AKT. Date was statistically analyzed using GraphPad Prism 5.
cMet and EGFR depletion assay
H441 cells were incubated with 100 nM antibodies at 37℃ overnight and then lysed with cell lysis buffer (Cell Signaling Technology). Lysates were cleared by centrifugation, and protein concentrations were determined by BCA Protein Assay (Pierce). Equal amounts of protein were separated by electrophoresis through NuPage Bis-Tris 4–12% gradient gel (Invitrogen), proteins were transferred onto PVDF membranes using the iBlot system and protocol from Invitrogen. Antibodies to cMet (Cat#4560), EGFR (cat# 2232), GAPDH (Cat#97166) were obtained from Cell Signaling Technology. All primary antibodies were used by 1:1000 dilution. Specific antigen-antibody interaction was detected with a HRP-conjugated secondary anti-rabbit IgG (Cell Signaling Technology), used by 1:5000 dilution.
FIT-Ig protein pharmacokinetic study in rats
PK properties of FIT-Ig protein were assessed in male Sprague-Dawley (SD) rats. The animals were cared for according to the National Institutes of Health guidelines; they were housed in rooms maintained at constant temperature and humidity under a 12-h light/dark cycle, were fed with normal rodent chow, and were allowed food and water ad libitum. The Institutional Animal Care and Use Committee of ChemPartner (Shanghai, China) approved all animal handing and experimental protocols. FIT-Ig proteins were administered to male SD rats at a single i.v. and s.c. dose of 5 mg/kg. Serum samples were collected at different time points over a period of 28 d and were analyzed by human cMet and EGFR capture ELISAs. Briefly, ELISA plates were coated with 1 ug/ml human cMet or EGFR protein (Sinobiological) overnight at 4℃, blocked with PBST plus 1% BSA. Serum samples were diluted and incubated on the plate for 1 hour at 37℃. Detection was performed with peroxidase labeled anti-human IgG (Fc specific) (Cat# A0293, Sigma), and concentrations were determined with the help of standard curve using the 4-parameter logistic fit. Values for the PK parameters were determined by 2-compartmental model and non-compartment model of WinNonlin software (Pharsight Corporation).
In vivo tumor xenograft models
All of the procedures relating to animal care, handling and treatment were performed according to the guidelines approved by Institutional Animal Care and Use Committee following the guideline of the Association for Assessment and Accreditation of Laboratory Animal Care. For the NCI-H1975-HGF model, NCI-H1975 cells (ATCC) were stably transfected with an expression vector containing the human HGF cDNA, and a stable clone producing 10 ng/mL HGF was selected for in vivo studies. A total of 5 × 106 cells were injected subcutaneously into the right flanks (0.2 mL/mouse) of Balb/c nude mice (female, 6–8 weeks). The LU2503 patient-derived xenograft (PDX) model was performed by Crown Biosciences. PDX tumor fragments were harvested from donor mice and subcutaneously inoculated into the right flanks of Balb/c nude mice (female, 8–10 weeks). Animals were treated with anti-cMet, anti-EGFR or cMet/EGFR FIT-Ig when tumors reached a mean tumor size of 100 to 140 mm3. All data was analyzed using GraphPad Prism software. Tumor volumes are plotted as mean ± standard error of the mean (SEM). A Two-way ANOVA combined with Bonferroni post-test was performed comparing treatment group with control groups. P < 0.05 was considered to be statistically significant. (*p < 0.05, **p < 0.01, ***p < 0.001)
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Materials.docx
Download MS Word (5.9 MB)References
- Park SS, Ryu CJ, Kang YJ, Kashmiri SV, Hong HJ. Generation and characterization of a novel tetravalent bispecific antibody that binds to hepatitis B virus surface antigens. Mol Immunol 2000; 37:1123-30; PMID:11451417; https://doi.org/10.1016/S0161-5890(01)00027-X
- Fischer JA, Hueber AJ, Wilson S, Galm M, Baum W, Kitson C, Auer J, Lorenz SH, Moelleken J, Bader M, et al. Combined inhibition of tumor necrosis factor alpha and interleukin-17 as a therapeutic opportunity in rheumatoid arthritis: Development and characterization of a novel bispecific antibody. Arthritis Rheumatol 2015; 67:51-62; PMID:25303306; https://doi.org/10.1002/art.38896
- Moores SL, Chiu ML, Bushey BS, Chevalier K, Luistro L, Dorn K, Brezski RJ, Haytko P, Kelly T, Wu SJ, et al. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res 2016; 76:3942-53; PMID:27216193; https://doi.org/10.1158/0008-5472.CAN-15-2833
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941-4; PMID:19509221; https://doi.org/10.1158/0008-5472.CAN-09-0547
- Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, Yoshihashi K, Okuyama-Nishida Y, Saito H, Tsunoda H, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med 2012; 18:1570-4; PMID:23023498; https://doi.org/10.1038/nm.2942
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974-7; PMID:18703743; https://doi.org/10.1126/science.1158545
- Sheridan C. Despite slow progress, bispecifics generate buzz. Nat Biotechnol 2016; 34:1215-7; PMID:27926715; https://doi.org/10.1038/nbt1216-1215
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs 2017; 9:182-212; PMID:28071970; https://doi.org/10.1080/19420862.2016.1268307
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol 2013; 31:753-8; PMID:23831709; https://doi.org/10.1038/nbt.2621
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108:11187-92; PMID:21690412; https://doi.org/10.1073/pnas.1019002108
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol 2007; 25:1290-7; PMID:17934452; https://doi.org/10.1038/nbt1345
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; https://doi.org/10.1016/j.molimm.2015.01.003
- Barrett A. Anti-IL-17 antibodies. International Publication Number: WO2007/070750A1 2006.
- Jesper P. Anti-human Interleukin-20 antibodies. International Publication Number: WO2010/000721 2009.
- Burmester GR, Feist E, Dörner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol 2014; 10:77-88; PMID:24217582; https://doi.org/10.1038/nrrheum.2013.168
- Guo A, Villén J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008; 105:692-7; PMID:18180459; https://doi.org/10.1073/pnas.0707270105
- Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17:77-88; PMID:20129249; https://doi.org/10.1016/j.ccr.2009.11.022
- Jakobovits A. Human monoclonal antibodies to epidermal growth factor receptor. 1997; Patent No.: US6235883.
- Michaud NR. Antibodies to cMet. 2004; Patent No.: US7498420.
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:0225-35; PMID:15737014; https://dx.doi/10.1371/journal.pmed.0020073
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005; 102:7665-70; PMID:15897464; https://doi.org/10.1073/pnas.0502860102
- Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol 2015; 8:130; PMID:26692321; https://doi.org/10.1186/s13045-015-0227-0
- Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, Wang L, Weiskopf K, Majeti R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs 2015; 7:946-56; PMID:26083076; https://doi.org/10.1080/19420862.2015.1062192
- Nagorsen D, Bargou R, Ruttinger D, Kufer P, Baeuerle PA, Zugmaier G. Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE antibody blinatumomab. Leuk Lymphoma 2009; 50:886-91; PMID:19455460; https://doi.org/10.1080/10428190902943077
- Wec AZ, Nyakatura EK, Herbert AS, Howell KA, Holtsberg FW, Bakken RR, Mittler E, Christin JR, Shulenin S, Jangra RK, et al. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 2016; 354:350-4; PMID:27608667; https://doi.org/10.1126/science.aag3267
- Bournazos S, Gazumyan A, Seaman MS, Nussenzweig MC, Ravetch J V. Bispecific Anti-HIV-1 antibodies with enhanced breadth and potency. Cell 2016; 165:1609-20; PMID:27315478; https://doi.org/10.1016/j.cell.2016.04.050
- Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, Torres C, Fine C, Batt MA, Jonathan R, et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. MAbs 2015; 862:470-82; PMID:25774965; https://doi.org/10.1080/19420862.2015.1022694
- Golay J, Choblet S, Iwaszkiewicz J, Cérutti P, Ozil A, Loisel S, Pugnière M, Ubiali G, Zoete V, Michielin O, et al. Design and validation of a novel generic platform for the production of tetravalent IgG1-like bispecific antibodies. J Immunol 2016; 196:3199-211; PMID:26921308; https://doi.org/10.4049/jimmunol.1501592
- Le Gall F, Reusch U, Little M, Kipriyanov SM. Effect of linker sequences between the antibody variable domains on the formation, stability and biological activity of a bispecific tandem diabody. Protein Eng Des Sel 2004; 17:357-66; PMID:15126676; https://doi.org/10.1093/protein/gzh039
- Davé E, Adams R, Zaccheo O, Carrington B, Compson JE, Dugdale S, Airey M, Malcolm S, Hailu H, Wild G, et al. Fab-dsFv: A bispecific antibody format with extended serum half-life through albumin binding. MAbs 2016; 8:1319-35; PMID:27532598; https://doi.org/10.1080/19420862.2016.1210747
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol 2014; 32:191-8; PMID:24463572; https://doi.org/10.1038/nbt.2797
- Wu C, Ying H, Bose S, Miller R, Medina L, Santora L, Ghayur T. Molecular construction and optimization of anti-human IL-1α/β dual variable domain immunoglobulin (DVD-Ig) molecules. MAbs 2009; 1:339-47; PMID:20068402; https://doi.org/10.4161/mabs.1.4.8755
- Fenn S, Schiller CB, Griese JJ, Duerr H, Imhof-Jung S, Gassner C, Moelleken J, Regula JT, Schaefer W, Thomas M, et al. Crystal structure of an Anti-Ang2 crossfab demonstrates complete structural and functional integrity of the variable domain. PLoS One 2013; 8:1-7; PMID:23613981; https://doi.org/10.1371/journal.pone.0061953
- Castoldi R, Ecker V, Wiehle L, Majety M, Busl-Schuller R, Asmussen M, Nopora A, Jucknischke U, Osl F, Kobold S, et al. A novel bispecific EGFR/Met antibody blocks tumor-promoting phenotypic effects induced by resistance to EGFR inhibition and has potent antitumor activity. Oncogene 2013; 32:5593-601; PMID:23812422; https://doi.org/10.1038/onc.2013.245
- Parton RG, Schrotz P, Bucci C, Gruenberg J. Plasticity of early endosomes. J Cell Sci 1992; 103(Pt 2):335-48; PMID:1478937
- St. Pierre CA, Leonard D, Corvera S, Kurt-Jones EA, Finberg RW. Antibodies to cell surface proteins redirect intracellular trafficking pathways. Exp Mol Pathol 2011; 91:723-32; PMID:21819978; https://doi.org/10.1016/j.yexmp.2011.05.011
- Weflen AW, Baier N, Tang Q-J, Van den Hof M, Blumberg RS, Lencer WI, Massol RH. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol Biol Cell 2013; 24:2398-405; PMID:23741050; https://doi.org/10.1091/mbc.E13-04-0174
- Wang Q, Villeneuve G, Wang Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep 2005; 6:942-8; PMID:16113650; https://doi.org/10.1038/sj.embor.7400491
- Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016; 8:1010-20; PMID:27285945; https://doi.org/10.1080/19420862.2016.1197457
- Schuurman J, Parren PWHI. Editorial overview: Special section: New concepts in antibody therapeutics: What's in store for antibody therapy? Curr Opin Immunol 2016; 40:vii-xiii; PMID:27083411; https://doi.org/10.1016/j.coi.2016.04.001
- Sheridan C. Amgen's bispecific antibody puffs across finish line. Nat Biotechnol 2015; 33:219-21; PMID:25748895; https://doi.org/10.1038/nbt0315-219
- Zlabinger GJ, Stuhlmeier KM, Eher R, Schmaldienst S, Klauser R, Vychytil A, Watschinger B, Traindl O, Kovarik J, Pohanka E. Cytokine release and dynamics of leukocyte populations after CD3/TCR monoclonal antibody treatment. J Clin Immunol 1992; 12:170-7; PMID:1400897; https://doi.org/10.1007/BF00918085
- Ferran C, Sheehan K, Dy M, Schreiber R, Merite S, Landais P, Noel LH, Grau G, Bluestone J, Bach JF, et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: Further evidence for transient in vivo T cell activation. Eur J Immunol 1990; 20:509-15; PMID:2138557; https://doi.org/10.1002/eji.1830200308
- Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 2010; 9:325-38; PMID:20305665; https://doi.org/10.1038/nrd3003