
ABSTRACT
The determination of the binding strength of immunoglobulins (IgGs) to targets can be influenced by avidity when the targets are soluble di- or multimeric proteins, or associated to cell surfaces, including surfaces introduced from heterogeneous assays. However, for the understanding of the contribution of a second drug-to-target binding site in molecular design, or for ranking of monovalent binders during lead identification, affinity-based assessment of the binding strength is required. Typically, monovalent binders like antigen-binding fragments (Fabs) are generated by proteolytic cleavage with papain, which often results in a combination of under- and over-digestion, and requires specific optimization and chromatographic purification of the desired Fabs. Alternatively, the Fabs are produced by recombinant approaches. Here, we report a lean approach for the functional assessment of human IgG1s during lead identification based on an in-solution digestion with the GingisKHAN™ protease, generating a homogenous pool of intact Fabs and Fcs and enabling direct assaying of the Fab in the digestion mixture. The digest with GingisKHAN™ is highly specific and quantitative, does not require much optimization, and the protease does not interfere with methods typically applied for lead identification, such as surface plasmon resonance or cell-based assays. GingisKHAN™ is highly suited to differentiate between affinity and avidity driven binding of human IgG1 monoclonal and bispecific antibodies during lead identification.
Introduction
Affinity describes the binding strength of a bimolecular interaction between a single antigenic determinant and a single epitope-binding region, whereas avidity equals the accumulated strength of multiple affinities between reaction partners. Hence, avidity requires 2 or more interaction sites and can be relevant for bivalent biotherapeutics binding to targets that have multiple binding sites (soluble di- or multimers), or are cell surface-associated.
The development of bivalent biotherapeutics like antibodies usually includes at least 2 assessments during lead identification: 1) the determination of the relevant in vivo interaction mode; and 2) identification of the candidate with the optimal interaction strength. In determining the interaction mode, it is important to understand the relevance of the second drug-to-target binding site. If the second binding site does not improve the binding strength of the molecule when analyzed with relevant targets (for soluble di- or multimeric targets), or relevant cells (for cell surface-associated targets), affinity-based binding reflects the relevant in vivo interaction mode. In this case, a contribution of the second drug-to-target binding site to the efficacy increase is stoichiometric and based on having a second valency for target binding. Otherwise, avidity-based binding drives the potency and the second drug-to-target binding site is required to achieve optimal binding strength, but at the cost of having a second valency.
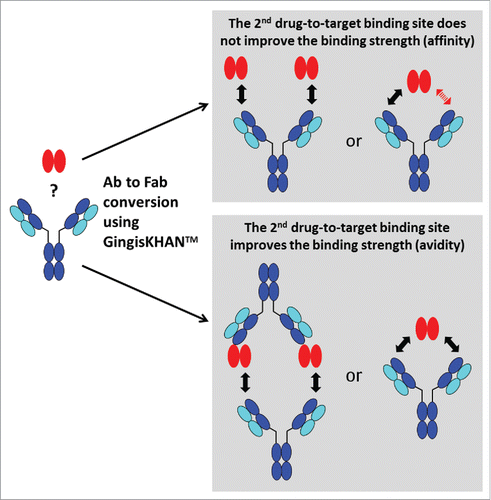
When identifying optimal candidates, stronger binders are typically preferred because their use may allow the reduction of the minimal effective dose or increased administration intervals, but binders with intermediate or even low interaction strength might be also desired. The same molecule, however, can display different binding strengths when analyzed as a monovalent antigen-binding fragment (Fab), with assessment of the binding strength based on affinity, or when analyzed as a bivalent mAb, which allows avidity-based binding (). If the determined in vivo relevant interaction mode is based on avidity, only an assay addressing avidity is reflective and relevant for selecting the best binder (, I). Assay results based on affinity will be too low, and depending on the desired drug interaction strength, do not allow the identification of the optimal candidate. The same correlation applies for affinity-based interaction in in vivo vs. in vitro assessments, i.e., assay results based on avidity will be to high compared with the desired monovalent interaction (see supplementary Fig. 1S). Default approaches commonly applied for both assessments involve the comparison of the monovalent construct with the corresponding bivalent construct for the determination of the in vivo relevant interaction mode. When the biotherapeutic has an IgG-like scaffold, the preparation of the monovalent construct requires the generation of single binding units like the Fab. Typically, this is achieved by either performing a proteolytic cleavage above the IgG hinge region and purification of the Fab (, II + III), or by recombinant expression of the Fab (, IV). Alternatively, di- or multimeric targets may be converted into monomeric structures if feasible, e.g., by recombinant methods, to allow assaying the bivalent antibody directly, but this approach is far less generic due to the more heterogeneous nature.
Figure 1. Avidity and affinity measurements by surface plasmon resonance (SPR) of antibodies and Fabs binding di- or multimeric targets. In contrast to IgGs targeting monomeric targets, it is often not possible to directly determine the affinity of IgGs binding di- or multimeric targets by SPR. In this case, and in both orientations of the SPR assay (immobilised/captured antibody or target, respectively), the affinity determinations are influenced by avidity, which refers to the accumulated strength of multiple affinities (I, only the assay format with the immobilized target protein is illustrated). To prevent avidity, IgGs are typically converted into Fabs, which are often obtained by endoprotease digest with a subsequent necessary Fab purification (II+III), or by recombinant Fab expression (IV). GingisKHAN™ is a specific and quantitative protease generating a homogenous pool of intact Fab and Fcs of human IgG1s without any over-digestion typical for unspecific proteolytic enzymes like papain. Here we tested if digestions with GingisKHAN™ allow direct high-throughput kinetic affinity determinations using SPR without any prior Fab purification needed (II).
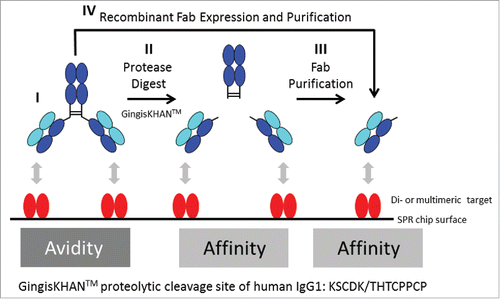
Fabs are often generated by partial proteolytic digestion of IgGs with unspecific proteases such as papain, which cleaves N-terminal of the hinge region containing the disulfide bonds joining the heavy chains, but below the site of the disulfide bond between the light and the heavy chain.Citation1-5 The hinge region is likely more susceptible to the episode of proteases because it is exposed and flexible. Successful in-solution digestions with the endoproteinase papain to obtain Fabs generally depend on IgG-specific optimizations and typically suffer from the presence of undigested IgG, over-digestion and lack of reproducibility. Consequently, each papain digest requires individual optimizations to obtain homogeneous fragments.Citation2,6-7 Using immobilized papain resins eliminates autolysis, protease contamination, and allows better control of the digestion reaction and chromatographic removal of the Fabs from the crude protease digests. Nevertheless, the proteolytic cleavage using immobilized papain still needs to be optimized, e.g., by the column flow rate.Citation8-9 Other ways to obtain Fabs or single binding units include partial proteolysis with endoproteinase Lys-CCitation10; mild reduction of F(ab')2 to Fab' fragments or IgGs to half-IgGsCitation11-16; and recombinant expression of the light chain and the heavy Fd chain.5 Generally, these approaches to obtain monovalent binders are not high-throughput methods. Use of unspecific proteases involves time-consuming IgG-specific optimizations and the inhibition of the protease or the purification of the Fabs to prevent their degradation. Recombinant expression is time consuming and laborious if several different Fabs are to be analyzed and compared in a high-throughput screen.
The cysteine protease gingipain KCitation17-20 (EC 3.4.22.47) of the pathogenic anaerobe bacteria Porphyromonas gingivalis has recently become commercially available as the recombinant protease GingisKHAN™ (KGP) from Genovis AB (Lund, Sweden). Gingipain K has been reported to hydrolyze human IgGs of the subclasses 1 and 3, whereby the heavy chains of IgG1s are cleaved at a single site above the hinge region. In contrast, heavy chains of IgG3s are hydrolysed at several sites around the CH2 region.Citation20 GingisKHAN™ is reported to digest human IgG1s with one single digestion site above the hinge region (proteolytic cleavage site: KSCDK/THTCPPCP) generating a homogenous pool of intact Fabs and crystallizable fragments (Fcs).Citation21
Here, we evaluated the use of GingisKHAN™ as a human IgG1 upper hinge region-cleaving protease during lead identification. We demonstrate by mass spectrometry that the GingisKHAN™ digest is specific and quantitative. Analysis of crude, non-purified GingisKHAN™ digests directly by surface plasmon resonance (SPR) demonstrates that the assessment of the relevant binding strength based on affinity is possible (, II only). In addition, direct assessment of crude digests by cell-based assays allow a lean determination of the in vivo relevant interaction mode, bypassing time-consuming Fab generation and eventual purification.
Results
Initially, we analyzed by mass spectrometry if indeed the mAb to Fab conversion of a total of 3 different monoclonal human IgG1s (mAb1, mAb2, mAb3) and 3 human IgG1-derived bispecific antibodies (mAb1 derived 1+1 CrossMabCH1-CL, mAb4 derived 2+1 CrossMabCH1-CL and mAb5 derived 2+1 CrossMabCH1-CL)Citation22 can be easily performed by GingisKHAN™ digestion. mAb1, mAb2 and mAb3 bind dimeric vascular endothelial growth factor A (VEGF-A),Citation23 multimeric angiopoietin-2 (Ang-2),Citation24 and cell surface-associated carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5),Citation25 respectively. The mAb1-derived 1+1 CrossMabCH1-CL, 22 is monovalent for VEGF-A and an undisclosed target, and the mAb4- and mAb5-derived trivalent 2+1 CrossMabsCH1-CL are bivalent for CEACAM5 and monovalent for an undisclosed target. The trivalent mAb4- and mAb5-derived 2+1 CrossMabsCH1-CL were included in this study because the bispecific antibody constructs should include at least 2 identical or biparatopic binding sites being monospecific for a given target to allow an avidity-based mechanism. MAb4 and mAb5 differ by their complex stability: the binding of mAb5 to CEACAM5 is very stable, whereas mAb4 displays a significant lower complex stability. Subsequently, we evaluated a direct use of non-purified GingisKHAN™-digests in different functional assays covering 2 assessments relevant in lead identification: 1) determination of the relevant in vivo interaction mode; and 2) identification of the candidate with the optimal binding strength. All used antibodies, and antibody constructs, and their valencies to targets are listed in the supplementary Table 1S.
Highly specific and quantitative digestions of human monoclonal and bispecific IgG1 antibodies with GingisKHAN™
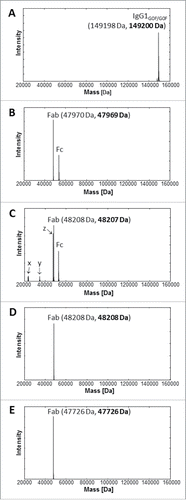
The quality of the intact mAb1, and GingisKHAN™ and papain digests of mAb1 were analyzed by ultra-high resolution electrospray ionization quadrupole time-of-flight (UHR-ESI-QTOF) mass spectrometry. The most intense signal with 149200 Da in the deconvoluted mass spectra of the IgG1 demonstrated the intact mAb1 with 2x agalacto-, core-fucosylated bi-antennary complex-type (G0F) Fc N-glycans (theoretical average mass: 149198 Da) (). Following an in-solution digest with GingisKHAN™, the deconvoluted mass spectrum of the digested mAb1 demonstrated only the presence of the 47969 Da Fab (theoretical average mass: 47970 Da) and the Fcs with its expected N-glycan heterogeneity (), confirming a highly quantitative and specific cleavage of mAb1. In contrast, the analysis of mAb1 digested with papain (1 h) determined not only the presence of the 48207 Da Fab (theoretical average mass: 48208 Da) and the multiple Fcs due to the heterogeneity of the Fc N-glycans, but also unassignable partly intense fragments corresponding to the masses 23422 Da, 23453 Da, 34587 Da, and 47607 Da (). Due to the lack of specificity, papain not only cleaves the upper hinge region, but also alternative sites depending on the structure and flexibility of the antibody being cleaved. To compare with the binding affinity of the mAb1 Fabs in the GingisKHAN™ digestion mixture, additional Fabs were purified following an in-solution papain digest of mAb1 and transient expression in human embryonic kidney (HEK) cells. Mass spectra of both purified Fabs revealed the high quality of both fragments. Only the intact Fabs corresponding to 48208 Da (theoretical average mass: 48208 Da) and 47726 Da (theoretical average mass: 47726 Da; heavy Fd chain C-terminal: VEPKSC), respectively, could be detected by mass spectrometry ().
Figure 2. UHR-ESI-QTOF mass spectrometry of mAb1 and Fabs thereof. Deconvoluted mass spectra of (A) mAb1, (B) mAb1 digested with GingisKHAN™, (C) mAb1 digested with papain (1 h), (D) a purified Fab following an in-solution papain digest of mAb1, and (E) a recombinant Fab. Undigested and single-digested IgG could not be detected in the digests with (B) GingisKHAN™ and (C) papain. The indefinite fragments x, y, z (C) due to the lack of specificity of papain correspond to the masses; x:23422 Da and 23453 Da, y:34587 Da, and z:47607 Da. Expected average and determined (bold) masses of the IgG1 with 2x G0F Fc N-glycans and the Fabs are stated in parentheses.
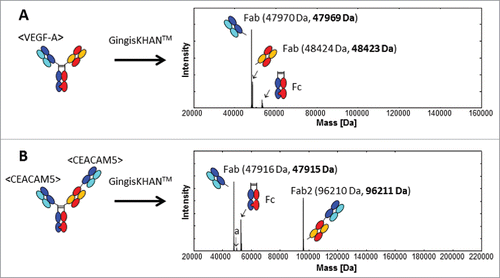
Mass spectra of mAb2 and mAb3 digested with GingisKHAN™ or undigested, a recombinant mAb2 Fab, and a purified mAb3 Fab obtained by digestion with papain in solution, demonstrated the same high quality and expected equivalent masses as in the case of mAb1 (data not shown). Also the quality of the intact and GingisKHAN™-digested mAb1-derived 1+1, mAb4-derived 2+1 and mAb5-derived 2+1 CrossMab bispecific antibodies were verified by mass spectrometry. The deconvoluted mass spectra of the GingisKHAN™ digested mAb1-derived and mAb5-derived CrossMabs are shown in and , respectively. The digests were complete without any undigested or single-digested IgGs or bispecific antibodies (missing one Fab) detectable by mass spectrometry. Nor could any unspecific digestion or further degradation of the fragments be detected in the crude digestion mixtures with GingisKHAN™. Next, we addressed the compatibility of the GingisKHAN™ digests with the methods relevant for lead identification.
Figure 3. UHR-ESI-QTOF mass spectrometry of (A) the mAb1 derived 1+1 and (B) mAb5 derived 2+1 CrossMabs digested with GingisKHAN™. Undigested and single-digested bispecific antibodies could not be detected in the deconvoluted spectra. The mass denoted a:50012 Da (B) was identified as the non-glycosylated Fc (expected average mass: 50012 Da). Expected average and determined (bold) masses of the Fabs are stated in parentheses.
Direct SPR of GingisKHAN™ digests of mAb1 and mAb2
To test whether the affinity of the mAb1 Fab obtained from the quantitative in-solution digest with the GingisKHAN™ protease could be reliably determined directly from the digestion mixture, we analyzed the binding affinity of the Fab to VEGF-A by SPR and compared it with the affinities of the recombinant mAb1 Fab and the Fab purified from the in-solution digest of mAb1 with papain. Papain digestion mixtures stopped by adding protease inhibitor antipain were also included in this analysis.
The binding to dimeric VEGF-A of mAb1, the papain and GingisKHAN™ digestion mixtures of mAb1, the purified mAb1 Fab from the papain digest, and the recombinant mAb1 Fab were analyzed by SPR as illustrated in ; I, II, II+III, and IV, respectively. A mouse anti-His-tag was immobilized to capture the dimeric VEGF-A-His on the sensor chip surface, and then the analytes binding to VEGF-A were injected. The derived sensorgrams were fitted to a 1:1 Langmuir binding model () and used to determine the association rate (ka), the dissociation rate (kd), and the binding (KD) constants. The rate and binding constants for the Fabs were determined to be comparable (). The binding constant of the Fab in the GingisKHAN™ digestion mixture was determined as 1.1 nM, and those of the recombinant Fab and the purified Fab after digestion with papain were 0.8 and 1.0 nM, respectively (). In comparison, the KD of the mAb1 IgG1 was determined to be 0.2 nM, demonstrating the avid binding to the dimeric VEGF-A. An optimized 1 hour papain digestion mixture stopped by adding antipain gave a binding constant of 1.9 nM (), which is comparable to the binding constants of the other Fabs. No binding to captured VEGF-A could be determined when the papain digestion was allowed to proceed for 3.5 h before adding antipain (). The additional digestion time with papain completely rendered the mAb1 Fab binding inactive, since no SPR signal could be obtained.
Figure 4. Affinity determinations by surface plasmon resonance of mAb1 and Fabs thereof. Sensorgrams of (A) mAb1, (B) mAb1 digested with GingisKHAN™, (C) mAb1 digested with papain (1 h, subsequently inhibited with antipain), (D) mAb1 digested with papain (3.5 h, subsequently inhibited with antipain), (E) a purified Fab following an in-solution papain digest of mAb1, and (F) a recombinant Fab binding to captured vascular endothelial growth factor A (VEGF-A121-His). The analyte was injected in a dilution series ranking from 2.2 to 1800 nM (colored sensorgrams). The black curves are the fitting curves using models from BIAevaluate. The arrows indicate the faster dissociation of the Fabs (B, C, E, and F) relative to the IgG1 (A).
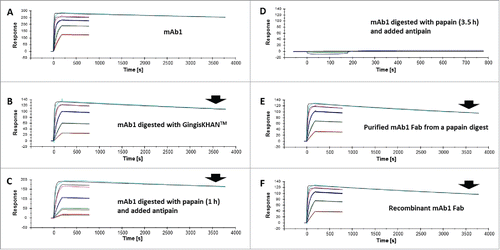
Table 1. Binding constants of mAb1 and Fabs thereof as determined by surface plasmon resonance.
With a second human IgG1 (mAb2 binding Ang-2), we confirmed the reliability of the direct affinity determination using the GingisKHAN™ digestion mixture. mAb2 and Fab thereof were tested in a reverse assay setup relative to the mAb1 SPR binding assay. Capturing the mAb2 monoclonal antibody or Fab with a mouse anti-human Fab antibody immobilized on the sensor chip surface and using monomeric Ang-2 receptor binding domainCitation26 fused to a human Fc (Ang-2-RBD-Fc) as analyte, we determined the rate and binding constants of the mAb2 antibody, the GingisKHAN™ digestion mixture, and the recombinant mAb2 Fab. The determined affinities were all comparable with KD values of 24, 25, and 28 nM, respectively. The monomeric Ang-2 bound to the captured mAb2 IgG without avidity (sensograms not shown).
To analyze the storage-stability of the GingisKHAN™ digests, the affinity determinations by SPR of the digested mAb1 and mAb2 stored at 4°C were repeated after 24 and 48 hours. The initially determined KD values of mAb1 and mAb2 were confirmed after storage (data not shown). Extended storage of the GingisKHAN™ digest of mAb1 and evaluation by mass spectrometry demonstrated the digest to be stable at 4°C for at least 4 weeks. After 4 weeks of storage at 4°C the KD was determined to be 0.8 nM (ka = 6.61E+04, kd = 5.19E-05), confirming the stability of the digest.
Cell-based assays of GingisKHAN™-digested mAb1 binding a soluble dimeric target and mAb2 binding a soluble multimeric target
The effect of a second drug-to-target binding site was analyzed via cell-based assays by comparing intact and GingisKHAN™-digested mAbs binding soluble di- or multimeric targets. The interaction is affinity-driven when the binding strength does not depend on the number of binding sites. If the interaction does depend on the number of binding sites, the interaction is avidity-driven ().
Figure 5. Analysis of the effect of a second drug-to-target binding site of a bivalent human IgG1 binding a soluble di-or multimeric target by comparing the intact and the GingisKHAN™-digested antibody in a cell-based assay. The interaction is affinity-driven when the binding strength does not depend on the number of binding sites. If the interaction does depend on the number of binding sites, the interaction is avidity-driven. The red arrow indicates that no drug-to-target binding occurs.
We tested the influence of mAb1 digested with GingisKHAN™ to inhibit VEGF-A (a soluble dimer) in a cell-based reporter gene assay measuring binding of VEGF-A to the VEGF receptor 2 and successive signal transduction. The bivalent mAb1 (2 binding sites), mAb1 digested with GingisKHAN™ (one binding site), the purified mAb1 Fab (one binding site) obtained by papain digestion, and the intact and GingisKHAN™-digested VEGF-A-monovalent bispecific mAb1-derived 1+1 CrossMabCH1-CL (both with one binding site) were tested and compared. Similar dose-response curves were obtained for all the molecules (), with one binding site with EC50-values of 10.3, 10.4, 12.1, and 12.5 nM for the GingisKHAN™-digested mAb1, the purified mAb1 Fab, the mAb1-derived 1+1 CrossMab, and the GingisKHAN™-digested mAb1-derived 1+1 CrossMab, respectively. The EC50 of the bivalent mAb1 (1.2 nM) is ∼9–10-fold lower than the EC50 of the Fabs and the VEGF-A-monovalent bispecific mAb1-derived 1+1 CrossMab. This demonstrates the increase of the binding strength (avidity) to the soluble dimeric VEGF-A by the 2 bindings sites of mAb1. The intact and GingisKHAN™-digested mAb1-derived 1+1 CrossMab, both with one VEGF-A-binding arm, demonstrated comparable efficacies and EC50 as the Fabs (12.1–12.5 versus 10.3–10.4 nM) (). The GingisKHAN™ protease alone did not inhibit VEGF-A-induced signal transduction and the luciferase signal.
Figure 6. (A) Inhibition of the binding of soluble dimeric vascular endothelial growth factor A (VEGF-A) to its receptor by the intact monoclonal bivalent antibody mAb1 (EC50 = 1.2 nM), mAb1 digested with GingisKHAN™ (EC50 = 10.3 nM), a purified mAb1 Fab obtained by an in-solution papain digest (EC50 = 10.4 nM), an intact (EC50 = 12.1 nM) and GingisKHAN™-digested (EC50 = 12.5 nM) bispecific VEGF-A-monovalent mAb1 derived 1+1 CrossMab, and the GingisKHAN™ protease alone (negative control), as measured by a VEGF-A-specific reporter gene assay. (B) Inhibition of the binding of multimeric angiopoitin-2 (Ang-2) to the tyrosine kinase receptor (Tie-2) by the intact monoclonal antibody mAb2 (EC50 = 3.0 nM), mAb2 digested with GingisKHAN™ (EC50 = 36 nM), and a bispecific Ang-2 monovalent mAb2 derived 1+1 CrossMab (EC50 = 57 nM), as measured by a cell-based Tie-2 receptor phosphorylation assay.
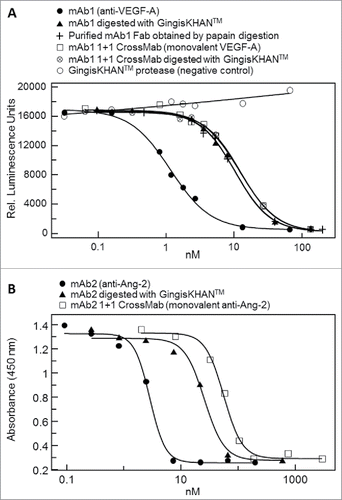
In addition, we tested the effect of digesting mAb2 binding the multimeric vascular growth factor Ang-2. The bivalent mAb2, mAb2 digested with GingisKHAN™, and an Ang-2-monovalent bispecific mAb2-derived 1+1 CrossMabCH1-CL were analyzed in a cell-based Tie-2 receptor phosphorylation assay where binding of Ang-2 to the tyrosine kinase receptor (Tie-2) induces autophosphorylation of the receptor. The grade of phosphorylation was determined by an ELISA. The method determines the capacity of the analyzed antibody or Fab to inhibit binding of Ang-2 to the Tie-2 receptor. The Tie-2 phosphorylation assay resulted in dose-response curves with EC50-values of 3.0 nM (mAb2; 2 binding sites), 36 nM (mAb2 digested with GingisKHAN™; one binding site) and 57 nM (mAb2-derived Ang2-monovalent CrossMab), demonstrating that the 2 bindings sites of mAb2 increase the binding strength to the soluble, multimeric target by a factor of 12 compared with the Fab (). The higher EC50 of the Ang-2-monovalent mAb2-derived CrossMab compared with the GingisKHAN™-digested mAb2 Fab is likely caused by better accessibility of the smaller Fab to the multimeric target.
Cell-based assays of GingisKHAN™-digested mAb3 and bispecific antibodies binding a cell surface-associated target
Other therapeutic targets like CEACAM5 are localized on the cell surface. To test the GingisKHAN™ digest involving this target class (), anti-CEACAM5 bivalent antibody mAb3 and the mAb4- and mAb5-derived trivalent 2+1 CrossMabsCH1-CL (all with 2 binding sites for CEACAM5) were analyzed in a flow cytometry assay measuring binding to CEACAM5-expressing MKN-45 cells via an Alexa Fluor 647-labeled anti-human kappa light chain detection antibody. The sigmoidal dose-response curve obtained with the bivalent mAb3 (EC50 of 5.9 nM) and the rather linear responses for the GingisKHAN™-digested mAb3 and a purified mAb3 Fab obtained by papain digest () demonstrate that both binding sites of the mAb3 are required to bind the cell surface-associated CEACAM5 with high binding strength (avidity). The presence of only one binding site results in affinity-driven binding demonstrated by the linear response. No binding was observed for the negative controls involving an anti-human fibroblast activation protein α27 antibody (anti-FAP), or the GingisKHAN™ protease alone ().
Figure 7. Analysis of the effect of a 2nd drug-to-target binding site of a trivalent human IgG1 derived bispecific antibody binding a cell-surface associated target by comparing the intact and the GingisKHAN™-digested antibody in a cell-based assay. The red arrow indicates that no drug-to-target binding occurs.
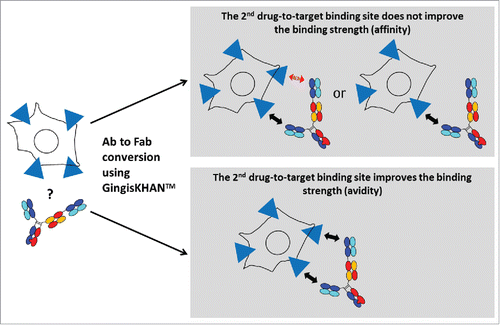
Figure 8. (A) Binding of the monoclonal bivalent antibody mAb3 (EC50 = 5.9 nM) and Fabs thereof (mAb3 digested with GingisKHAN™, and a purified mAb3 Fab obtained from an in-solution papain digest) to cell surface-associated carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5) on the human MKN-45 cell line, as measured by flow cytometry. Negative controls included an anti-human fibroblast activation protein α (anti-FAP) and the GingisKHAN™ protease alone. (B and C) Binding to CEACAM5 on the human MKN-45 cell line of the intact (bivalent CEACAM5-binding) and GingisKHAN™-digested (monovalent CEACAM5-binding) bispecific (B) mAb4 (intact: EC50 = 6.8 nM) and (C) mAb5 derived 2+1 CrossMabs (intact/digested: EC50 = 9.3 nM/10.1, respectively), as measured by flow cytometry. Negative controls with the GingisKHAN™ protease alone were included.
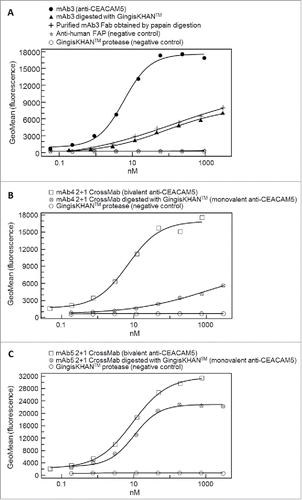
The mAb4- and mAb5-derived trivalent 2+1 CrossMabs were analyzed using methods similar to those used for mAb3. The intact mAb4-derived 2+1 CrossMab also leads to a complete sigmoidal dose-response curve (EC50 of 6.8 nM), whereas the GingisKHAN™-digested 2+1 CrossMab (one binding site) caused a linear response (). In contrast, both the mAb5-derived 2+1 CrossMab (2 binding sites, EC50 of 9.3 nM) and the GingisKHAN™ digested sample (one binding site, EC50 of 10.1 nM) gave sigmoidal dose response curves, although with different upper asymptotes (). Compared to the mAb5-derived 2+1 CrossMab, the contribution of the second drug-to-target binding site of the mAb4-derived 2+1 CrossMab to the overall binding strength is much more pronounced. With the mAb5-derived 2+1 CrossMab, the second drug-to-target binding site contributes only a small increase in binding strength and the avidity effect is not so pronounced.
Discussion
In this study, we observed a highly specific and quantitative cleavage of monoclonal and bispecific antibodies of the human IgG1 isotype by GingisKHAN™. On the contrary, we did not observe any cleavage of the mouse capturing antibodies, nor of captured target protein by GingisKHAN™ in the SPR assays (data not shown). This is consistent with the observation that GingisKHAN™ has only been reported to cleave human IgG1s at a single site above the hinge region and human IgG3 at several sites in the Fc domain.Citation20-21 Furthermore, the requirement of mild reducing conditions (i.e., 2 mM cysteine) inactivates the protease in standard SPR running buffers. Moreover, the mild reducing conditions do not reduce the IgG interchain disulfide bonds.Citation21 With GingisKHAN™, we neither observed any unspecific fragments of the human IgG1s or the bispecific antibodies analyzed, which would eventually influence the active concentration and necessitate a Fab purification, nor any undigested product, which would contaminate affinity-based binding by avidity.
For lead identification, it is particularly important to understand how the drug is interacting with the target in vivo in the case of a soluble di- or multimeric or cell surface-associated target. We have shown, based on mAb1, mAb2, mAb3, and the mAb4-derived and mAb5-derived 2+1 CrossMabs, that the determination of the putative in vivo interaction mode is possible based on the GingisKHAN™ digests. To elucidate the binding strengths of the Fab arms to their targets and the contribution of a second drug-to-target binding site, we used undigested mAbs and bispecific antibodies targeting soluble di- or multimeric targets (mAb1 and mAb2) or cell surface-associated targets (mAb3, mAb4-derived and mAb5-derived 2+1 CrossMabs) in comparison to their respective GingiKHAN™ digests. mAb1-mAb3-, and the mAb4-derived CrossMab require the second drug-to-target binding site to increase the binding strength by approximately one order of magnitude or more, whereas the mAb5-derived CrossMab is much less affected by measurement as monovalent binder. Two hypotheses can explain to these observations. The first hypothesis is that the second drug-to-target binding site does not add to binding increase because the second target is not bound. This can be caused by either steric hindrance or spatial constraints, e.g., by the target being present in cell-surface densities that do not allow simultaneous binding of both arms ( and , upper panels). The second hypothesis is that the second drug-to-target binding site does not increase the binding strength significantly since the complex stability of the monovalent binder is very high. Avidity-based increase of the binding strength is only significant when the complex stability is low. Further differentiation of both hypotheses was out of scope of this work and requires, for example, comparison of cell lines having different cell-surface densities, or an assessment of the involved paratopes of the antibodies.
Another important parameter for lead identification is characterization in terms of processability, physicochemical properties, and function. During functional characterization, determination of the binding strength, or eventually the kinetic parameters, is crucial. In cases were affinity-based interaction is relevant, it is necessary to measure the binders in a monovalent form, e.g., as Fabs. We demonstrated that the determined affinity and kinetic rate constants of Fabs in GingisKHAN™ digestion mixtures correspond to those of purified Fabs obtained by recombinant expression or purified from papain digests. The presence of GingisKHAN™ in the digestion mixtures did not influence the SPR-capturing of the human IgGs or Fabs by mouse antibodies or their binding to captured target protein.
Understanding the difference of avidity- and affinity-based binding is important in lead identification. In , we suggest a decision tree for the lead identification of bivalent and multivalent antibody formats with check points involving the conversion to Fabs. As previously discussed, a first decision point is the determination of the putative in vivo interaction mode. When it is known whether this is driven by avidity or not, this information not only supports drug design as such, but also allows the adequate setup for screening and the subsequent identification of the candidate with the desired binding strength (). A limitation of using GingisKHAN™ is that only antibodies containing a human IgG1 hinge region can be converted to Fabs. Also, the determination of the relevant in vivo interaction mode is limited to assays not dependent on a functional Fc-domain because this is generally separated from the Fabs by GingisKHAN™.
Figure 9. Suggested decision tree during lead identification of bivalent and multivalent antibodies (Ab) for the determination of the relevant in vivo interaction mode and the identification of the candidate with the optimal binding strength. Check points involving the conversion of the bivalent and multivalent antibodies to Fabs are highlighted in green. *In the case of a monovalent target and an available assay excluding avidity (e.g., using the soluble monovalent target in solution), the Ab to Fab conversion may be omitted.
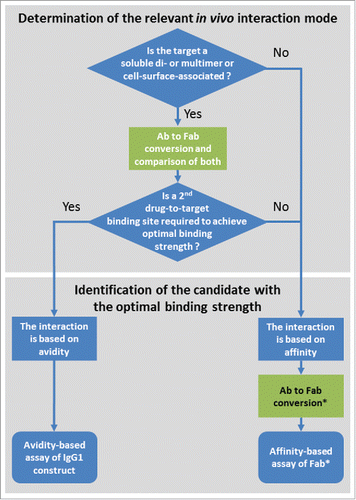
GingisKHAN™ can be used for bispecific antibody formats based on the human IgG1 framework, as demonstrated by the digestion of the mAb1-derived 1+1 CrossMab, the mAb4- and mAb5-derived 2+1 CrossMabs. With regard to the GingisKHAN™-digests of these bispecific antibodies, we observed that the ‘CH-CL crossed’ heavy chains (hinge region amino acid sequence: …GECDKTHTCPPCP; amino acid changes relative to the wild-type heavy chain are underlined)Citation28 were not digested equally efficient as the wild-type heavy chains in the same molecules. However, a repeated incubation with additional GingisKHAN™ and reducing agent ensured the complete digests of the CrossMabs. Based on the cleavage site of human IgG1s, many other bispecific antibody formatsCitation29-30 are expected to be digestible by GingisKHAN™.
In summary, we have shown that the GingisKHAN™ protease is highly suited to differentiate between affinity- and avidity-driven binding of human IgG1s monoclonal antibodies and bispecific antibody formats in a lean and efficient manner. This reported method is advantageous to the current widely used approaches, as it allows a high-throughput and precise kinetic characterization of human IgG1s for their dimeric, multimeric or cell-surface-associated targets without the need for extended antibody-specific optimizations and subsequent Fab purification. Using GingisKHAN™, only minor optimization might be needed to digest different antibodies, which significantly affects high throughput evaluation in lead identification. Also, a complete and specific conversion into intact Fabs using GingisKHAN™ eliminated the need for a correction of the active concentration. In comparison, a less-specific protease like papain will eventually completely digest any antibody, and the digest consequently needs to be carefully controlled. Thus, the papain digest of each antibody needs specific optimization to minimize the influence on the active concentration. In fact, it is very difficult to control papain digestion such that only the upper hinge region is cleaved, and not also alternative sites depending on the structure and flexibility of the antibody being cleaved. Digesting the bispecific 2+1 CrossMabs with papain proved to be particularly challenging, as several indefinite fragments were generated (data not shown). Beside the use of GingisKHAN™ for the determination of binding strengths of human IgG1-type frameworks, the protease can also be used in cases where IgG1s binding monomeric targets are difficult to immobilize on the SPR surface. Other applications of GingisKHAN™ may include the analysis of the molecular chain assembly of bispecific antibodies, focused analysis of the Fcs or the generation of high-quality Fabs for the structural analysis and structure-function relationships of human IgG1-target bindings at atomic resolution, e.g., by X-ray crystallography.Citation5,31
Materials and methods
Target proteins, antibodies and proteases
All target proteins, monoclonal and bispecific antibodies were obtained from Roche Innovation Center Munich. Papain in suspension was obtained from Roche Applied Science. GingisKHAN™ was purchased from Genovis AB.
Transient fab expression and purification
The antibody light and half-heavy chain (Fd) coding regions were ordered as gene syntheses and cloned via unique restriction sites into separate expression vectors enabling secretory expression in HEK cells growing in suspension. Transfection (1:1 plasmid ratio) of HEK293 cells (Thermo Fisher Scientific) was performed according to the cell suppliers instructions using Maxiprep (Qiagen) preparations of the antibody vectors, Opti-MEM I medium, 293fectin, and a cell density of 1–2 × 106 viable cells/mL in serum-free FreeStyle 293 expression medium (all reagents from Thermo Fisher Scientific). The Fab-containing cell culture supernatant was harvested after 7 d of cultivation in shake flasks by centrifugation at 14,000 × g for 30 min and filtered through a 0.22 μm sterile filter. The filtered supernatant was stored at −80°C until purification of the Fab finally formulated in 20 mM histidine, 140 mM NaCl, pH 6.0.
Papain digest and Fab purification
The antibodies were diluted in 20 mM histidine, 140 mM NaCl, pH 6.0 to a final concentration of 1 mg/mL, then added 5 mM L-cysteine (Sigma-Aldrich) and 0.08 unit papain in 20 mM histidine, 140 mM NaCl, pH 6.0, and incubated at 37°C for 1 hour (optimized conditions for mAb1) if not otherwise stated. Papain was inhibited by adding 100 µM antipain (Sigma-Aldrich). To isolate the Fabs from non-cleaved antibody, Fcs and papain, the mixtures were applied to CaptureSelect IgG-CH1 and MabSelect SuRe affinity chromatography (GE Healthcare). Finally, size-exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) was performed using 20 mM histidine, 140 mM NaCl, pH 6.0 as running buffer. The concentrations of the Fabs were determined by measuring the optical densities at 280 nm and using the molar extinction coefficients calculated on the basis of the amino acid sequences.
GingisKHAN™ digests
2000 U GingisKHAN™ was reconstituted in 200 µL ddH2O, and the 10x reducing agent (supplied by the vendor) was freshly prepared in 50 µL ddH2O (final concentration: 20 mM cysteine) before each digestion. 100 µg monoclonal or bispecific antibody was diluted to a final concentration of 1 mg/mL in 100 mM Tris, pH 8.0 and subsequently digested with 10 µL reconstituted GingisKHAN™ and 11 µL of freshly prepared 10x reducing agent at 37°C for ∼1 hour. The digests were controlled by mass spectrometry. If partially digested, the samples were incubated again with additional protease and reducing agent.
UHR-ESI-QTOF mass spectrometry
The samples were desalted by HPLC on a Sephadex G25 5 × 250 mm column (Amersham Biosciences) using 40% acetonitrile with 2% formic acid (v/v). The total mass was determined by UHR-ESI-QTOF mass spectrometry on a maXis 4G UHR-QTOF MS system (Bruker Daltonik) equipped with a TriVersa NanoMate source (Advion). Calibration was performed with sodium iodide (Tof G2-Sample Kit 2; Waters). For the recombinant and purified Fabs, data acquisition was done at 900–2600 m/z (ISCID: 0.0 eV), for the human IgG1s, bispecific antibodies, and digests, data acquisition was done at 900–4000 m/z (ISCID: 0.0 eV). The raw mass spectra were evaluated and transformed into individual relative molar masses using an in-house developed software tool.
Surface plasmon resonance
Binding affinities and kinetics were investigated by SPR using a Biacore T200 instrument (GE Healthcare). All experiments were performed at 25°C using PBS-T (10 mM Na2HPO4, 140 mM NaCl, 0.05% Tween 20, pH 7.4) as running and dilution buffer. Mouse anti-His-tag or mouse anti-human Fab antibodies (GE Healthcare; Cat. No. 28995056 and BR100839, respectively) were immobilized on Series S C1 Sensor Chips (GE Healthcare) using standard amine coupling chemistry. VEGF-A121-His (amino acids VEGF-A with His-tag, Roche Innovation Center Munich) or mAb2 IgG/Fabs, respectively, were captured on the surface, leading to a response between 10 and 50 RU. The analytes (mAb1 binders or monomeric Ang-2 receptor binding domainCitation26 fused to a human Fc (Ang2-RBD-Fc; Roche Innovation Center Munich)) were injected for 180 s (with mAb2: 90s) at concentrations from 2.2 up to 1800 nM (with mAb2: 3.7 up to 900 nM) onto the surface (association phase) at a flow rate of 30 µL/min. The dissociation phase was monitored for up to 3600 s (with mAb2: 180 s) by washing with running buffer. The surface was regenerated by injecting 10 mM glycine, pH 1.5 for 60 sec at a flow rate of 5 µL/min. Bulk refractive index differences were corrected by subtracting the response obtained from a mock surface and by subtracting blank injections (double referencing). The derived curves were fitted to a 1:1 Langmuir binding model using the BIAevaluation software.
VEGF-A-specific reporter gene assay
All media and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific. A reporter gene cell line GloResponse™ NFAT-RE-luc2P/KDR HEK293 expressing KDR (KDR = VEGF receptor 2) harbouring a NFAT responsive element in front of the firefly luciferase gene was purchased from Promega Corporation. The cells were cultivated in suspension in FreeStyle™ 293 Expression Medium with 100 µg/ml HygromycinB (Sigma-Aldrich) and 250 µg/mL geneticin (Calbiochem). Upon binding of VEGF-A to KDR a signal transduction via calcineurin results in activation of NFAT, translocation to the nucleus, binding to the NFAT responsive element and subsequently expression of the luciferase gene. VEGF-A121 (Roche Innovation Center Munich; end concentration in well 10.7 nM; 40 µL/well) was incubated with anti-VEGF-A binders (antibody or Fabs) or a negative control with GingisKHAN™ only (diluted in DMEM, 1% FBS, 40 µL/well) for ∼30 min at room temperature. 5 × 104 (in 40 µL DMEM, 1% FBS) GloResponse™ NFAT-RE-luc2P/KDR HEK293 cells were added and the plate incubated for 5 hours at 37°C with 5% CO2. The plate was equilibrated at room temperature for ∼15 minutes before the luminescence substrate (Promega Corporation, ONE-Glo™ EX, 60 µL/well) was added. The contents were mixed on an orbital shaker for about 1–3 minutes at 600 rpm. The signal intensity was measured using a luminescence reader.
Tie-2 receptor phosphorylation assay
All media and FBS were purchased from Thermo Fisher Scientific. All steps were performed at room temperature if not otherwise indicated. HEK293 cells were stably transfected with an expression vector for the human angiopoietin receptor Tie-2 (Roche Innovation Center Munich). A monoclonal anti-Tie-2 antibody (10 µg/mL in DPBS, 100 µL/well; R&D Systems) was immobilized overnight in a Nunc MaxiSorp microtiter plate (Thermo Fisher Scientific) at 4°C. The plate was washed (3 × 300 µL PBS, 0.05% polysorbate 20; Sigma-Aldrich) and blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich) in DPBS (200 µL/well) for one hour. In an U-bottom polypropylene microtiter plate (Greiner bio-one) Ang-2 (R&D Systems, 40 µL/well, 70 nM in DMEM/F12 ) and Ang-2 binders (40 µL/well, in DMEM/F12) were mixed and incubated for 30 min. 2 × 105 HEK293 Tie2 cells in 40 µl DMEM/F12 were added, mixed and incubated for 8 min at room temperature. 60 µL ice-cold lysis buffer (3-fold concentrated, 450 mM sodium chloride, 75 mM MES sodium salt, 6 mM sodium orthovanadate, 6% Triton-X-100, cOmplete protease inhibitor (Sigma-Aldrich)) was added per well, mixed and incubated for 15 minutes at room temperature. 100 µL of the cell lysate was transferred to the blocked and washed MaxiSorb plate and incubated for 90 minutes. The washing step was repeated. 100 µL biotinylated anti-phosphotyrosine monoclonal antibody (Merck Millipore, 0.3 µg/mL in DPBS, 1% BSA) was added and incubated for 60 minutes. After washing, 100 µL horseradish peroxidase-conjugated streptavidin (Sigma-Aldrich, 100 mU/mL) was added and incubated for 30 minutes. After a final washing step, 100 µL of TMB substrate (Sigma-Aldrich) was added and incubated for 15 minutes. The substrate reaction was stopped with 100 µL 2 N sulfuric acid (Carl Roth GmbH & Co) and the absorbance was measured at 450 nm.
CEACAM5 cell surface binding assay
All media and FBS were purchased from Thermo Fisher Scientific. 1 × 105 MKN-45 gastric adenocarcinoma cells (DSMZ no.: ACC 409) cultured in RPMI1640, 20% FBS, 1x GIBCO GlutaMAX were washed twice with PBS, 5% FBS, resuspended in PBS, 5% FBS and incubated with the anti-CEACAM5 binders or negative controls (an anti-FAP antibody (Roche Innovation Center Munich) or GingisKHAN™ only) for one hour at 4°C. Bound antibodies/Fabs were detected using a mouse anti-human kappa light chain antibody (150 µg/mL, Roche Innovation Center Munich) labeled by the Alexa Fluor 647 Protein Labeling Kit according to the instructions of the manufacturer (Molecular Probes, Thermo Fisher Scientific). The mixture was incubated in the dark at 4°C for 30 min and analyzed after a washing step with DPBS, 2% FBS by flow cytometry using a BD FACSCanto II and the FACSDiva Software (BD Biosciences). The bound anti-CEACAM5 molecules were detected by measuring the fluorescence signal. Gating of viable cells was done using forward and sideward scatter based on size and granularity. The specificity was verified by an isotype control (Alexa Fluor 647-labeled mouse IgG2a, BD Biosciences).
supplemental_material.zip
Download Zip (193.2 KB)Acknowledgments
We wish to thank all colleagues from Roche Diagnostics GmbH, who kindly provided input to the generation of Fabs.
References
- Porter RR. The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem J. 1959;73:119-26. PMID:14434282
- Mage M, Lamoyi E. Preparation of Fab and F(ab’) fragments from monoclonal antibodies. In: Schook LB, editor. Monoclonal antibody production techniques and applications. New York (US): Marcel Dekker Inc; 1987. p. 79-97
- Akita EM, Nakai S. Production and purification of Fab' fragments from chicken egg yolk immunoglobulin Y (IgY). J Immunol Methods 1993; 162(2):155-64. PMID:8315286
- Andrew SM, Titus JA. Fragmentation of Immunoglobulin G. Curr Protocols Cell Biol. Hoboken, New Jersey, USA: John Wiley & Sons Inc; 2003. p. 16.4.1-16.4.10. doi:10.1002/0471143030.cb1604s17. PMID:18228421
- Zhao Y, Gutshall L, Jiang H, Baker A, Beil E, Obmolova G, Carton J, Taudte S, Amegadzie B. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif. 2009;67(2):182-89. doi:10.1016/j.pep.2009.04.012. PMID:19442740
- Parham P. On the fragmentation of monoclonal IgG1, IgG2a, and IgG2b from BALB/c mice. J Immunol. 1983;131(6):2895-902. PMID:6358356
- Parham P. Preparation and purification of active fragments from mouse monoclonal antibodies. In: Weir DM, editor. Handbook of Experimental Immunology. Oxford (UK): Blackwell Scientific Publications; fourth ed; 1986. p. 14.1-14.23
- Tischer W, Kasche V. Immobilized enzymes: crystals or carriers? Trends Biotechnol. 1999;17(8):326-35. PMID:10407405
- Luo Q, Mao X, Kong L, Huang X, Zou H. High-performance affinity chromatography for characterization of human immunoglobulin G digestion with papain. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776(2):139-47. PMID:12137995
- Gadgil HS, Bondarenko PV, Pipes GD, Dillon TM, Banks D, Abel J, Kleemann GR, Treuheit MJ. Identification of cysteinylation of a free cysteine in the Fab region of a recombinant monoclonal IgG1 antibody using Lys-C limited proteolysis coupled with LC/MS analysis. Anal Biochem. 2006;355(2):165-74. doi:10.1016/j.ab.2006.05.037. PMID:16828048
- von Pawel-Rammingen U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 2002;21(7):1607-15. doi:10.1093/emboj/21.7.1607. PMID:11927545
- von Pawel-Rammingen U, Björck L. IdeS and SpeB: immunoglobulin-degrading cysteine proteinases of Streptococcus pyogenes. Curr Opin Microbiol. 2003;6(1):50-5. PMID:12615219
- Ishikawa E, Yoshitake S. A general and mild procedure for the purification of rabbit Fab' antibodies. J Immunol Methods. 1980;38(1–2):117-23. PMID:7452000
- Billah MM, Hodges CS, Hays HC, Millner PA. Directed immobilization of reduced antibody fragments onto a novel SAM on gold for myoglobin impedance immunosensing. Bioelectrochemistry. 2010;80(1):49-54. doi:10.1016/j.bioelechem.2010.08.005. PMID:20880761
- DeSilva BS, Wilson GS. Solid phase synthesis of bifunctional antibodies. J Immunol Methods. 1995;188(1):9-19. PMID:8551042
- Makaraviciute A, Jackson CD, Millner PA, Ramanaviciene A. Considerations in producing preferentially reduced half-antibody fragments. J Immunol Methods. 2016;429:50-6. doi:10.1016/j.jim.2016.01.001. PMID:26779832
- Scott CF, Whitaker EJ, Hammond BF, Colman RW. Purification and characterization of a potent 70-kDa thiol lysyl-proteinase (Lys-gingivain) from Porphyromonas gingivalis that cleaves kininogens and fibrinogen. J Biol Chem. 1993;268(11):7935-42. PMID:8385128
- Okamoto K, Kadowaki T, Nakayama K, Yamamoto K. Cloning and sequencing of the gene encoding a novel lysine-specific cysteine proteinase (Lys-gingipain) in Porphyromonas gingivalis: structural relationship with the arginine-specific cysteine proteinase (Arg-gingipain). J Biochem. 1996;120(2):398-406. PMID:8889827
- Fujimura S, Hirai K, Shibata Y, Nakayama K, Nakamura T. Comparative properties of envelope-associated arginine-gingipains and lysine-gingipain of Porphyromonas gingivalis. FEMS Microbiol Lett. 1998;163(2):173-9. PMID:9673019
- Vincents B, Guentsch A, Kostolowska D, von Pawel-Rammingen U, Eick S, Potempa J, Abrahamson M. Cleavage of IgG1 and IgG3 by gingipain K from Porphyromonas gingivalis may compromise host defense in progressive periodontitis. FASEB J. 2011;25(10):3741-50. doi:10.1096/fj.11-187799. PMID:21768393
- Genovis AB. Lund: Sweden. https://www.genovis.com/products/gingiskhan/
- Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs 2016;8(6):1010-20. doi:10.1080/19420862.2016.1197457. PMID:27285945
- Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161(2):851-8. PMID:2735925
- Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, Xu A, Leidich R, Radziejewska E, Rafique A, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol. 2003;10(1):38-44. doi:10.1038/nsb880. PMID:12469114
- Lisowska E, Krop-Watorek A, Sedlaczek P. The dimeric structure of carcinoembryonic antigen (CEA). Biochem Biophys Res Commun. 1983;115(1):206-11. PMID:6615527
- Barton WA, Tzvetkova D, Nikolov DB. Structure of the angiopoietin-2 receptor binding domain and identification of surfaces involved in Tie2 recognition. Structure. 2005;13(5):825-32. doi:10.1016/j.str.2005.03.009. PMID:15893672
- Aertgeerts K, Levin I, Shi L, Snell GP, Jennings A, Prasad GS, Zhang Y, Kraus ML, Salakian S, Sridhar V, et al. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J Biol Chem. 2005;280(20):19441-4. doi:10.1074/jbc.C500092200. PMID:15809306
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187-92. doi:10.1073/pnas.1019002108. PMID:21690412
- Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015;20(7):838-47. doi:10.1016/j.drudis.2015.02.008. PMID:25728220
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2 Pt A):95-106. doi:10.1016/j.molimm.2015.01.003. PMID:25637431
- Kovari LC, Momany C, Rossmann MG. The use of antibody fragments for crystallization and structure determinations. Structure. 1995;3(12):1291-3. PMID:8747455