
ABSTRACT
Asthma affects more than 300 million people worldwide and poses a large socioeconomic burden, particularly in the 5% to 10% of severe asthmatics. So far, each entry of new biologics in clinical trials has led to high expectations for treating all severe asthma forms, but the outcome has only been successful if the biologic, as add-on treatment, targeted specific patient subgroups. Indeed, we now realize that asthma is a heterogeneous disease with multiple phenotypes, based on distinct pathophysiological mechanisms, called endotypes. Thus, asthma therapy is gradually moving to a personalized medicine approach, tailored to individual's asthma endotypes identified through biomarkers. Here, we review the clinical efficacy of antibody-related therapeutics undergoing clinical trials, or those already approved, for the treatment of severe type 2 asthma. Biologics targeting type 2 cytokines have shown consistent efficacy, especially in patients with evidence of type 2 inflammation, suggesting that the future of asthma biologics is promising.
Introduction
Asthma is described in the 2016 Global Initiative for Asthma report as “a heterogeneous disease, usually characterized by chronic airway inflammation. It is defined by the history of respiratory symptoms such as wheeze, shortness of breath, chest tightness and cough that vary over time and in their occurrence, frequency and intensity, together with variable expiratory airflow limitation”.Citation1 To better understand its heterogeneity, which is highlighted by the variety of clinical presentations, physiologic characteristics, pathogenic pathways and outcomes, the concept of asthma phenotyping has emerged.
An asthma phenotype is defined by the International European Respiratory Society/American Thoracic Society guidelines, “as the composite, observable characteristics of an organism, resulting from interaction between its genetic make-up and environmental influences, which are relatively stable – but not invariable – with time”.Citation2 Asthma phenotypes were initially focused on combinations of clinical, physiologic and hereditary characteristics, but they have evolved to link biology to phenotype.Citation3 Interestingly, these phenotypes are now evolving into asthma endotypes, wherein a specific biological pathway is identified that explains the observable properties of a phenotype, with the goal to improve therapy.Citation3 Endotypes would differ in terms of genetic susceptibility, environmental risk factors, age of onset, airway inflammation, clinical presentation, prognosis and response to standard and new therapies, but definitive clustering of these characteristics or their relation to pathobiology remains uncertain (Supplementary Figure 1).Citation4
With almost 1 in 8 children and 1 in 12 adults affected, asthma is one of the most common chronic diseases, resulting in up to 300 million people affected worldwide.Citation4 In many patients, the disease can be controlled by a combination of nonspecific drugs, an inhaled corticosteroid (ICS) and a short- or long-acting β2-adrenergic agonist (LABA). Nevertheless, in 5 to 10% of patients, the disease runs a severe course. In such cases, the patient will require treatment with high-dose ICS plus a second controller medication (such as LABAs or leukotriene receptor antagonists) or systemic steroids to prevent the disease from becoming ‘uncontrolled’, or the disease may remain ‘uncontrolled’ despite these treatments.Citation2 This loss of control manifests as frequent severe exacerbations that require systemic steroids or hospitalization. However, responses to these treatments can vary and do not modify the course of the disease, requiring an urgent need for new and more effective drugs to prevent the occurrence of potentially life-threatening episodes.
Biologics (i.e., drugs produced by living cells through biological processes, and mimic natural biological substances such as antibody-related therapeuticsCitation5) targeting specific inflammatory pathways have emerged as promising personalized medicines in the treatment of severe asthma. Their use is explained by the fact that the disease is characterized by inflammatory responses involving multiple pathways and triggers. Increased levels of inflammatory molecules, cytokines in particular, have been identified in clinical samples and their role in disease pathogenesis and pathophysiology have been demonstrated in preclinical studies using asthma mouse models, leading to their extensive investigation as potential targets.Citation4 Consequently, there are currently a dozen biologics, mainly antibody-related therapeutics, in clinical studies for patients with moderate-to-severe asthma.
The economic and healthcare benefits of treating asthma are considerable. Most costs for asthma come from the severe asthmatics that require frequent hospital admissions due to exacerbations, which are often caused by virus infection. More specifically, the total cost of asthma has been estimated to be approximatively €17.7 billion in Europe, of which €9.8 billion is accounted for by the indirect costs of loss of productivity.Citation6 In the US, the annual costs amount to $18 billion.Citation7 Although the costs of biologics are expensive compared to current treatment options, effective biologics for a well-defined endotype may be cost effective in the long-term, by preventing hospital admissions due to severe asthma exacerbations and by reducing systemic side effects of ICS.Citation8
In this review, we focus on the importance of a personalized medicine using a biomarker-driven approach, the immunological basis of type 2 and non-type 2 inflammations in asthma, the development of biologics that interrupt specific pathways involved in type 2 inflammation, the evidence of efficacy of these personalized treatments in recent clinical trials, their limitations, and the emergence of novel approaches.
Personalized medicine to treat severe asthma using a biomarker-driven approach
The increased use of biologics in clinical trials has facilitated the understanding of asthma heterogeneity and the subsequent development of asthma endotypes (Supplementary Figure 1). It underscored the importance of selecting the appropriate patient subsets with the correct target, dosing and mode of delivery during the clinical trials of these new biologics, by using the adapted outcome measures to the biological pathway(s) being targeted. From this comes the need for a personalized medicine using a biomarker-driven approach for development of biologics in severe asthma treatments (Supplementary Table 1).
Table 1. Asthma biomarkers used in clinical trials to predict the response to biologics directed at mediators of type 2 asthma.
One important tool in the development of personalized medicine is the application of biomarkers to stratify patients.Citation9 A biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes or pharmacologic responses to a therapeutic intervention”.Citation10 Targeted therapy benefits mostly from a biomarker that encompasses both high diagnostic, theragnostic (i.e., the ability to predict treatment effect) and prognostic capacities. Ideally, the biomarker might be the pathophysiological therapeutic target itself (i.e., the ‘maker’ of disease in a specific endotype).Citation11 Importantly, biomarkers are critical in the design and implementation of efficient and cost-effective clinical trials.Citation12
An early asthma biomarker was the induced sputum cell count (eosinophils and neutrophils). Indeed, the presence of high sputum eosinophil count was found to be predictive of response to corticosteroid therapy.Citation13 Since then, asthma biomarkers have expanded to include mainly blood eosinophil counts, total serum immunoglobulin E (IgE) levels, fraction of exhaled nitric oxide (FENO) in the exhaled air and serum periostin ().Citation14–17
The technique of induced sputum cell count (eosinophils and neutrophils) has been pivotal in the emergence of the concept of asthma endotyping. Although it is technically demanding and time-consuming, several centers have applied this technique to characterize airway inflammation.Citation18 Based on sputum cell count analysis, in addition to clinical phenotyping (including allergen skin-prick tests and/or allergen-specific serum IgE) and type 2 biomarkers (), two groups of airway inflammations in asthma have been described: type 2 (allergic eosinophilic and nonallergic eosinophilic asthma) and non-type 2 (neutrophilic, paucigranulocytic and mixed granulocytic asthma).
Type 2 and non-type 2 airway inflammations in asthma
Type 2: allergic and non-allergic eosinophilic asthma
Most children and roughly 50% of adults have allergic eosinophilic asthma, in which the disease coincides with allergic sensitization (atopy) defined by the presence of serum IgE antibodies and/or a positive skin-prick test to the (lipo)proteins of common inhaled allergens such as Derp 1 from the house dust mite Dermatophagoides pteronissinus.Citation4,19 In contrast, nonallergic eosinophilic asthma often develops later in life (i.e., late onset asthma) and, per its definition, has neither IgE reactivity to allergens in the serum nor any obvious involvement of the adaptive immune system such as T-helper type 2 cells (TH2) cells.Citation4 This form of the disease is often associated with chronic rhinosinusitis and nasal polyps, and is difficult to treat, often requiring long-term treatment with systemic steroids ().Citation4
Table 2. Characteristics of type 2 and non-type 2 airway inflammations in asthma.
TH2 cells and type 2 innate lymphoid cells (ILC2) are master drivers of type 2 immunity by expressing the transcription factor GATA3, which controls type 2 cytokine production ().Citation4 Interleukin (IL)-4 and IL-13 are central cytokines to the type 2 inflammation induced by TH2 cells and ILC2s, but also by other type 2 inflammatory cells such as eosinophils, alternatively activated macrophages, basophils and mast cells.Citation20 In chronic severe asthmatics, basophils are also a prominent source of IL-13 and IL-5, and might sustain the disease process in an IL-33-dependent manner ().Citation21 IL-4 receptor alpha (IL-4Rα) is the common receptor subchain for IL-4 and IL-13, which can be dimerized with γc (expressed on cells restricted to hematopoietic cell lineages) or with IL-13Rα1 (expressed fairly ubiquitously, e.g., airway epithelial cells). IL-4 activates both type of receptors, whereas IL-13 only activates the receptor dimerized with IL-13Rα1. Thus, whilst both interleukins can promote IgE isotype switching in B cells, only IL-4 activates TH2 effector cells ().Citation22–24
Figure 1. Simplified schematic representation of four different types of airway inflammation in asthmatic patients. (A) Type 2 consists of allergic and nonallergic eosinophilic asthma. (a) In allergic eosinophilic asthma, T-helper type 2 (TH2) cell lymphocytes and mast cells drive eosinophilic airway inflammation in an allergen-specific, immunoglobulin E (IgE)-dependent manner. (b) In nonallergic eosinophilic asthma, innate lymphocytes such as natural killer T cells (NKT cells) and innate lymphoid cells type 2 (ILC2) cells might contribute to airway eosinophilia via the production of interleukin (IL)-5, in response to pollutants or infectious agents. (B) Non-type 2 consists of neutrophilic and paucigranulocytic asthma. (c) The mechanisms underlying neutrophilic asthma need to be elucidated, but the IL-17 pathway and CXCL8 have been associated with the airway neutrophilia. More precisely, IL-17A and IL-17F play important roles in host responses to extracellular pathogens via the upregulation of antimicrobial proteins and induction of cytokines and chemokines involved in neutrophil expansion (e.g., GM-CSF) and recruitment (e.g., CXCR ligands). (d) Paucigranulocytic asthma has been poorly studied. It is thought to be not inflammatory and is characterized by the absence of increased numbers of inflammatory cells, suggesting the involvement of non-inflammatory mechanisms mediated by airway remodeling responses that lead to extensive airway narrowing. The biologics being evaluated in clinical trials or already approved as add-on treatment, on top of high-doses inhaled corticosteroid (ICS) and a short- or long-acting β2-adrenergic agonist (LABA), for (C) allergic and (D) nonallergic eosinophilic asthma are depicted in light grey. CRTH2: prostaglandin D2 receptor 2; CXCL8: C-X-C motif chemokine ligand 8; CXCR: C-X-C chemokine receptor; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; FcϵRI: Fc epsilon receptor I; GM-CSF: granulocyte-macrophage colony-stimulating factor; ICS: inhaled corticosteroid; IgE: immunoglobulin epsilon; ILC2: innate lymphoid cell type 2; IL: interleukin; IL-4Rα: interleukin-4 receptor alpha; IL-5Rα: interleukin-5 receptor alpha; IL-9R: interleukin-9 receptor; IL-25R: interleukin-25 receptor; IL-33R: interleukin-33 receptor; LABA: long-acting β2-adrenergic; MHC: major histocompatibility complex; NKT: natural killer T; PGD2: prostaglandin D2; TCR: T-cell receptor; TH2: T-helper type 2 cell; TH17: T-helper type 17 cell; TSLP: thymic stromal lymphoietin; TSLPR: thymic stromal lymphoietin receptor.
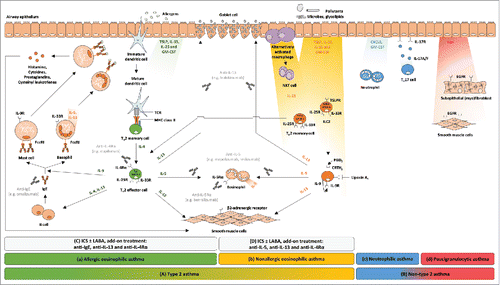
IL-13 signalling leads to the differentiation of bronchial epithelial cells into mucus-producing goblet cells and may potentiate airway smooth muscle cell contraction ().Citation20 IL-4 is required for TH2 priming and maturation, whereas TH2 differentiation, promoted by the dendritic cells, is enhanced by cytokines made by epithelial cells, such as thymic stromal lymphoietin (TSLP), IL-33, IL-25 and granulocyte-macrophage colony-stimulating factor (GM-CSF).Citation25 Those cytokines also exert an important tissue checkpoint for full activation of primed TH2 cells and ILC2s that enter target organs such as the lungs ().Citation26 It is also now clear that TH2 memory cells can secrete cytokines even in the absence of T-cell receptor ligation in nonallergic eosinophilic asthma, and could react to non-specific stimuli imposed on the airway epithelium ().Citation26,27
IgE and IL-5 are two other important proteins in type 2 asthma. IgE, produced by B cells, binds to the type I high affinity IgE receptor (FcϵRI), which activates mast cells and basophils associated with IgE-mediated hypersensitivity when cross-linked by allergen ().Citation4 IL-5 is produced, like IL-13, by multiple cell types implicated in severe asthma, including TH2 cells, ILC2s, eosinophils and basophils. IL-5 binds to IL-5 receptor complex principally expressed on eosinophils, and is involved in the differentiation and maturation of eosinophils in the bone marrow and in their survival and migration to tissues ().Citation4
Non-type 2: noneosinophilic asthma
Although asthma is classically associated with eosinophilia and type 2 cytokines, some asthmatic patients show a neutrophil-predominant disease with an absence of TH2 cytokines and their downstream signatures.Citation28–30 These non-type 2 asthma patients have generally adult-onset disease, and are less likely to be atopic ().Citation31–34 The underlying causes and triggers are not well understood, but are heterogeneous and might encompass obesity, respiratory infections, smoking and air pollution. Some patients with non-type 2 asthma seem to have neutrophilic inflammation with less severe reversible airway obstruction and a TH17 cytokine milieu ().Citation35–37 The cytokine production by TH17 cells and other IL-17-producing cells is resistant to inhibition by corticosteroids, which explains why neutrophil-rich inflammation driven by IL-17 is the pathological correlate of a subgroup of patients with steroid-resistant asthma ().Citation4 Additionally, other patients have mixed granulocytic asthma when both eosinophils and neutrophils are increased or paucigranulocytic asthma when both these inflammatory cells are below the thresholds ( and ).Citation18,38 These non-type 2 asthma groups remain poorly defined, clinically heterogeneous and without specific biomarkers, making molecular endotyping and targeted therapy approaches difficult.
Importantly, the identification of the type 2 and non-type 2 airway inflammations has fostered the concept of targeted biologics and patient's stratification, introducing personalized medicine in severe asthma treatment.
Type 2 biologic approaches
Immunoglobulin E
Omalizumab is a monoclonal IgG1κ antibody with a human framework and complementarity-determining regions from a humanized anti-IgE murine antibody (MAE11), produced with hybridoma technology ( and ).Citation39 Omalizumab inhibits IgE effector functions by blocking IgE binding FcϵRI on mast cells, but does not cause mast cell activation because it cannot bind to IgE on cell surfaces where the FcϵRI receptor already masks the anti-IgE epitope ().Citation40 With prolonged treatment, omalizumab also reduces the expression of FcϵRI on mast cells and basophils.Citation41 Omalizumab became the first monoclonal antibody approved by the Food and Drug Administration (FDA, 2003) and European Medicines Agency (EMA, 2005) to treat asthma patients of 12 years and older ().
Table 3. Biologics evaluated for the treatment of moderate-to-severe type 2 asthma.
Nevertheless, the use of omalizumab has been limited by its expense (US wholesale prices for omalizumab average almost $1300 per patient-month for an asthmatic patient who weighs less than 90 kg42), the need for multiple injections that may then lead to injection-site reactions, a black box warning on anaphylaxis, and new warnings on cardiovascular risk.Citation43 Thus, an improved ability to predict responsive patients with high certainty was important, requiring new clinical trials or prospective observational cohort studies (e.g., registries). Although omalizumab has not been prospectively studied in patients identified based on other type 2 biomarkers, a retrospective analysis of the Phase 3 study by Hanania et al. (EXTRA trial)Citation44 divided patients into those with and without type 2 inflammation based on median splits of blood eosinophil counts, serum periostin and FENO levels (Supplementary Table 2). Patients with biomarker levels greater than the median had greater reductions in asthma exacerbations with omalizumab therapy compared with those with levels lower than the median (Supplementary Table 2).Citation45 Although no other outcomes were affected, this approach should be prospectively validated.
Continuation of omalizumab after 5-year treatment resulted in continued benefit, as evidenced by improved symptom control and reduced exacerbation risk in the XPORT trial (Supplementary Table 2).Citation46 Approximately half of the patients remained free of exacerbations during the one-year study period despite withdrawal of omalizumab. Although this study suggested that omalizumab might have beneficial disease-modifying effects, this positive outcome could also be due to the natural history of the disease (e.g., spontaneous evolution from severe asthma towards mild-moderate asthma in a subgroup of patients after about 5 years or improvement in asthma control thanks to removal from persistent allergen exposure such as domestic animals, e.g., cats and dogs).
In 2016, FDA and EMA approved an expanded age range for omalizumab to include children six to 11 years of age with moderate-to-severe persistent asthma, having a positive skin test or in vitro reactivity to an airborne allergen and symptoms that are inadequately controlled with ICS ( and Supplementary Table 2). This approval followed successful pediatric clinical trials such as one conducted by Lanier et al. 2009 (Supplementary Table 2).Citation47 Nevertheless, to date, strong biomarkers to identify responders are still lacking. These issues, as well as further cost-effectiveness analyses, are still open and need to be investigated in future pediatric studies.
Interleukin-13
Lebrikizumab is a humanized IgG4κ antibody that binds IL-13 with high affinity at an epitope that strongly overlaps with the binding site of IL-4Rα and inhibits its activity ( and ).Citation48 In previous studies, lebrikizumab treatment was associated with significantly and substantially decreased serum periostin, FENO and serum IgE, and modestly increased peripheral blood eosinophil count.Citation17,49 Notably, the extent of the pharmacodynamic effect was greater in subjects who had high periostin levels at baseline.Citation17 Nevertheless, lebrikizumab did not consistently show significant reduction in asthma exacerbations in biomarker-high patients (periostin ≥50 ng/mL or blood eosinophils ≥300 cells per μL) in more recent replicate Phase 3 studies (LAVOLTA I and II),Citation50 leading to the discontinuation of this biologic in asthma (Supplementary Table 2). The discontinuation could be due to the non-optimal patient selection through the use of serum periostin, which has only a weak association with airway periostin level and is therefore not the best biomarker for IL-13 activity in the airways. In contrast, FENO is a good biomarker of IL-13 activity in the airway and is responsive to treatment with anti-IL-13 antibodies ().
Fourteen weeks of treatment with tralokinumab, another human monoclonal IgG4λ antibody targeting IL-13 ( and ), was associated with only modest improvement in FEV1 and some decrease in β2-agonist use (Supplementary Table 2).Citation51 In a large Phase 2 study, tralokinumab did not significantly reduce asthma exacerbation rates in patients with severe uncontrolled asthma. Improvement in FEV1 with tralokinumab given every 2 weeks and results of post-hoc subgroup analyses suggested a possible treatment benefit in a defined population of patients with severe uncontrolled asthma (Supplementary Table 2). This effect is being further investigated in ongoing Phase 3 trials, along with the potential utility of periostin and dipeptidyl peptidase-4 (DPP-4, a gene whose expression is induced by IL-13) as biomarkers of interleukin-13 pathway activation.Citation52 Nevertheless, tralokinumab did not meet the primary endpoint of a significant reduction in the annual asthma exacerbation rate in the overall population of severe, uncontrolled asthma patients, compared with placebo in STRATOS I, the first of two pivotal Phase 3 trials (Supplementary Table 2). Nevertheless, in a planned analysis, a clinically-relevant reduction in annual asthma exacerbation rate was observed in a sub-population of patients with an elevated biomarker associated with increased IL-13 activity.Citation53 Thus, this sub-group of patients will now be the focus for the future analysis of STRATOS II, the second ongoing pivotal Phase 3 trial. Indeed, STRATOS I explored the potential use of biomarkers to identify patients with an enhanced response to tralokinumab, whereas STRATOS II is designed to validate the biomarker population identified in STRATOS I. Overall, these recently published clinical trials on therapeutic antibodies targeting IL-13, have highlighted relevant limitations with partial effects that could be due to overlapping biological actions of IL-4 and IL-13. The combination approach inhibiting both IL-4 and IL-13 is likely to be more effective.
Interleukin-4 receptor alpha
Dupilumab, a human monoclonal IgG4κ antibody targeting IL-4Rα and inhibiting both IL-4 and IL-13 signalling pathways, increased lung function and reduced severe exacerbations in patients with uncontrolled persistent asthma irrespective of baseline eosinophil count (, and Supplementary Table 2).Citation54 Dupilumab is currently being studied in Phase 3 studies and the results are expected end of 2017 (Supplementary Table 2). Dupilumab is FDA-approved for atopic dermatitis and a proof-of-concept study in nasal polyposis was clearly positive.Citation55 Altogether, these results implicate an added benefit for treatment with dupilumab in patients with severe asthma and co-morbidities such as atopic dermatitis (e.g., in younger patients) or nasal polyposis (e.g., in late-onset asthmatics). Notably, three other approaches (a recombinant human IL-4 variant, a recombinant extracellular portion of the human IL-4Rα, and an antibody) targeting IL-4Rα were discontinued due to lack of efficacy ().
Interleukin-5
The importance of targeting the appropriate patient subsets was highlighted by the clinical development of treatments targeting IL-5. These treatments did not significantly improve lung function in an unselected population whilst they improved asthma control and reduced exacerbations in selected patients with severe asthma exhibiting an eosinophilic phenotype.Citation56,57 The importance of IL-5 in driving the persistent systemic and airway eosinophilic inflammation has been demonstrated by the efficacy of mepolizumab, a humanized monoclonal IgG1κ anti-IL-5 antibody approved by the FDA and EMA ( and ), to further decrease eosinophilic inflammation in those patients with refractory asthma despite high dose of inhaled or oral corticosteroids.Citation58,59 The clinical relevance of the persistent eosinophilic inflammation was demonstrated by the reduction in exacerbation rate and the improvement in quality of life observed in severe eosinophilic asthma patients receiving mepolizumab, even if only a modest effect was observed on FEV1.Citation58 The DREAM study, a large intravenous mepolizumab trial in patients with severe asthma receiving high-dose ICS/LABA treatment with evidence of eosinophilic/type 2-high inflammation (≥1 in the prior year: sputum eosinophils >3%, >300 eosinophils per µL peripheral blood, or FENO >50 ppb) identified blood eosinophil counts of 300 cells per µL or greater as a highly predictive biomarker of treatment response (Supplementary Table 2).Citation56 Three different doses of mepolizumab (75, 250 or 750 mg at 4-week intervals) were equally effective in decreasing clinically significant asthma exacerbations compared with placebo, with the greatest reductions seen in those with the highest blood eosinophil counts and greatest prior exacerbation history. No effect on other asthma outcomes were observed, including symptoms and FEV1 due to impressive placebo effects on patient-reported outcomes.Citation56 Similarly, in the MENSA trial, a large Phase 3 study of patients with eosinophilic asthma (based on peripheral blood eosinophilia) with recurrent exacerbations despite high-dose ICS (monthly 75 mg-intravenous or 100 mg-subcutaneous), mepolizumab significantly decreased exacerbations by 47% to 53%, increased FEV1 and modestly affected symptoms and asthma control scores compared with placebo (Supplementary Table 2).Citation60
Like mepolizumab, reslizumab is another humanized monoclonal IgG4κ anti-IL-5 antibody approved by the FDA and EMA ( and ). Two recent duplicate trials compared reslizumab (3 mg/kg dose, intravenous administration) to placebo in patients with poorly controlled asthma using medium-to-high doses ICS, a blood eosinophil count of ≥400 cells per µL, and at least one severe asthma exacerbation in the previous year.Citation61 In both Phase 3 studies, patients receiving reslizumab had a significant reduction in the frequency of asthma exacerbations compared with those receiving placeboCitation61 (Supplementary Table 2), reinforcing the role of eosinophils in several asthma outcomes.
Interleukin-5 receptor alpha
Benralizumab, a humanized afucosylated monoclonal IgG1κ antibody that binds to IL-5Rα, blocks IL-5 receptor signalling and induces antibody-directed cell-mediated cytotoxicity leading to depletion of IL-5Rα-expressing target cells (eosinophils and basophils, and ).Citation62 In the recent Phase 3 SIROCCO trial, the results confirm the efficacy and safety of benralizumab for patients with severe asthma and elevated blood eosinophils, which are uncontrolled by high-dosage ICS plus LABA, and provide support for benralizumab as an additional option to treat this disease in this patient population (Supplementary Table 2).Citation63 In another recent Phase 3 study (CALIMA trial, Supplementary Table 2), benralizumab significantly reduced annual exacerbation rates and was generally well tolerated in patients with severe, uncontrolled asthma with blood eosinophils 300 cells per μL or greater.Citation64
Overall, approaches targeting the IL-5 pathway have been efficacious, with prominent effects on exacerbations in adult patients with uncontrolled severe eosinophilic asthma. Nevertheless, it remains unclear whether approaches to block the receptor IL-5Rα and opsonize eosinophils, will differ in efficacy, safety or target endotype from those targeting the ligand IL-5.Citation43 However, the optimal dosing interval between injections appears to be different and advantageous for benralizumab (8 weeks) versus for mepolizumab and reslizumab (4 weeks) (Supplementary Table 2).
Limitations of monotherapies: emergence of combination therapies for severe asthma?
Translation of discoveries into treatments is challenging, time-consuming and expensive as illustrated by the 10-year gap between the development of omalizumab, the first biologic for the treatment of severe allergic asthma, and its approval by FDA in 2003.Citation12 Generally, only about 1 in 10 therapeutic candidates that enter Phase 1 trials becomes a marketed product, and this high failure rate contributes substantially to the high costs of drug development.Citation65
Implicit in this observation is the fact that preclinical models do not easily or perfectly simulate the vast heterogeneity of the human disease, and that correlation of pathophysiology with clinically measurable and meaningful outcomes such as symptoms, asthma control and exacerbations, is difficult.Citation66 Indeed, although the testing of biologics might begin in animal models, the immunologic response in human is more complex and heterogeneous, encompassing multiple endotypes and regulated by distinct biological pathways, implicating that asthma is a syndrome rather than a single disease (Supplementary Figure 1).Citation32,67 Moreover, it is known that endotypes may somewhat vary over timeCitation29,68 and also overlap, because there is currently no clear demarcation between these groupings. Therefore, patients may exhibit clinical or pathologic features of multiple groups, emphasizing the limitations in the current understanding of endotypes and the difficulty to use them routinely in clinical practice at this stage.Citation13 Consequently, the concept of treating severe asthma by targeting a single molecule has had limited success.
ICS, which is currently the mainstay therapy, is thought to act by suppressing a range of pro-inflammatory pathways.Citation69,70 However, long-term use of high-dose ICS therapy has potential to cause systemic side effects, such as impaired growth in children, decreased bone mineral density, skin thinning and bruising, and cataracts.Citation71 The emergence of the heterogeneity and the different endotypes of the disease processes where many different inflammatory mediators, cytokines and cells are dysregulated further highlights that new approaches are required to treat asthma effectively. Many studies show that airway inflammation, remodelling and airway hyperresponsiveness are dissociated and may be mediated by different mechanisms under different conditions.Citation72 Thus, combination therapies, targeting reciprocally regulated inflammatory and potentiating pathways in asthma, may be a more effective therapeutic approach for severe asthma.
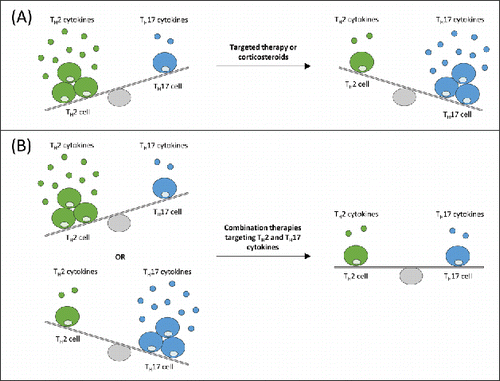
Interestingly, in a cross-sectional study of asthmatics of varying severity (n = 51), gene expression of endobronchial tissue analysis revealed three major patient clusters: type 2, type 17, and type 2/type 17-low.Citation73 Type 2 and type 17 patterns were mutually exclusive in individual patient samples, and their gene signatures were inversely correlated and differentially regulated by IL-13 and IL-17A. The dichotomous pattern of TH2 and TH17 signatures was explored in a preclinical model of allergen-induced asthma, which showed that type 2 cytokine suppression promoted TH17 responses. Neutralization of IL-4 or IL-13 resulted in increased TH17 cells and neutrophilic inflammation in the lung. However, neutralization of IL-13 and IL-17 protected mice from eosinophilia, mucus hyperplasia, airway hyperreactivity and abolished the neutrophilic inflammation, which suggests that combination therapies targeting both pathways may maximize therapeutic efficacy across a patient population comprising both type 2 and type 17 endotypes ().Citation73
Figure 2. Simplified schematic representation of targeted TH2 and TH17 cytokines therapies that can lead to amplification of activity of the opposing pathway in a murine house dust mite model of asthma.Citation73 (A) With suppression of TH2 activity by targeted therapy or corticosteroids, a TH17-permissive environment exists. A direct relationship between TH17 and TH2 disease exists, whereby, through mutual cross regulation, TH17 asthma may represent a transition or switch away from TH2-mediated disease. Thus, by treating TH2 patients with corticosteroids, TH17 asthma may have been promoted. (B) Combination therapy targeting TH2 cytokine, such as interleukin-13, and TH17 cytokine, such as interleukin-17, in patients expressing either a TH2 or TH17 signature may provide additional efficacy over single TH2 or TH17 inhibition. TH2: T-helper type 2 cell; TH17: T-helper type 17 cell.
Conclusion
The treatment of severe asthma in both adults and children still relies heavily on the use of high-dose ICS plus a second controller medication (such as LABAs or leukotriene receptor antagonists), or systemic steroids.Citation4 However, responses to these treatments can vary and do not modify the course of the disease, requiring an urgent need for new and more effective drugs to prevent the occurrence of potentially life-threatening episodes.
The addition of omalizumab as the first targeted biologic approved for asthma treatment has led to renewed optimism of improvements in outcomes in patients with type 2 severe asthma. Biologic approaches targeting type 2 inflammation have since emerged as promising new personalized medicines in patients with evidence of type 2 inflammation based on specific biomarker profiles ( and ). Indeed, whereas the monoclonal anti-IL-5 antibodies mepolizumab and reslizumab were not beneficial in unselected adult patients with moderate asthma, they decreased asthma exacerbations and improved symptoms and lung function in severe asthma patients with persistent sputum eosinophil counts or increased blood eosinophil levels (Supplementary Table 2). The first clinical trials using type 2-targeting biologics have highlighted the importance of determining the optimal biomarkers necessary to identify and understand which endotypes are responsive to specific biologics to identify which patients will benefit from which biologics at which optimal dose (Supplementary Table 1), thereby allowing a better prediction of responses to biologics. Progress in this field will not only allow better diagnosis and targeted treatment, but will also provide feedback on the fundamental research questions that need to be addressed. Interestingly, there is a potential for other biologics to provide benefit in treatment of severe asthma, such as anti-TSLP or anti-IL-33 monoclonal antibodies,Citation4 especially if the key elements for successful biologic development are applied (Supplementary Table 1), such as the identification of the endotypes who will respond to these biologics identified through biomarkers (Supplementary Figure 1).
Nevertheless, predicting response to therapy remains problematic. Because the results of the clinical trials summarized in Supplementary Table 2 varied even for biologics directed toward the same signalling pathway, it is appreciated that the response to a biologic can be confounded by multiple factors, including treatment duration, dose, asthma severity and endotype and differing outcome measures assessed.Citation43 Moreover, much remains to be understood regarding their long-term efficacy and safety, their comparative therapeutic efficacy, and, finally, their cost-effectiveness. Additionally, with only omalizumab approved for children of six to 11 years of age (), accurate biomarkers to identify responders are still currently lacking and cost-utility analyses need to be investigated for pediatric studies. Otherwise, development of biologics in severe asthma patients lacking type 2 biomarkers remains in its infancy and will require greatly improved molecular understanding of their underlying pathologies.
It is known that endotypes may somewhat vary over timeCitation29,68 and also overlap, because there is currently no clear demarcation between these groupings. Consequently, the concept of treating severe asthma by targeting a single molecule has been successful only in highly selected patient subgroups. Combination therapies, targeting reciprocally regulated inflammatory and potentiating pathways in asthma, may be a more effective therapeutic approach for a broader population of patients with severe asthma.
Disclosure of potential conflicts of interest
C.B and H.D.H. are employees at argenx. G.B. has, within the last 5 years, received honoraria for lectures from AstraZeneca, Boehringer-Ingelheim, Chiesi, GlaxoSmithKline, Novartis, Pfizer, Teva, UCB and Zambon; he is a member of advisory boards for AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Novartis, Sanofi/Regeneron and Teva. The other authors disclosed no potential conflicts of interest.
Author contributions
All authors have contributed to the development and writing of the manuscript, and approved the final version.
SUPPLEMENTARY_MATERIAL.docx
Download MS Word (119.5 KB)Funding
This work was supported by the Agentschap voor Innovatie door Wetenschap en Technologie (IWT, grant #IWT130849).
Related Research Data
References
- Reddel HK, Bateman ED, Becker A, Boulet LP, Cruz AA, Drazen JM, Haahtela T, Hurd SS, Inoue H, de Jongste JC, et al. A summary of the new GINA strategy: a roadmap to asthma control. Eur Respir J. 2015;46:622–39. doi:10.1183/13993003.00853-2015. PMID:26206872.
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, Adcock IM, Bateman ED, Bel EH, Bleecker ER, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2013;43(2):343–73. doi:10.1183/09031936.00202013. PMID:24337046.
- Ray A, Oriss TB, Wenzel SE. Emerging molecular phenotypes of asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308:L130–L40. doi:10.1152/ajplung.00070.2014. PMID:25326577.
- Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56. doi:10.1038/ni.3049. PMID:25521684.
- Kumar R, Singh J. Biosimilar drugs: current status. Int J Appl Basic Med Res. 2014;4:63–6. doi:10.4103/2229-516X.136774. PMID:25143877.
- Braido F. Failure in asthma control: reasons and consequences. Scientifica. 2013;2013:549252. doi:10.1155/2013/549252. PMID:24455432.
- Sullivan PW, Ghushchyan VH, Slejko JF, Belozeroff V, Globe DR, Lin SL. The burden of adult asthma in the United States: evidence from the Medical Expenditure Panel Survey. J Allergy Clin Immunol. 2011;127:363–9. doi:10.1016/j.jaci.2010.10.042. PMID:21281868.
- Chipps BE, Peters SP. Biologic Therapies of Immunologic Diseases. Immunol Allergy Clin North Am. 2017;37:xvii–xviii. doi:10.1016/j.iac.2017.02.001. PMID:28366489.
- Arron JR, Townsend MJ, Keir ME, Yaspan BL, Chan AC. Stratified medicine in inflammatory disorders: from theory to practice. Clin Immunol. 2015;161:11–22. doi:10.1016/j.clim.2015.04.006. PMID:25934386.
- Colburn W, DeGruttola VG, DeMets DL, Downing GJ, Hoth DF, Oates JA, et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Biomarkers Definitions Working Group. Clinical Pharmacology & Therapeutics 2001; 69:89–95. doi:10.1067/mcp.2001.113989. PMID:11240971
- de Roos E, Braunstahl GJ, Lahousse L, Brusselle G. Targeted Therapy for Older Patients with Uncontrolled Severe Asthma: Current and Future Prospects. Drugs & Aging. 2016;33:619–28. doi:10.1007/s40266-016-0397-7.
- Doyle R. Drug Development and Biologics in Asthma. A New Era. Annals of the American Thoracic Society 2016; 13:S83–S4. doi:10.1513/AnnalsATS.201509-573MG. PMID:27027958
- Gauthier M, Ray A, Wenzel SE. Evolving concepts of asthma. Am J Respir Crit Care Med. 2015;192:660–8. doi:10.1164/rccm.201504-0763PP. PMID:26161792.
- Zissler U, Esser‐von Bieren J, Jakwerth C, Chaker A, Schmidt‐Weber C. Current and future biomarkers in allergic asthma. Allergy. 2016;71(4):475–94. doi:10.1111/all.12828. PMID:26706728.
- Bayes HK, Cowan DC. Biomarkers and asthma management: an update. Curr Opin Allergy Clin Immunol. 2016;16:210–7. doi:10.1097/ACI.0000000000000263. PMID:27057795.
- Staton TL, Choy DF, Arron JR. Biomarkers in the clinical development of asthma therapies. Biomarkers 2016; 10:165–76. doi:10.2217/bmm.15.116. PMID:26764286
- Hanania NA, Noonan M, Corren J, Korenblat P, Zheng Y, Fischer SK, Cheu M, Putnam WS, Murray E, Scheerens H, et al. Lebrikizumab in moderate-to-severe asthma: pooled data from two randomised placebo-controlled studies. Thorax. 2015;70:748–56. doi:10.1136/thoraxjnl-2014-206719. PMID:26001563.
- Demarche S, Schleich F, Henket M, Paulus V, Van Hees T, Louis R. Detailed analysis of sputum and systemic inflammation in asthma phenotypes: are paucigranulocytic asthmatics really non-inflammatory? BMC Pulm Med. 2016;16:46. doi:10.1186/s12890-016-0208-2. PMID:27044366.
- Kauffman HF, Tamm M, Timmerman JAB, Borger P. House dust mite major allergens Der p 1 and Der p 5 activate human airway-derived epithelial cells by protease-dependent and protease-independent mechanisms. Clin Mol Allergy. 2006;4:5. doi:10.1186/1476-7961-4-5. PMID:16569217.
- Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454:445–54. doi:10.1038/nature07204. PMID:18650915.
- Gordon ED, Simpson LJ, Rios CL, Ringel L, Lachowicz-Scroggins ME, Peters MC, et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proceedings of the National Academy of Sciences 2016; 113:8765–70. doi:10.1073/pnas.1601914113. PMID:27432971
- Grünig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–3. doi:10.1126/science.282.5397.2261. PMID:9856950.
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi:10.1126/science.282.5397.2258. PMID:9856949.
- Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med. 2004;10:493–9. doi:10.1016/j.molmed.2004.08.004. PMID:15464449.
- Allakhverdi Z, Comeau MR, Jessup HK, Yoon BRP, Brewer A, Chartier S, Paquette N, Ziegler SF, Sarfati M, Delespesse G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–8. doi:10.1084/jem.20062211. PMID:17242164.
- Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, Gate RE, Haliburton GE, Ye CJ, Marson A, et al. A tissue checkpoint regulates type 2 immunity. Nature Immunol. 2016;17:1381–7. doi:10.1038/ni.3582. PMID:27749840.
- Guo L, Huang Y, Chen X, Hu-Li J, Urban JF Jr, Paul WE. Innate Immune Function of TH2 Cells in vivo. Nature Immunol. 2015;16:1051–9. doi:10.1038/ni.3244. PMID:26322482.
- Dweik RA, Sorkness RL, Wenzel S, Hammel J, Curran-Everett D, Comhair SA, Bleecker E, Busse W, Calhoun WJ, Castro M, et al. Use of exhaled nitric oxide measurement to identify a reactive, at-risk phenotype among patients with asthma. Am J Respir Crit Care Med. 2010;181:1033–41. doi:10.1164/rccm.200905-0695OC. PMID:20133930.
- McGrath KW, Icitovic N, Boushey HA, Lazarus SC, Sutherland ER, Chinchilli VM, Asthma Clinical Research Network of the National Heart, Lung, and Blood Institute. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med. 2012;185:612–9. doi:10.1164/rccm.201109-1640OC. PMID:22268133.
- Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2–driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–95. doi:10.1164/rccm.200903-0392OC. PMID:19483109.
- Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181:315–23. doi:10.1164/rccm.200906-0896OC. PMID:19892860.
- Wu W, Bleecker E, Moore W, Busse WW, Castro M, Chung KF, Calhoun WJ, Erzurum S, Gaston B, Israel E, et al. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J Allergy Clin Immunol. 2014;133:1280–8. doi:10.1016/j.jaci.2013.11.042. PMID:24589344.
- Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol. 2004;113:101–8. doi:10.1016/j.jaci.2003.10.041. PMID:14713914.
- Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, Garudathri J, Raymond D, Poynter ME, Bunn JY, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. Journal of Allergy and Clinical Immunology. 2011;128:508–15. doi:10.1016/j.jaci.2011.06.009. PMID:21782230.
- McKinley L, Alcorn JF, Peterson A, DuPont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–97. doi:10.4049/jimmunol.181.6.4089. PMID:18768865.
- Shaw DE, Berry MA, Hargadon B, McKenna S, Shelley MJ, Green RH, et al. Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. CHEST Journal 2007; 132:1871–5. doi:10.1378/chest.07-1047. PMID:17925424
- Manni ML, Trudeau JB, Scheller EV, Mandalapu S, Elloso MM, Kolls JK, Wenzel SE, Alcorn JF. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunology. 2014;7:1186–98. doi:10.1038/mi.2014.8. PMID:24549277.
- Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial–mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc. 2004;1:93–8. doi:10.1513/pats.2306034. PMID:16113419.
- Belliveau PP. Omalizumab: a monoclonal anti-IgE antibody. Med Gen Med. 2005;7:27. PMID:16369332.
- D'Amato G, Liccardi G, Noschese P, Salzillo A, D'Amato M, Cazzola M. Anti-IgE monoclonal antibody (omalizumab) in the treatment of atopic asthma and allergic respiratory diseases. Current Drug Targets-Inflammation & Allergy 2004; 3:227–9. doi:10.2174/1568010043343615. PMID:15379589
- Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fc [gamma] receptors in dendritic cells and macrophages. Nat Rev Immunol. 2014;14:94–108. doi:10.1038/nri3582. PMID:24445665.
- Wu AC, Paltiel AD, Kuntz KM, Weiss ST, Fuhlbrigge AL. Cost-effectiveness of omalizumab in adults with severe asthma: results from the Asthma Policy Model. J Allergy Clin Immunol. 2007;120:1146–52. doi:10.1016/j.jaci.2007.07.055. PMID:17904628.
- Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol. 2015;135:299–310. doi:10.1016/j.jaci.2014.12.1871. PMID:25662302.
- Hanania NA, Alpan O, Hamilos DL, Condemi JJ, Reyes-Rivera I, Zhu J, Rosen KE, Eisner MD, Wong DA, Busse W. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154:573–82. doi:10.7326/0003-4819-154-9-201105030-00002. PMID:21536936.
- Hanania NA, Wenzel S, Rosén K, Hsieh HJ, Mosesova S, Choy DF, Lal P, Arron JR, Harris JM, Busse W. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med. 2013;187:804–11. doi:10.1164/rccm.201208-1414OC. PMID:23471469.
- Ledford D, Busse W, Trzaskoma B, Omachi TA, Rosén K, Chipps BE, et al. A Randomized, Multicenter Study Evaluating Xolair® Persistency Of Response After Long-Term Therapy (XPORT). J Allergy Clin Immunol. 2016;140:162–9. doi:10.1016/j.jaci.2016.08.054. PMID:27826098.
- Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF. Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol. 2009;124:1210–6. doi:10.1016/j.jaci.2009.09.021. PMID:19910033.
- Ultsch M, Bevers J, Nakamura G, Vandlen R, Kelley RF, Wu LC, Eigenbrot C. Structural basis of signaling blockade by anti-IL-13 antibody lebrikizumab. J Mol Biol. 2013;425:1330–9. doi:10.1016/j.jmb.2013.01.024. PMID:23357170.
- Corren J, Lemanske RF Jr, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–98. doi:10.1056/NEJMoa1106469. PMID:21812663.
- Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, Yokoyama A, Olsson J, Gray S, Holweg CT, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med. 2016;4:781–96. doi:10.1016/S2213-2600(16)30265-X. PMID:27616196.
- Piper E, Brightling C, Niven R, Oh C, Faggioni R, Poon K, She D, Kell C, May RD, Geba GP, et al. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. 2013;41:330–8. doi:10.1183/09031936.00223411. PMID:22743678.
- Brightling CE, Chanez P, Leigh R, O'Byrne PM, Korn S, She D, May RD, Streicher K, Ranade K, Piper E. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3:692–701. doi:10.1016/S2213-2600(15)00197-6. PMID:26231288.
- Panettieri RA Jr, Brightling C, Sjobring U, Péterffy A, Tornling G, Daoud SZ, et al. STRATOS 1 and 2: considerations in clinical trial design for a fully human monoclonal antibody in severe asthma. J Clin Invest. 2015;5:701–11. doi:10.4155/cli.15.38.
- Wenzel S, Castro M, Corren J, Maspero J, Wang L, Zhang B, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β 2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. The Lancet 2016; 388:31–44. doi:10.1016/S0140-6736(16)30307-5. PMID:27130691
- Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, Gevaert P, Hellings P, Jiao L, Wang L, Evans RR, et al. Effect of subcutaneous dupilumab on nasal polyp burden in patients with chronic sinusitis and nasal polyposis: a randomized clinical trial. JAMA. 2016;315:469–79. doi:10.1001/jama.2015.19330. PMID:26836729.
- Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. The Lancet 2012; 380:651–9. doi:10.1016/S0140-6736(12)60988-X. PMID:22901886
- Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, Young J, Wilkins HJ, Henkel T, Nair P, Res-5-0010 Study Group. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184:1125–32. doi:10.1164/rccm.201103-0396OC. PMID:21852542.
- Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–84. doi:10.1056/NEJMoa0808991. PMID:19264686.
- Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O'Byrne PM. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–93. doi:10.1056/NEJMoa0805435. PMID:19264687.
- Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198–207. doi:10.1056/NEJMoa1403290. PMID:25199059.
- Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, Murphy K, Maspero JF, O'Brien C, Korn S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3:355–66. doi:10.1016/S2213-2600(15)00042-9. PMID:25736990.
- Kolbeck R, Kozhich A, Koike M, Peng L, Andersson CK, Damschroder MM, Reed JL, Woods R, Dall'acqua WW, Stephens GL, et al. MEDI-563, a humanized anti–IL-5 receptor α mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344–53. doi:10.1016/j.jaci.2010.04.004. PMID:20513525.
- Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β 2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. The Lancet 2016; 388:2115–27. doi:10.1016/S0140-6736(16)31324-1. PMID:27609408
- FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet 2016; 388:2128–41. doi:10.1016/S0140-6736(16)31322-8. PMID:27609406
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51. doi:10.1038/nbt.2786. PMID:24406927.
- Arron JR, Scheerens H, Matthews JG, David R. Redefining approaches to asthma: developing targeted biologic therapies. Adv Pharmacol. 2013;66:1–49. doi:10.1016/B978-0-12-404717-4.00001-9. PMID:23433454.
- Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–25. doi:10.1038/nm.2678. PMID:22561835.
- Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. 2006;11:54–61. doi:10.1111/j.1440-1843.2006.00784.x. PMID:16423202.
- Barnes P, Adcock I, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–63. doi:10.1183/09031936.05.00117504. PMID:15738302.
- Umland SP, Schleimer RP, Johnston SL. Review of the molecular and cellular mechanisms of action of glucocorticoids for use in asthma. Pulm Pharmacol Ther. 2002;15:35–50. doi:10.1006/pupt.2001.0312.
- Dahl R. Systemic side effects of inhaled corticosteroids in patients with asthma. Respir Med. 2006;100:1307–17. doi:10.1016/j.rmed.2005.11.020. PMID:16412623.
- Hansbro PM, Kaiko GE, Foster PS. Cytokine/anti‐cytokine therapy–novel treatments for asthma? Br J Pharmacol. 2011;163:81–95. doi:10.1111/j.1476-5381.2011.01219.x. PMID:21232048.
- Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, Jia G, Ohri CM, Doran E, Vannella KM, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7:301ra129. doi:10.1126/scitranslmed.aab3142. PMID:26290411.