
ABSTRACT
TL1A, a tumor necrosis factor-like cytokine, is a ligand for the death domain receptor DR3. TL1A, upon binding to DR3, can stimulate lymphocytes and trigger secretion of proinflammatory cytokines. Therefore, blockade of TL1A/DR3 interaction may be a potential therapeutic strategy for autoimmune and inflammatory diseases. Recently, the anti-TL1A monoclonal antibody 1 (mAb1) with a strong potency in blocking the TL1A/DR3 interaction was identified. Here, we report on the use of hydrogen/deuterium exchange mass spectrometry (HDX-MS) to obtain molecular-level details of mAb1′s binding epitope on TL1A. HDX coupled with electron-transfer dissociation MS provided residue-level epitope information. The HDX dataset, in combination with solvent accessible surface area (SASA) analysis and computational modeling, revealed a discontinuous epitope within the predicted interaction interface of TL1A and DR3. The epitope regions span a distance within the approximate size of the variable domains of mAb1′s heavy and light chains, indicating it uses a unique mechanism of action to block the TL1A/DR3 interaction.
Introduction
TL1A, a tumor necrosis factor (TNF) superfamily member, is a TNF-like homotrimeric cytokine that is expressed predominantly by endothelial cells and monocytes.Citation1 TL1A is upregulated by the proinflammatory cytokines TNF and interleukin (IL)-1 and by immune complexes (IC).Citation2 On activated T cells, TL1A functions specifically via its surface-bound receptor, death domain receptor DR3, to promote cell survival and secretion of proinflammatory cytokines, including interferon (IFN)-γ and granulocyte-macrophage colony-stimulating factor.Citation1,3,4 The action of TL1A can be blocked by its interaction with the secreted decoy receptor 3 (DcR3), a soluble protein of the TNF receptor superfamily.Citation5 In the presence of DcR3, TL1A/DR3- mediated cellular responses could be completely abrogated, indicating that the local level of soluble decoy receptor can affect the overall outcome of TL1A effects.Citation6 Recently, several DR3 mutants have been found to efficiently inhibit TL1A-induced cell death and secretion of IFN-γ.Citation7 Novel therapies that block TL1A/DR3 interaction may thus be efficacious for diverse inflammatory and immune diseases.
An IgG4 monoclonal antibody, mAb1, with high affinity binding (nM) to human TL1A was recently identified in house. A cell-based assay indicated that mAb1 functions as a DR3 blocker upon binding to TL1A, suggesting its unique epitope potentially overlaps the binding interface in TL1A/DR3 complex. These properties prompted us to conduct detailed epitope characterization of mAb1/TL1A interaction to better understand its unique mechanism of action.
Epitope mapping of an antibody can be carried out by different methods, including binding assessment of different segments of the antigen to the antibody via surface plasmon resonance (SPR) or enzyme-linked immunosorbent assay (ELISA).Citation8,9 The segments can be generated chemically or through yeast expression,Citation10 characterization of which can be done in a high-throughput manner, but the information provided by these methods is limited to the linear epitope. Site-directed mutagenesis coupled to a binding assay offers residue-level epitope information for both linear and conformational epitopes.Citation11 However, without the complementary information provided by other orthogonal approaches, mutagenesis can be laborious.
In this study, we used a mass spectrometry-based protein footprinting strategy, hydrogen/deuterium exchange mass spectrometry (HDX-MS), to obtain molecular details of the mAb1 binding epitope. HDX-MS is widely used for higher order structure characterization of protein therapeutics.Citation12–16 This approach provides comprehensive information of protein conformational dynamics throughout the amino acid sequence of the entire protein, except proline, due to its universal labeling property. Within the protein amide backbone, HDX rates depend on the local solvent accessibility and hydrogen bond network, and can be compared across conditions, including the presence or absence of antibodies in the antigens undergoing evaluation in epitope mapping experiments.Citation17–20 The deuterium content can be analyzed at different spatial resolution from intact protein to amide residues via electron-based MS gas phase fragmentation.Citation21–24 The HDX results for the epitope mapping of mAb1/TL1A, combined with solvent-accessible surface area (SASA) analysis of TL1A antigen, revealed two peptide regions in TL1A, which are composed mainly of the loop-type structures, as the primary mAb1 binding sites. HDX coupled with MS gas phase fragmentation electron-transfer dissociation (ETD) provided residue-level information. The epitope regions obtained by HDX were further evaluated by antibody-antigen docking studies. The combined results pinpoint the key mAb1 binding residues in the two distinct loop regions in TL1A, which are located in the potential DR3/TL1A binding interface and are within the approximate size of the variable domains of mAb1′s heavy and light chains. Our study demonstrates the unique mechanism of action of mAb1′s interaction with TL1A and blockade of DR3 binding either directly at the DR3 binding interface or through steric hindrance. It also demonstrates the practical utility of the combination approach of HDX-MS with computational analysis for the detailed molecular investigation of the conformational epitope of protein therapeutics.
Results
Conformational dynamics of TL1A
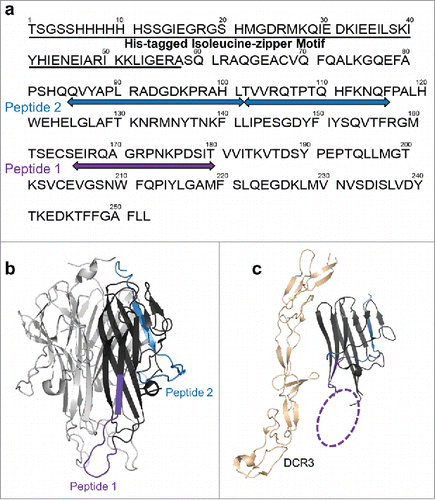
Each monomer of the trimeric human TL1A consists of 251 amino acids, including 35 residues in the cytoplasmic domain, 24 residues in the transmembrane region, and 192 residues in the extracellular domain. The crystal structure of the extracellular domain of TL1A homotrimer (PDB: 2RE9)Citation25 reveals a protein core that is composed of a β-sandwich structure. The hydrophobic core stabilizes the trimeric complex of TL1A. Prior to epitope mapping experiments, we applied HDX-MS to better understand the conformational dynamics of TL1A in solution. Using HDX, protein regional conformational dynamics in solution can be monitored by following the HDX kinetics, which provides a means to characterize the protein's conformational stability and can be used to rationalize the antibody binding mechanism upon the availability of the epitope information. The TL1A construct used in this study contains a His-tagged isoleucine-zipper motif in the N-terminal region that facilitates the trimer formation, and the presence of trimeric TL1A was confirmed by size-exclusion chromatography (SEC) (Figure S1).
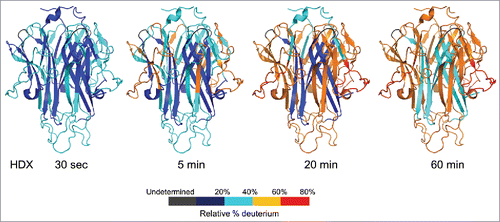
In the HDX-MS study of TL1A, exchange was followed into 59 peptides covering 97% of the TL1A sequence. The deuterium uptake was monitored for each peptic peptide across 30 sec, 5 min, 20 min, and 60 min exchange periods. displays the structural mapping of HDX data onto the extracellular domain of TL1A homotrimer crystal structure (PDB: 2RE9). The HDX kinetic data indicate that the core of TL1A's trimeric structure, which is part of the monomer's β-sandwich, is very solvent protective and structurally stable as it exhibited only ∼30% deuterium incorporation after 60 min exchange. The central region of this interface is composed predominantly of hydrophobic residues from each monomer positioned to contribute to the hydrophobic core of the trimer. The outer β-loop-β regions showed relatively faster exchange rates compared to the core region, indicating that the outer loop regions are solvent exposed, dynamic, or the hydrogen-bonding network is weak in these regions. In general, current HDX kinetics data on the conformational dynamics of TL1A's extracellular domain are consistent with its crystal structure and support the trimeric structure of TL1A under current HDX conditions. In addition, our HDX results suggest that the trimeric form of TL1A is structurally stable based on the overall slow conformational dynamics in the core regions.
Figure 1. HDX kinetics mapped onto TL1A trimer crystal structure (PDB code: 2RE9). The structure only represents the extracellular domain. Regions were colored from blue to red based on the extent of deuterium uptake (%).
HDX revealed discontinuous epitope of anti-TL1A mAb
HDX-MS affords molecular-level details of an antibody's epitope while preserving both the antigen's and antibody's native states in solution. In this study, HDX epitope mapping was conducted on mAb1, an IgG4 monoclonal antibody with high affinity binding to human TL1A that was identified in house. Based on the SPR analysis, mAb1 exhibits strong binding to human TL1A, with Kd measured at 0.38 nM (Figure S2). This strong affinity ensures stable TL1A/mAb1 complex formation under our HDX conditions.
In the HDX epitope mapping of mAb1, exchange was followed into 34 TL1A peptides covering 80% of the TL1A sequence and the deuterium uptake was monitored for each peptic peptide across 30 sec, 5 min, 20 min, 60 min, and 240 min exchange periods. The slight decrease of sequence coverage in the epitope mapping experiment compared to the antigen-only HDX is likely the outcome of the presence of mAb1, which introduced mAb1′s peptides and thus hampered identification of the TL1A peptides. HDX epitope mapping experiments were also conducted for mAb1′s antigen-binding fragment (Fab1) for epitope evaluation.
The differential HDX results of the TL1A and TL1A/mAb1 complex exhibited the same profile as the TL1A and TL1A/Fab1 complex ( and ), indicating that there was no potential non-specific interaction of mAb1′s Fc domain with TL1A in the TL1A/mAb1 HDX experiment. The HDX data revealed two regions (peptide 1, composed of residues 166–180; and peptide 2, composed of residues 85–101 and 102–116) as the potential binding epitope (, and ). Among them, peptide 1 showed a total >10 Da reduction across five HDX time points upon mAb1 binding, whereas peptide 2 showed a lower extent of HDX reduction. The level of HDX protection in peptide region 1 was greater in the TL1A/mAb1 complex than in the TL1A/Fab1 complex, which was likely caused by the larger size of mAb1 as compared to Fab1 that greatly reduced the solvent accessibility of this region upon binding. Although overlapping peptic peptides in region 116–127 demonstrated HDX reduction upon mAb1 binding, the levels were low and not significant, with a total < 2 Da reduction across five HDX time points upon Fab1 binding. Therefore, region 116–127 was not considered part of the epitope. Both peptide 1 and peptide 2 regions exhibited stable solvent protection or hydrogen bond formation across sec to 4 hr periods based on the HDX kinetics (), in which at least one amide hydrogen in regions 85–101 and 102–116 each was occluded from the solvent in the presence of mAb1, and at least two amide hydrogens in region 166–180 were affected by mAb1 binding. The HDX kinetics data are in accord with the SPR analysis of TL1A/mAb1 complex, which exhibited stable complex \formation based on the slow binding off rate (Figure S2). Both peptide regions are mainly composed of the long loop regions outside of TL1A (), which is not unexpected as the preferential sites for mAb1 binding, given that these loops are solvent exposed and dynamic based on HDX kinetics.
Figure 2. Differential HDX of TL1A upon (a) mAb1 binding and (b) Fab1 binding. Accumulated HDX reduction of peptic peptides across different exchange time periods: 30 sec (orange), 5 min (red), 20 min (light blue), 60 min (dark blue), and 240 min (black). The total levels of HDX reduction were color coded in three categories: D uptake difference 2.5–4.5 D (transparent green); D uptake difference 4.5–10 D (transparent yellow); D uptake difference >10 D (transparent red). (c) HDX kinetics curves of 85QVYAPLRADGDKPRAHL101, 102TVVRQTPTQHFKNQF116 and 166EIRQAGRPNKPDSIT180. TL1A upon mAb1 binding (blue) showed significant HDX reduction compared to TL1A alone (red). The non-deuterated peptide mass is shown in the upper left-hand corner of each plot.
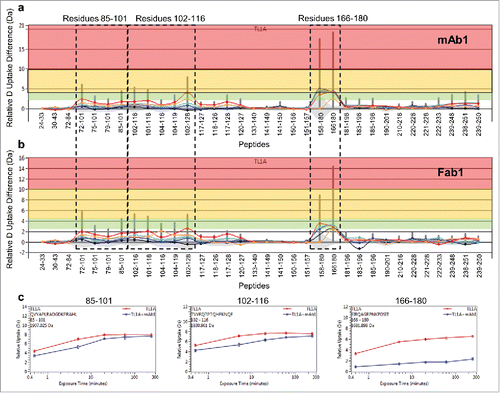
Figure 3. HDX revealed two major epitope regions of mAb1, peptide 1 and peptide 2. (a) Epitope mapped onto the TL1A protein sequence. The TL1A construct is composed of the His-tagged isoleucine-zipper motif followed by the extracellular domain; (b) TL1A trimer crystal structure (PDB code: 2RE9); and (c) TL1A/DcR3 crystal structure (PDB code: 3K51).
Epitope evaluation by SASA analysis and computational docking
HDX epitope mapping monitors antibody binding-induced solvent protection on the antigen's amide backbone hydrogens. Protein-protein interaction that involves solvent-exposed side chains with minimum protection on the amide hydrogens may not be fully captured by HDX. To obtain complementary information on the mAb1′s epitope, we calculated SASA to assess the solvent exposed residues in TL1A trimer. Studies have shown that selected residues located at protein surfaces can contribute significantly to protein–protein interaction energies,Citation26,27 in particular, aromatic amino acids (Tyr, Phe, Trp), basic amino acids (Arg, Lys), acidic amino acids (Asp, Glu) and polar amino acids (Gln, Asn, Ser, Thr). These residues are often important contributors to antigen/mAb interactions, and were considered functional residues in this study. We performed SASA analysis for all the residues in TL1A based on the TL1A crystal structure and calculated their percent side chain exposure (Table S1). Not surprisingly, among all the residues in TL1A, functional residues comprise a significant portion of amino acids in peptide regions 1 and 2. and contain the computational assessment of exposed amino acids corresponding to peptide regions 1 and 2, respectively. The tables show percent side chain exposure, which can be used to assess the functional residues in these peptide regions. Protein-protein interactions are stabilized by van der Waals dispersion forces, hydrophobic interactions, electrostatic interactions, and hydrogen bonds resulting from interactions between the side chains of the interacting proteins. For peptide region 1 (), it is important to note that E166, R168, Q169, R172, and K175 have functional amino acid side chains that are extremely exposed (50 – 97%).
Table 1. Computational assessment of exposed amino acids corresponding to peptide region 1. Residues that are solvent exposed and prone to protein-protein interactions are considered functional residues.
Table 2. Computational assessment of exposed amino acids corresponding to peptide region 2. Residues that are solvent exposed and prone to protein-protein interactions are considered functional residues.
For peptide region 2 (), the most exposed functional residues are T109, Q110. H111, F112, K113, N114, and Q115, with 62–93% solvent exposure, and these form the second bulk of the discontinuous epitope. Residues in region 85–101, within peptide 2, and region 116–127, which is adjacent to peptide 2, exhibited a lesser extent of solvent exposure compared to the loop region 102–116 based on both SASA analysis (Table S1) and the HDX kinetics dataset. In addition, region 85–101 comprises a β-sheet that is part of the trimer core and is not in the same interface with peptide 1 based on the X-ray crystal structure. The HDX protection observed in both regions upon mAb1 binding was likely due to the binding-induced conformational changes or hydrogen bond formation. The fast conformational dynamics of loop region 102–116 potentially contributed to this outcome.
The HDX epitope data of mAb1, when mapped on to the TL1A/DcR3 structure (PDB: 3K51),Citation28 illustrated that peptide 2 is structurally closer to the TL1A/DcR3 binding interface (). Although the structure of peptide 1 region was not resolved in the TL1A/DcR3 crystal structure, the flexible loop region is located in the same interface with peptide 2, and could be structurally close to the TL1A/DcR3 binding interface as well.
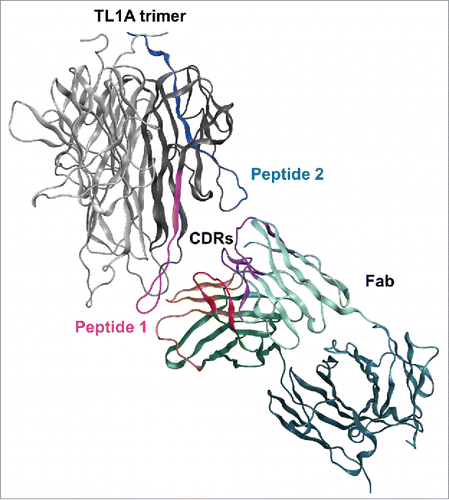
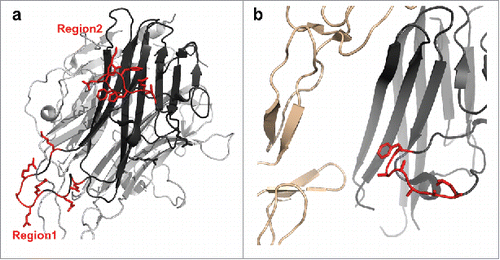
To further rationalize the mAb1 binding mechanism in TL1A, constrained protein-protein docking was conducted using constraints between the paratope of mAb1 and the proposed TL1A epitope comprising both peptide 1 and peptide 2. The binding epitope of mAb1, peptide regions 1 and 2, are about 35Å apart in the TL1A crystal structure. As exhibited in , TL1A's two peptide regions can contact complementarity-determining region (CDR) loops in VH and VL. The CDRs of mAbs span a similar distance of about 30–40 Å which is well within the approximate size of the variable domains of mAb1.
Figure 4. Molecular docking of TL1A with Fab. Epitope regions, peptide 1 and peptide 2, can contact CDR loops in VH and VL of Fab.
Epitope refinement by HDX-ETD
HDX-MS provides information about protein conformational dynamics at the peptide level,Citation12 the spatial resolution of which depends on the experimental conditions. In epitope mapping, further improvement of HDX-MS spatial resolution to the amide-level should allow one to narrow the potential regions that are involved in antibody-antigen interaction, which is extremely useful in understanding the detailed antibody binding mechanism and for the design of mutagenesis if one wants to modulate the antibody binding event.
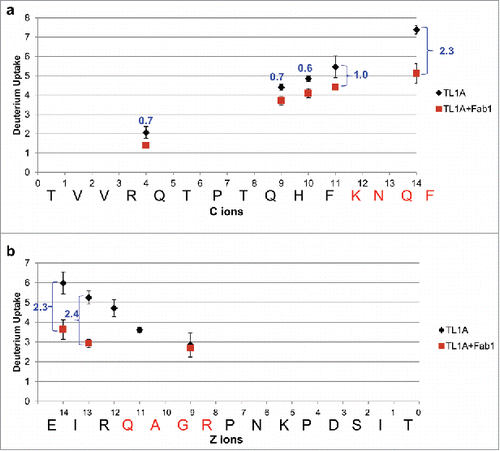
To refine the spatial resolution of mAb1, we conducted additional HDX-MS experiments coupled with ETD on the epitope peptide regions. ETD allows fragmentation of peptides in the gas phase with minimum deuterium scrambling as compared to the traditional MS-based fragmentation strategy, i.e., collisional-induced dissociation.Citation21 The level of deuterium scrambling was monitored and optimized using a synthetic peptide, HHHHHHIIKIIK, following the procedure described previouslyCitation21 (Figure S3). In the HDX-ETD epitope mapping, we used Fab1 instead of mAb1 to enhance signal-to-noise ratios in the experiment. HDX-ETD was conducted in a similar way as in the peptide-level HDX experiment, but the proteins were only exchanged for 5 min, a time point that exhibited a sufficient level of deuterium uptake in TL1A and significant HDX reduction upon Fab1 binding. The triply charged ions of the major epitope regions, 102TVVRQTPTQHFKNQF116 and 166EIRQAGRPNKPDSIT180, were then isolated and fragmented by ETD in the gas phase.
displays the deuterium uptake plots of ETD fragments for 102TVVRQTPTQHFKNQF116 and 166EIRQAGRPNKPDSIT180. ETD on 102TVVRQTPTQHFKNQF116 produced C-type fragment ions, whereas Z-type ions were observed for 166EIRQAGRPNKPDSIT180. The deuterium uptake for the Cn fragment ion of peptide region 102–116 represents the deuterium uptake in the region of its N-terminus to (n+1)th amino acid, whereas the deuterium uptake for the Zn fragment ion of peptide 166–180 represents the deuterium uptake in the region of its C-terminus to the (n-1)th amino acid. The gaps in sequence coverage were caused by the weak fragment ions signals. It is worth noting that the ETD resolution depends on the intensity and quality of the fragment ions, which could be compromised upon the optimization of ionization conditions to minimize the deuterium scrambling. For peptide region 102–116 (), although C9 and C10 showed around 0.7 Da HDX reduction with good precision upon Fab1 binding, the information regarding the deuterium uptake from C5 to C8 ions was missing. Therefore, the deuterium uptake difference of 0.7 Da could be contributed by any residue or all residues in 106QTPTQH111, which is not significant. Residues 113KNQF116, on the other hand, exhibited a total of 1.7 Da HDX reduction, ΔD of C14 – ΔD of C10, upon Fab1 binding. Residues in this region are thus likely the major contact residues in peptide region 102–116 for Fab1 binding. For peptide region 166–180 (), no significant HDX reduction was observed on Z9 ion, indicating that residues in 173PNKPDSIT180 were not affected upon Fab1 binding. The HDX reduction observed on Z13 and Z14 ions indicates that 169QAGR172 could be the major contact residues in peptide region 166–180 for mAb1 binding. Interestingly, there is good correlation between the data from HDX-ETD spatial epitope and SASA analysis. For peptide region 1, the most exposed residue Q169 is within epitope 169QAGR172. For peptide region 2, K113, N114, and Q115 within epitope 113KNQF116 were all identified as functional residues, with N114 being the most exposed residue. Our results demonstrate the correlation of HDX experiments, three-dimensional structure, and computational analysis.
Figure 5. Deuterium uptake plots of ETD fragment ions between TL1A alone (black) and TL1A/Fab1 (dark red). (a) 102TVVRQTPTQHFKNQF116 and (b) 166EIRQAGRPNKPDSIT180. HDX-ETD was conducted in triplicate and the average deuterium uptake of each fragment ion was reported with standard deviation. The level of Fab1 binding-induced HDX reduction on each fragment ion was labeled in blue.
Discussion
Epitope mapping is important to the development of new therapeutic antibodies, and is crucial for the protection of intellectual property in biopharmaceutical industries. We present here the application of HDX-MS together with computation modeling for the epitope mapping of an IgG4 anti-TL1A mAb, mAb1, which exhibited strong potency in blocking the TL1A/DR3 interaction. TL1A is a homotrimer with an average molecular weight of 90 kDa. Considering a stoichiometry of one antibody binding to each monomeric TL1A, the addition of antibodies to TL1A results in a ∼550 kDa protein complex that could require significant efforts for high resolution epitope characterization via X-ray crystallography or nuclear magnetic resonance (NMR) analysis. In addition, epitope mapping of anti-TL1A antibodies using protein fragment-based screening strategies could provide incomplete information due to potential conformational epitopes of these antibodies in this well-structured antigen. HDX-MS, on the other hand, allows conformational characterization of native protein in solution. Although structural resolution is low in the HDX-MS epitope mapping experiment, it affords faster throughput compared to X-ray crystallography or NMR analysis. These properties make HDX a suitable technique in epitope mapping of complex protein systems such as TL1A.
TL1A contains a predicted hydrophobic transmembrane region near the N-terminus and an extracellular carboxyl domain that can be cleaved from the cell membrane by proteases.Citation5 The X-ray crystal structure of the soluble extracellular domain of TL1A reveals that each monomer forms a jellyroll β-sandwich fold and is surrounded by multiple loops (, PDB: 2RE9).Citation25 The subunit interface of TL1A is formed by interactions between the edges of the β-sandwich in one monomer and the inner sheet of the neighboring monomer. Our HDX kinetics data indicated that the TL1A subunit interface is very solvent protected and conformationally stable, whereas the β sheets and loops located outside the protein core are relatively solvent exposed.
In the epitope mapping experiments of mAb1/TL1A, two regions including residues 102–116 and residues 166–180 in the two long loop regions of TL1A were identified as the potential binding epitope. The epitope assignment was based on the differential HDX experiments, the SASA analysis of the functional residues throughout the TL1A sequence, and the molecular docking of TL1A and Fab1. SEC analysis of the TL1A/Fab1 complex (Figure S1) revealed a 3:3 binding stoichiometry, i.e., one Fab1 binds to one TL1A monomer. Based on the binding stoichiometry, one could also propose an alternative binding mechanism in which mAb1 bound to residues 166–180, causing significant HDX reduction in that region, and induced an allosteric effect on residues 102–116. However, residues 166–180 in each of the TL1A monomer are about 15Å apart in the TL1A crystal structure, which is too close for binding with three Fab1s. Therefore, both regions 102–116 and 166–180 should be directly involved in mAb1 binding.
In the HDX-MS methodology, the analysis of overlapping peptides released from multiple proteolytic digestions can provide residue-level information.Citation29–31 However, different peptides could have different propensities for interacting with reversed-phase liquid chromatography columns, leading to different levels of deuterium back-exchange that makes deuterium level subtraction problematic.Citation32 Therefore, we applied HDX-ETD on selected peptide regions, i.e., residues 102–116 and 166–180, for epitope refinement. HDX-ETD results suggest that the amide backbone hydrogens in 113KNQF116 and 169QAGR172 were stabilized either by solvent protection or the changes of hydrogen bonding network upon Fab1 binding to the solvent-exposed functional residues' side chains in the two loop regions as revealed by SASA analysis. Taken together the computational analysis and HDX-ETD data define a discontinuous epitope on the surface of TL1A that is in contact with mAb1 (). In addition, these residues are structurally close to the TL1A/DcR3 binding interface (). An orthogonal approach such as mutagenesis in these epitope regions could further pinpoint the key epitope residues and provide additional insight into the mAb binding mechanism.
Figure 6. Residues in TL1A that are involved in mAb1 binding as determined by both HDX-ETD and computational modeling. (a) All residues are solvent exposed as shown in the TL1A monomer of its trimer crystal structure (PDB code: 2RE9). (b) A zoom-in view of the binding residues in peptide region 2 of TL1A in the TL1A/DcR3 crystal structure (PDB code: 3K51).
Based on the flow cytometry analyses, mAb1 efficiently inhibits TL1A binding to human DR3 Chinese hamster ovary (CHO) cells, with an IC50 of 0.524 nM (Figure S4). It is well established that DcR3 binding to TL1A abrogates the TL1A/DR3-mediated cellular responses.Citation6 Thus, DcR3 and DR3 could share similar binding interfaces in TL1A. The combined results indicate a unique mechanism of mAb1 interaction with the flexible outer loops of TL1A, which creates the steric hindrance to block the DR3 binding. In addition, our study demonstrates the practical utility of HDX-MS methodology in combination with orthogonal computational analysis for detailed investigation of conformational epitopes of therapeutics.
Materials and methods
Materials
Trimeric human TL1A and anti-hTL1A IgG4 mAb, mAb1, were expressed from CHO cells in house. The TL1A construct contains a His-tagged isoleucine-zipper motif in the N-terminal region that facilitates the trimer formation.Citation33 N-glycan sites were identified at N135 and N231. Trimetric TL1A was purified by Ni-affinity chromatography, buffer exchanged into phosphate-buffered saline (PBS), and analyzed by SEC (Figure S1). The concentrations of both hTL1A and mAb1 were determined by UV 280 nm absorbance. Dipotassium hydrogen phosphate, monopotassium phosphate, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP.HCl) and deuterium oxide were purchased from Sigma-Aldrich (St. Louis, MO). P1 synthetic peptide, HHHHHHIIKIIK, was from Anaspec (Fremont, CA).
Hydrogen-deuterium exchange
Prior to HDX experiments, non-deuterated experiments were performed to generate a list of common peptic peptides for the recombinant TL1A trimer (4 µM) and protein complex of TL1A trimer with mAb1 or TL1A trimer with mAb1′s Fab (1:3 molar ratio). The stoichiometry of one TL1A trimer binding to three Fabs was characterized by SEC (Figure S3). In the HDX-MS experiment, 5 µL of each sample (TL1A or TL1A with mAb/Fab) was diluted into 55 µL of D2O buffer (10 mM phosphate buffer, D2O, pD 7.0, pD = pH + 0.4) to start the labeling reactions. The reactions were carried out for different periods of time: 30 sec, 5 min, 20 min, 60 min and 240 min. By the end of each labeling reaction period, the reaction was quenched by adding quenching buffer (100 mM phosphate buffer with 4 M GdnCl and 0.4 M TCEP, pH 2.5, 1:1, v/v) and 50 µL of quenched sample was injected into Waters nanoACQUITY UPLC HDX Manager™.Citation34
Chromatography and mass spectrometry
For peptide-level HDX, the protein samples were digested online using a Poroszyme™ immobilized pepsin cartridge, 2.1 mm x 30 mm (Thermo Fisher, San Jose, CA) at 20 °C and for 3 min. The deuterated peptides were trapped on an ACQUITY UPLC BEH C18 VanGuard Pre-column, 130Å, 1.7 µm, 2.1 mm x 5 mm and desalted for 3 min at 100 µL/min using 0.1% formic acid in water. The deuterated peptides were eluted from the trapping and separation columns, 1.0 × 50.0 mm ACQUITY UPLC C18 BEH (Waters Corp.) in 7.5 min by an 8% – 85% acetonitrile:water gradient containing 0.1% formic acid at 65 µL/min. All the chromatographic elements were held at 0.0 ± 0.1 °C. Mass spectra were obtained with a Waters Synapt G2si Q-TOF equipped with standard ESI source (Waters Corp.). The instrument configuration was the following: capillary was 3.5 kV, sampling cone at 35 V, source temperature of 80 °C and desolvation temperature of 175 °C. Mass spectra were acquired over an m/z range of 260 to 2000. For HDX-ETD experiments, 1,3-dicyanobenzene as electron carrier was used for gas-phase fragmentation of selected peptic peptide with an isolation window of 6 (+/- 3 Da). A synthetic peptide, HHHHHHIIKIIK, was used for deuterium scrambling test as described previously by Jørgensen and co-workers.Citation21 To minimize the deuterium scrambling the sampling cone was set at 20 V in the HDX-ETD experiment. All experiments were performed in triplicate.
HDX data analysis
Peptic peptides identification was accomplished through a combination of exact mass analysis and MSE using ProteinLynx Global Server 3.0.2 (Waters Corp., Milford, MA, USA). Deuterium uptake was calculated and plotted versus the exchange time using Waters DynamX 3.0™ software. The deuterium uptake level was calculated based on the difference between the centroid of the isotopic distribution for peptide ions from undeuterated protein and the centroid of the isotopic distribution for peptide ions from the deuterium-labeled sample. The % deuterium uptake was calculated using the theoretical maximum number of exchangeable amide hydrogens, excluding the two N-terminal residues in each peptide, with the consideration of ∼92% D2O in each incubation. For HDX-ETD, the ETD fragment ions were assigned using ProteinProspector version 5.18.1 and MagTran version 1.03 (Amgen, Thousand Oaks, CA)Citation35 was used to obtain the centroid of the deuterium-containing isotopic distribution for each fragment ion. All assignments, deuterated spectra, and data processing were manually checked and verified.
Computational analysis
The SASA calculations, including side chain exposure, were performed using MOE (Molecular Operating Environment, 2016.0801; Chemical Computing Group, Montreal, QC, Canada). SASA is defined as the surface traced by the center of a sphere with the radius of a water molecule as it is rolled over the surface of a structure or molecular model using the solution first developed by Lee and Richards.Citation36 The side chain exposure of residue X is compared to the ideal surface area as determined from Gly-X-Gly tripeptide with the main chain in an extended conformation.Citation37 The exposure is then normalized by dividing the SASA by the maximum SASA for a given amino acid type as reported by Miller et al.Citation37 The side chain exposure is reported as a percentage where the side chain exposure = residue SASA/maximum SASA.
Antibody's fab homology modeling
The structure for the Fab domain of mAb1 was obtained by homology modeling using well-defined protocols of BioLuminate, version 1.9 (Schrödinger, LLC, New York, NY, USA), which includes refinement of the heavy chain CDR3. Antibody modeling begins with identification of suitable antibody framework templates derived from a database of structurally diverse and comprehensive antibody structures. Using the identified templates, the next step initiates the building of the antibody framework for the conserved “scaffold” regions of the antibody (regions excluding the antibody CDRs). The final part of model building is to generate conformations for the antibody CDR loops. For heavy chain CDR1 and CDR2 and light chain CDR1, CDR2 and CDR3, templates based on antibody structures that are available from the PDB are used.Citation38 Accurate models of heavy chain CDR3 require additional conformation sampling. We used PRIME loop modeling refinement as implemented in BioLuminate.Citation39 Following model-building, refinement of the model was performed using the protein preparation workflow within the Schrödinger software MAESTRO, version 10.2 (Schrödinger, LLC, New York, NY, USA).
Fab-antigen protein-protein docking
The modeled antibody's Fab was docked onto the TL1A trimer taken from the crystal structure (PDB code: 3K51). Protein docking was carried out using MOE. The protein docking procedure consists of three major steps: 1) protein model representation, 2) sampling, and 3) scoring. Model representation includes simplifying the all-atom protein representation into a coarse-grain bead model. Each protein residue is replaced with 1 to 3 beads that encode both van der Waals parametersCitation40 and applied effective charge. The energy model is comparable to force field parameters and includes implicit solvation. The bead charges approximate the all-atom field, and are derived from the protein itself. The energy function consists of a Lennard-Jones 8–6 VdW term plus a Coulomb interaction. The final coarse-grained refinement also includes a bead-based GBVI implicit solvation term.Citation41 The sampling step requires global pose exploration to enumerate potential starting poses. For a given protein, a set of orientations are generated such that the movement of the surface atoms between nearest-neighbor rotations corresponds to the translational grid spacing. This is done by taking a uniform distribution of rotation axes and scaling the rotation angles by molecular radius. Fast Fourier Transform is then applied to a grid representation of each orientation to simultaneously evaluate the energy of all discrete translations. For each orientation, the top two translations are retained, and the top two orientations are retained for each translation. Additional refinement is carried out using truncated-Newton minimization of the continuous rigid-body coordinates (translations and rotation). The outcome of the starting point enumeration produces approximately 100,000 starting poses. The remaining protein poses post-sampling are then refined and scored using a full-atom potential (AMBER). The final scoring phase is computationally more expensive, and is therefore reserved for the top 200 poses. CDR restraints were applied by identifying exposed regions of the CDR loops and using an energy penalty to require that all poses contain a minimum number of residue contacts within these regions. The top poses were evaluated for surface complementarity based upon the complementarity score and visual inspection of surfaces as implemented in the protein_contact_surfaces script implemented in MOE.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
SUPP_MAT.zip
Download Zip (831.1 KB)Acknowledgments
The authors thank Drs. Adrienne Tymiak, Sharon Cload and Luisa Salter-Cid from Bristol-Myers Squibb Company for their support of this project. Drs. Yingru Zhang and Jun Dai from Bristol-Myers Squibb Company for technical assistance in SEC analysis. John Gunn from Chemical Computing Group, Montreal, QC, Canada for his assistance with protein docking protocols.
References
- Migone T-S, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, et al. TL1A Is a TNF-like Ligand for DR3 and TR6/DcR3 and Functions as a T Cell Costimulator. Immunity. 2002;16:479–92. doi:10.1016/S1074-7613(02)00283-2. PMID:11911831.
- Tang C-H, Hsu T-L, Lin W-W, Lai M-Z, Yang R-S, Hsieh S-L, Fu WM. Attenuation of Bone Mass and Increase of Osteoclast Formation in Decoy Receptor 3 Transgenic Mice. Journal of Biological Chemistry. 2007;282:2346–54. doi:10.1074/jbc.M603070200. PMID:17099218.
- Tougaard P, Zervides KA, Skov S, Hansen AK, Pedersen AE. Biologics beyond TNF-α inhibitors and the effect of targeting the homologues TL1A-DR3 pathway in chronic inflammatory disorders. Immunopharmacology and Immunotoxicology. 2016;38:29–38. doi:10.3109/08923973.2015.1130721. PMID:26810853.
- Facco M, Cabrelle A, Calabrese F, Teramo A, Cinetto F, Carraro S, Martini V, Calzetti F, Tamassia N, Cassatella MA, et al. TL1A/DR3 axis involvement in the inflammatory cytokine network during pulmonary sarcoidosis. Clinical and Molecular Allergy. 2015;13:16. doi:10.1186/s12948-015-0022-z. PMID:26240517.
- Kim S, Zhang L. Identification of naturally secreted soluble form of TL1A, a TNF-like cytokine. J Immunol Methods. 2005;298:1–8. doi:10.1016/j.jim.2004.12.019. PMID:15847792.
- Yang C-R, Hsieh S-L, Teng C-M, Ho F-M, Su W-L, Lin W-W. Soluble Decoy Receptor 3 Induces Angiogenesis by Neutralization of TL1A, a Cytokine Belonging to Tumor Necrosis Factor Superfamily and Exhibiting Angiostatic Action. Cancer Res. 2004;64:1122–9. doi:10.1158/0008-5472.CAN-03-0609. PMID:14871847.
- Levin I, Zaretsky M, Aharoni A. Directed evolution of a soluble human DR3 receptor for the inhibition of TL1A induced cytokine secretion. PLOS ONE. 2017;12:e0173460. doi:10.1371/journal.pone.0173460. PMID:28278297.
- Kashiwase H, Ishimura M, Ishikawa Y, Nishigaki T. Characterization of one monoclonal antibody against feline immunodeficiency virus p24 and its application to antigen capture ELISA. J Virol Methods. 1997;68:183–92. doi:10.1016/S0166-0934(97)00121-3. PMID:9389408.
- Kang M, Kim SY, An SSA, Ju YR. Characterizing affinity epitopes between prion protein and [beta]-amyloid using an epitope mapping immunoassay. Exp Mol Med. 2013;45:e34. doi:10.1038/emm.2013.63. PMID:23907583.
- Cho YK, Chen I, Wei X, Li L, Shusta EV. A yeast display immunoprecipitation method for efficient isolation and characterization of antigens. J Immunol Methods. 2009;341:117–26. doi:10.1016/j.jim.2008.11.005. PMID:19041873.
- Jin L, Fendly BM, Wells JA. High resolution functional analysis of antibody-antigen interactions. J Mol Biol 1992;226:851–65. doi:10.1016/0022-2836(92)90636-X. PMID:1380563.
- Huang RY, Chen G. Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry. Anal Bioanal Chem. 2014;406:6541–58. doi:10.1007/s00216-014-7924-3. PMID:24948090.
- Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discovery Today. 2014;19:95–102. doi:10.1016/j.drudis.2013.07.019. PMID:23928097.
- Pan LY, Salas-Solano O, Valliere-Douglass JF. Antibody Structural Integrity of Site-Specific Antibody-Drug Conjugates Investigated by Hydrogen/Deuterium Exchange Mass Spectrometry. Analytical Chemistry. 2015;87:5669–76. doi:10.1021/acs.analchem.5b00764. PMID:25938577.
- Zhang J, Banks DD, He F, Treuheit MJ, Becker GW. Effects of Sucrose and Benzyl Alcohol on GCSF Conformational Dynamics Revealed by Hydrogen Deuterium Exchange Mass Spectrometry. J Pharm Sci. 2015;104:1592–600. doi:10.1002/jps.24384. PMID:25693946.
- Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. mAbs. 2015;7:84–95. doi:10.4161/19420862.2014.985494. PMID:25524268.
- Prądzińska M, Behrendt I, Astorga-Wells J, Manoilov A, Zubarev RA, Kołodziejczyk AS, Rodziewicz-Motowidło S, Czaplewska P. Application of amide hydrogen/deuterium exchange mass spectrometry for epitope mapping in human cystatin C. Amino Acids. 2016;48:2809–20. doi:10.1007/s00726-016-2316-y. PMID:27573935.
- Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY, et al. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Analytical Chemistry. 2017;89:2250–8. doi:10.1021/acs.analchem.6b03058. PMID:28193005.
- Malito E, Faleri A, Lo Surdo P, Veggi D, Maruggi G, Grassi E, Cartocci E, Bertoldi I, Genovese A, Santini L, et al. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proceedings of the National Academy of Sciences. 2013;110:3304–9. doi:10.1073/pnas.1222845110.
- Malito E, Biancucci M, Faleri A, Ferlenghi I, Scarselli M, Maruggi G, Lo Surdo P, Veggi D, Liguori A, Santini L, et al. Structure of the meningococcal vaccine antigen NadA and epitope mapping of a bactericidal antibody. Proceedings of the National Academy of Sciences. 2014;111:17128–33. doi:10.1073/pnas.1419686111.
- Zehl M, Rand KD, Jensen ON, Jørgensen TJD. Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J Am Chem Soc. 2008;130:17453–9. doi:10.1021/ja805573h. PMID:19035774.
- Rand KD, Zehl M, Jensen ON, Jørgensen TJD. Protein Hydrogen Exchange Measured at Single-Residue Resolution by Electron Transfer Dissociation Mass Spectrometry. Analytical Chemistry. 2009;81:5577–84. doi:10.1021/ac9008447. PMID:19601649.
- Huang RYC, Garai K, Frieden C, Gross ML. Hydrogen/Deuterium Exchange and Electron-Transfer Dissociation Mass Spectrometry Determine the Interface and Dynamics of Apolipoprotein E Oligomerization. Biochemistry. 2011;50:9273–82. doi:10.1021/bi2010027. PMID:21899263.
- Landgraf RR, Chalmers MJ, Griffin PR. Automated Hydrogen/Deuterium Exchange Electron Transfer Dissociation High Resolution Mass Spectrometry Measured at Single-Amide Resolution. Journal of The American Society for Mass Spectrometry. 2012;23:301–9. doi:10.1007/s13361-011-0298-2. PMID:22131230.
- Jin T, Guo F, Kim S, Howard A, Zhang Y-Z. X-ray crystal structure of TNF ligand family member TL1A at 2.1Å. Biochem Biophys Res Commun. 2007;364:1–6. doi:10.1016/j.bbrc.2007.09.097. PMID:17935696.
- Hoskins J, Lovell S, Blundell TL. An algorithm for predicting protein–protein interaction sites: Abnormally exposed amino acid residues and secondary structure elements. Protein Science. 2006;15:1017–29. doi:10.1110/ps.051589106. PMID:16641487.
- Xue LC, Dobbs D, Bonvin AMJJ, Honavar V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015;589:3516–26. doi:10.1016/j.febslet.2015.10.003. PMID:26460190.
- Zhan C, Patskovsky Y, Yan Q, Li Z, Ramagopal U, Cheng H, Brenowitz M, Hui X, Nathenson SG, Almo SC. Decoy Strategies: The Structure of TL1A:DcR3 Complex. Structure. 2011;19:162–71. doi:10.1016/j.str.2010.12.004. PMID:21300286.
- Mayne L, Kan Z-Y, Sevugan Chetty P, Ricciuti A, Walters B, Englander S. Many Overlapping Peptides for Protein Hydrogen Exchange Experiments by the Fragment Separation-Mass Spectrometry Method. Journal of The American Society for Mass Spectrometry. 2011;22:1898–905. doi:10.1007/s13361-011-0235-4. PMID:21952777.
- Kan Z-Y, Walters BT, Mayne L, Englander SW. Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis. Proceedings of the National Academy of Sciences. 2013;110:16438–43. doi:10.1073/pnas.1315532110..
- Hu W, Walters BT, Kan Z-Y, Mayne L, Rosen LE, Marqusee S, Englander SW. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proceedings of the National Academy of Sciences. 2013;110:7684–9. doi:10.1073/pnas.1305887110..
- Sheff J, Rey M, Schriemer D. Peptide–Column Interactions and Their Influence on Back Exchange Rates in Hydrogen/Deuterium Exchange-MS. J Am Soc Mass Spectrom. 2013;24:1006–15. doi:10.1007/s13361-013-0639-4. PMID:23649779.
- Hicks MR, Holberton DV, Kowalczyk C, Woolfson DN. Coiled-coil assembly by peptides with non-heptad sequence motifs. Fold Des. 1997;2:149–58. doi:10.1016/S1359-0278(97)00021-7. PMID:9218952.
- Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-Speed and High-Resolution UPLC Separation at Zero Degrees Celsius. Analytical Chemistry. 2008;80:6815–20. doi:10.1021/ac8008862. PMID:18672890.
- Zhang Z, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. Journal of the American Society for Mass Spectrometry 1998;9:225–33. doi:10.1016/S1044-0305(97)00284-5. PMID:9879360.
- Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55:379–IN4. doi:10.1016/0022-2836(71)90324-X. PMID:5551392.
- Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol. 1987;196:641–56. doi:10.1016/0022-2836(87)90038-6. PMID:3681970.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi:10.1093/nar/28.1.235. PMID:10592235.
- Zhu K, Day T. Ab initio structure prediction of the antibody hypervariable H3 loop. Proteins: Structure, Function, and Bioinformatics. 2013;81:1081–9. doi:10.1002/prot.24240..
- Basdevant N, Borgis D, Ha-Duong T. A Coarse-Grained Protein–Protein Potential Derived from an All-Atom Force Field. J Phys Chem B. 2007;111:9390–9. doi:10.1021/jp0727190. PMID:17616119.
- Labute P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem. 2008;29:1693–8. doi:10.1002/jcc.20933. PMID:18307169.