
ABSTRACT
Monoclonal antibody (mAb)-based blockade of programmed cell death 1 (PD-1) or its ligand to enable antitumor T-cell immunity has been successful in treating multiple tumors. However, the structural basis of the binding mechanisms of the mAbs and PD-1 and the effects of glycosylation of PD-1 on mAb interaction are not well understood. Here, we report the complex structure of PD-1 with toripalimab, a mAb that is approved by China National Medical Products Administration as a second-line treatment for melanoma and is under multiple Phase 1-Phase 3 clinical trials in both China and the US. Our analysis reveals that toripalimab mainly binds to the FG loop of PD-1 with an unconventionally long complementarity-determining region 3 loop of the heavy chain, which is distinct from the known binding epitopes of anti-PD-1 mAbs with structural evidences. The glycan modifications of PD-1 could be observed in three potential N-linked glycosylation sites, while no substantial influences were detected to the binding of toripalimab. These findings benefit our understanding of the binding mechanisms of toripalimab to PD-1 and shed light for future development of biologics targeting PD-1. Atomic coordinates have been deposited in the Protein Data Bank under accession code 6JBT.
Introduction
Monoclonal antibody (mAb)-based immune checkpoint therapy (ICT), which involves blocking immune checkpoint receptor-ligand interactions to restimulate antitumor T-cell immunity for tumor immunotherapy, has gain particular interest since the approval of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-targeting ipilimumab in 2011.Citation1,Citation2 As a critical inhibitory molecule in modulation of T-cell reactivity, programmed cell death 1 (PD-1) plays pivotal roles in immune suppression within the tumor microenvironment.Citation3–Citation6 The blockade of the interaction between PD-1 and its ligand, PD-L1, to interrupt the inhibitory signaling in T cells could release the preexisting antitumor T-cell activity to kill tumor cells.Citation7,Citation8 To date, seven immune checkpoint-blocking mAbs have been approved by US Food and Drug Administration (FDA), i.e. CTLA-4-targeting ipilimumab (2011, Bristol Myers-Squibb); PD-1-targeting nivolumab (2014, Bristol Myers-Squibb), pembrolizumab (2014, Merck & Co., Inc.) and cemiplimab (2018, Sanofi and Regeneron); and PD-L1-targeting atezolizumab (2017, Genentech), durvalumab (2017, AstraZeneca) and avelumab (2017, Merck KGaA and Pfizer).Citation1,Citation9 Clinical studies reveal that these mAbs not only induce durable tumor suppression in multiple tumors as monotherapy, but also play important roles in improving antitumor efficacy in combination with chemotherapy, targeted therapy, and other immunotherapies, e.g. chimeric antigenic receptor engineered T cells (CAR-T) or oncolytic virus.Citation9–Citation11
The structural basis of the binding of PD-1/PD-L1-blocking mAbs to their targets has been reported, which facilitates our understanding of the blocking mechanisms and differential binding characteristics of these mAbs.Citation12–Citation18 The binding of nivolumab is mainly located on an unexpected N-terminal loop of PD-1, which is out of the immunoglobulin (Ig)-like domain that contributes binding to PD-L1.Citation15 On the other side, pembrolizumab binds to the flexible C’D loop of PD-1, which is also not involved in the interaction with PD-L1.Citation16 Although these two PD-1-targeting mAbs bind to different regions on PD-1 to block the PD-1/PD-L1 interaction, they compete with each other with stereospecific hindrance when binding to PD-1.Citation15 PD-L1-targeting mAbs bind to PD-L1 with distinct orientations, and mainly target the loops of PD-L1 to interrupt PD-1/PD-L1 interaction.Citation12,Citation13,Citation17 Nanobodies targeting PD-L1 that block the binding of PD-1/PD-L1 have also been reported.Citation19 However, whether these mAbs are representative of all the binding modes of PD-1/PD-L1 blocking mAbs is unknown. Nevertheless, there are more mAbs targeting PD-1/PD-L1 in clinical trials and investigations of the binding mechanisms of these mAbs would provide better understanding of mAb-based PD-1/PD-L1 blockade for tumor ICT.Citation20
Glycosylation modifications of PD-1 or PD-L1 and the effects of these glycosylations to the interaction with therapeutic mAbs, which may be correlated with the clinical responsiveness of these mAbs, are concerns in this field.Citation15,Citation21 Both PD-1 and PD-L1 have glycosylation modifications; in particular, PD-1 is highly glycosylated.Citation15 Li et al. recently reported that glycan modifications of PD-L1 influence the interaction with PD-1, and subsequent immunosuppression in triple-negative breast cancer.Citation22 In addition to the expression in T cells, PD-1 is also reported to exist in tumor cells, which commonly have dysregulated glycosylation modifications.Citation23 It is reported that the binding of the two FDA-approved mAbs, pembrolizumab and nivolumab, with PD-1 is independent of PD-1 glycosylation.Citation15,Citation16 However, the glycan modifications of PD-1 and the effects of binding to other PD-1 targeting mAbs are less understood.
Toripalimab, a humanized IgG4 mAb targeting PD-1, was approved by the China National Medical Products Administration in 2018 as a second-line treatment for melanoma, and it is also undergoing multiple Phase 1-Phase 3 clinical trials in numerous indications, including nasopharyngeal carcinoma, and nonsmall cell lung cancer. Here, we determined the complex structure of toripalimab and PD-1 to elucidate the structural basis of the interaction between toripalimab and PD-1. The dominant binding to the FG loop of PD-1 with a long complementarity-determining region 3 (CDR3) loop of heavy chain (H chain) is substantially different compared to that of nivolumab and pembrolizumab. Glycosylation modifications of PD-1 could be observed in three potential N-linked glycosylation sites, and the binding to toripalimab was investigated. These findings improve our understanding of the binding mechanisms of toripalimab, and broaden our knowledge about mAb-based tumor ICT.
Results
Tumor suppression of the PD-1 targeting toripalimab
Toripalimab could block the interaction between PD-1 and its ligands. The ability of toripalimab to compete with PD-L1 or PD-L2 for PD-1 binding was assessed in enzyme-linked immunosorbent assay (ELISA) and flow cytometry-based competition experiments (). In ELISA-based assays, recombinant human PD-1 (hPD-1) extracellular domain (ECD) protein was immobilized to microtiter plates. The results showed that toripalimab could efficiently block the binding of ligands PD-L1 and PD-L2 to PD-1 (). Toripalimab was also confirmed to block interactions between PD-1 and PD-L1 and PD-L2 in flow cytometry-based competition assays, which involved 293T cells stably transfected with human PD-1 (). Half-maximal inhibitory concentrations (IC50) of toripalimab in ELISA-based PD-1/PD-L1 inhibition assays were determined to be 0.8 nM for PD-L1 and 1.3 nM for PD-L2, while the IC50 of toripalimab was determined to be 1.3 nM for PD-L1 and 3.7 nM for PD-L2 in flow cytometry-based assays. The results indicated that toripalimab could block the interaction of PD-1 with its ligands, PD-L1 and PD-L2, and showed a higher blocking efficacy to PD-1/PD-L1 than PD-1/PD-L2.
Figure 1. Antitumor efficacy of toripalimab in a MC38 bearing mouse model. (a,b) Blocking of the binding of PD-1 to PD-L1 or PD-L2 using protein-based ELISA assay (a) or cell-based flow cytometry assay (b). (c) Flow chart of the animal study. MC38 was inoculated into human PD-1 knock-in mice of the C57BL/6 background. Toripalimab was administered via intraperitoneal (i.p.) injection every 3 or 4 d from day 7 (D7) after MC38 tumor inoculation. The size of the tumor was monitored every 3 or 4 d after injection of toripalimab or control mAb. Saline and control mAb were enrolled as negative control. (d) Mice bearing subcutaneous MC38 palpable tumors for 7 d were treated i.p. with four doses of toripalimab, 0.3, 1, 3, and 10 mg/kg, or saline or control IgG. The data with each dot show the average tumor volume of the group while the SE was presented as longitudinal bars. (e–j) Individual follow-up of tumor sizes is presented for each experimental group with each line showing the changes of the tumor size of each mouse.
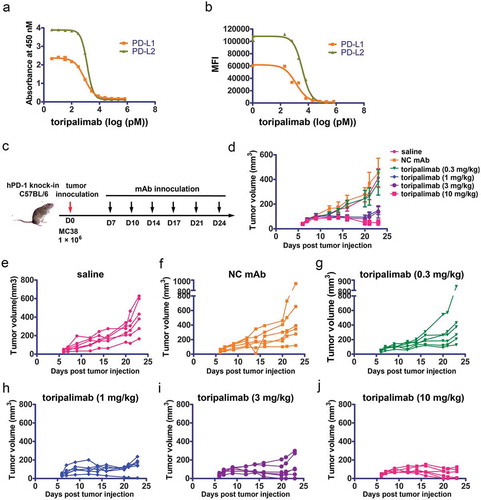
The in vivo tumor suppression efficacy of toripalimab was examined in hPD-1 knock-in mice of C57BL/6 background (C57/hPD-1) by inoculation of the syngeneic tumor cell line MC38. The C57/hPD-1 mice were subcutaneously inoculated with 1 × 10Citation6 MC38 cells and the size of the tumor was monitored after injection of the toripalimab or negative control IgG4 (antikeyhole limpet hemocyanin (KLH) IgG4) (). The results showed that inhibition of tumor growth was observed in a dose-dependent manner with substantial antitumor efficacy in 1, 3, and 10 mg/kg treatment groups with toripalimab (). Compared with the negative control IgG4-treated group, the tumor sizes in the toripalimab-treated groups decreased significantly at the end of the observation period (day 23), with P values being less than 0.05 in the 1 and 3 mg/kg groups, and P < 0.01 in the 10 mg/kg group. The low dose group (0.3 mg/kg) showed no significant change in tumor size compared to control Ig (P > 0.05). The EC50 dose for toripalimab in this MC38 tumor model likely falls between 0.3 and 1 mg/kg. Therefore, the PD-1 targeting toripalimab exhibits substantial tumor suppressive efficacy in a dose-dependent manner.
FG loop of PD-1 dominates the binding to toripalimab
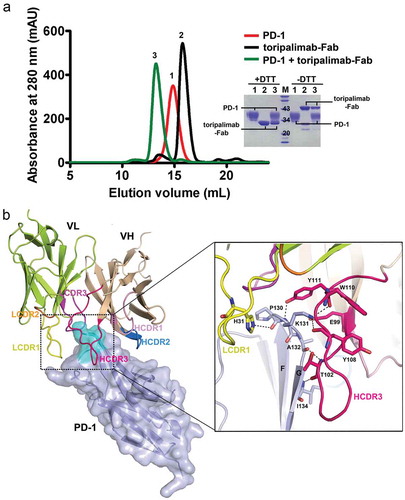
To elucidate the binding characteristics of toripalimab to PD-1 and the blocking mechanisms of toripalimab to PD-1/PD-L1 interaction, the complex structure of toripalimab and PD-1 was determined at a resolution of 2.6 Å after screening of crystals of toripalimab-antigen-binding fragment (Fab)/PD-1 complex proteins (Table S1 and ). The toripalimab binds to PD-1 with a total buried surface of 2011 ÅCitation2, while H chain and light (L) chain contributes comparable buried surfaces to PD-1, with a buried surface of 961 ÅCitation2 and 1, 049 ÅCitation2, respectively. Overall, all three CDRs of the heavy chain (HCDRs) of toripalimab are involved in the interaction with PD-1, while CDR1 and CDR3 of its light chain (LCDR1 and LCDR3) are engaged in recognition to PD-1 (). The binding of toripalimab to PD-1 is mainly located on the FG loop of PD-1, which is mainly contributed by HCDR3 and LCDR1 of toripalimab, with multiple hydrogen bond interactions. Toripalimab possesses a long HCDR3 loop with 18 amino acids, which forms multiple contacts with the FG loop of PD-1. Specifically, the amino acids of HCDR3 (E99, T102, Y108, W110, and Y111) contributed major hydrogen bond interactions with amino acids from FG loop of PD-1 (P130, K131, A132, and I134) ((b)). The H31 of LCDR1 of toripalimab also forms hydrogen bond interactions with P130 of the FG loop. Additionally, amino acids from HCDR1, HCDR2, and LCDR1 contact with FG loop of PD-1 with multiple van der Waals’ forces (). Taken together, the binding of toripalimab to PD-1 is mainly contributed by the long HCDR3 loop of toripalimab, while FG loop of PD-1 contributed most of the interactions with toripalimab.
Table 1. Residues contributed interaction between toripalimab and PD-1.
Figure 2. The complex structure of toripalimab and PD-1. (a) Gel filtration profiles of PD-1 (red), toripalimab-Fab (black), and the PD-1/toripalimab-Fab complex (green) were analyzed by size-exclusion chromatography as indicated. The SDS-PAGE analyses are shown in reducing (+DTT) or nonreducing (-DTT) conditions, one for PD-1, two for toripalimab-Fab, and three for toripalimab-Fab/PD-1 complex. (b) The complex structure of toripalimab and PD-1. The V fragment of toripalimab is shown as cartoon (heavy chain (VH), wheat; light chain (VL), lemon), and PD-1 is shown as surface representation (light blue). The CDR1, CDR2, and CDR3 loops of the heavy chain (HCDR1, HCDR2, and HCDR3) are colored in light pink, marine, and hot pink, respectively. The CDR1, CDR2, and CDR3 loops of light chain (LCDR1, LCDR2, and LCDR3) are colored in yellow, orange, and magenta, respectively. The FG loop of the PD-1 molecule is highlighted in cyan. The right panel showed the detailed binding of toripalimab to the FG loop of PD-1. Residues involved in the hydrogen bond interaction are shown as sticks and labeled. Hydrogen bonds are shown as dashed black lines.
Blocking mechanisms of toripalimab to PD-1/PD-L1 interaction
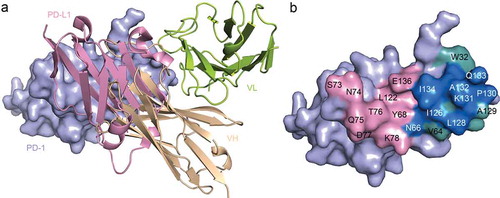
To explore the blocking mechanisms of toripalimab to the interaction of PD-1/PD-L1, the structure of toripalimab/PD-1 complex was superimposed with PD-1/PD-L1 complex (PDB code: 4ZQK). Overall, the binding of toripalimab to PD-1 exhibited stereospecific hindrance to that of PD-L1. Specifically, the H chain of toripalimab provides major conflicts with PD-L1, while L chain is away from the binding interface of PD-1/PD-L1 (). Further analysis of the binding surface of toripalimab on PD-1, in comparison with that of PD-L1, revealed that the overlapped binding surface mainly locates on FG loop (P130, K131, A132, and I134), which is also the major target for toripalimab binding (). Taken together, these findings suggest that the binding of toripalimab to PD-1 would abrogate the binding of PD-L1, which is mainly induced by H chain-derived stereo clash.
Figure 3. Competitive binding of toripalimab and PD-L1 with PD-1. (a) Superposition of the toripalimab/PD-1 complex structure with PD-1/PD-L1 complex structure (PDB code: 4ZQK). PD-L1 is shown in light pink while VH of toripalimab is colored in wheat and VL in lemon. (b) Binding surface of PD-1 with PD-L1 or toripalimab. The residues in contact with PD-L1 are colored in light pink, whereas residues in contact with toripalimab are colored in light teal, and the overlapping residues bound by both PD-L1 and toripalimab are colored in marine.
Glycosylation-independent binding of toripalimab to PD-1
Previous studies revealed that PD-1 is highly glycosylated.Citation15 Therefore, the influences of PD-1 glycosylation to the binding of toripalimab were further evaluated. PD-1 protein has four potential N-linked glycosylation sites, N49, N58, N74, and N116. The structural analysis of PD-1 showed that glycosylation modifications could be observed in three of these N-glycosylation sites, N49, N58, and N116 (). Similar glycosylation modifications were found on N49 and N58, which consist of two N-acetylglucosamines (NAG) and one fucose (FUC), while only one NAG was visible on N116. Of note, N58 is located near the interface of toripalimab and PD-1. Glycosylation modifications might also affect the overall structure of PD-1, and further affect the binding to toripalimab. Therefore, the binding affinity of toripalimab to PD-1 was evaluated with PD-1 proteins obtained from 293T expressing system, which enables full glycosylation modification of PD-1, or refolded from inclusion bodies expressed in E. coli, which is deficient in any glycosylation modifications.
Figure 4. Glycan modifications of PD-1 and glycosylation independent binding of toripalimab. (a) Complex structure of toripalimab-Fab and PD-1 with glycan depicted as sticks in orange. Four potential N-linked glycosylation sites, N49, N58, N74, and N116, were shown as sticks in light teal. (b). SPR assay characterization of the binding between toripalimab and PD-1 proteins using a BIAcoreT100 system. The refolded PD-1 protein (L25-R147), which is expressed in E. coli and refolded in vitro, and PD-1 protein obtained from 293T cells were analyzed for binding affinity with toripalimab, with toripalimab immobilized on the chip. The binding characteristics (Ka, Kd, and KD) were labeled accordingly. The data presented here are a representative of three independent experiments with similar results.
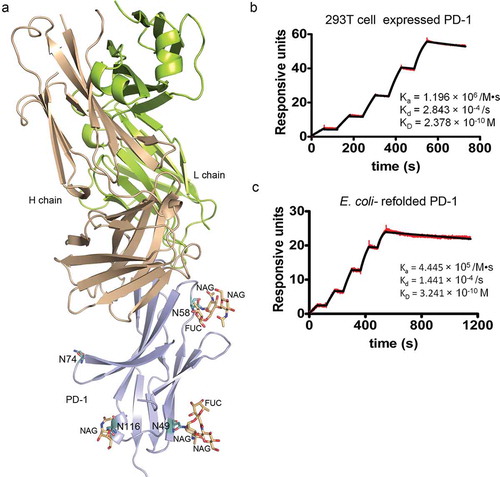
The binding profiles of toripalimab with PD-1 proteins obtained from 293T or E. coli expression system were analyzed using a surface plasmon resonance (SPR) assay with toripalimab immobilized on the chip. The results revealed that the binding affinity (KD) of toripalimab with PD-1 proteins from E. coli (KD = 0.324 nM) showed no substantial difference with that from 293T cells (KD = 0.238 nM) (). This finding suggests that the binding of toripalimab to PD-1 is independent of any glycosylation modifications.
Comparative binding characteristics of PD-1 targeting mAbs
The complex structures of two US-FDA approved anti-PD-1 mAbs, nivolumab and pembrolizumab, have been reported, which enabled us to investigate the binding characteristics of these mAbs, which have promising efficacy as tumor immunotherapy.Citation15,Citation16
The PD-1 extracted from the complex with PD-L1, nivolumab or pembrolizumab was superimposed with that from the toripalimab/PD-1 complex (). We found that these mAbs bind mainly on the loops of PD-1. The pembrolizumab mainly binds to the C’D loop, while the binding of nivolumab mainly involves the N-terminal loop. In the meanwhile, the binding of toripalimab to PD-1 mainly locates on the FG loop, as also described above ( and ). The C’D loop and N terminal loop of PD-1 could only be visible upon binding with the mAbs, indicating the flexibility of these loops. Of note, the FG loop exhibits varied conformations upon binding to different partners (). The FG loop is invisible in the PD-1/pembrolizumab complex, which indicates the flexibility of this loop. A substantial shift of 8.19 Å was observed with the FG loop when PD-1 binds to nivolumab or toripalimab. The binding of nivolumab is mainly contributed by the N-terminal loop, though multiple contacts were also observed with the FG loop.Citation15 This finding suggests that the FG loop of PD-1 adopts different conformations upon binding to different mAbs.
Figure 5. Comparative binding of PD-1 targeting mAbs. (a) Superimposition of the PD-1 from complex structures of PD-1/PD-L1 (cyan) (PDB code: 4ZQK), PD-1/pembrolizumab (light pink) (PDB code: 5JXE), PD-1/nivolumab (lemon) (PDB code: 5WT9), and PD-1/toripalimab (light blue) (PDB code: 6JBT). The loops contributed major binding to the mAbs were highlighted in dashed circles. (b) Comparison of the FG loop of the PD-1s from the complex structures. The FG loop of PD-1 shifted 8.19 Å upon the binding to nivolumab or toripalimab. (c) Superimposition of the complex structures of PD-1/pembrolizumab, PD-1/nivolumab and PD-1/toripalimab with the mAbs shown as ribbon in red, blue, and lemon, respectively. The PD-1 from PD-1/toripalimab complex is shown as surface in light blue with the FG loop highlighted in cyan.
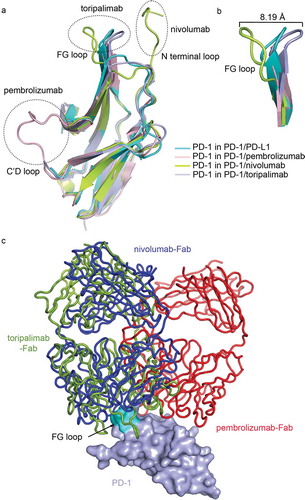
Structures of the nivolumab/PD-1 and pembrolizumab/PD-1 complexes were superimposed with toripalimab/PD-1 complex, with the structure of PD-1 fixed to investigate the binding mode of different mAbs. Comparative analysis revealed that the binding orientation of toripalimab to PD-1 is similar to that of nivolumab, while pembrolizumab adopts a distinct binding orientation (). Though similar binding orientation is observed between toripalimab and nivolumab, the major binding regions on PD-1 are substantially different between these two mAbs, as also described above. These findings suggest that therapeutic mAbs targeting PD-1 adopt distinct binding modes to interrupt the interaction of PD-1 and PD-L1.
Discussion
In this study, we report the antitumor efficacy of toripalimab in a mouse model and the structural basis of the interaction between toripalimab and PD-1. Toripalimab has been reported to be competent in decreasing the expression of PD-1 in both CD4+ and CD8+ T cells.Citation24 Here, we show that toripalimab could significantly reduce the tumor burden in the mouse model in a dose-dependent manner. The structural analysis revealed that the binding of toripalimab to PD-1 induced stereo hindrance for the binding of PD-L1, and the overlapping binding surface has indicated the competitive binding to PD-L1. We found that binding of toripalimab to PD-1 mainly locates on the FG loop, a loop that also contributes multiple interactions with PD-L1.Citation25 Moreover, the glycosylation-independent binding of toripalimab to PD-1 also provided useful information for our understanding of the antitumor mechanisms, especially when targeting PD-1 in tumor cells, which are usually accompanied with deficiency in glycosylation modifications.Citation23 All these findings broaden our understanding of the binding mechanisms of toripalimab, and may facilitate its clinical applications.
PD-1 binds to PD-L1 or PD-L2 with the front β-sheet faces consisting of CC’FG strands.Citation25,Citation26 In our study, we found that the binding of toripalimab to PD-1 is mainly located on the FG loop of PD-1, while amino acids from FG loop of PD-1 (L128, P130, K131, and A132) also contributed multiple interactions with PD-L1.Citation25 The toripalimab contains an unconventional, long HCDR3 loop consisting of 18 amino acids, which provided major hydrogen bond interactions with the FG loop of PD-1. It could be deduced that this HCDR3 loop would be responsible for the high binding affinity of toripalimab to PD-1, as observed in the SPR assay. Of note, the FG loop of PD-1 is highly flexible, such that it adopts different conformations to bind to different counterparts, e.g. nivolumab and toripalimab. The binding of toripalimab to PD-1 has induced substantial stereo clash with the binding of PD-L1, which is mainly mediated through the H chain of toripalimab. Therefore, toripalimab’s blockade of PD-1/PD-L1 interactions is due to both the overlapping binding regions of toripalimab and PD-L1 on PD-1, and stereo hindrance of the H chain of toripalimab to PD-L1. Meanwhile, nivolumab and pembrolizumab mainly bind to the N-terminal loop and the C’D loop of PD-1, indicating that the flexible loops of PD-1 are more prone to be targeted by therapeutic mAbs targeting PD-1.
Disordered glycosylation modifications of the proteins in tumor cells have played critical roles in the development and progression of multiple tumors.Citation21 PD-1 protein is highly glycosylated, and all of the four potential N-linked glycosylation sites are suggested to be glycosylated.Citation15 Intrinsic expression of PD-1 in tumor cells has been reported to play critical roles in promotion of tumor growth, and mAbs targeting tumor cell-intrinsic PD-1 would induce substantially decreased progression of the tumors.Citation23 Therefore, PD-1-specific mAbs not only restimulate the exhausted T cells to evoke antitumor immunity, but also suppress tumor growth by directly targeting tumor cell-intrinsic PD-1. The dysregulated glycan transferase expression in tumor cells raises the concern that the binding efficacy of PD-1 specific mAbs might be affected by the disordered glycosylation modifications of PD-1 in tumor cells, and thereafter affects the treatment efficacy.Citation23,Citation27 The structure of PD-1 reported here, which is expressed in 293T cells, revealed that glycosylation modifications could be observed at three of four N-linked glycosylation sites. The N-linked glycosylation sites are mainly located on the loops of PD-1, namely BC loop (N58), CC’ loop (N74), and EF loop (N116), except the N49 at the beginning of B strand. All the visible glycosylation modifications of PD-1 are away from the interface of PD-1/PD-L1, and the binding of PD-1 to PD-L1 is not directly affected by these glycosylation modifications, as is reported previously by our group and the others.Citation15,Citation16 However, Li et al. recently reported that N-linked glycosylation of PD-L1 would affect the contact between PD-1 and PD-L1, suggesting that glycosylation modifications might be involved in modulation of PD-1 signaling.Citation22 Through the determination of the binding affinity of toripalimab to fully glycosylated or nonglycosylated PD-1 proteins using SPR assay, no substantial differences were observed, indicating the independence of glycosylation modification to toripalimab binding to PD-1. These results suggest that clinical application of toripalimab may be less likely to be affected by dysregulated glycosylation modifications of PD-1.
In summary, we report the molecular basis of toripalimab binding to PD-1 for tumor ICT. Toripalimab binds to the FG loop of PD-1 with an unconventional, long HCDR3, which is distinct from the currently FDA-approved nivolumab and pembrolizumab. The binding of toripalimab to PD-1 is independent of glycosylation modifications of PD-1. All these findings facilitate our understanding of the binding mechanisms of toripalimab, and may benefit future development of biologics targeting PD-1 for tumor immunotherapy.
Materials and methods
Plasmid construction and protein purification
The DNA encoding the ectodomain of human PD-1 (residues M1-Q167, including signal peptide) with six histidines at the C terminus of the sequence was cloned into pCAGGS vector (Addgene) with EcoRI and BglII restriction sites as previously described.Citation15 Plasmid pCAGGS-PD-1 was transiently transfected into 293T cells for protein expression; the cells were cultured with 5% CO2 at 37oC. The supernatant was collected after 72 h, and the protein was purified by sequentially His-Trap HP column (GE Healthcare) and SuperdexTM 200 10/300 GL (GE Healthcare) in a buffer containing 20 mM Tris and 150 mM NaCl (pH 8.0).
The DNA encoding the ectodomain of human PD-1 (residues L25-R147) was cloned into the pET-21a vector (Novagen) with NdeI and XhoI restriction sites and transformed into Escherichia coli strain BL21 (DE3) for protein expression.Citation15 The inclusion bodies of recombinant proteins were purified and then refolded as previous described.Citation15 Soluble proteins were further purified by a SuperdexTM 200 10/300 GL (GE Healthcare) with a running buffer of 20 mM Tris (pH 8.0) and 150 mM NaCl.
Full-length toripalimab proteins were obtained from Shanghai Junshi Biosciences. The purified mAbs were digested with the Human IgG Fab and F(ab’)2 Preparation Kits (Thermo Scientific) according to the manufacturer’s instructions. The protein fragments were purified by HiTrap Protein A FF (GE Healthcare), and then exchanged to the buffer of 20 mM Tris (pH 8.0), 150 mM NaCl.
Blocking of PD-1 and its ligands with ELISA and flow cytometry
The ability of toripalimab to compete with PD-L1 or PD-L2 for PD-1 binding was assessed in ELISA and flow cytometry-based competition experiments. In ELISA-based assays, recombinant human PD-1 ECD fusion protein was immobilized to microtiter plates, washed, and blocked using 1% bovine serum albumin (BSA). Biotin-labeled PD-L1 and PD-L2 Fc fusion (R&D Systems) were added to the plate, together with a titration of toripalimab antibody. The bound PD-L1 and PD-L2 Fc fusion proteins were detected by incubation with horseradish peroxidase-labeled streptavidin secondary antibody (Jackson ImmunoResearch, catalog no. 016-030-084). Half-maximal inhibitory concentrations (IC50) of toripalimab were determined using a log(inhibitor) vs. response—variable slope curve fit (GraphPad Prism).
In flow cytometry-based competition assay, biotin-labeled PD-L1 and PD-L2 Fc fusion were incubated with 293T cells stably transfected with human PD-1 (293T-hPD1). Cell surface-bound PD-L1 and PD-L2 Fc fusion protein was subsequently detected by incubation with PE-labeled Streptavidin secondary antibody (eBioscience, catalog no. 12-4317-87). Half-maximal inhibitory concentrations (IC50) of toripalimab were determined using a log (inhibitor) vs. response—variable slope curve fit (GraphPad Prism).
In vivo antitumor activity in a syngeneic tumor model
A genetically modified mouse strain with human PD-1 knocked-in was employed in our study. For the MC38 syngeneic model, mice were subcutaneously inoculated with 1 × 10Citation6 MC38 cells in 100 uL phosphate-buffered saline on day 0. On day 6, the inoculated mice were randomized into six groups (tumor volume averages 20–70 mm3) and treated with either control Ig (anti-KLH, Junshi Biosciences, lot no. 20160929), saline, or toripalimab at 0.3, 1, 3, and 10 mg/kg via intraperitoneal injection twice a week. The tumor growth was monitored twice a week and the volume of the tumors was calculated by the formula: ½ length × widthCitation2.
Complex preparation and crystallization
The mammalian cell-expressed PD-1 protein and toripalimab-Fab were mixed at a molar ration of 1:1. The mixture was incubated on ice for 30 min and further purified by SuperdexTM 200 10/300 GL (GE Healthcare). A total of 10 mg/mL of toripalimab/PD-1 proteins were used for crystal screening by vapor-diffusion sitting-drop method at 4°C. Diffractable crystals were obtained in a condition consisting of 0.09 M halogens consisting of NaF, NaBr, and NaI additives, 0.1 M Tris-Base (pH 8.5), 37.5% (v/v) MPD-P1K-P3350 consisting of MPD (racemic), PEG 1K and PEG 3350 (Morpheus® MD1-46 kit, Molecular Dimensions).
Data collection and structure determination
To collect the diffraction data, all crystals were flash-cooled in liquid nitrogen after incubating in reservoir solution containing 20% (v/v) glycerol. The diffraction data were collected at Shanghai Synchrotron Radiation Facility (SSRF) BL17U, and all data were processed with HKL2000.Citation28 The complex structure was solved by molecular replacement method using phase with the reported PD-1 structure (PDB: 5WT9) and Fab structure (PDB: 5WT9) as the search models.Citation29,Citation30 Subsequent model building and refinement were performed using coot and phenix to refine the results, respectively.Citation31,Citation32 The stereochemical qualities of the final model were assessed with MolProbity.Citation33s Data collection and refinement statistics are summarized in Table S1. All structural figures were generated using Pymol (http://www.pymol.org).
SPR analysis
The SPR analysis was performed at room temperature using a BIAcore®T100 system with CM5 chips (GE Healthcare). For all the analyses, an HBS-EP buffer consisting of 10 mM HEPES (pH 7.4), 150 mM NaCl and 0.005% (v/v) Tween-20 was used as running buffer, and all proteins were exchanged to the same buffer in advance via gel filtration. The blank channel of the chip was used as the negative control. To detect the toripalimab binding to different forms of PD-1 proteins, toripalimab protein was immobilized on the chip by antihuman IgG at about 70 response units. Gradient concentrations of PD-1 (0.975, 1.95, 3.9, 7.81, and 15.625 nM) were then flowed over the chip surface. After each cycle, the sensor surface was regenerated with 3M MgCl2. The binding kinetics were all analyzed with the software of BIA evaluation® Version using a 1:1 Langmuir binding model.
Data deposition
Atomic coordinates have been deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb) under accession code 6JBT.
Abbreviations
| CAR-T | = | chimeric antigenic receptor engineered T cells |
| CDR | = | complementarity-determining region |
| CTLA-4 | = | cytotoxic T-lymphocyte-associated protein 4 |
| ELISA | = | enzyme-linked immunosorbent assay |
| ECD | = | extracellular domain |
| Fab | = | antigen-binding fragment |
| Fv | = | variable fragment domain |
| H chain | = | heavy chain |
| ICT | = | immune checkpoint therapy |
| IC50 | = | Half-maximal inhibitory concentrations |
| Ig | = | immunoglobulin |
| KLH | = | keyhole limpet hemocyanin |
| mAb | = | monoclonal antibody |
| NAG | = | N-acetylglucosamines |
| FUC | = | fucose |
| PD-1 | = | programmed cell death 1 |
| SPR | = | surface plasmon resonance. |
Conflict of interests
The authors declare that they have no conflict of interests.
Supplemental Material
Download MS Word (13.4 KB)Acknowledgments
This study was supported by National Major Science & Technology Major Project (2015ZX09102017, 2017ZX09302009). Strategic Priority Research Program of Chinese Academy of Sciences (CAS) (XDA12020358, XDB29040201). G.F.G. and J.Y. are supported by the NSFC Innovative Research Group (grant no. 81621091). We thank the staff of BL17U and BL19U beamline at the Shanghai Synchrotron Radiation Facility for assistance with data collection. We also thank Yuanyuan Chen, Bingxue Zhou, and Zhenwei Yang from the Institute of Biophysics, CAS, for their technical support in the SPR assay.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Tan S, Gao GF. New hope for cancer treatment: cancer immunotherapy. Chinese Sci Bull. 2015;60:3155–57. In Chinese.
- Callahan MK Postow MA and Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–78. doi:10.1016/j.immuni.2016.04.023.
- Ishida Y, Agata Y, Shibahara K and Honjo H. Induced Expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell-death. EMBO J. 1992;11:3887–95.
- Nishimura H, Nose M, Hiai H, Minato N and Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T and Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–97. doi:10.1073/pnas.192461099.
- Curiel TJ, Wei S, Dong HD, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–67. doi:10.1038/nm863.
- Hirano F, Kaneko K, Tamura H, Dong HD, Wang SD, Ichikawa, Rietz C, Flies DB, Lau JS, Zhu GF, Tamada K, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96.
- Passaro C, Alayo Q, DeLaura I, McNulty JJ, Grauwet K, Ito H, Bhaskaran V, Mineo M, Lawler SE, Shah K, et al. Arming an oncolytic herpes simplex virus Type 1 with a single chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res. 2018. doi:10.1158/1078-0432.CCR-18-2311.
- Mahoney KM, Rennert PD and Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84. doi:10.1038/nrd4591.
- Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36:847–56. doi:10.1038/nbt.4195.
- Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, Lim H, Won Heo T, Park KY, Lee YJ, et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat Commun. 2016;7:13354. doi:10.1038/ncomms13354.
- Liu K, Tan S, Chai Y, Chen D, Song H, C W Z, Shi Y, Liu J, Tan W, Lyu J, et al. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 2016;27:151–53. doi:10.1038/cr.2016.102.
- He M, Chai Y, Qi J, C W Z, Tong Z, Shi Y, Yan J, Tan S and Gao GF. Remarkably similar CTLA-4 binding properties of therapeutic ipilimumab and tremelimumab antibodies. Oncotarget. 2017;8:67129–39. doi:10.18632/oncotarget.18004.
- Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, Qi J, Wong G, Zhu X, Liu WJ, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8:14369. doi:10.1038/ncomms14369.
- Na Z, Yeo SP, Bharath SR, Bowler MW, Balikci E, Wang CI and Song H. Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 2017;27:147–50. doi:10.1038/cr.2016.77.
- Tan S, Liu K, Chai Y, Zhang CW, Gao S, Gao GF and Qi J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell. 2018;9:135–39. doi:10.1007/s13238-017-0412-8.
- Tan S, Zhang CW and Gao GF. Seeing is believing: anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct Target Ther. 2016;1:16029. doi:10.1038/sigtrans.2016.29.
- Zhang F, Wei H, Wang X, Bai Y, Wang P, Wu J, Jiang X, Wang Y, Cai H, Xu T, et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. 2017;3:17004. doi:10.1038/celldisc.2017.4.
- Tan S, Chen D, Liu K, He M, Song H, Shi Y, Liu J, Zhang CW, Qi J, Yan J, et al. Crystal clear: visualizing the intervention mechanism of the PD-1/PD-L1 interaction by two cancer therapeutic monoclonal antibodies. Protein Cell. 2016;7:866–77. doi:10.1007/s13238-016-0337-7.
- Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55. doi:10.1038/nrc3982.
- Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201. doi:10.1016/j.ccell.2018.01.009.
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic pd-1 receptor functions promote tumor growth. Cell. 2015;162:1242–56. doi:10.1016/j.cell.2015.08.052.
- Fu J, Wang F, Dong LH, Zhang J, Deng CL, Wang XL, Xie XY, Zhang J, Deng RX, Zhang LB, et al. Preclinical evaluation of the efficacy, pharmacokinetics and immunogenicity of Toripalimab, a programmed cell death protein-1 (PD-1) monoclonal antibody. Acta Pharmacol Sin. 2017;38:710–18. doi:10.1038/aps.2016.161.
- Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N and Garboczi DN. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A. 2008;105:3011–16. doi:10.1073/pnas.0712278105.
- Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG and Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A. 2008;105:10483–88. doi:10.1073/pnas.0804453105.
- Xu C, Ng DT. Glycosylation-directed quality control of protein folding. Nat Rev Mol Cell Biol. 2015;16:742–52. doi:10.1038/nrm4073.
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26.
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–82.
- Collaborative computational project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–63. doi:10.1107/S0907444994003112.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi:10.1107/S0907444904019158.
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi:10.1107/S0907444909052925.
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi:10.1107/S0907444909042073.