
ABSTRACT
We report a case study in which liquid-liquid phase separation (LLPS) negatively impacted the downstream manufacturability of a therapeutic mAb. Process parameter optimization partially mitigated the LLPS, but limitations remained for large-scale manufacturing. Electrostatic interaction driven self-associations and the resulting formation of high-order complexes are established critical properties that led to LLPS. Through chain swapping substitutions with a well-behaved antibody and subsequent study of their solution behaviors, we found the self-association interactions between the light chains (LCs) of this mAb are responsible for the LLPS behavior. With the aid of in silico homology modeling and charged-patch analysis, seven charged residues in the LC complementarity-determining regions (CDRs) were selected for mutagenesis, then evaluated for self-association and LLPS properties. Two charged residues in the light chain (K30 and D50) were identified as the most significant to the LLPS behaviors and to the antigen-binding affinity. Four adjacent charged residues in the light chain (E49, K52, R53, and R92) also contributed to self-association, and thus to LLPS. Molecular engineering substitution of these charged residues with a neutral or oppositely-charged residue disrupted the electrostatic interactions. A double-mutation in CDR2 and CDR3 resulted in a variant that retained antigen-binding affinity and eliminated LLPS. This study demonstrates the critical nature of surface charged resides on LLPS, and highlights the applied power of in silico protein design when applied to improving physiochemical characteristics of therapeutic antibodies. Our study indicates that in silico design and effective protein engineering may be useful in the development of mAbs that encounter similar LLPS issues.
Introduction
Liquid-liquid phase separation (LLPS) of protein solutions is a thermodynamically driven phenomenon that typically occurs at high concentration under refrigerator temperatures (2–8°C), disrupting the homogeneity of protein solutions.Citation1 During LLPS, a homogeneous protein solution spontaneously separates into two co-existing liquid phases: a protein-poor light phase and a protein-rich heavy phase. This phenomenon is fully reversible, with the proteins maintaining their native structure.Citation2 In recent years, LLPS challenges during the formulation development of multiple mAbs or mAb derivatives have been described in the literature.Citation1–Citation6 Several of the reports propose that the progression to, and final equilibrium state of, LLPS are dictated by physicochemical conditions, such as temperature, pH, ionic strength, buffering agents, and the type and concentration of other excipients.Citation3–Citation5
MAbs are effective therapeutics for numerous pathological conditions, including cancer, inflammation, infectious diseases, autoimmune diseases, and neurological disorders.Citation7 Platformed purification process strategies that aim to standardize manufacturing processes and expedite process development for these molecules now dominate the biopharmaceutical industry.Citation8,Citation9 During large-scale manufacturing, purification intermediates are often kept at ambient temperature for short periods of time, usually not exceeding 7 days; lower temperatures of 2–8°C are preferred for extended storage to maintain product quality. LLPS is often considered a manageable challenge for downstream processing with the majority of the reported LLPS cases today arising from formulation development, particularly for mAbs requiring high concentration formulation.Citation1–Citation6 Luo et. al. reported that LLPS caused transient turbidity during the low pH elution process in Protein A chromatography, where instantaneous effluent protein concentration can exceed 50 mg/mL.Citation10 Many excipients with diverse physio-chemical properties, such as citrate, phosphate, polyols, sugars, amino acids, and polymers have been found to be highly effective at preventing LLPS.Citation4 In light of these findings, LLPS became better understood and easier to manage during formulation development, yet occasionally mitigation solutions for the biophysical property at one stage in drug manufacturing may not be compatible with downstream process steps.
It has been proposed that LLPS behaviors may be caused by the propensity of a protein for reversible self-association (RSA).Citation2,Citation10,Citation11 Self-association of an antibody can occur between antigen-binding fragment (Fab) domains,Citation12,Citation13 or between Fab domains and the crystallizable fragment (Fc) domains.Citation14,Citation15 The driving forces that underlie antibody self-association are diverse, with both electrostatic and hydrophobic interactions likely playing key roles.Citation16,Citation17 Mutational substitutions of hydrophobic residues (such as tryptophanCitation18 and phenylalanineCitation12) or charged residues (such as histidineCitation16 and lysineCitation6) have proven to be effective at reducing self-associations. Self-associations leading to LLPS can often be dominated by electrostatic interactions. For example, Casaz et. al reported that the single substitution of a negatively charged glutamate residue with a positively charged lysine residue effectively mitigated the LLPS by reducing electrostatic interactions between molecules.Citation6 While there is a clear link between electrostatic interactions and RSA, a more detailed understanding that includes contributions of charged residues and differences due to amino acid type, especially those in the complementarity-determining regions (CDRs), is limited.
In this work, we carried out a systematic investigation to gain mechanistic insight into LLPS behavior and developed mitigation strategies based on those insights. First, we show electrostatic interaction-driven self-associations and the formation of high-order complexes correlate with LLPS behaviors. Second, we identify light chain in the CDR as the key domain involved in self-association and LLPS. Third, the contribution of seven charged residues from the Lc in the CDR were evaluated by mutagenesis and an LLPS-free mutant was obtained based on the implementation of the prior learnings. Last, we propose an integrated process optimization and protein engineering strategy for mAbs with similar LLPS issues.
Results
LLPS of several purification intermediates provoke challenges to the drug substance manufacturing process of mAb-X
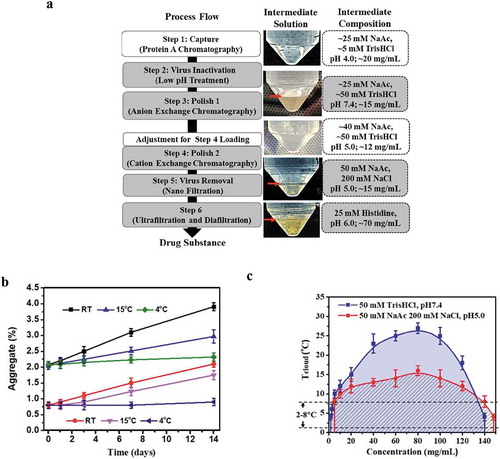
(left panel) depicts the purification process developed for mAb-X, a human IgG1-λ antibody. The mAb-X drug substance produced by the manufacturing process met product-quality specifications suitable for use in clinical studies. However, when the purification process intermediates were stored at 2 to 8°C, several of the intermediates showed distinct layers resulting in two clear liquid phases (, middle panel). After these intermediates were equilibrated to room temperature and mixed thoroughly, the intermediates became a single homogeneous optically clear phase, and no longer exhibited two liquid phases (data no shown). Once returned to 2 to 8°C, these intermediates initially became turbid, and then again separated into two phases, which is behavior indicative of LLPS. Both liquid phases contained mAb-X, but the upper phase contained mAb-X at concentrations of 1 to 3 mg/mL, whereas the lower phase contained the antibody at concentrations of ~ 150 mg/mL. These observations were consistent with published literature pertaining to LLPS.Citation1,Citation2,Citation10,Citation19 In large-scale production of therapeutic mAbs, purification intermediates are often kept at ambient temperature for brief intervals (e.g. up to 7 days), but a lower temperature (2–8°C) is often preferred for longer duration (e.g. more than 7 days). During storage, the aggregate level of mAb-X in the purification intermediates increased quickly when stored at ambient temperature; in contrast, the aggregation rate was reduced dramatically at refrigerator temperatures (). To maintain product quality, storing these intermediates at a refrigerated temperature is preferable, but, as detailed above, resulted in LLPS.
Figure 1. LLPS observed with mAb-X and interfered with the downstream manufacturing process. (a) Downstream process flow for mAb-X, purification intermediate solution (at 4oC), and the corresponding solution composition. mg/mL concentration is for mAb-X. The purification intermediates from the grey-shaded steps showed LLPS under the refrigerated conditions. (b) Aggregation rate for mAb-X purification intermediates at different temperatures. (c) Liquid-liquid coexistence curves for mAb-X under the two indicated conditions.
When LLPS occurred in these intermediate process pools during storage, the lower phases were found to be highly viscous. The viscosity of the lower (heavy) phases remained exceptionally high (>100 cP) even after the solutions were warmed to room temperature. Mixing these highly viscous heterogeneous intermediates at large-scale and achieving solution homogeneity was time-consuming. Mixing with an impeller for an extended time generates shear stress and air-liquid interfacial stress, which can result in protein aggregation.Citation20 Insufficient mixing also led to product loss and was associated with high column back-pressure (due to high viscosity) during chromatography load steps. In-process samples obtained from manufacturing for analytical testing are often kept refrigerated or frozen prior to testing. Misleading results (e.g., significantly lower protein concentrations) were observed during early development of mAb-X. As a result, a special sample-handling procedure was implemented to address the LLPS issue. Taken together, the LLPS seen with mAb-X caused substantial challenges to large-scale manufacturing and sample testing.
Recent studies have reported that sucrose and arginine-glutamate excipients can prevent mAb solutions from undergoing LLPS at high protein concentrations.Citation4,Citation10,Citation19 Indeed, we found that the addition of 200 mM sucrose effectively prevented the LLPS behavior of mAb-X, and therefore sucrose was used as an excipient for the product formulation (data not shown). However, the addition of sucrose during the purification steps was not suitable because buffers containing 200 mM sucrose caused high column back-pressures, introduced the risk endotoxin contamination, and increased risk of microbial growth. Addition of 100 mM arginine-glutamate also effectively suppressed LLPS, but using substantial amounts of arginine-glutamate in downstream buffers could increase the cost of manufacturing and compromise chromatography performance. Therefore, we did not consider the addition of arginine-glutamate for the mAb-X manufacturing process.
LLPS is traditionally evaluated via liquid-liquid coexistence curves (also called phase diagrams).Citation1,Citation2,Citation10,Citation19 We generated liquid-liquid coexistence curves () for mAb-X in two solution conditions that led to LLPS during downstream processing (after Steps 1 and 5). At 2 to 8°C, LLPS began to occur at relatively low protein concentrations, 1.0 mg/mL in the pH 7.4 condition and 3.0 mg/mL in the pH 5 .0 condition. Thus, to maintain these low concentrations and avoid LLPS in process intermediates, dilution factors ranging from 3–10 would be required. However, such dilution factors are typically not practical in large-scale manufacturing because of facility fit limitations and tank volume constraints. Based on aggregation rates and our liquid-liquid coexistence curves, we determined that storing purification intermediates at 15°C could prevent LLPS and minimize aggregation.
According to buffer compositions of the intermediates (, right panel), we also identified additional characteristics contributing to the LLPS behavior of mAb-X. Neutral pH and low ionic strength buffers (Steps 2 and 6), and acidic pH with 200 mM sodium chloride (Steps 4 and 5) led to LLPS, whereas acidic pH and low sodium chloride concentrations (Step 1 intermediate and Step 4 loading material, both were also evaluated at 4°C as other intermediates) led to optically clear solutions without LLPS. These results suggest that the LLPS behavior of mAb-X was governed by pH and sodium chloride concentration, indicative of an electrostatic interaction-driven mechanism for mAb-X’s LLPS behaviors.
Optimization of process conditions could alleviate LLPS with limitations for large-scale manufacturing
We tested whether the addition of sodium chloride could mitigate the LLPS seen at 4°C for Step 2 and Step 3 intermediates, which were in solution at low ionic strength and at a neutral pH of 7.4. The addition of sodium chloride concentrations of 200 mM or greater under neutral pH conditions effectively prevented the occurrence of LLPS (). However, during the flow-through anion exchange chromatography of Step 3, where host cell proteins (HCPs) are retained on the column while mAb-X passes through, the 200 mM sodium chloride inhibited HCPs retention and subsequently resulted in poor HCP removal. Conversely, removal of HCPs could be achieved using hydrophobic interaction/anion exchange mixed mode chromatography (data not shown), although the presence of 200 mM sodium chloride still decreased the binding capacity on the subsequent cation exchange chromatography (CEX) step. An approximately two-fold dilution to lower the sodium concentration in the solution restored the CEX binding capacity, albeit at the expense of creating a new volume challenge for manufacturing. This approach is thus unattractive for facility fit reasons, as previously described.
Figure 2. The effects of pH and sodium chloride concentration on the LLPS seen with mAb-X purification intermediates (a) Solution behaviors at pH 7.4 with varying sodium chloride concentrations. (b) Solution behaviors at pH 5.0 with varying sodium chloride concentrations. (c) Solution behavior of mAb-X at different pH values without sodium chloride.

Similarly, the addition of extra 300 mM (500 mM in total) sodium chloride to the CEX (Step 4) intermediate effectively prevented LLPS occurrence in solution (). However, using 500 mM sodium chloride for elution on the CEX column negatively impacted aggregate removal. Although the additional 300 mM sodium chloride can be added to the CEX elution pool after the completion of elution, a high concentration sodium chloride stock solution (2 ~ 4 M, to avoid large dilution factor) and a mixing step are needed, complicating the manufacturing process. As an alternative to sodium chloride addition, protein bound on CEX resins can be eluted by increasing pH with little or no salt. Therefore, we prepared mAb-X in buffers of varying pH and evaluated the solution appearance. Despite this mitigation attempt, LLPS still occurred under most of the pH conditions tested except pH5.0. Therefore, higher pH elution was not a viable option.
In summary, it was possible to mitigate the observed LLPS behavior for mAb-X through process optimization, but the optimal conditions for suppression of LLPS were suboptimal for other aspects of the process, and therefore unattractive to implement in a large-scale biopharmaceutical manufacturing process. Exhausting all reasonable mitigation strategies throughout the manufacturing process, we embarked on a molecular engineering strategy to first identify the protein interactions contributing to this undesirable biophysical property, then to modify mAb-X in an effort to eliminate LLPS behavior without sacrificing antibody binding and activity.
Identification of the mAb-X VL region responsible for LLPS
To determine the relative contributions of different domains on the mechanisms underlying LLPS, F(ab’)2 and Fab were expressed, purified, and buffer exchanged into solutions with conditions similar to Steps 1–6. LLPS was also observed on the F(ab′)2 after the fragment solutions were stored at 4°C for 24 hours (), indicating that the Fc domain was not critical to the LLPS of mAbX. Remarkably, LLPS was not seen with the single Fab domain, suggesting the bivalent double-arm structure is necessary for LLPS.
Figure 3. Mapping the domain of mAb-X associated with LLPS. (a) Solution behavior of full-length mAb-X IgG, F(ab′)2, and Fab. (b) Solutions for mAb-X formatted with different IgG subclasses or light-chain isotypes. (c) Solutions for mAb-X, mAb-Y, and chimeric mAbs. Samples were prepared in 50 mM Tris-HCl, pH 7.4, at approximately 12 mg/mL, and stored at 4ºC for 24 hours.
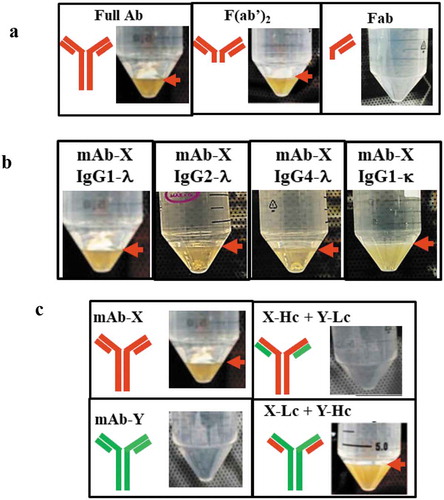
To determine whether different IgG subclasses or light-chain isotypes were more prone to LLPS, the VL and VH regions of mAb-X (lambda light and IgG1 heavy chains, respectively) were cloned into the formats of IgG2-λ, IgG4-λ, and IgG1-κ. LLPS was observed at 4°C for both the IgG2 and IgG4 subclasses (), suggesting that IgG subclass did not influence propensity for LLPS. These subclasses shared 90% sequence homology and differed in their constant regions, disulfide bond positions between the heavy and light chain, amino acids in the Fc region.Citation21 Our data suggest these differences are not critical for the LLPS of mAb-X. Notably, LLPS was also observed for the κ light chain isotype, indicating the major contribution to mAb-X-associated LLPS is likely due to differences in variable region of the Fab arm.
To determine whether the light chain or the heavy chain was responsible for the LLPS observed with mAb-X, we exploited another antibody, mAb-Y, which binds to the same antigen as mAb-X but does not exhibit LLPS under similar storage conditions, to study switched variant combinations (). We generated the chimeric mAbs X-Hc+Y-Lc, which contained the heavy chain from mAb-X and the light chain from mAb-Y, and Y-Hc+X-Lc, which contained the light chain from mAb-X. LLPS was observed with Y-Hc+X-Lc, but not with X-Hc+Y-Lc (), further narrowing the likely cause of LLPS to the mAb-X light chain, and in particular the variable region (VL) of the mAb-X light chain based on the totality of the data.
Correlation of mAb-X self-association propensity with LLPS
Several reported studies on mAbs proposed that LLPS behaviors correlate with self-association propensity.Citation2,Citation10,Citation11 To determine whether such a correlation exists for mAb-X, the F(ab′)2, and the Fab were evaluated by dynamic light scattering (DLS) under various conditions where mAb-X showed LLPS (i.e., 50 mM Tris-HCl, pH7.4). Using DLS, the interaction parameter (kD) can be obtained by plotting the free solution diffusion coefficient as a function of protein concentrations. Self-association slows down molecular movement (corresponding to a lower diffusion coefficient), resulting in a negative kD value. Therefore, the more negative the kD value, the stronger the self-association. As shown in , the diffusion coefficients for mAb-X decreased with increasing protein concentration. This decrease led to a highly negative kD value of −42.8 mL/g, indicating strong self-associations for mAb-X in solution. The F(ab′)2 had a slightly less negative kD value (−38.0 mL/g) compared to the full-length antibody.
Figure 4. Correlation of LLPS with mAb-X self-association. (a) Plot of self-diffusion coefficients against protein concentration to obtain the kD for full mAb-X IgG, F(ab′)2, and Fab. The samples were prepared in 50 mM Tris-HCl, pH 7.4. (b) The effects of sodium chloride on the kD of mAb-X in 50 mM Tris-HCl, pH 7.4. (c) Correlation of kD (obtained by DLS) and plasma shift (obtained by AC-SINS) for seven mAbs.
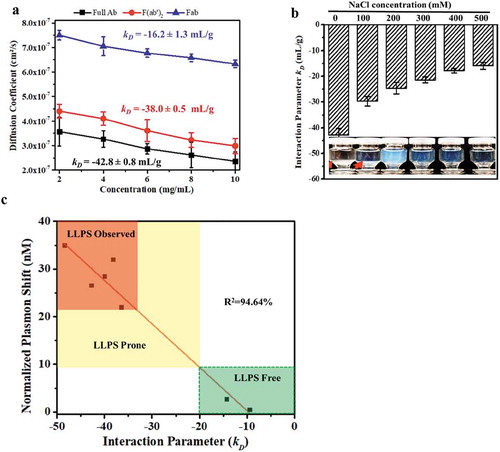
As discussed above, the full-length antibody and the F(ab′)2 both showed LLPS, confirming the correlation between LLPS and strong self-association. In contrast, the Fab self-association (kD = −16.2 mL/g) was much weaker than the full-length antibody or the F(ab′)2. The Fab did not show LLPS under this condition, which led us to speculate that weak self-association does not correlate with LLPS. To confirm this hypothesis, we evaluated the self-association of mAb-X under conditions that promote, as well as inhibit, LLPS. As shown in , addition of NaCl (ranging from 100 to 500 mM) gradually weakened the self-association, with the kD increasing from −42.8 to −17.3 mL/g, and correspondingly inhibited LLPS. In essence, mAb-X exhibited relatively strong self-association (kD <-23 mL/g) in solutions that displayed LLPS, and weaker interactions (kD values >-23 mL/g) in solutions that did not show LLPS.
Under the conditions studied, for mAb-X, DLS and a threshold kD value of <-23 mL/g derived from it is a good guide for process optimization to proactively screen for strong self-association conditions, allowing LLPS to be prevented. However, DLS requires purified protein samples, limiting its application in early antibody development screening. Affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS) is a well-established assay for evaluating protein self-associations that does not require purified sample. This assay can be employed to evaluate antibodies contained in unpurified samples, such as harvested cell culture supernatants.Citation22 In AC-SINS studies, a greater shift in lambda wavelength is indicative of stronger self-association. To ensure AC-SINS can be used as a screening method in our subsequent study, the kD values from DLS and the AS-SINS results for the mAb variants studied in were compared. As represented in , there is a good correlation between the results from these two assays where the values in the plot indicated by the green box correlate with the formation of LLPS, signaling when preventative measures should be taken to mitigate the liability.
In summary, LLPS was highly correlated with a propensity for strong self-association, and both DLS and AC-SINS were found to be useful tools for process development and optimization. These tools are likely also to be useful for protein engineering and structure optimization, as explored in the remainder of this work.
Homology modeling for selecting residues targeted for mutagenesis
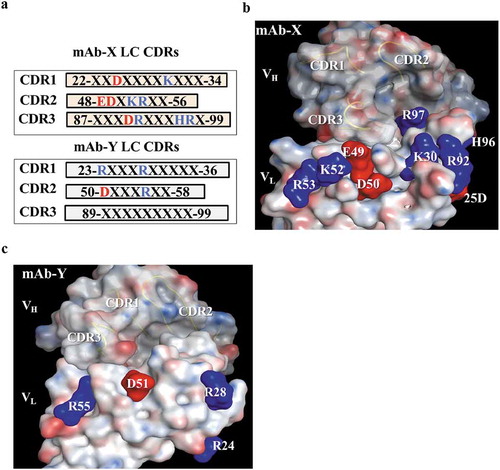
Our data suggest that the VL region of mAb-X is responsible for the LLPS properties, which are primarily driven by electrostatic interactions. Thus, we investigated the charged residues in the VL domain. All of the VL framework residues in mAb-X are fully human germline amino acids, which, by design, minimize antibody immunogenicity. Therefore, framework residues were not our primary focus. Although some could contribute to self-association, we sought to make changes elsewhere, focusing on residues involved in antibody folding, structure, or binding. We compared the VL amino acid sequence of mAb-X with the VL sequence of the well behaved mAb-Y. Interestingly, mAb-X has 10 charged residues within the VL CDRs while mAb-Y has only four charged residues in the VL CDRs. This inspired the hypothesis that the charged residues in the CDRs of mAb-X VL are responsible for self-association leading to LLPS. We particularly wanted to study surface-exposed charged residues because they are more likely to be involved in intermolecular interactions than the buried ones.
Typically, the distribution and configuration of these residues is evaluated using a resolved protein structure, but, at the time of our study, the mAb-X crystal structure had not been solved. We conducted homology modeling on the Fv region of mAb-X to study the distribution of these charged residues on the Fab protein surface (). We applied the Protein Patch Analyzer function from the Molecular Operating Environment (MOE) software onto the modeled structure to identify where the patches of charged residues reside. We identified three potential charged patches in the VL CDR regions: positive Patch 1 formed by amino acids K30 and R92; positive Patch 2 formed by residues K52 and R53; and negative Patch 3 formed by E49 and D50. In the modeled structure for mAb-Y (), the four charged residues (R24, R28, D51 and R55) appeared to reside far away from each other, and therefore did not form charged patches. The solvent-accessible surface areas (ASAs) for the charged residues in the VL CDRs of mAb-X were generated using the Protein Property function in MOE software; the values are summarized in . Five charged residues, D25, K30, R92, K52, and R53, that are highly exposed were selected for further investigation. Although the negatively charged residues E49 and D50 are not highly exposed in the modeled structure, they were also targeted for mutagenesis. These two residues were considered potentially important because long-range electrostatic interactions can still be effective when compared to hydrophobic interactions. H96 was ruled out for further evaluation because of the pKa value of histidine (not charged at pH7.4). Amino acid residues R97 and D91 were not considered because of their low surface exposures and the excessive distances from the identified patches.
Table 1. Mutation design for the selected charged CDR residues.
Figure 5. Identification of surface-exposed charged amino acids in the CDRs, based on the homology model. (a) Protein sequences of light chain CDRs for mAb-X and mAb-Y, showing positively (blue) and negatively (red) charged amino acids. (b-c) Molecular surface of mAb-X (b) and mAb-Y (c) Variable fragment homology models generated using the MOE.2016 software package. VL CDR charge amino acids are illustrated on the surface. VH CDR loops are also illustrated.
Identification of residues responsible for LLPS
The seven charged residues were first changed individually to alanine by substitution mutations to disrupt potential charge-charge interactions, and these alanine-substituted variants were evaluated using AC-SINS and DLS. As shown in , these mutations had dramatically different effects on self-association. The alanine variants on five of the charged residues D25, K30, E49, D50 and R92 resulted in a significant shift in plasmon wavelength as measured by AC-SINS, and subsequently evaluated by DLS where they also had reduced kD values. Mutation of the other two residues, K52 and R53, had minimal effect on the plasmon wavelength shift and were not evaluated further. The five mutants that showed a significant shift in AC-SINS plasmon shift were further evaluated by DLS. These results suggest these five charged residues (D25, K30, E49, D50 and R92) in the VL CDRs play a significant role in self-associations of mAb-X. Among the five residues, D50 and K30 appeared to have the greatest effects on self-association, with each variant exhibiting a profound effect on reducing self-association as evaluated by both DLS and AC-SINS.
Table 2. Molecular properties and self-association behaviors of mAb-X mutants.
Ranking of the five influential charged residues according to their importance toward driving self-association was determined by their kD values is: D50> K30> R92> D25> E49. These five variants were prepared at 10 mg/mL in 50 mM Tris, pH 7.4 (a condition where mAb-X was previously observed to undergo LLPS), and their solution behaviors were evaluated. The mutant D50A did not show LLPS. K30A still showed LLPS, but the extent was substantially reduced; for example, a very small dense phase and longer time (48 h) to form LLPS were observed. In contrast, the R92, D25 and E49 mutants still showed LLPS within 2 h as was seen with the wildtype. These data are consistent with their AC-SINS and DLS results, and suggested that D50 and K30 are the most critical residues responsible for self-association and further LLPS. Notably, K30 and D50 are also critical to antigen-binding affinity since mutating these two residues significantly reduced the mAb-X affinity to the antigen. This finding illuminated the challenges of LLPS mitigation, and signaled that resolution of the liability without losing mAb-X activity would involve engineering more than these two residues. Importantly, despite the significance of D50 driving LLPS, mutating this influential residue to other amino acids is expected to significantly decrease the antigen binding activity, ruling out the possibility of targeting this residue for protein engineering.
For therapeutic mAbs, when protein engineering is required, substituting targeted amino acid positions with germline encoded at the same positions are often performed because such a mutation is less likely to introduce immunogenicity. Here, the uncharged germline residues (i.e., N, Q and S) from several different closely matched germlines are encoded at the six positions, as listed in . Since their size and side chain are similar to the residues being targeted, the influence of charge alone can be evaluated. In light of the fact that the most influential position of D50 could not be substituted without loss of binding affinity, all other charged residue positions were mutated again, this time by exchanging germline residues rather than encoding alanine substitutions. As measured by AC-SINS (), mutations on K30, E49, and R53 resulted in reduced self-association, while mutations on D25, K52 and R92 had minimal impact on self-association. Based on the results for mutation to D25, this position is deemed to not be critical to self-association and LLPS, and was therefore dropped from further studies. DLS results also confirmed that the three variants K30Q, E49Q and R53Q had less self-association, revealing their contribution, but when the solution behaviors of these three mutants were also evaluated in 50 mM Tris, pH 7.4, they still exhibited LLPS.
To better understand these results, we assessed whether these mutations altered the net charge of mAb-X, and determined the pI for the mutants using a capillary isoelectric focusing immunoassay (cIEF). As shown in , the mutations did not alter pI significantly, and therefore the differential impacts on LLPS as seen among the 6 positions tested were not caused by the subtle changes in pI.
Our results confirmed that electrostatic interactions are crucial for the LLPS properties of mAb-X. Residues K30 and D50 are the most critical, and E49 in the charged patch 1 and R92 in the charged patch 3 adjacent to D50 and K30, respectively, also demonstrate significant contributions to the self-association and LLPS of mAb-X. These data also reveal mitigating self-association does not invariably solve LLPS problems.
Mitigation of LLPS while maintaining antigen binding affinity
Mutating critical residues that contribute to electrostatic interactions to germline residues proved to be ineffective at mitigating LLPS; however, the data demonstrated how significant these charged residues are to LLPS. To achieve effective repression of the electrostatic (attractive) interactions, we encoded additional mutations on the identified residues to introduce repulsion, substituting amino acids with opposing charged residues, such as from E to K or from R to E. As shown in , other than variants E49K and R52E, several of the variants (K30E, R53E and R92E) had significantly reduced self-association. As anticipated, mutating them to opposingly charged residues was more effective than changing them to the uncharged germline residues in reducing self-association. Additionally, we also compared the effectiveness of D and E substitution at position K30 with the intention of studying whether the length of the side chain plays a role, where glutamate (E) has a slightly longer side chain than aspartate (D). As shown in the , comparing mutations K30E and K30D, no significant difference the absolute magnitude of self-association was seen as determined by kD when we interchanged these two residues. In fact, we observed a bigger impact on antigen-binding affinity of mutation when substituting residue K30 to D30.
Evaluation of the solution behaviors of these charge-altered variants showed that K30E and K30D did not exhibit LLPS, but both substitutions significantly decreased the antigen-binding affinities, again demonstrating the key role residue K30 played in both LLPS and target binding. Three other charge variants K52E, R53E, and R92E still exhibited LLPS, despite having reduced self-association, but had no effect on binding. Therefore, with the goal of reducing self-association and LLPS without reducing affinity to the target, we combined those charged-altering mutations that reduced self-association without significantly affecting affinity, while the residues (K30 and D50), which are important for target binding, remain unchanged. Employing this novel mutagenesis strategy, the dual variant R53E/R92E was engineered, tested, and exhibited no self-association. To confirm this dual variant was completely free of LLPS, R53E/R92E protein samples were prepared at 30, 50 and 110 mg/mL in the process buffers where mAb-X demonstrated LLPS and incubated at 4ºC for 24 hours. LLPS no longer occurred under these conditions. Combining dual mutations that together suppressed self-association without having a negative impact on antigen-binding affinity was an effective protein engineering approach for mAb-X: the resulting double-mutation variant R53E/R92E did not affect antigen-binding affinity and abolished LLPS.
Discussion
In this work, the LLPS behaviors that posed significant challenges to large-scale bioprocessing for mAb-X were investigated and mitigated through process optimization and protein engineering. The LLPS behaviors of mAb-X were governed by pH, ionic strength, protein concentration, and temperature, and similar to LLPS behaviors of mAbs previously reported.Citation1–Citation6 Sucrose, glutamate-arginine, and sodium chloride were found to be effective to suppress the LLPS of mAb-X during downstream processing. However, the amount of these excipients required for the processing buffers to prevent LLPS either compromised process performance or were suboptimal for other reasons (e.g., high ionic strength). Our mutagenesis strategy solved these issues at the molecular level by targeting residues that drove self-association. A strategy that included exploring substitutions that addressed side chain length, refining the variant and combining charge-altered substitutions mitigated mAb-X LLPS. The findings and the resulting impacts on manufacturing identified in this work may be translatable and provide a general framework for how to approach other mAbs with similar LLPS issues.
LLPS-elicited manufacturing challenges can be alleviated by process optimization, albeit with tradeoffs. From a process development perspective, in our view the ideal strategy for prevention of LLPS is to systematically identify LLPS propensities at an early lead optimization stage, and take mitigation action through protein engineering approaches. Issues early in drug development can be identified by evaluating solution behaviors of the mAb of interest in typical downstream processing buffers. Many excipients have been found to inhibit LLPS effectively, making LLPS more manageable for formulation development, where there are often fewer constraints around use of costly excipients at high concentrations. However, in a large-scale bioprocessing context, use of excipients is far less attractive, and therefore preventing the problem by understanding and selectively modifying the intrinsic properties of the protein that cause LLPS is more practical. Our study highlights the critical role of understanding the underlining driving forces responsible for LLPS in determining the appropriate mitigation strategy. Protein engineering offers not only an illuminating understanding of the root cause, it can simultaneously remedy the manufacturing challenge by eliminating the problem.
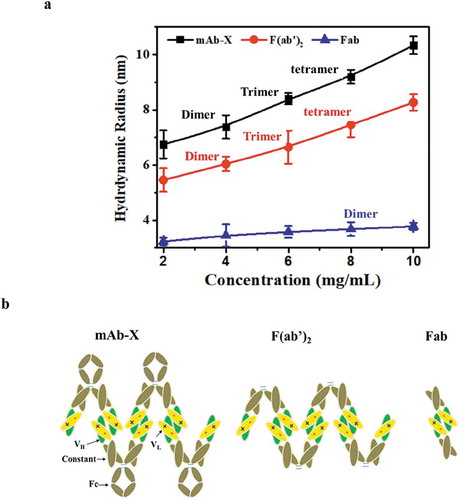
Several recent published reports proposed that self-association correlates with LLPS propensity for mAbs.Citation2,Citation10,Citation11 Such correlations were also observed in our work, yet despite this propensity, we discovered that this relationship was complex and did not always result in LLPS for mAb-X. Here, weak self-association as determined by DLS usually did not lead to LLPS. For example, under the conditions tested on mAb-Y, mAb-X and their mutant variants, a DLS-measured kD value of less than −23 mL/g ( and ) and a plasmon shift greater than 5 nm as measured by AC-SINS defines the threshold self-association values for onset of LLPS. We also found that the F(ab′)2 version of mAb-X had LLPS behaviors very similar to the full-length antibody, ruling out involvement of the Fc domain. According to the protein sizes estimated from DLS data (), the F(ab′)2 was able to form higher-order complexes more similar to full-length mAb-X, but the Fab was only able to form dimers, and interestingly did not have an LLPS issue. Moreover, for mAb-X, we observed that the CDR regions in the light chain are predominantly responsible for inter-molecule interactions. Based on these observations, we proposed simplified self-association models for the full-length mAb, F(ab′)2 and Fabs (). Interactions between Fab and Fc regions were excluded from the models for simplicity purpose; however, weak interactions between Fab and Fc likely exist, although they are not critical to the LLPS of mAb-X. In addition to strong self-association, we proposed that the formation of higher-order complexes, or an extended self-association network, is another prerequisite for LLPS.
Figure 6. Proposed model of mAb-X self-interaction. (a) Hydrodynamic size as a function of concentration for full mAb-X, F(ab′)2 and Fab. Size (as a dimer, trimer, or tetramer) of the self-associated complexes was estimated by DLS data. (b) Proposed model for self-interactions.
LLPS properties of mAbs have been extensively studied recently, yet there remains a limited understanding of how amino acid attributes such as charge property and surface distribution drive the underlining mechanism. Chow et al. recently proposed that, according to their mAb data, phase separation was mainly driven by charge and not hydrophobicity (from the CDRs).Citation11 Casaz et al. reported substituting a single glutamate residue (in the CDR) with lysine effectively mitigated the LLPS by reducing electrostatic interactions.Citation6 In our study, electrostatic interactions played a key role in driving LLPS for mAb-X. In addition to the reported findings that pH and ionic strength govern LLPS behaviors,Citation2–Citation5,Citation10 we hypothesize that solvent accessible charged residues play a critical role in LLPS. We compared the sequences and modeled structure of mAb-X with another mAb that binds to the same antigen but had no LLPS issue, which revealed that mAb-X had more charged residues in the CDR regions. Interestingly, the topographic surface distribution revealed these charged residues formed two positively and one negatively charged patches, as illustrated in (wildtype).
Figure 7. Illustration of proposed CDR charge distributions of the mAb-X wild-type (a) and LLPS-free mutants (b-d). Blue and red shades represent positively and negatively charged patches, respectively. Green arrows represent potential salt bridges.
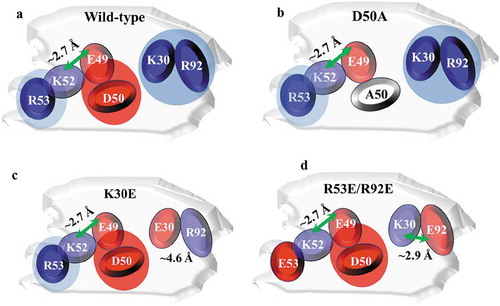
Our mutagenesis study proved the charged residues R53, D50, K30 and R92 in the CDRs of the VL are prominent in the self-association and LLPS. E49 and K52 affect LLPS to a lesser extent. The distance between the E49 side-chain carbonyl oxygen and the K52 side-chain nitrogen atoms measured in the homology model is about 2.7 Å, which is within the threshold distance (4Å) required for formation of a salt bridge.Citation23 This suggests that E49 and K52 may form an intra-molecular salt bridge, neutralizing their charges and reducing their involvement in the inter-molecule electrostatic interactions. We propose that the charge-charge interactions mainly occur between the three positively charged residues K30, R92 and R53, and the negatively charged residue D50, which was identified as the most critical charged residue in the interaction interface. The mutation of D50 to A50 significantly reduced the self-associations and effectively inhibited LLPS. As illustrated in (mutant D50A), replacing the negatively charged D50 with A50 resulted in only one remaining negatively charged CDR residue in the interface, E49. Hence, the attraction between E49 and other positively charged residues is likely weaker than the repulsion produced by the four residues.
Mutating K30 to A30 effectively reduced self-association and LLPS, but was unable to eliminate LLPS. In contrast, changing it to a negatively charged glutamate effectively prevented LLPS from happening. We speculate such a mutation disrupts the original K30-D50 charge-charge interaction, and the introduced E30 likely neutralizes the nearby R92. According the homology model, the distance between the E30 side-chain carbonyl oxygen and the R92 side-chain nitrogen atoms is about 4.6 Å, slightly longer than the threshold distance (4Å) for formation of a salt bridge. A salt bridge may not form between E30 and R92, but it seems that the local charge distribution is balanced. Although the inter-molecular interaction between R53 and D50 remained undisturbed, their interactions are not strong enough to cause LLPS. We showed that mutating either of the two most crucial charged residues, K30 and D50, resulted in significant reduction of antigen-binding affinity, while mutating R53E or R93E reduced self-associations, but most importantly not target binding. Therefore, we engineered a double mutant (R53E/R92E) with K30 and D50 unchanged. Combining the two mutations at R53 and R92 significantly reduced mAb-X self-association and eliminated LLPS while slightly improving target binding. As depicted in (R53E/R92E), the introduced E92 mutation likely neutralizes K30, breaking the original inter-molecular K30-D50 interaction. The distance between the E92 side-chain carbonyl oxygen and the K30 side-chain nitrogen atoms is about 2.9 Å; therefore, a salt bridge between E92 and K30 may form.
Mutating R53 to E53 introduced a negatively charged glutamate residue generating inter-molecular repulsions. The difference in the number of charged residues in the CDRs of mAb-X compared to the well-behaved mAb-Y attracted our attention. Intriguingly, in variants K30E and the final dual variant R53E/R92E where LLPS is resolved, the number of charged residues remain the same as the wildtype. Based on these findings, we conclude that the formation of both negatively and positively charged patches and a sufficient number of charged residues in the CDRs contingent on their distribution on the protein surface are the two dominant factors that drive LLPS. Therefore, our protein engineering strategy can be practiced by disrupting these charge patches or introducing charge repulsion. For mAb-X, we conclude that the inter-molecular charge-charge interactions resulted in LLPS. These charged patches likely constitute the self-association interface, with D50 and K30 forming a salt bridge. Indeed, comparison of the charge distribution of the wildtype exhibiting LLPS to the evenly distributed charged residues of the mutants () shows that the impact of charged patches on the mAb surface is the predominant force driving protein-protein interactions. With these results, we hypothesize reducing the number of charged residues in the mAb is paramount to avoiding or disrupting charged patches in the CDRs.
There is no effective a priori approach to predict whether a mAb will exhibit LLPS during downstream processing. Our study illuminates steps in the screening cascade to evaluate the potential for LLPS. Moving forward, we propose to evaluate the surface charge of a mAb destined for development, considering the number and position on the protein structure. For instance, when there are several charged residues in the CDRs (e.g., 10 in mAb-X vs. 4 in mAb-Y), the mAb’s behavior in solution at pH close to the pI at a low ionic strength (a condition favoring LLPS) should be evaluated. For this purpose, use of in silico graphics modeled on the protein structure resolved by crystal X-ray diffraction or NMR would be ideal. Others have reported antibody engineering guided by crystal structures that successfully reduced viscosity and self-association.Citation6,Citation24 When a crystal structure is not available, the combination of hydrogen-deuterium exchange mass spectrometry (HDX-MS) and homology modelling is also an effective approach.Citation16,Citation24 Generally, HDX-MS is time consuming, and tight development timelines often do not allow such an in-depth study for a therapeutic mAb at an early development phase.
In our work, the modeled structure used for analyzing the charge distribution of the CDR residues led to protein engineered variants and meaningful data. Furthermore, the modeled structure was consistent with the crystal structure of the Fab for mAb-X that was fully resolved (data not shown) after the completion of this work, supporting the strategy of using homology modeling. Here, we used in silico protein analysis and an engineering strategy for mitigating a critical manufacturing LLPS issue. We identified the number of charged residues in the CDRs, carried out homology modelling and charged patch analysis on the CDR residues, used protein engineering to break potential charge-charge interactions, and then evaluated the impact of those variants on LLPS and antigen-binding affinity. With the success of our methods, we propose to use them in progression to mitigate protein self-association and LLPS early in the drug development path. Resolving LLPS challenges with molecular engineering improves overall product stability and simplifies formulation development and downstream manufacturing.
Materials and methods
Chemicals
The chemicals used in this study were obtained from J.T. Baker (Phillipsburg, NJ). Buffers and cleaning solutions were prepared in-house. Citrate-stabilized 20 nm gold nanoparticles were obtained from Ted Pella Inc. (Redding, CA). Goat anti-human IgG (Fcγ specific, 109–005-098) and goat whole IgG (nonspecific, 005–000-003) antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Poly (ethyleneglycol) methyl ether thiol (PEG, 2000 MW) was obtained from Sigma-Aldrich (St. Louis, MO).
Antibody generation
The mAbs evaluated in this study are human monoclonal antibodies comprising two identical heavy chains and two identical light chains, with an overall molecular weight of approximately 150 kDa. Unless noted otherwise, the mAbs were pre-purified with greater than 95% monomer purity with MabSelect™ SuRe™ (MSS) Protein A resins from GE Healthcare LifeSciences (Marlborough, MA) or with Capture selectTM CH1 reins from ThermoFisher Scientific (Grand Island, NY).
To generate mAb-X, DNA encoding the variable heavy (VH) and the variable light (VL) chains of mAb-X were synthesized by GeneART (Life Technologies, Carlsbad, CA) and cloned into in-house mammalian expression vectors as IgG1, IgG2, or IgG4 subclasses. These subclasses differed primarily in their constant region, and particularly in their hinges and upper CH2 domains, which are involved in binding Fc receptors.Citation21 Single or multiple mutations were introduced into the variable region by using PCR overlap extension with mutagenic primers. DNA encoding the mutated variable regions were cloned into the IgG1 expression vector. DNA encoding chimeric mAb-X/mAb-Y variants were constructed by restriction enzyme digestion and ligation-based molecular cloning.
The expression vectors were transfected transiently into human embryonic kidney 293 (HEK293) cells using 293Fectin (Life Technologies) or into G22 Chinese hamster ovary (CHO) cells using polyethylenimine according to the manufacturer’s instructions. Following transfection, medium was cultured from 6 or 10 days, filtered through a 0.22 μm sterile filter, and purified as described above.
To generate Fab or F(ab)′2, DNA encoding the VH and VL of wildtype or mutant mAb-X was cloned into an in-house mammalian expression vector encoding a human lambda chain and a human IgG CH1 and hinge regions truncated at position C233 (for Fab) or P243 [for F(ab)′2] according to Kabat numbering.Citation25 The expression vectors were transfected transiently into CHO cells as described above.
Chromatography
Chromatographic experiments were carried out on an ÄKTA Avant controlled by Unicorn software version 6.4 (GE Healthcare LifeSciences). The resins were packed to a bed height of 19 ± 3 cm in Vantage L laboratory columns (Millipore, Billerica, MA), which had an inner diameter of 1.15 cm. Sample protein concentration was measured using a NanoDrop™ 2000 (Thermo Fisher Scientific, Wilmington, DE).
Liquid-liquid coexistence curve
Opalescence and turbidity of the solution were measured as percent transmittance using an 8453 UV system from Agilent Technologies (Santa Clara, CA). The 8453 UV system was equipped with a temperature control module connected to a water bath. The measurement was carried out at 600 nm (where the proteins in this study have no absorbance) in a quartz cuvette with a path length of 1 cm. Samples were incubated at 35°C for 10 min, then the temperature was decreased gradually to 2°C at the rate of 0.5°C per minute. The temperature for the onset of phase separation (Tcloud), as indicated by a rapid decrease in transmittance, was then determined and plotted against protein concentration to generate a liquid–liquid coexistence curve (or liquid–liquid phase diagram). Triplicate measurements were made for each sample, and the averaged values were used to generate liquid-liquid coexistence curves. All measurements are triplicates.
Dynamic light scattering for interaction parameter determination
Each sample was prepared at concentrations of 2, 4, 6, 8 and 10 mg/mL in 50 mM Tris-HCl buffer, pH 7.4, and then filtered using a 0.2 µm filter. Each sample was then centrifuged at 2000 rpm for 2 minutes to remove any potential air bubbles before analysis. Samples were analyzed with a high-throughput, 384-well plate on DynaPro DLS instrument (Wyatt Technology, Santa Barbara, CA) equipped with a 633 nm laser. The scattered light was monitored at 173° to the incident beam and auto correlation functions were generated using DYNAMICS software version 7.1.9.3. The mutual diffusion coefficient (Dm) obtained by DLS was plotted against the protein concentration (g/mL) to obtain the kD from the following equation: Dm = Ds (1+ kD × C). Ds is the self-diffusion coefficient (the value of Dm at infinite dilution), and C is the protein concentration. Ds is obtained from the intercept of a plot of Dm versus C, and kD was obtained from the slope. Three samples were prepared for each condition and all measurements are triplicates.
Affinity capture self-interaction nanoparticle spectroscopy
The AC-SINS assay was performed as described previously.Citation16,Citation22 Briefly, citrate-stabilized 20 nm gold nanoparticles were coated with 80% anti-human Fcγ-specific antibody (capturing) plus 20% nonspecific goat IgG. Non-capture nanoparticles were generated by coating only with nonspecific goat IgG. To measure self-association, mAb samples (50 µg/mL) in phosphate-buffered saline (PBS) pH 7.4 were incubated with either capture or non-capture nanoparticles for 2 h at room temperature. Absorbance was measured on a SPECTROstar Nano UV/Vis plate reader (BMG Labtech, Cary, NC) from 490 to 700 nM. The wavelength of peak absorbance (plasmon wavelength) was identified by using the Find Maximum calculation tool in the MARS data analysis software (BMG Labtech). For each antibody, the normalized plasmon shifts were calculated by subtracting the plasmon wavelength of the non-capture nanoparticles from those of the capture particles. All measurements are duplicates.
In silico homology modeling
The homology model of the mAb-X and mAb-Y variable domains (Fv) was constructed with Molecular Operation Environment (MOE) version 2016.08 (Chemical Computing Group, Inc., [CCG], Montreal, Canada). The MOE numbering scheme (CCG), which combines the loop definitions of the Kabat scheme with the residue numbering of the Chothia scheme,Citation26,Citation27 was used for antibody numbering. Following homology model construction, the VH and VL sequences of mAb-X were used to search the Fab Database from the Protein Data Bank (PDB) structures for suitable templates. Sequence alignment scores for both the framework and the CDR were calculated according to the BLOSUM62 amino acid substitution matrix.Citation27 CDR loops were calculated as similarity, and the structure score (geometric mean) of an antibody subdomain was calculated as follows: structure score = topology × geometry × (0.7 × B-Factors) × (0.65 × occupancy) × (0.75 × Resolution) × (0.65 R/Rfree).Citation27
Structure 4QF1, which had an identity to mAb-X of 100% for VL and 94.4% for VH to mAb-X and a structure score of 91.6% for VL and 91.2% for VH, was chosen as the framework template. CDRs were modeled using the MOE Model Build Assign Loops function. The similarity and overall structure scores of each CDR loop were as follows (% similarity, % structure): L1 (86.6, 97.7), L2 (100, 99.3), L3 (93.1, 92), H1 (95.1, 94.7), H2 (91.3, 95.6), H3 (48.1, 92.4). The MOE Protein Patch Analyzer was used to predict which residues would contribute to positive, negative, or hydrophobic patches. An analysis with MOE Protein Property was used to characterize residue properties, such as charge, water-accessible surface area, in square Angstroms (ASA A2), and surface exposure percentage.
Binding affinity assay
The binding affinities of mAb-X and its mutant variants to recombinant human antigen were measured by Biolayer Interferometry on an Octet384 instrument (ForteBio, Menlo Park, CA). To determine intrinsic binding affinity, 2 μg/mL antibodies mixed in 1× Kinetics Buffer (PBS pH 7.2, 3 mg/mL bovine serum albumin, 0.05% (v/v) Tween 20, ForteBio) were captured by anti-human IgG Fc biosensors (ForteBio). Captured antibodies were washed, and association and dissociation measurements were carried out by using serial dilutions of the antigen protein. A nonlinear fit of the experimental data was carried out in Octet384 software v.7.2 to obtain the dissociation constants.
Capillary isoelectric focusing immunoassay
Antibody samples were analyzed on a PeggySue instrument (ProteinSimple, San Jose, CA) according to the manufacturer’s instructions.Citation28 Briefly, mAbs were diluted to 4 µg/mL in Simple Dilute (ProteinSimple) and mixed with Premix G2 pH separation gradient containing fluorescence-labeled isoelectric point (pI) standards (pI 5.5, 7.3, 8.4 and 9.7, ProteinSimple), 0.25% tetramethylethylenediamine (Amresco, Solon, OH), 50% Premix G2 pH 4 to 10 (ProteinSimple) and 4% of Pharmalytes pH 8 to 10.5 (GE Healthcare Life Sciences). Horseradish peroxidase-conjugated mouse antihuman IgG Fc antibody (Jackson ImmunoResearch) and Peroxide-XDR/Luminol (ProteinSimple) were used for detection. Data analysis was performed using Compass software (ProteinSimple).
Abbreviations
| AC-SINS | = | affinity-capture self-interaction nanoparticle spectroscopy |
| CDR | = | complementarity-determining region |
| DLS | = | dynamic light scattering; |
| Fab | = | antigen-binding fragment |
| Fc | = | crystallizable fragment |
| IgG | = | immunoglobulin G |
| kD | = | interaction parameter |
| LC | = | light chain |
| LLPS | = | liquid-liquid phase separation |
| mAb | = | monoclonal antibody |
| RSA | = | reversible self-association |
Disclosures of potential conflicts of interest
All authors were current or former employees of AstraZeneca at the time of this research study, with stock and/or stock options in AstraZeneca.
Acknowledgments
We would like to thank Lu Shan and Vaheh Oganesyan for their contributions and discussions. Special thanks to Albert Schmelzer for his careful review of the article. Editorial support was provided by Frances McFarland (funded by AstraZeneca).
References
- Wang Y, Lomakin A, Latypov RF, Benedek GB. Phase separation in solutions of monoclonal antibodies and the effect of human serum albumin. Proc Natl Acad Sci U S A. 2011;108:16606–11. doi:10.1073/pnas.1112241108.
- Mason BD, Zhang-van Enk J, Zhang L, Remmele RL Jr., Zhang J. Liquid-liquid phase separation of a monoclonal antibody and nonmonotonic influence of Hofmeister anions. Biophys J. 2010;99:3792–800. doi:10.1016/j.bpj.2010.10.040.
- Ahamed T, Esteban BN, Ottens M, van Dedem GW, van der Wielen LA, Bisschops MA. 2007. Phase behavior of an intact monoclonal antibody. Biophys J. 93:610–19. doi:10.1529/biophysj.106.098293.
- Raut AS, Kalonia DS. Effect of excipients on liquid-liquid phase separation and aggregation in dual variable domain immunoglobulin protein solutions. Mol Pharm. 2016;13:774–83. doi:10.1021/acs.molpharmaceut.5b00668.
- Reiche K, Hartl J, Blume A, Garidel P. Liquid-liquid phase separation of a monoclonal antibody at low ionic strength: influence of anion charge and concentration. Biophys Chem. 2017;220:7–19. doi:10.1016/j.bpc.2016.08.003.
- Casaz P, Boucher E, Wollacott R, Pierce BG, Rivera R, Sedic M. 2014. Resolving self-association of a therapeutic antibody by formulation optimization and molecular approaches. MAbs. 6:1533–39. doi:10.4161/19420862.2014.975658.
- Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–38. doi:10.1080/19420862.2018.1556465.
- Shukla AA, Hubbard B, Tressel T, Guhan S, Low D. Downstream processing of monoclonal antibodies–application of platform approaches. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:28–39. doi:10.1016/j.jchromb.2006.09.026.
- Shukla AA, Wolfe LS, Mostafa SS, Norman C. Evolving trends in mAb production processes. Bioeng Transl Med. 2017;2:58–69. doi:10.1002/btm2.10061.
- Luo H, Lee N, Wang X, Li Y, Schmelzer A, Hunter AK. 2017. Liquid-liquid phase separation causes high turbidity and pressure during low pH elution process in Protein A chromatography. J Chromatogr A. 1488:57–67. doi:10.1016/j.chroma.2017.01.067.
- Chow CK, Allan BW, Chai Q, Atwell S, Lu J. Therapeutic antibody engineering to improve viscosity and phase separation guided by crystal structure. Mol Pharm. 2016;13:915–23. doi:10.1021/acs.molpharmaceut.5b00817.
- Bethea D, Wu SJ, Luo J, Hyun L, Lacy ER, Teplyakov A 2012. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 25:531–37. doi:10.1093/protein/gzs047.
- Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J Pharm Sci. 2008;97:4219–27. doi:10.1002/jps.21322.
- Chaudhri A, Zarraga IE, Kamerzell TJ, Brandt JP, Patapoff TW, Shire SJ. 2012. Coarse-grained modeling of the self-association of therapeutic monoclonal antibodies. J Phys Chem B. 116:8045–57. doi:10.1021/jp301140u.
- Arora J, Hu Y, Esfandiary R, Sathish HA, Bishop SM, Joshi SB. 2016. Charge-mediated Fab-Fc interactions in an IgG1 antibody induce reversible self-association, cluster formation, and elevated viscosity. MAbs. 8:1561–74. doi:10.1080/19420862.2016.1222342.
- Geoghegan JC, Fleming R, Damschroder M, Bishop SM, Sathish HA, Esfandiary R. Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. MAbs. 2016;8:941–50. doi:10.1080/19420862.2016.1171444.
- Esfandiary R, Parupudi A, Casas-Finet J, Gadre D, Sathish H. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatic and hydrophobic interactions. J Pharm Sci. 2015;104:577–86. doi:10.1002/jps.24237.
- Pepinsky RB, Silvian L, Berkowitz SA, Farrington G, Lugovskoy A, Walus L. 2010. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci. 19:954–66. doi:10.1002/pro.372.
- Kheddo P, Bramham JE, Dearman RJ, Uddin S, van der Walle CF, Golovanov AP. Investigating liquid-liquid phase separation of a monoclonal antibody using solution-state NMR Spectroscopy: effect of Arg.Glu and Arg.HCl. Mol Pharm. 2017;14:2852–60. doi:10.1021/acs.molpharmaceut.7b00418.
- Bekard IB, Asimakis P, Bertolini J, Dunstan DE. The effects of shear flow on protein structure and function. Biopolymers. 2011;95:733–45. doi:10.1002/bip.21646.
- Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi:10.3389/fimmu.2014.00520.
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T. 2014. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 6:483–92. doi:10.4161/mabs.27431.
- Kumar S, Nussinov R. Close-range electrostatic interactions in proteins. Chembiochem. 2002;3:604–17. doi:10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X.
- Dobson CL, Devine PW, Phillips JJ, Higazi DR, Lloyd C, Popovic B. 2016. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 6:38644. doi:10.1038/srep38644.
- Kabat EA. National Institutes of H, Columbia U. Sequences of proteins of immunological interest. Bethesda (MD): U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health; 1991.
- Chothia C, Lesk AM. 1987. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 196:901–17.
- Maier JK, Labute P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins. 2014;82:1599–610. doi:10.1002/prot.24576.
- Michels DA, Tu AW, McElroy W, Voehringer D, Salas-Solano O. Charge heterogeneity of monoclonal antibodies by multiplexed imaged capillary isoelectric focusing immunoassay with chemiluminescence detection. Anal Chem. 2012;84:5380–86. doi:10.1021/ac3008847.