
ABSTRACT
Asthma is characterized by airway hyperresponsiveness and inflammation, as well as underlying structural changes to the airways. Interleukin-4 (IL-4) is a key T-helper type 2 (Th2) cytokine that plays important roles in the pathogenesis of atopic and eosinophilic asthma. We developed a novel humanized anti-IL-4Rα antibody that can potently inhibit IL-4/IL-13-mediated TF-1 cell proliferation. Using monocytes isolated from human peripheral blood mononuclear cells (PBMCs), we revealed a critical role of CD32 in modulating the immune responses of monocytes in response to blockade of IL-4Rα signaling pathway. We, therefore, devised a new strategy to increase the efficacy of the anti-IL-4Rα monoclonal antibody for the treatment of asthma and other atopic diseases by co-engaging CD32 and IL-4Rα on monocytic cells by choosing IgG classes or Fc mutations with higher affinities for CD32. The antibody with selectively enhanced affinity for CD32A displayed superior suppression of IL-4-induced monocytes’ activities, including the down-regulation of CD23 expression. Intriguingly, further analysis demonstrated that both CD32A and CD32B contributed to the enhancement of antibody-mediated suppression of CD23 expression from monocytes in response to blockade of IL-4Rα signaling. Furthermore, inhibition of IgE secretion from human PBMC by the antibody variants further suggests that the complex allergic inflammation mediated by IL-4/IL-4Rα signaling might result from a global network where multiple cell types that express multiple FcγRs are all involved, of which CD32, especially CD32A, is a key mediator. In this respect, our study provides new insights into designing therapeutic antibodies for targeting Th2 cytokine-mediated allergic pathogenesis.
Introduction
Asthma is characterized by airway hyper - responsiveness (AHR) and inflammation, as well as underlying structural changes to the airways. Asthma affects an estimated 241 million children and adults worldwide, of which approximately 5–10% is severe asthma.Citation1,Citation2
Interleukin-4 (IL-4) and interleukin-13 (IL-13) are major T-helper type 2 (Th2) cytokines that play important roles in the pathogenesis of atopic and eosinophilic asthma. IL-4 plays a key role in regulating naïve T cells to develop into Th2-like cells capable of producing cytokines such as IL-4, IL-13, and IL-5. IL-4 also determines the class switch of human B cells to expression of IgE and IgG4 and mouse B cells to IgE and IgG1. In addition, IL-4 upregulates major histocompatibility complex class II (MHCII) and CD23 expression, as well as the expression of the IL-4 receptor on B cells and myeloid lineage cells, such as monocytic cells.Citation3 IL-4 receptors (IL-4R) are present in a broad spectrum of tissues, including hematopoietic, endothelial, epithelial, muscle, fibroblast, hepatocyte, and brain tissues. The receptor consists of a 140 kDa IL-4Rα chain that binds IL-4 with high affinity (KD 20 to 300 pM); this chain is shared with IL-13. There are two types of receptor complexes centered on IL-4Rα, type I: IL-4Rα/γc and type II: IL-4Rα/IL-13Rα1. The binding of IL-4 to IL-4Rα, in the type I complex, induces dimerization with the common γ chain (γc), which then activates JAK1 and JAK3. Activated JAKs result in the phosphorylation and activation of STAT-6. Phosphorylated STAT-6 dimers translocate to the nucleus and activate the transcription of genes, including CD23 and MHCII in B cells, IL-4, and IL-13 in T cells, and eotaxin in fibroblasts.Citation3
Multiple lines of evidence support the role of IL-4/IL-13 pathway in promoting asthma and the atopic syndrome. In animal models, blockade of the IL-4/IL-13 pathway through the use of antibodies or antagonists of IL-4/IL-13 and IL-4Rα have been shown to reduce AHR, airway inflammation, mucus overproduction, eosinophil recruitment, and airway remodeling.Citation1 In clinical studies, patients on dupilumab, an anti-IL-4Rα monoclonal antibody (mAb) that targets IL-4Rα, thereby inhibiting the binding of both IL-4 and IL-13, showed an 87% reduction of the asthma exacerbation, improved forced expiratory volume in 1 s (FEV1) and improvements in quality of life.Citation2,Citation4 Taken together, the results suggest that targeting the IL-4/IL-13 signaling pathway, especially with antibodies, could provide additional therapeutic means to treat asthma and other atopic diseases.
The cytotoxic functions mediated by the Fc domain of therapeutic antibodies via binding to Fcα receptors, for instance, antibody-dependent cell-mediated cytotoxicity, and antibody-dependent cellular phagocytosis, are crucial for their therapeutic efficacies.Citation5-Citation8 Four human subclasses of IgG (IgG1, IgG2, IgG3, IgG4) are produced in response to various antigens. Correspondingly, there are two classes of FcγRs exhibiting opposite effector functions: 1) the activating receptors (hFcγRI, hFcγRIIA, hFcγRIIC, FcγRIIIA, and FcγRIIIB), which possess immunoreceptor tyrosine-based activation motifs (ITAMs) in their intracytoplasmic domain, and 2) the inhibitory receptor (hFcγRIIB), which possesses an immunoreceptor tyrosine-based inhibitory motif (ITIM).Citation9 Most cells express several FcγRs and the effector responses mediated by different IgG subclasses are the sum of their differential affinities for activating or inhibitory hFcγRs.Citation9-Citation12
Human monocytic cells, including monocytes, monocyte-derived macrophages and dendritic cells (DCs) express all three major FcγRs, CD64 (FcγRI), CD32 (FcγRII), and CD16 (FcγRIII). These cells constitute an important component of both normal and asthmatic airways, and play a number of critical roles in the regulation of asthmatic airway inflammation.Citation13,Citation14 CD32A is the most abundant and prevalent activating FcγR on monocytes and macrophages.Citation12,Citation15 Indeed, CD32A was found to promote secretion of several pro-inflammatory cytokines including tumor necrosis factor, IL-1β, and IL-8 from monocytes/macrophages isolated from patients with rheumatoid arthritis.Citation16 By contrast, CD32B, the inhibitory counterpart of CD32A, is expressed at relatively low levels on monocytes and macrophages. As the only FcγR expressed on B cells, the inhibitory functions of CD32B on B cell activities are well documented.Citation17-Citation19 However, CD32B is expressed and inducible by IL-4 on human monocytes and can inhibit CD32A-mediated macrophage activation.Citation20,Citation21 Furthermore, it was shown that co-expression of CD32A and CD32B on monocyte-derived DCs reveals a homeostatic checkpoint for inducing tolerance or immunity by immune complexes (ICs).Citation22 These studies suggest that CD32 might be crucial in modulating the allergic immune responses in asthma via regulating monocytes activities.
In this study, we reveal a critical role of CD32, especially CD32A, in modulating the immune responses of monocytes in response to blockade of IL-4Rα signaling pathway by a novel anti-IL-4α antibody. Based on this finding, we devised a new strategy to increase the efficacy of the anti-IL-4Rα mAb for treatment of asthma and other atopic diseases by co-engaging CD32 and IL-4Rα on monocytic cells by choosing IgG subclasses and Fc mutations with higher affinities for CD32. Both CD32A and CD32B contributed to the enhancement of antibody-mediated suppression of CD23 expression from monocytes in response to blockade of IL-4Rα signaling. Indeed, inhibition of IgE secretion from human peripheral blood mononuclear cells (PBMCs) by the antibody variants further suggest that the complex allergic inflammation mediated by IL-4/IL-4Rα signaling might result from a global network of multiple cell types that express multiple FcγRs, of which CD32, especially CD32A, is a key mediator. In this respect, our study provides insights into designing therapeutic antibodies for targeting Th2 cytokine-mediated allergic pathogenesis.
Results
Characterization of the humanized anti-hIL-4Rα mAb
Mouse mAbs against IL-4Rα were obtained by screening a large panel of hybridomas created from IL-4Rα immunized mice. Clone mAb4-2 exhibiting the highest affinity for IL-4Rα extracellular domains (ECDs) was selected and subsequently humanized. The humanized mAb4-2 (referred as mAb4-2) showed a binding affinity for IL-4Rα ECDs comparable to that of dupilumab ()). Biacore further confirmed that the equilibrium dissociation constant (KD) of both antibodies are within 10−10M ()). Furthermore, the ability of mAb4-2 to block the interaction of IL-4 with IL-4Rα was also found to be comparable to that of dupilumab ()).
Figure 1. Characterization of the anti-IL-4Rα monoclonal antibody clone mAb4-2. (a) Binding abilities of mAb4-2 to IL-4Rα were measured by ELISA. IL-4Rα antigen was coated on the plate. Antibodies, mAb4-2, dupilumab, and control mAb were serially diluted and added to the plate; (b) the equilibrium dissociation constants (KD) of dupilumab and mAb4-2 for IL-4Rα were measured by Biacore; (c) The ability of mAb4-2 to block the interaction of IL-4Rα and IL-4 was assayed by competition ELISA. mAb4-2, dupilumab, and control mAb were serially diluted and pre-incubated with IL-4Ra. The sample mixtures were then added to the plate coated with IL-4.
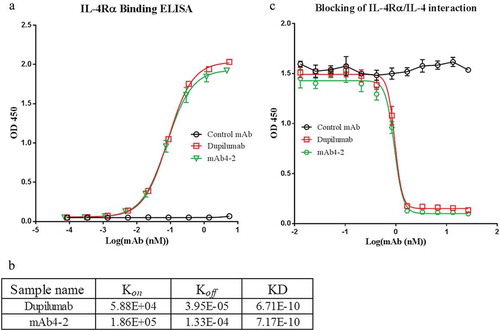
Next, we tested the bioactivity of mAb4-2 in IL-4-dependent TF-1 cell proliferation assays. Addition of mAb4-2 effectively inhibited IL-4-mediated TF-1 cell proliferation with an IC50 of 1.38nM, which is on par with dupilumab, whose IC50 is 1.93 nM ()). Given that dupilumab can inhibit the binding of IL-13 to the receptor complexes by binding to IL-4Rα, we stimulated TF-1 cell proliferation using IL-13 instead of IL-4 and tested the ability of mAb4-2 to inhibit IL-13 dependent cell growth. As expected, mAb4-2 also effectively inhibited IL-13-mediated TF-1 proliferation at an IC50 of 0.23 nM, compared to that of dupilumab (IC50: 0.49 nM). These data demonstrate that mAb4-2, like dupilumab, is a powerful dual blocker that simultaneously inhibits IL-4- and IL-13-mediated cell proliferation ()).
Figure 2. MAb4-2 possesses abilities to inhibit TF-1 cell proliferation mediated by either IL-4 or IL-13. (a) mAb4-2 inhibited IL-4-induced TF-1 proliferation in a dose-dependent manner. Dupilumab, a marketed anti-IL-4Ra mAb, serves as a control mAb; (b) mAb4-2 inhibited IL-13-induced TF-1 proliferation in a dose-dependent manner. Dupilumab serves as a control mAb.
In addition to bioactivities, biophysical and chemical analysis also demonstrated that mAb4-2 has favorable properties as a drug candidate, including purity, thermal stability, and pharmacokinetics (Fig S1A, B, C). Therefore, mAb4-2 was chosen for further characterization and optimization.
Characterization of FcγRs on TF-1 cells that interact with mAb4-2 antibody variants of various IgG subclasses
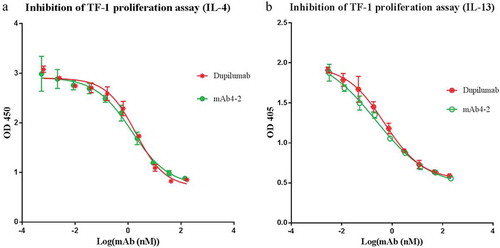
During our initial optimization for the mAb4-2 backbone structure, we discovered that mAb4-2 of an IgG1 subclass (referred as mAb4-2-IgG1) showed a fourfold increase of potency (IC50: 3.8 nM) in inhibiting IL-4-mediated TF-1 cell proliferation compared with that of mAb4-2 of an IgG4 subclass (IC50: 13.3nM; referred as mAb4-2-IgG4) ()). The difference of activities of two IgG subclasses was surprising, as Biacore showed that switching Fc subclasses of mAb4-2 did not change the binding affinities of the two antibodies ()). The Biacore result led us to think that the Fc effects on inhibition of cell proliferation might be due to the interaction of IgG subclasses with FcγR on TF-1 cells.
Figure 3. The mAb4-2 potency of suppressing TF-1 proliferation depends upon the antibody’s Fc. (a) after serial dilution, mAb4-2-IgG1 and mAb4-2-IgG4, the antibody with either IgG1 or IgG4 heavy chain were added to TF-1 cells in the presence of IL-4; (b) the equilibrium dissociation constants (KD) of mAb4-2-IgG1 and mAb4-2-IgG4 for IL-4Rα were obtained using Biacore; (c) FcγRs on the surface of TF-1 cells were identified by FACS using FITC-labeled anti-CD16, anti-CD32, and anti-CD64 specific antibodies. FITC-labeled anti-CD19 serves as negative control.
To test this hypothesis, we first made an N297A mutant (mAb4-2-IgG1-N297A) that abolishes the ability of IgG1 binding to FcγRs owing to inhibition of glycosylation,Citation9 while maintaining its affinity for the IL-4Rα ECD intact. Proliferation assay demonstrated that the potency of mAb4-2-IgG1 was significantly impaired by the N297 mutation (Fig S2), suggesting that the interaction of the antibody Fc domain with FcγRs does significantly affect the antibody’s activity. To further identify which receptors are involved in these events, we investigated the Fc gamma receptors expressed on TF-1 cells by fluorescence-activated cell sorting (FACS). We found that TF-1 cells only express CD32 (FcγRII), but not CD16 and CD64 ()), suggesting that CD32 is the receptor with which the Fc domain of mAb4-2 antibodies interacts. Because the commercial anti-CD32 antibody cannot distinguish CD32A (FcγRIIA) and CD32B (FcγRIIB) isoforms, we expressed in-house anti-CD32A and anti-CD32B antibodies, which showed high selectivity for CD32 isoforms expressed on TF-1 cells. FACS conducted with the two antibodies confirmed that CD32A is more abundant than CD32B on TF-1 cells, thus showing that CD32A is the major FcγR in TF-1 cells with which anti-IL-4 antibodies interact (Fig S3A, B).
CD32 (FCγRII) provides additional inhibitory signals for suppressing TF-1 cell proliferation in response to blockade of anti-Il-4Rα signaling
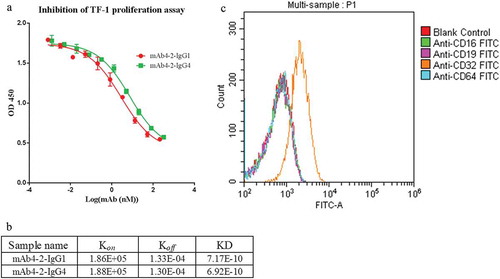
To confirm that the affinity of IgG subclasses for CD32 plays a role in modulating the antibody activities, we introduced S267E/L328F (SELF) double mutations on the Fc portion of mAb4-2-IgG1, named mAb4-2-IgG1-SELF.Citation19 We then tested this antibody’s ability to bind TF-1 cells engineered to express different CD32 constructs. In agreement with the previous study, mAb-IgG1-SELF whose affinity for CD32B is enhanced by up to 430-fold showed increased binding to TF-1 cells overexpressing CD32B, as well as cells overexpressing CD32A-R131, but not cells overexpressing CD32A-H131 (Fig S4).
Next, the potency of mAb4-2 variants in inhibition of TF-1 cell proliferation was assessed in response to IL-4. As shown earlier, mAb4-2-IgG1 showed a fourfold increase of potency in inhibiting IL-4-mediated TF-1 cell proliferation relative to the IgG4 counterpart, suggesting that CD32A, which, unlike CD32B, has a tenfold higher affinity for IgG1 relative to IgG4, played a key role in the event, in agreement with its dominating expression pattern on TF-1 cells. Furthermore, mAb4-2-IgG1-SELF showed another fourfold increase in potency (IC50: 0.9 nM) relative to that of mAb4-2-IgG1. In contrast, IgG1-SELF, a non-targeting antibody with the S267E/L328F mutations, showed no effects on IL-4-induced TF-1 cell proliferation, indicating that the increased affinity for CD32A (IgG1/IgG4 > 10-fold) as well as for CD32B (IgG1-SELF/IgG1 > 430-fold) facilitated the inhibitory function of the antibodies following the ligation of the antibody with IL-4Rα ()).
Figure 4. CD32 plays an important role in modulating antibody-mediated inhibition of TF-1 proliferation in response to blockade of IL-4Rα. (a) mAb4-2-IgG4, mAb4-2-IgG1, and mAb4-2-IgG1-SELFCitation19 were added to TF-1 cells in the presence of IL-4, following serial dilution; (b) an agarose gel showed the full-length CD32A and CD32B cDNA amplified by PCR using specific primers, following the reverse transcription from the extracted mRNA of TF-1 cells (left). Left lane: CD32A cDNA; middle Lane: molecular weight ladder; right Lane: CD32B cDNA; the sequencing result of CD32A cDNA (right), “A”, the single-nucleotide polymorphism (SNP), was highlighted, which results in H131 (bold) in the amino acid sequence; (c) a surrogate antibody of mAb4-2-IgG1, 4C6-IgG1, which can bind to IL-4Rα, but cannot block the signaling of IL-4Rα, was added to TF-1 cells in the presence of IL-4 after serial dilution. mAb4-2-IgG1 was used in the same assay as a positive control.
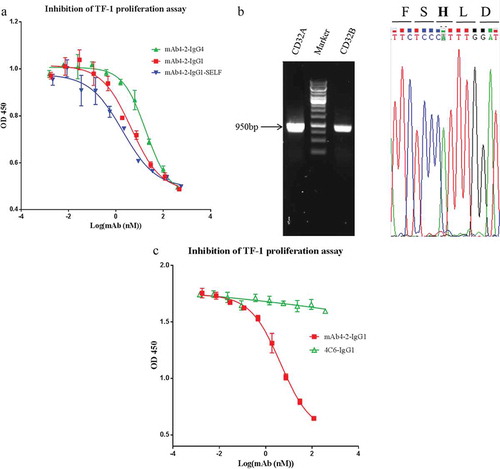
Given CD32A has two isotypes, CD32A-R131 and CD32A-H131, we next identified which isotype was expressed on TF-1 cells by genotyping. Interestingly, we found that CD32A-H131 and CD32B mRNAs were both transcribed in TF-1 cells, although the amount of CD32B mRNA was about half of that of CD32A mRNA ()). This result confirmed our FACS finding in that CD32A is abundantly expressed from TF-1 cells, while CD32B is expressed to a much lesser degree. The fact that the IgG1 antibody introduced with the SELF mutation increased the inhibitory potency of the antibody compared to the parental antibody supports the role of CD32B in this event. Since TF-1 expresses CD32A-H131, an isotype whose affinity for SELF mutations is not enhanced, the increased potency by the SELF mutant can be only explained by its binding to CD32B. Taken together, this observation demonstrated that CD32A, possibly in conjunction with CD32B, plays critical roles in modulating the inhibitory functions of the anti-IL-4 antibodies.
Intriguingly, a surrogate of mAb4-2-IgG1, 4C6, which exhibits the same affinity for IL-4Rα but cannot block IL-4Rα signal transduction (Fig S5A, B), failed to inhibit IL-4R signaling ()). This finding suggests that blockade of the IL-4Rα by the mAb4-2 antibody is a prerequisite for CD32A that is ligated with the antibody Fc to send out additional inhibitory signals.
CD32, especially CD32A, plays critical roles in reinforcing the antibody-mediated suppression of IL-4-induced CD23 expression on CD14+ monocyte
The expression level of CD23, the IgE low-affinity receptor II, on peripheral blood monocytes is IL-4 dependentCitation23 and appears to be associated with asthma severity and exacerbation.Citation24 We then tested mAb4-2 variants for their ability to block IL-4-induced CD23 expression of CD14+ monocytes from human PBMC. As predicted, mAb4-2-IgG4 showed a potent activity to inhibit CD23 expression from CD14+ monocytes, when compared to controls. Interestingly, when mAb4-2-IgG1 was used, the ability of blocking IL-4-induced CD23 expression was significantly improved (). Since the affinity of CD32A for the IgG1 subclass is tenfold higher than that of the IgG4 counterpart, we reasoned that CD32A plays an important role in this event. In agreement with this finding, CD32A is the most abundant FcγR expressed on monocytes as well as monocyte-derived macrophages and dendritic cells.Citation12
Figure 5. CD32A, together with CD32B, plays a key role in modulating antibody-mediated suppression of CD23 expression on monocytes in response to IL-4Rα blockade. Monocytes were isolated from six donor’s PBMCs. The level of CD23 expression from the blood monocytes treated with a variety of reagents in the presence of IL-4 was accessed by FACS. IL-4 control: cells treated with IL-4 in the absence of antibodies; IgG1-SELF control: an irrelevant IgG1 mAb with the SELF mutation; mAb4-2-N297A: an aglycosylation form of mAb4-2, owing to the point mutation of N297; mAb4-2-IgG4: the IgG4 mAb4-2; mAb4-2-IgG1: the IgG1 mAb4-2; mAb4-2-IgG1-SELF: the IgG1 mAb4-2 with the SELF mutation; mAb4-2-IgG1-SELF+Anti-CD32A: monocytes were treated with mAb4-2-IgG1-SELF and anti-CD32A antibody simultaneously; mAb4-2-IgG1-SELF+Anti-CD32B: monocytes were treated with mAb4-2-IgG1-SELF and anti-CD32B antibody simultaneously; mAb4-2-IgG1-SELF+Anti-CD32A+Anti-CD32B: three antibodies, mAb4-2-IgG1-SELF, anti-CD32A, and anti-CD32B were added to monocyte samples simultaneously.
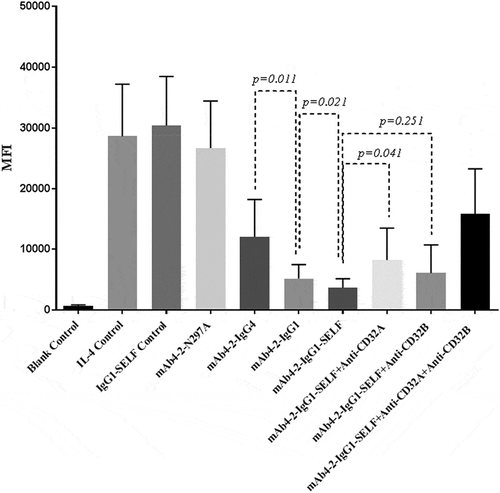
Next, we wanted to test if enhancing the binding affinity of the antibody for CD32 can further improve the inhibition of CD23 expression from CD14+ monocytes. As seen previously, the addition of mAb4-2-IgG1-SELF showed increased inhibition relative to mAb4-2-IgG1, however, to a lesser degree (p= 0.02) when compared to that of mAb4-2-IgG1 from the IgG4 counterpart (p= 0.01) (). To confirm that CD32A indeed contributed to the enhancement of the inhibitory activity of the antibody, we, therefore, examined IL-4-induced CD23 expression on isolated blood monocytes in the presence of anti-IL-4Rα antibodies and the two anti-CD32A- and CD32B-specific antibodies. Addition of anti-CD32A antibody into samples in the presence of mAb4-2-IgG1-SELF significantly increased the CD23 expression relative to that of mAb4-2-IgG1-SELF treatment alone (p < .05). By contrast, the addition of anti-CD32B antibody into samples in the presence of mAb4-2-IgG1-SELF did not significantly increase the CD23 expression relative to that of mAb4-2-IgG1-SELF treatment alone (p > 0.05). However, anti-CD32A antibody could not fully rescue the CD23 expression to the level comparable to that of mAb4-2-IgG4 treatment. Interestingly, adding both anti-CD32A and anti-CD32B together fully rescued the expression of CD23 compared to that of adding mAb4-2-IgG4 alone (). Taken together, these results demonstrated that, although CD32A appeared to be more important for mAb4-2 antibody variants mediated suppression of CD23 expression than CD32B, both CD32 isotypes worked coordinately in aiding antibodies to deliver extra inhibitory signals in monocytes in addition to blockade of IL-4Rα.
Both CD32A and CD32B play critical roles in inhibition of Il-4-induced IgE secretion from PBMC
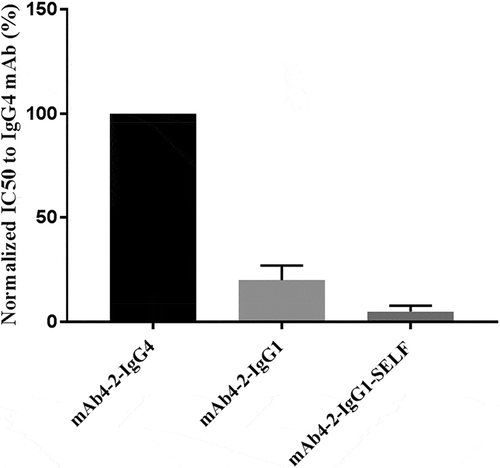
As IgE levels in asthma patients are associated with increased frequency of exacerbations under the control of IL-4,Citation25 we tested mAb4-2 antibodies for suppression of IL-4-induced IgE secretion using human PBMC. MAb4-2-IgG1 antibody showed a nearly fivefold stronger suppression of IL-4-mediated IgE secretion from PBMC relative to the IgG4 counterpart, once again suggesting that CD32A contributed to the mAb4-2 antibody-mediated suppression of IgE secretion. Introduction of SELF mutations provided an extra fourfold inhibition relative to the IgG1 counterpart (). Since IgE is secreted from B cells that only express CD32B, this result implies that CD32B is also involved in modulating the antibody activity in the suppression of IgE secretion. Interestingly, the roles of CD32A in this event may reflect a more complex networking amongst different immune cells, since CD32A is not expressed on B cells but on nonlymphoid cells.
Figure 6. Both CD32A and CD32B play important roles in modulating antibody-mediated suppression of IgE secretion from human PBMCs. The IC50 of each antibody, mAb4-2-IgG4, mAb4-2-IgG1, and mAb4-2-IgG1-SELF in the suppression of IgE secretion from blood B cells were measured in the presence of IL-4. The calculated IC50 for each antibody was normalized to and expressed as a percentage of that of mAb4-2-IgG4. The lower the percentage, the higher the potency is relative to the IgG4 antibody.
Discussion
In this study, we developed a humanized anti-IL-4Rα mAb, mAb4-2, which can efficiently block IL-4/IL-13-mediated cell signaling in a Th2 cytokine-mediated proliferation model. Using this molecule, we studied the effects of the interaction of the Fc portion of the antibody with FcγRs on the effector cells on modulating IL-4-induced immune responses. We found that the binding affinity of the antibody Fc for CD32, especially the activating CD32A, has profound effects on modulating the immune responses mediated by IL-4/IL-4Rα signaling.
IL-4 has a variety of effects in various tissues. In keeping with the broad range of actions of this cytokine, IL-4 receptors are also expressed in a broad spectrum of tissues. Analysis using IL-4Rα variants demonstrated that IL-4Rα chain cytoplasmic region regulates two distinct activities. One is required for activation of proliferative pathways, and the other is required for IL-4-induced expression of proteins such as CD23 and IgE.Citation3 In the TF-1 cell proliferation model, we investigated the effectiveness of mAb4-2 antibody variants in inhibiting the IL-4-dependent proliferation of this cell line. The following variants, mAb4-2-IgG1-SELF > mAb4-2-IgG1 > mAb4-2-IgG4, exhibited increased suppression of TF-1 cell proliferation in a manner that depended on the affinity for FcγR. Further analysis demonstrated that TF-1 cells express CD32 only, indicating that the enhanced potency is due to improved binding of the Fc portion of the antibody to CD32. FACS analysis using anti-CD32A and anti-CD32B specific antibodies as well as genotyping showed that CD32A is more abundantly expressed on TF-1 cells than CD32B. Consistent with this observation, mAb4-2-IgG1 exhibited much stronger inhibitory activity relative to mAb4-2-IgG4, which may be explained by the difference of binding affinity of the two Fc subclasses for CD32A, but not for CD32B.Citation11 In support of the CD32B expression pattern, the SELF mutations significantly improved the potency of both IgG1 and IgG4 due to its increased affinity for CD32B (although SELF exhibits strong affinity for CD32A-R131, this variant is not expressed in the cells tested). Our studies demonstrated that CD32, both CD32A and CD32B, plays a key role in modulating the activities of anti-IL-4 antibodies in suppressing the IL-4-induced TF-1 proliferation.
Bruhns et al.Citation11 have conducted a comprehensive comparison of the affinities of human FcγRs for human IgG subclasses, where they showed that IgG1 and IgG4 have similar affinities for CD32B (KA = 2x105 M−1), whereas, compared to IgG4, IgG1 binds tenfold stronger to CD32A. These data are in alignment with our results, and suggest that CD32A might be the major FcγRII that regulates the activities of anti-IL-4Rα antibodies for the following reasons: 1) IgG1 subclass significantly improved the antibody potency when compared to the IgG4 counterpart, and the increased potency was due to the stronger affinity between IgG1 and CD32A; 2) CD32A proteins are much more abundant and prevalent on the surface of TF-1 cells relative to CD32B proteins; and 3) although the differences in affinities between the IgG1/IgG4 antibodies and between IgG1-SELF/IgG1 antibodies are 10-fold and 430-fold for CD32A and CD32B, respectively, the two pairs show the same 4-fold increase in potency.
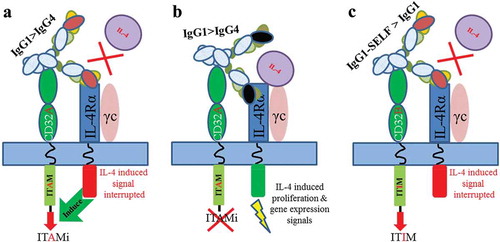
Since CD32A is an activating receptor, the inhibitory activity associated with this receptor was initially surprising to us. We, therefore, set out to investigate the phenomenon further by using a surrogate of mAb4-2, 4C6, which has the same affinity for IL-4Rα but cannot block IL-4Rα signal transduction. We found that 4C6 failed to block IL-4-induced TF-1 proliferation. This finding raises a possibility that CD32A is only activated in response to the blockade of IL-4Rα pathways, and that cross-linking the IL-4Rα with CD32A by the antibody positively upregulates the FcγR activity to reinforce the inhibitory signals of IL-4Rα blockade. Indeed, the Elson’s groupCitation26 demonstrated that CD32A was involved in regulation of the inhibitory function of an anti-TLR4 antibody. The not-yet-well-defined phenomenon, known as inhibitory ITAM (ITAMi), in terms of activating FcγRs sending out inhibitory signals, relies on a so-called “scorpion effect”, where an antibody interacts simultaneously with a target and an FcγR on the same cell in cis.Citation12 Although ITAMi remains to be further elucidated, recent research from the Monteiro lab,Citation27 where they showed that tyrosine 304 within CD32A ITAM is critical for ITAMi signaling, may shed light on the mechanism. However, in our study, we demonstrated that blocking the targeted receptor by antibodies is a prerequisite for CD32A-mediated ITAMi signaling. Whether or not the process is target specific, i.e., specific for IL-4Rα, remains to be further characterized. More importantly, it is plausible that the status of the targeted receptor either activated or inhibited by an antagonist or an agonist may dictate the CD32A activating or inhibitory responses, respectively ().
Figure 7. Schematic of the model of IL-4Rα blockade dependent ITAMi. (a) mAb4-2 binds to IL-4Rα and blocks the interaction of IL-4 with IL-4Rα while cross-linking the CD32A (the binding affinity:IgG1 > IgG4). The disruption of the IL-4-mediated signaling allows IL-4Rα to signal the Fc bound CD32A, resulting in activation of the ITAMi signaling; (b) 4C6 binds to IL-4Rα but cannot block the interaction of IL-4 with IL-4Rα, while cross-linking the CD32A. The undisturbed IL-4/IL-4Rα signaling does not send out signals to the Fc-bound CD32A, and thus no ITAMi is activated; (c) In the case of CD32B, mAb4-2 binds to IL-4Rα and blocks the interaction of IL-4 with IL-4Rα, while cross-linking the CD32B (the binding affinity:IgG1-SELF > IgG1), resulting in activation of the ITIM signaling.
Upregulation of CD23 expression, the low-affinity IgE receptor IgɛRII, is one of the hallmarks of IL-4-induced immune responses and is believed to have important roles in the pathogenesis of asthma.Citation28 Thus, we assessed the activity of our antibodies in suppressing the upregulation of CD23 expression using isolated monocytes from human PBMC. We discovered that antibodies of IgG1 subclass rendered much stronger suppression than did antibodies of IgG4 subclass, indicating that an FcγR that has a much stronger affinity for IgG1 than IgG4 might contribute to this effect. Therefore, the data from this ex-vivo model aligned reasonably well with our in-vitro cell assays. Since CD32A is the most abundant FcγR on the monocytic cells, we reasoned that CD32A also plays a crucial role in modulating the expression of CD23 on blood monocytes alongside the mAb4-2 antibodies. In line with this finding, using an anti-CD32A antibody, we demonstrated that CD32A is indeed responsible for enhancing the inhibitory function of the mAb4-2 antibodies, since the addition of anti-CD32A antibody partially rescued the suppression of CD23 expression rendered by mAb4-2-IgG1-SELF and mAb4-2-IgG1, respectively. Although adding anti-CD32B antibody did not rescue the CD23 expression from the suppression by the SELF mutant (p > .05), addition of both CD32A and CD32B completely rescue the CD23 expression to levels comparable to that of mAb4-2-IgG4 from the SELF mutant. The importance of CD32A in the regulation of CD23 on monocytes may simply reflect the expression level of the receptor when compared to CD32B. Further studies need to be done to tease out the roles of the two CD32 isoforms in this event.
The above result, however, cannot rule out the possibility that FcγRs on monocytes, other than CD32, can also bind to the Fc portion of the antibodies, and thus transmit intracellular signals accordingly. In fact, the inhibition of mAb4-2-IgG4 alone relative to controls might reflect the blockade of IL-4Rα plus the net inhibitory signals from all FcγRs bound to the IgG4 Fc. In the case of CD23 expression, the extra impact of FcγRs on the antibody potency above the IgG4 antibody appears to be the sum of activities of both FcγRIIs, of which CD32A was more dominant. Therefore, the observed activity might be the combination of the affinities and densities of all FcγRs together, in that FcγRs on monocytes, both activating and inactivating, contribute to the reinforcement of the inhibitory signals in response to the inhibition of IL-4Rα signaling.
In the case of IgE secretion, our anti-IL-4Rα antibodies displayed a strong inhibitory ability in a manner that appears to associate with the affinity for CD32A, since the IgG1 antibody provides much stronger inhibition than the IgG4 counterpart. Moreover, introducing SELF mutations to the IgG1 antibody further increased the inhibitory potency of the parental IgG1 antibody. These results indicate that, in addition to CD32A, CD32B may also modulate an antibody’s ability to suppress IgE secretion, given that CD32B is the only FcγR expressed on B cells.Citation29 Recently, CD32C, whose extracellular region is homologous to CD32B while the cytoplasmic tail is homologous to CD32A with the presence of an ITAM, was found to be expressed on B cells in people who possess an open reading frame (ORF) allele, and could counterbalance the negative feedback of CD32B.Citation30 As expected, CD32C and CD32B indeed have similar binding affinities for both IgG1 and IgG4, given the high homology of their extracellular domains.Citation11 Thus, the net negative signaling observed in this event might reflect the possibilities that: 1) none or few hPBMCs that we tested have the functional CD32C expressed, as less than 15% of healthy individuals have the ORF allele; 2) CD32B may be more abundantly expressed on B cells compared to CD32C with the ORF allele; 3) more intriguingly, it is plausible that CD32C, when engaged with anti-IL-4Ra antibodies, might execute ITAMi signaling as CD32A did. However, the fact that CD32A has a role in this event may reflect a more complex situation, where the amount of soluble IgE in PBMCs can be affected by multiple factors and immune cells. Indeed, IL-4 simultaneously induces IgE production and CD23 expression.
Multiple lines of evidence demonstrated that IgE production from B cells is negatively regulated by CD23.Citation28 There are two isoforms of CD23, CD23a and CD23b. CD23a is exclusively expressed on human B cells, whereas CD23b is upregulated by IL-4 on a variety of hematopoietic cells, including B cells and monocytes. Using CD23 transgenic mice, Payet-Jamroz M et al. demonstrated that CD23 expressed on non-lymphoid cells provided B cells with signals that resulted in the decrease of IgE production.Citation31 More recently, the Chan lab showed that signaling through CD23 on B cells differs from that of the monocytic lineage cells.Citation32 They proposed that CD23 plays roles in the regulation of IgE synthesis in human B cells, whereas CD23 may regulate the phagocytosis of IgE and allergen in monocytes. These data suggest that, as a principal mediator of most type I allergic reactions, IgE production is tightly controlled by comprehensive cross-talk between B cells and surrounding non-lymphoid cells, in which CD23 plays key roles in response to IL-4. Indeed, conflicting observations were also seen for the roles of CD23 and IgE in the regulation of inflammatory responses in asthma, probably due to the complexity of the disease environment. The Borish lab showed that individuals with asthma displayed a much higher level of CD23 expression on monocytes relative to that of normal individuals,Citation24 whereas the Weiss’ group demonstrated that the elevated IgE levels seen in asthmatic patients were associated with reduced CD23 expression.Citation25 Therefore, the amount of IgE observed in our studies was most likely a net result from the manipulation of the global signaling of IL-4Rα, activating and inhibitory FcγRs on B cells and monocytes by anti-IL-4Rα antibodies.
Nevertheless, we revealed an important role of the activating CD32, in particular, CD32A, in modulating anti-IL-4Rα antibody-mediated suppression of IL-4-induced inflammatory responses. Cross-linking of IL-4Rα and CD32A by antibodies allows CD32A to send out extra inhibitory signals in response to IL-4Rα blockade. It appears that the situation of the targeted receptor affected by antibodies tells CD32A what to do, since the blockade of IL-4Rα is prerequisite for activating CD32A-mediated ITAMi signaling. Furthermore, in a Th2 cytokine-mediated allergic inflammatory environment, monocytes, as a key modulator of allergic inflammation, express various FcγRs, of which CD32A is the most abundant and prevalent. It is plausible that such similar regulations could also be seen in IL-13-mediated immune responses, as IL-13 not only shares IL-4Rα in its receptor complexes but also shares some of the effector functions with IL-4, including regulation of IgE production and CD23 expression.Citation33 Further studies need to be done to verify this hypothesis. Therefore, design and application of therapeutic antibodies to intervene Th2 cytokine-mediated atopic diseases, including asthma, should take CD32A activities into consideration, as well as the contribution of other activating and inhibitory FcγRs, in modulating the inflammatory responses in addition to blockade of receptors.
Materials and methods
Cell culture
TF-1 cells, a factor – dependent human erythroleukemic cell line, were purchased from the American Type Culture Collection (ATCC® CRL-2003™). TF-1 cells were cultured in RPMI 1640 medium (Gibco™), supplemented with 10% heat-inactivated fetal bovine serum (Gibco™), 1% GlutaMAX™ Supplement (Gibco™), 1% Penicillin-Streptomycin (Gibco™) and 5 ng/ml recombinant human GM-CSF (Amoytop Biotech) in a humidified CO2 incubator at 37°C. PBMCs were separated by a Histopaque-1077 (Sigma) gradient method using blood from healthy donors (after informed consent, Changhai Hospital, Shanghai, China) and cultured in RPMI 1640 complete medium. HEK293E, purchased from the NRC Biotechnology Research Institute, were cultured in freestyle 293 medium (Life technologies) with 1% Penicillin-Streptomycin.
Protein expression and purification
Constructs expressing antibody variants were generated using pTT5 vector (NRC Biotechnology Research Institute). The resulting expression vectors were transiently transfected into HEK293E cells using 1 μg/mL 25-kDa linear polyethylenimine (Polysciences, Inc.). One day after transfection, valproic acid (Sigma) was added to cell culture at a final concentration of 3 mM. On day 2 post-transfection, medium comprising 10% GlutaMAX, 10% 400 g/L glucose and 80% freestyle 293 medium was added to the cell culture at 10% of the total volume. Conditioned medium was collected 5–6 days after transient transfection. Antibodies in the culture media were purified by MabSelect SuRe (GE) affinity columns using an Akta Avant 25 fast protein liquid chromatography (FPLC) purification system. After equilibrating the column with buffer A (25 mM sodium phosphate, 150 mM sodium chloride, pH = 7.0), the culture media containing anti-IL-4R antibodies were applied to the column, which was then eluted with buffer B (100 mM sodium citrate, pH = 3.5) to collect the desired proteins. The eluted antibodies were further polished by gel filtration on a SuperdexTM 200 column (GE) if necessary.
Humanization of mAb4-2 and antibody engineering
Humanization of mAb4-2 was conducted by grafting complementarity determining regions of the parental murine anti-human IL-4Rα antibody (Clone mAb4-2) into human heavy and light-chain frameworks as described previously.Citation34 PCR site-directed mutagenesis was used to generate IgG variants of the humanized mAb4-2 following the manufacturer’s instructions (Cat#R010B, Takara). The sequences of all variants were verified by sequencing at Genewiz (Shuzou, China). MAbs capable of discriminating the human inhibitory Fcγ-receptor IIB (CD32B) from the activating Fcγ-receptor IIA (CD32A) were prepared in-house. The sequence information of 2B6 (specific for CD32B and designated Anti-CD32B) and IV.3 (specific for CD32A and designated Anti-CD32A) were extracted from US20160159908Citation35 and US20160145336,Citation36 respectively. Gene synthesis and sequencing were all performed by Genewiz (Shuzou, China). Genes for all antibodies were subcloned into the mammalian expression vector pTT5 and transiently expressed from HEK293E cells. Antibodies in the cell culture media were purified with protein A affinity columns (Mabselect, GE) by FPLC.
Enzyme-linked immunosorbent assay
Ninety-six-well plates (Cat#9018, Costar) were coated with 10 ng/well of FLAG-tagged IL-4Rα (produced in-house) overnight. The plates were washed with phosphate-buffered saline (PBS) containing 0.05% Tween-20 (PBS-T), blocked for 1 h with PBS containing 1% bovine serum albumin (BSA) and incubated with serial dilutions of the purified anti-IL-4Rα antibodies for 1 h at room temperature. The plates were washed with PBS-T for 5 min three times and incubated with horseradish peroxidase (HRP)-conjugated goat-anti-human IgG (Fc-specific and highly cross-adsorbed) antibody (Cat#SAB3701283, Sigma) for 1 h at room temperature. The plates were washed, and the reaction developed with chromogenic substrates for 2-3 min. The plates were then read on a SpectraMax 190 reader (Molecular Devices) at 450 nm. The same protocol was used for competition ELISA, except that the plates were coated with 200 ng/well of IL-4 followed by incubation of serial dilutions of the purified antibodies in the presence of 200 ng/ml biotinylated IL-4Rα. HRP-Streptavidin was then used to detect bound competing IL-4Rα.
TF-1 cell proliferation assay
Prior to use, TF-1 cells were harvested in logarithmic phase and washed 2 times in PBS. Cells were resuspended in the assay medium (the growth medium without GM-CSF) and added to a 96-well cell culture plate at 1 × 105 cells/150 μl/well, and placed for an hour in a humidified chamber at 37°C and 5% CO2. After 1 h, serial dilutions of anti-IL-4Rα antibodies were added to each well in the presence of 80 ng/ml rhIL-4 (Cat#204-IL-050, R&D systems) or rhIL-13 (Cat#213-ILB-005, R&D systems) for a final reaction volume of 200 μl, and the plates were incubated for 96 h at 37°C and 5% CO2. At 90 h, CCK-8 reagent (Cat#CK04, Dojindo) was added as per manufacturer’s instructions. The plates were read at 405/450 nm on a SpectraMax 190 reader (Molecular Devices). The IC50 values were calculated using GraphPad Prism 6 software (GraphPad Software).
Flow cytometric analysis of CD23 expression on monocytes
PBMCs were separated from the blood of healthy donors by the Histopaque-1077 (Sigma) gradient method. Isolated PBMCs in complete RPMI-1640 medium were added to round-bottom 96-well plates (Cat#3799, Costar®) at 2 × 105 cells/150 μl/well. Fifty microliters of serial dilutions of anti-IL-4Rα antibodies were added to each well in the presence of 80 ng/ml rhIL-4 for a final reaction volume of 200 μl, and the plates were incubated for 48 h at 37°C and 5% CO2. After 2 days, PBMCs were stained with phycoerythrin mouse anti-human CD23 antibody (Cat#555711, BD biosciences), and recombinant human IgG1 (in-house) was added to block non-specific binding to Fc receptors at 1mg/ml. After fixation with 4% paraformaldehyde for 10 min at room temperature, PBMCs were analyzed using CytoFLEX Cytometer System (Beckman Coulter). Monocytes were identified based on forward scatter/side scatter, as well as CD14 staining.
In the case of identification of FcγRs expressed from TF-1 cells, TF-1 cells were incubated with fluorescein isothiocyanate-(FITC)-conjugated mAbs (FITC-isotype control, Cat#555748; FITC-CD19, Cat#555412; FITC-CD16, Cat#555406; FITC-CD32, Cat#555448; FITC-CD64, Cat#555527; all from BD Biosciences) for 1 h at room temperature in PBS containing 1% BSA. Flow cytometry was performed as described above.
ELISA analysis of IgE secretion from PBMCs
PBMCs in complete RPMI-1640 medium were added to round-bottom 96-well plates (Cat#3799, Costar®) at 2 × 105 cells/150 μl/well. Fifty microliters of serial dilutions of anti-IL-4Rα antibodies were added to each well in the presence of 80 ng/ml rhIL-4 for a final reaction volume of 200 μl. The plates were incubated for 14 days at 37°C and 5% CO2. After 14 days, cell culture supernatants were collected and IgE concentrations were determined by a sandwich ELISA. Briefly, IgE in the culture medium was captured by anti-human IgE antibody (Cat#I6284-1MG, Sigma) coated plates after 1 h incubation at room temperature. The plates were washed and incubated with biotinylated anti-Human IgE antibody (A18803, Thermo Fisher Scientific) for 1 h at room temperature. The plates were washed and incubated with HRP-conjugated streptavidin (Cat#554066, BD Biosciences) for 1 h at room temperature. The plates were washed, and the reaction developed with the addition of chromogenic substrates for 2-3 min. The plates were then read on a SpectraMax 190 reader (Molecular Devices) at 450 nm. IgE levels from the culture medium were quantitated with an IgE standard curve.
Binding affinity analysis by surface plasmon resonance (SPR)
The interaction of antibody variants with IL-4Rα was monitored using Biacore T200 (GE Healthcare). Briefly, antibody variants were captured by protein A/G (Cat#21186, Thermo Scientific) immobilized on the CM5 sensor chip (Cat#BR-1005–30, GE Healthcare). Varying concentrations of IL-4Rα were injected at a flow rate of 30 μl/min. Sensorgrams were obtained at each concentration, and kinetic analysis was performed using Biacore T200 Evaluation Software (GE Healthcare).
CD32A genotyping of TF-1 cell line
mRNA of TF-1 cells was extracted, and complementary DNA was synthesized using PrimeScriptTM 1st Strand cDNA Synthesis Kit (Takara). The cDNA of CD32A was amplified by PCR using specific primers (Forward primer: 5- CACTGGACGTTGGCACAGTGCTGGG −3, reverse primer:5- ATGACCACATGGCATAACGTTACTC −3). The CD32A genotype of TF-1 cell line was determined by sequencing the PCR product (Genewiz).
Abbreviations
| AHR | = | airway hyper-responsiveness |
| DCs | = | dendritic cells |
| FPLC | = | fast protein liquid chromatography |
| FACS | = | fluorescence-activated cell sorting |
| ITAM | = | immunoreceptor tyrosine-based activation motif |
| ITIM | = | immunoreceptor tyrosine-based inhibitory motif |
| ICs | = | immune complexes |
| IL-4 | = | interleukin-4 |
| IL-4R | = | IL-4 receptor |
| IL-13 | = | interleukin-13 |
| ITAMi | = | inhibitory ITAM |
| MHCII | = | major histocompatibility complex class II |
| mAb | = | monoclonal antibody |
| ORF | = | open reading frame |
| PBMCs | = | peripheral blood mononuclear cells |
| Th2 | = | T-helper type 2. |
Disclosure of Potential Conflicts of Interest
None. All authors are employees of 3sBio when the study was carried out.
Supplemental Material
Download Zip (9.2 MB)Acknowledgments
We thank Shengtao Rui and Qing Gao for excellent technical assistance. We would also like to acknowledge the expert services provided by the protein chemistry and analytical department at 3sBio.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s publisher’s website.
Additional information
Funding
References
- Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19(115):46–54. doi:10.1183/09059180.00007609.
- Barranco P, Phillips-Angles E, Dominguez-Ortega J, Quirce S. Dupilumab in the management of moderate-to-severe asthma: the data so far. Ther Clin Risk Manag. 2017;13:1139–49. doi:10.2147/TCRM.S125964.
- Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38. doi:10.1146/annurev.immunol.17.1.701.
- Wenzel S, Castro M, Corren J, Maspero J, Wang L, Zhang B, Pirozzi G, Sutherland ER, Joish VN, Eckert L, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: a randomized double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016:1–14. doi:10.1016/S0140-6736(16)30307-5.
- Stewart R, Hammond SA, Oberst M, Wilkinson RW. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J Immunother Cancer. 2014;2:29. doi:10.1186/s40425-014-0029-x.
- Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol. 2015;67:171–82. doi:10.1016/j.molimm.2015.03.255.
- Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013;6:1. doi:10.1186/1756-8722-6-1.
- Kenkre VP, Hong F, Cerhan JR, Lewis M, Sullivan L, Williams ME, Gascoyne RD, Horning SJ, Kahl BS. Fc gamma receptor 3A and 2A polymorphisms do not predict response to rituximab in Follicular lymphoma. Clin Cancer Res. 2016;22(4):821–26. doi:10.1158/1078-0432.CCR-15-1848.
- Nimmerjahn F, Ravetch JV. Fcg receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi:10.1038/nri2206.
- Dahan R, Emanuela S, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28:285–95. doi:10.1016/j.ccell.2015.08.004.
- Bruhns P, Iannascoli B, England P, Mancardi DA, Femandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcα receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25. doi:10.1182/blood-2008-09-179754.
- Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov. 2012;11:311–31. doi:10.1038/nrd2909.
- Grubczak K, Moniuszko M. The role of different monocyte subsets and macrophages in asthma pathogenesis. Prog Health Sci. 2015;5:176–84.
- Zaslona Z, Przybranowski S, Wilke C, Rooijen NV, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193:4245–53. doi:10.4049/jimmunol.1400580.
- Alevy YG, Tucker J, Mohanakumar T. CD32A (FcgRIIa) mRNA expression and regulation in blood monocytes and cell lines. Mol Immunol. 1992;29:1289–97.
- Mullazehi M, Mathsson L, Lampa J, Ronnelid J. Surface-bound anti-type II collagen-containing immune complexes induce production of tumor necrosis factor a, interleukin-1b, and interleukin-8 from peripheral blood monocytes via Fcg receptor IIa. Arthritis Rheumatism. 2006;54:1759–71. doi:10.1002/art.21892.
- Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IWL, Jacob N, Zalevsky J, Desjarlais JR, Stohl W, et al. Antibody-mediated coengagement of FcαRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol. 2011;186:4223–33. doi:10.4049/jimmunol.1003412.
- Crowley JE, Stadanlick JE, Cambier JC, Cancro MP. FcgRIIB signals inhibit BLyS signaling and BCR-mediated BLyS receptor up-regulation. Blood. 2009;113:1464–73. doi:10.1182/blood-2008-02-138651.
- Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JR. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45:3926–33. doi:10.1016/j.molimm.2008.06.027.
- Tridandapani S, Siefker K, Teillaud J, Carter JE, Wewers MD, Anderson CL. Regulated expression and inhibitory function of FcγRIIb in human monocytic cells. J Biol Chem. 2002;277:5082–89. doi:10.1074/jbc.M110277200.
- Roghanian A, Stopforth RJ, Dahal LN, Cragg MS. New revelations from an old receptor: immunoregulatory functions of the inhibitory Fc gamma receptor, FcgRIIB (CD32B). J Leukoc Biol. 2018;1–12. DOI:10.1002/JLB.2MIR0917-354R
- Boruchov AM, Heller G, Veri M, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–23. doi:10.1172/JCI24772.
- Vercelli D, Leung DYM, Jabara HH, Geha RS. Interleukin 4 dependent induction of IgE synthesis and CD23 expression by the supernatants of a human helper T cell clone. Int Arch Allergy Appl Immunol. 1989;88:119–21.
- Williams J, Johnson S, Mascali JJ, Smith H, Rosenwasser LJ, Borish L. Regulation of low affinity IgE receptor (CD23) expression on mononuclear phagocytes in normal and asthmatic subjects. J Immunol. 1992;149:2823–29.
- Tantisira KG, Silverman ES, Mariani TJ, Xu J, Richter BG, Klanderman BJ, Litonjua AA, Lazarus R, Rosenwasser LJ, Fuhlbrigge AL, et al. FCER2: a pharmacogenetic basis for severe exacerbations in children with asthma. J Allergy Clin Immunol. 2007;120:1285–91. doi:10.1016/j.jaci.2007.09.005.
- Dunn-Siegrist I, Leger O, Daubeuf B, Poitevin Y, Depis F, Herren S, Kosco-Vilbois M, Dean Y, Pugin J, Elson G. Pivotal involvement of Fc gamma receptor IIA in the neutralization of LPS signaling via a potent novel anti-TLR4 monoclonal antibody, 15C1. J Biol Chem. 2007;282:34817–27. doi:10.1074/jbc.M706440200.
- Mkaddem SB, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, Benna JE, Launay P, Goujon J, Benhamou M, et al. Shifting FcγRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J Clin Invest. 2014;124:3945–59. doi:10.1172/JCI74572.
- Conrad DH, Ford JW, Sturgill JL, Gibb DR. CD23: an overlooked regulator of allergic disease. Curr Allergy Asthma Rep. 2007;7:331–37.
- Rankin CT, Veri M, Gorlatov S, Tuaillon N, Burke S, Huang L, Inzunza HD, Li H, Thomas S, Johnson S, et al. CD32B, the human inhibitory Fc-g receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood. 2006;108:2384–91. doi:10.1182/blood-2006-05-020602.
- Li X, Wu J, Ptacek T, Redden DT, Brown EE, Alarcon GS, Ramsey-Goldman R, Petri MA, Reveille JD, Kaslow RA, et al. Allelic-dependent expression of an activating Fc Receptor on B cells enhances humoral immune responses. Sci Transl Med. 2013;5:1–10. doi:10.1126/scitranslmed.3007097.
- Payet-Jamroz M, Helm SL, Wu J, Kilmon M, Fakher M, Basalp A, Tew JG, Szakal AK, Noben-Trauth N, Conrad DH. Suppression of IgE responses in CD23-Transgenic animals is due to expression of CD23 on nonlymphoid cells. J Immunol. 2001;166:4863–69.
- Chan MA, Gigliotti NM, Matangkasombut P, Gauld SB, Cambier JC, Rosenwasser LJ. CD23-mediated cell signaling in human B cells differs from signaling in cells of the monocytic lineage. Clin Immunol. 2010;137:330–36. doi:10.1016/j.clim.2010.08.005.
- Hershey GKK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–90.
- Hwang WYK, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods. 2005;36:35–42. doi:10.1016/j.ymeth.2005.01.004.
- Johnson LS, Huang L U.S. Patent No. 9,284,375. 2016. Washington, DC: U.S. Patent and Trademark office.
- Francis J, Amirkhosravi A, Meyer T, Robles-Carrillo L U.S. Patent No. 9,382,321. 2016. Washington, DC: U.S. Patent and Trademark office.