
ABSTRACT
Monoclonal antibodies (mAbs) have revolutionized the treatment landscape in many disciplines of human medicine. To continue this exciting trend, sustained identification of new, validated and preferably functional targets are needed. However, this is the precise bottleneck in today’s development of the next generation of therapeutic mAbs. Failures in translating a target into a successful therapeutic mAb are much more frequent than successes. Labex MAbImprove is a French-led consortium of academic laboratories jointly working on several aspects of the development of next-generation mAbs. The network organizes annual international meetings gathering academia and industry to discuss the different challenges faced in the therapeutic mAbs field. The 2018 symposium (also called AIS2018 and co-organized with MabDesign, the immunotherapy French industrial sector) focused on the discovery and validation of new targets for therapeutic mAbs. Key players from industry and academia outlined a number of exciting contributions, notably dealing with new innovations in the target discovery area, but also lessons learned from failures in the past. This report summarizes the talks presented at the AIS2018. We aim at broad dissemination of the most relevant, unpublished findings presented during the meeting, and hope to inspire all the contributors in this field to take new directions and bring about improvements.
KEYWORDS:
Introduction
Over 80 approved and marketed therapeutic antibodies define today’s biologics landscape, which is worth over 100 billion US dollars in revenue. While overall this figure is impressive, it is worth noting that these 80 or so antibodies only target about 50 targets.Citation1,Citation2 Of these, five antigens (tumor necrosis factor alpha [TNFα], CD20, epidermal growth factor receptor, human epidermal growth, and vascular endothelial growth factor alpha) feature as targets for a total of 23 different antibodies ()). In 2018, regulatory bodies received market approval applications for another seven antibodies directed against these same targets ()). Finally, it is worth noting that these five targets are also responsible for three-fourths of the total therapeutic antibody business revenue in 2018 ()). Following the rationale that the market never lies, one could conclude that the quest for new targets has been often more painful than rewarding. Large biopharmaceutical companies are thus extremely prudent, investing in targets that are likely (more than certainly) to work. A good example is the surge in programmed cell death protein 1 (PD1)- and programmed cell death ligand 1 (PD-L1)-targeting antibodies (six products on the market, six in Phase 3 clinical trials) and the overall interest in developing other immune checkpoint inhibitors. In contrast to this, examples such as NOTCH inhibitor antibodies brontictuzumab and tarextumab, or CD33-targeting antibody-drug conjugate (ADC) vadastuximab talirine, show how both new and old targets can quickly lead to costly failures. While these examples make the overall market strategy understandable, it is clear that novel targets are needed for the development of more disease-specific antibodies, bringing us closer to realizing the personalized medicine dream. To learn from past mistakes, a detailed analysis of the entire process is required, starting from target discovery to antibody validation.
Figure 1. Market overview of targets for therapeutic mAbs. a) Number of therapeutic antibodies directed against different targets since 2015. b) Targets of mAbs that are currently in Phase 3 clinical trials. c) MAb therapeutic market share per target in billion US dollars (data from 2017).
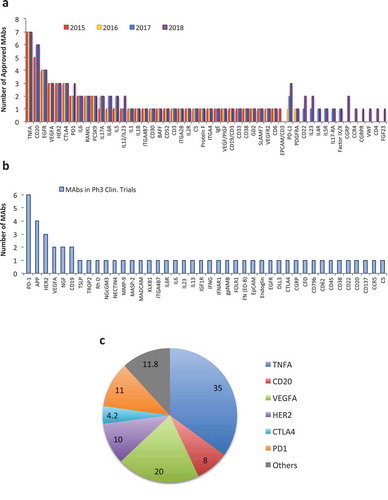
Motivated by these ideas, on June 25–26, 2018, in Montpellier France, a scientific meeting was organized around Targets for MAbs and Innovative Approaches For Their Discovery & Validation. Participants from pharma companies and academia aimed at answering several key questions: What are the characteristics of “good” antibody targets? Which techniques can be used for their discovery and selection? Which path should one select for validating targets and bringing them to the clinic? What mode of action (MOA)/function is preferred for a given target? Apart from these very relevant questions, the meeting aimed in particular at discussion of both success stories as well as failures, and exploration of these from different angles. As the first to begin addressing these questions, Dr. Roy Baynes (Merck Sharp and Dohme) delivered a Keynote Lecture on pembrolizumab, a PD-1 blocking antibody, particularly stressing milestones that led to a successful development of Keytruda®. The next key point in the AIS 2018 meeting was dedicated to discovery approaches and target identification. There, new technologies were introduced, such as single-cell sequencing approaches that are bound to revolutionize our understanding of complex diseases, including cancer. The meeting also hosted a Pitch Session where new approaches for discovery and validation of new targets for monoclonal antibodies (mAbs) were discussed. Particularly interesting were the novel phage display libraries that were presented, going beyond antibody selection into target discovery and screening tool. The fourth session addressed the pre-clinical validation phase, including the newest methods, models and companion tests required for successful entry of mAbs into early clinical testing. The final session was particularly dedicated to new and emerging targets. As outlined in ), 42 therapeutic mAbs are currently in Phase 3 testing; however, only 10% of these are likely to obtain market approval in 2019 (additionally to another 10% that represent biosimilars and new mAbs for “old” targets). The success of biologics will, therefore, necessitate the constant identification of novel targets and their development into products ready to enter clinical testing. We hope that, with this meeting report, we can contribute to better understand of this early discovery and development process, thereby leading to better targets and antibodies that will have higher success in the clinic.
Plenary keynote: pembrolizumab – a transformative backbone anti-cancer therapy
Dr. Roy Baynes from Merck Sharp and Dohme (MSD) opened the meeting with his detailed analysis of pembrolizumab’s (Keytruda®) development and future plans. He highlighted that many companies are addressing PD-1 as a target, and that the field is highly competitive. Clinical data are currently being generated more rapidly than the scientific underpinnings are being defined. The strategic goal at MSD was to improve long-term disease control and survival in a wide range of cancers with PD-1 interdiction. The strategic imperatives have included establishing pembrolizumab monotherapy as a foundational element of cancer treatment.
From early on, they took into consideration that only a subpopulation of patients generally responds. Consequently, a second key strategic imperative has been the use of precision medicine to enrich for responders. In the initial screening of cancers for PD-1 antibody therapeutic activity, they evaluated major cancers that had either significant PD-L1 expression or large numbers of mutations. They screened a matrix of 30 tumor types, and over 25 of them showed responses. Since then, investigator-sponsored research has identified activity in additional tumor types. This has led to a broad monotherapy program, and marketing approvals in melanoma, non-small-cell lung cancer (NSCLC), chronic myeloid leukemia, primary mediastinal B-cell lymphoma, as well as head and neck, urothelial, gastric, cervical, colorectal cancer (CRC) with high level microsatellite instability (MSI-Hi), and any MSI-Hi cancer. Approval in the US has been granted for 13 indications. Pembrolizumab has received 12 Breakthrough Therapy Designations from the Food and Drug Administration.
Monotherapy has demonstrated improvements in overall survival (OS) as first line for NSCLC, melanoma, ipilimumab-refractory melanoma and second line for bladder cancer. Long-term follow-up of the Keynote 001 trial showed 5-year OS of 34% in the total population and 41% in treatment-naïve melanoma patients. Keynote-006 was a registration trial that compared anti-PD1 with anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) treatments for ipilimumab-naïve melanoma patients. Anti-PD-1 mAb treatment proved superior to anti-CTLA4 therapy. Long-term follow-up demonstrated that this advantage in OS is maintained. Recent results from Keynote-054, studying pembrolizumab in the adjuvant setting in melanoma, indicated a 43% reduction in risk of recurrence or death, regardless of disease stage and PD-L1 expression. Keynote-042 Phase 3 trial studying pembrolizumab monotherapy in metastatic non-squamous and squamous NSCLC expressing PD-L1 with a tumor proportion score >1% showed a 20% reduction in the risk of death and a 4 months OS benefit over standard chemotherapy. Long-term follow-up of Keynote-001NSCLC front line patients, showed that, in those who were PD-L1 strongly positive, a 50% survival was observed at 5 years. In second-line head and neck cancer patients, monotherapy showed a directionally favorable OS benefit.
The PD-L1 immunohistochemistry (IHC) test is now a companion diagnostic for the selection of responsive patients in certain cancer types. Gene expression profiling, initially using some 700 genes and then reduced to an 18-gene inflammation signature, is also highly predictive of responsiveness, but does not appear to add much over PD-L1 status by IHC. Defective DNA mismatch repair (MMR), as demonstrated by MSI-Hi status, is also predictive of anti-PD-1 mAb responsiveness in CRC and all other cancer types evaluated. MMR-deficient CRC and other cancer types had higher response rates to pembrolizumab compared to DNA MMR-proficient cancers.
The third imperative of pembrolizumab development strategy was to explore the combination of pembrolizumab with other therapies. One can organize thinking about combinations in a number of ways. For example, a mechanistic approach might include priming the immune effector arm, reducing the immunosuppressive microenvironment or increasing the presentation of tumor antigens by a process of immunogenic cell death. Another way of thinking about combinations is through the lens of response to anti-PD-1 therapy. Analysis of a number of internal data sets has shown that “non-inflamed” tumors rarely respond to pembrolizumab. Almost all responders have baseline tumor inflammation. However, some inflamed tumors were also non-responders. MSD is exploring resistance biology markers as the possible basis of this non-responsiveness. Such resistance biology may be addressable with combination therapy. A third way of thinking about combinations might be the types of combinations. For example, pembrolizumab might be combined with standard therapies, with targeted therapies, with other immunologic modulators, with cancer vaccines or with oncolytic viruses.
The experience with pembrolizumab and chemotherapy provides an example of mAb and standard therapy combinations. In NSCLC, chemotherapy combination with pembrolizumab was explored initially in Phase 2 screening studies. Impressive data were generated in combination with carboplatinum and pemetrexed in a randomized Phase 2 study known as Keynote 21G. This study allowed cross-over from the chemotherapy control arm to pembrolizumab at progression. At the initial presentation at the 2016 European Society for Medical Oncology Congress in 2016 with about 9 months of follow-up data, highly significant improvement in progression-free survival was observed, but no real difference in OS. However, with a longer follow-up, the survival Kaplan–Meier curves have progressively diverged in favor of pembrolizumab, thereby indicating the initial combination is critical and favored over a sequential approach as occurred in those crossing-over from the control arm. This study supported accelerated approval of chemotherapy-pembrolizumab combination in the US. To confirm this accelerated approval, Keynote-189, a much larger version of Keynote 21G was conducted. This Phase 3 combination study of carboplatinum, pemetrexed and pembrolizumab combination vs. the same chemotherapy that allowed crossover showed 51% reduction in the risk of death when pembrolizumab was combined with chemotherapy. Keynote-407 evaluated a platinum, taxane, pembrolizumab combination vs. platinum-taxane chemotherapy allowing for cross-over in squamous cell lung cancer. The study showed a 36% reduction in risk of death at a relatively early analysis.
Encouraging activity of chemotherapy-pembrolizumab combinations has been seen in early studies in a number of cancer types, including gastric cancer, triple negative breast cancer, human epidermal growth factor-2 negative breast cancer, estrogen-receptor-positive (ER+) breast cancer, as well as head and neck cancer. Many of these applications are now the subject of ongoing randomized studies. Considering targeted therapies, combinations of tyrosine kinase inhibitors with pembrolizumab have shown encouraging activity in early studies in melanoma, based upon BRAF-MEK inhibitor combination with pembrolizumab. In kidney cancer, combinations with axitinib or lenvatinib have shown increased activity over the already impressive activity seen with pembrolizumab monotherapy. These observations led to a number of randomized studies with registration intent. A number of studies are ongoing exploring combinations with other immunologic-modulating therapies. An example of a combination study involves talimogene laherparepvec, an oncolytic virus, in melanoma where response rates in the order of 60% led to a Phase 3 trial being conducted with this combination vs. pembrolizumab monotherapy. In terms of ongoing biomarker research, MSD continues to explore ligand expression, inflammatory and other gene signatures, resistance biology, mutational load, defects in DNA repair mechanisms and MMR deficiency as potential approaches to optimizing patient selection and identifying promising combination strategies.
Following this exciting talk, the audience asked several questions, of which the question of toxicity was the first. Dr. Baynes suggested that generally toxicities should be explored for each combination. A generalization is that, thus far, non-overlapping toxicities of agents have usually been additive, whereas overlapping toxicities may in certain circumstances lead to exaggerated toxicity. The next question concerned the performance of pembrolizumab in comparison to other anti-PD-1 or anti-PD-L1 antibodies. Dr. Baynes pointed out that there have been no head-to-head comparisons between the anti-PD-1 mAbs or between anti-PD-1 and anti-PD-L1 mAbs. Thus, any inferences are being drawn from cross-treatment comparisons, but these can be misleading. There are clearly significant differences between molecular characteristics of the well-characterized anti-PD-1 mAbs. A question of particular interest concerned the PD-L2 target. Dr. Baynes highlighted that MSD has evaluated PDL-2 expression in certain datasets, for example, in their head and neck cancer studies. While PD-L1 and PD-L2 expression are highly correlated, PD-L2 expression does appear to be an independent predictive marker. In connection to this, the next question concerned the predictive power of PD-L1 expression? Dr. Baynes stressed that there are some PD-L1 positive patients who do not respond and some PD-L1 negative patients who do. However, PD-L1 status by IHC is an important and clinically very useful biomarker, with the exception of tumors where the marker is almost uniformly present (e.g., melanoma). Thus, PD-L1 has been approved as a companion diagnostic in certain tumor types; additional biomarker work may well reveal important additional precision medicine approaches to define responder populations and identify resistance biology. Finally, the audience asked about the clinical failure when Keytruda® was used with an indoleamine-pyrrole 2,3-dioxygenase inhibitor. Dr. Baynes acknowledged that a large, well-conducted randomized study showed no benefit of this combination in melanoma compared to pembrolizumab alone. However, he emphasized that there could be a number of possible explanations, such as: 1) the mechanism may not be therapeutically important in this tumor type; 2) it may not be therapeutically important more broadly; or 3) it may not be sufficient in terms of inhibiting the formation of tryptophan-derived intermediates. He pointed out that further work is ongoing in other tumor types, exploring precision medicinal tools to identify responding subsets and other possible mechanisms aimed at more complete inhibition of intermediate production.
SESSION I – VALIDATED TARGETS: lessons learned from success & failures
This session was opened by the talks of Dr. Hervé Perron (Geneuro SA) and Dr. Hans Martin Schneble (Servier) who described a new neutralizing antibody GNbAC1, which abrogates human endogenous retrovirus (HERV)-W envelope protein-mediated oligodendrocyte maturation blockade and inhibits toll-like receptor 4 (TLR4)-driven innate immune inflammation. Dr. Peron explained at the beginning of his talk that MSRV are members of the HERV-W family and a newly discovered endogenous retrovirus. This family of viruses entered the germline millions of years ago, representing about 8% of human genome sequences. It is now known that different viruses play the role of the environmental trigger that activates HERV-elements, and thus becomes associated with multiple sclerosis (MS). MSRV-envelope proteins are detectable in MS patients (evidenced by increased RNA expression and DNA copy number). In brain, macrophages and microglial cells express MSRV-Env, few astrocytes and CD3 T-cells in lymphoid infiltrates may have membrane-bound secreted Env. HERV-Env protein is expressed in newly forming lesions, but is limited to perivascular macrophages and microglia in the areas of effective demyelination. Env is expressed in active and expanding lesions, but is limited to the rim of microglia. This active microglial frontier is most probably involved in plaque expansion. Env can induce MS-like symptoms in mice, demyelination, and autoimmunity against myelin antigens.Citation3
Dr. Peron and his team identified the MSRV-Env-neutralizing antibody GNbAC1, and showed a strong decrease of clinical score, weight stabilization and survival in mice treated with the mAb. Non-treated mice showed clinical progression. GNbAC1 rescues maturation and myelination, while Env interferes with maturation of oligodendrocyte precursor cells when migrated to lesions. It is worth noting that the Env protein is not expressed in healthy individuals, while abnormal expression is triggered by environmental factors, such as Epstein–Barr virus, human herpes virus 6, varicella virus and herpes simplex virus infections, especially in MS. Functionally, HERV-W Env will activate TLR4 pathways, thus activating the immune system and impairing functions of non-immune cells.
Following these fundamental explanations by Dr. Peron, Dr. Schneble continued by describing the clinical trial involving the GNbAC1 antibody and MS patients. He highlighted the fact that MS is one of the most frequent neurological diseases, with two mechanisms driving the disease: inflammation and neurodegeneration. Concerning inflammation, about a dozen anti-inflammatory treatments are available. However, there is a high need for treating neurodegeneration in all forms of MS. Brain atrophy is today the best-studied marker of neurodegeneration, and occurs early in MS at a high annual rate of 0.5–1.35% per year.Citation4 Anti-inflammatory treatments are not effective on neurodegeneration and do not stimulate remyelination. There is, therefore, an urgent need for new agents that will tackle the neurodegenerative changes in the brain, and thus help patients with MS. Servier in collaboration with Geneuro started a CHANGE-MS dose-finding and proof-of-concept Phase 2b study evaluating GNbAC1. The results showed that GNbAC1 was not effective in lowering the systemic inflammation. The primary endpoint of preventing (or lowering) new inflammatory lesions (as evaluated by magnetic resonance imaging (MRI)) was not reached. However, promising results on secondary endpoints of neurodegeneration with the highest dose (18 mg/kg) were observed. Namely, the mAb had consistent effects on regional and global brain atrophy, significant reduction of black holes as another marker of neurodegeneration (independent on atrophy) and a good safety profile. Overall, the study showed promising results that will be explored in more detail in the future.
The talks given by Drs. Peron and Schneble prompted several questions from the audience. One of these was the rationale to give a combination with several anti-inflammatory treatments. Dr. Schneble acknowledged that this might be a good option for MS patients who still have a high inflammatory activity. Another relevant question concerned the CHANGE-MS clinical trial. Namely, it was of interest to understand if there was any significant clinical effect observed for the MS patients. Dr. Schneble explained that in this study the patient population exhibited high inflammatory activity and clinical symptoms mainly due to relapses. As GNbAC1 was not effective against systemic inflammation, no clinical effect was observed. Moreover, as the patient population was clinically very stable, it was difficult to evidence any treatment effect on MS-caused disability. Dr. Schneble noted that seeing the translation of effects on neurodegeneration-related MRI-markers (as observed with GNbAC1 treatment) into a clinically meaningful effect would require studies over 2 or 3 years in patients with progressive MS.
The next speaker in this session was Dr. Stephanie Cornen (Innate Pharma) who gave a talk on new natural killer (NK) cell immune checkpoint inhibitors. Dr. Cornen reiterated that immuno-oncology is changing cancer treatment by harnessing the power of the body’s immune system to recognize and kill cancer cells. This approach aims at restoring immune surveillance and can lead to durable responses in some patients who have failed all available conventional therapies. Innate Pharma focuses on three strategic key pillars, immune checkpoints, tumor antigens, and the tumor microenvironment, and in accordance with that Dr. Cornen proceeded to outline the individual projects: anti-NKG2A (IPH2201), anti-Siglec 9, and anti-CD39 (IPH5201)/anti-CD73 (IPH5301) antibodies.
Monalizumab (IPH2201) is a first-in-class humanized IgG4 targeting NKG2A (NK Group 2A), which is expressed as a heterodimer with CD94 on subsets of NK cells, γδ T cells and tumor-infiltrating CD8 + T cells. This inhibitory receptor binds to HLA-E molecules, which are frequently upregulated on cancer cells, providing a negative regulatory signal to tumor-infiltrating lymphocytes (TILs). Monalizumab blocks binding of CD94/NKG2A to HLA-E, reducing inhibitory signaling, and thereby unleashing NK and T cell responses. Several combinations are being explored in preclinical studies. First, combined NKG2A and PD-L1 blockade increase complete response rate in a CD8 + T and NK cell-dependent manner in vivo. Secondly, NKG2A blockade enhances cetuximab-mediated ADCC towards head and neck tumor cell lines in a dose-dependent manner. Monalizumab is currently in Phase 2 development in various cancer indications and combinations. Innate Pharma and its partner AstraZeneca are jointly developing monalizumab according to their commercialization agreement. The development plan includes a Phase 2 combination clinical trial with durvalumab (MEDI4736), an anti-PD-L1 immune checkpoint inhibitor developed in solid tumors by MedImmune/AstraZeneca. Updated clinical data show preliminary anti-tumor activity in patients with recurrent/metastatic microsatellite-stable CRC, a population historically unresponsive to PD-1/L1 blockade.Citation5 Parallel to this, Innate Pharma has conducted a Phase 2 combination clinical trial with cetuximab in head and neck cancers. Preliminary data presented at AACR 2018 suggest promising anti-tumor activity of the combination.Citation6
The second project focused on Siglecs, a family of 15 members of sialic acid-binding receptors that vary in their expression on immune cells and their specificity for sialic acid–containing ligands. Siglec-9, which is an inhibitory receptor expressed on NK and myeloid cells (including dendritic cells (DC), monocytes and neutrophils), preferentially binds to α2,3-linked sialic acids. Sialic acids are involved in tumor cell malignancy, and reportedly act as part of a mechanism of escape from immune surveillance. Siglec-9 can interact with sialic acids of malignant and normal cells, and dampen immune cell functions. Thus, Siglec-9-sialic acid interaction disruption may promote anti-tumor immunity. Innate Pharma generated high-affinity anti-Siglec-9 antibodies that block the interaction between Siglec-9 and its ligands. These antibodies potently enhance NK cell cytotoxicity in vitro by blocking interactions with sialic acid expressed on tumor target cells. They notably showed that anti-Siglec-9 antibodies improve anti-tumor response induced by the blockade of the immune checkpoint NKG2A. Using flow cytometry analyses, they demonstrated that Siglec-9 is expressed on several immune cell types, including lymphocytes and myeloid cells, pointing to potential multiple MOA. Finally, Siglec-9 expression is maintained on tumor-infiltrated immune cells as demonstrated by IHC, and Siglec-9 is upregulated on circulating T cells in cancer patients, suggesting a putative role on adaptive immunity.
The last project outlined by Dr. Cornen concerned the adenosine pathway. Blockade of CD73 enzymatic activity has recently been reported to improve immune checkpoint inhibitor anti-tumor activity. In addition, Innate Pharma showed that in vivo blockade of adenosine triphosphate (ATP)/Ado pathway in CD39 knock-out mice resulted in improved anti-tumor efficacy of immune checkpoint therapies (i.e., anti-PD-1, anti-CTLA-4) and chemotherapy such as oxaliplatin. IHC and flow cytometry evaluation showed that CD73 is expressed by tumor cells, and that CD39 is frequently up-regulated on TILs compared to peripheral blood mononuclear cells (PBMC) or adjacent non-tumor tissue. They generated anti-human CD39 (IPH52) and anti-human CD73 (IPH53) blocking antibodies with unique properties for cancer immunotherapy. These mAbs potently inhibit the enzymatic activity and reverse adenosine-mediated T-cell suppression in vitro in the presence of ATP and CD39- and CD73-expressing immune cells. The anti-CD39 IPH52 mAb enhances DC activation and subsequent T-cell proliferation in vitro, probably by maintaining high concentrations of ATP in the extracellular compartment. The anti-CD73 IPH53 mAb is more potent than benchmark mAbs currently in Phase 1 clinical development for the blockade of soluble and membrane-associated CD73 enzymatic activity and for adenosine monophosphate-mediated T-cell suppression reversion. Finally, Dr. Cornen showed results demonstrating that combining IPH52 and IPH53 mAbs at sub-optimal doses leads to a strong reversion of immune cell inhibition in the presence of ATP.
The final speaker of this session was Dr. Nicolas Poirier (OSE Immunotherapeutics) who delivered a talk on development of anti-interleukin (IL)-7 receptor mAb for the treatment of inflammatory bowel disease (IBD). Dr. Poirier highlighted at the start of his talk that IL-7 is a homeostatic cytokine and an upstream mediator fueling immune response. IL-7 fuels chronic inflammation by controlling human memory T-cell persistence. Because IL-7 receptor is expressed at low levels in Tregs, targeting IL-7 receptor could be a way to specifically target pathologic memory cells. Indeed, it has been shown that IL-7 receptor blockade causes T-cell depletion and prolongs survival. This was the rationale behind OSE Immunotherapeutics’ goal of developing a novel therapeutic IL-7 receptor-targeting antibody. They have performed the antibody screening based on STAT5 signaling, which is down-stream IL-7R. The isolated clone OSE127, emerging from selection studies, showed no internalization, in contrast to other antibodies, but experiments performed in monkeys demonstrated that the epitope targeted by OSE127 is key to the activity of the antibody. OSE127 is efficient in significantly reducing inflammation in a long-lasting manner. The antibody-blocked antigen engaged memory T cell survival and had no significant impact on quiescent human T cells. Possible indications for the antibody are therefore MS, type 1 diabetes and other immune system-driven diseases. Dr. Poirier also pointed out that, in IBD patient treated with anti-TNF-α, IL-7R mucosal over-expression was identified as a factor associated with non-responsiveness to treatment. The same effects were observed for the anti-α4β7 integrin therapy, suggesting that OSE127 might represent a natural choice for rescuing patients who develop resistance or do not respond to anti-TNF-α and anti-α4β7 integrin therapies. This exciting hypothesis should be validated in future clinical studies.
SESSION II – DISCOVERY APPROACHES: clinical material & target identification
The second session was opened by Dr. Björn Frendéus (BioInvent) who delivered a talk on a patient-centric approach to cancer immunotherapy discovery. At the beginning of his talk, Dr. Frendéus stressed that there is a great need to develop new immunomodulatory therapies with novel and complementary MOA. Current understanding suggests that reasons why available immunotherapies work in only a fraction of cancer patients include lack of or inadequate immune infiltration in tumors. BioInvent aims to complement currently available immunotherapies by discovering and developing new immunomodulatory targets and antibody-based products. The company focuses on targeting and overcoming key immune suppressive mechanisms and cells in the tumor microenvironment; mechanisms of mAb resistance, tumor-associated myeloid cells, and cancer-associated regulatory T cells. BioInvent has developed a patient-centric function-first platform for discovery of novel oncology targets and mAbs (F.I.R.S.T™); the company characterizes immune infiltrates in tumors and assesses tumor-infiltrating lymphocytes (TIL) phenotype and function. This allows them to validate TILs for use in their mAb discovery approach, as well as to identify candidate targets for generation of mAb with new functions. F.I.R.S.T™ discovery process uses primary patient-derived and mouse immune competent cancer model-derived cells. The functional screening of mAb-target pairs is conducted in complex experimental models both ex vivo and in vivo, notably such as syngeneic, transgenic and humanized mice. This allows BioInvent to identify clinically relevant targets and antibodies.
In their Treg program, BioInvent aims to identify antibodies and target receptors that are broadly specific to cancer Treg vs. effector T cells, and potentially associated with improved tolerability compared with currently available Treg-targeting drugs. Treg are very important immune regulatory cells, and emerging evidence indicates that Treg depletion may contribute to efficacy and MoA of both immune agonist and T cell checkpoint antibodies. In their F.I.R.S.T™-Treg program BioInvent has found that cancer-associated Tregs are phenotypically distinct, and that immune checkpoint receptors, e.g., CTLA4, OX40, and ICOS, are over-expressed on cancer-associated Tregs. Importantly, cancer-associated Tregs also showed enhanced suppression of proliferation of CD8 + T responder cells. F.I.R.S.T™ screening of the phage display library n-CoDeR (1010 human scFv) generated antibodies that had in vivo antitumor activity in models responsive (CT26) or resistant to immune checkpoint inhibitors (MC38). Antibody clones with in vivo antitumor activity differed in their ability to modulate the tumor microenvironment; some clones showed profound Treg depletion and modulation of CD8:Treg ratios, whereas other showed a general enhanced T cell influx with no apparent change in CD8:Treg ratios.
In the next step of the Treg program, they looked at identifying targets. Known targets included those currently pursued in clinical development, e.g., ICOS, OX40, 4-1BB, glucocorticoid-induced TNFR-related protein, and CTLA4, validating the F.I.R.S.T™ approach. Other antibodies did not bind to the “usual suspects” or previously developed targets, indicating the discovery potential of the F.I.R.S.T™ approach. An example of an undisclosed target/antibody pair that synergized with anti-PD-1 to enhance in vivo antitumor activity in partially responsive, and checkpoint resistant, MC38, and B16 tumor xenografts, respectively, was presented.
Finally, a new strategy to overcome the resistance to immunotherapies in a potentially more efficacious and better-tolerated manner was presented. This strategy builds on tumor-restricted production of immunomodulatory full-length human antibodies, following introduction of mAb coding sequences into a tumor-tropic oncolytic Vaccinia virus. In a first collaborative program, BioInvent and their oncolytic virus’ expert partner Transgene aim to boost tumor immune infiltration, prime innate immunity, enhance antigen presentation and achieve tumor-localized Treg depletion. The strategy consists of incorporating the sequences of a full-length human anti-CTLA-4 antibody into an oncolytic Vaccinia virus vector and use of the resulting agent in the context of dual checkpoint inhibition (anti-CTLA-4 and anti-PD-1/PD-L1). Virally mediated local expression of anti-CTLA-4 is expected to improve tolerability compared to the systemically delivered ipilimumab, whilst retaining local Treg depletion and T effector cell boosting effects in the tumor microenvironment.
The second Keynote talk presented by Prof. Mark Cragg (University of Southampton, UK), outlined the challenges and opportunities of targeting the inhibitory Fc gamma receptor IIB (FcγRIIB). Prof. Cragg reminded attendees that rituximab was the first antibody that demonstrated clinical efficacy in oncology, which boosted the belief that antibodies could be successful anti-cancer therapeutics. Fc receptors are key mediators of mAb efficacy, expressed on different immune cell types at different levels. Malignant B cells can also express FcγRIIB. This can cause the rapid internalization of rituximab from the tumor cell surface, resulting in its degradation. High expression of FcγRIIB, therefore, limits rituximab immunotherapy, and is one potential resistance mechanism. Increase in FcγRIIB and suppression of activating Fc receptors is also observed on macrophages during tumor development, again resulting in therapy resistance. One strategy to overcome these issues is to block FcγRIIB. Although there is high homology with the activatory FcγRIIA, they were able to generate, in collaboration with BioInvent, a novel antibody using phage display that does not activate the receptor, blocks its activity and prevents rituximab internalization. Combination of the novel antibody against FcγRIIB with rituximab showed prominent deletion of B cells in a hCD20, hFcγRIIB transgenic mouse model, which lacks mouse FcγRII. Treg depletion is also influenced by FcγR interactions, and can elicit tumor control. Blocking FcγRIIB with mAb has different effects on the target and effector cells. In this case, wild type IgG is not optimal, but rather an Fc-inert FcγRIIB blocker works better.
The next talk in this session was delivered by Dr. Marlon Stoeckius (NY Genome Center) who outlined one of the most exciting and rapidly expanding target discovery approaches today, namely single-cell sequencing. In line with this, Dr. Stoeckius presented two new single-cell RNAseq approaches called CITE-seq and Cell Hashing. The first approach, CITE-seq aims at getting the best out of RNAseq and fluorescence-activated cell sorting (FACS) analysis. Namely, FACS can measure only a few proteins simultaneously due to known difficulties of spectral overlapping. Single cell RNAseq is powerful in quantifying gene expression in single cells, but the technique inherently does not reveal data about the protein expression levels, which can be sometimes markedly different. RNAseq is therefore limited in depth, and low-level transcripts are frequently omitted or not reliably measured due to PCR amplification biases. CITE-seq uses oligonucleotide tagged antibodies that mimic mRNA oligo, which can be integrated in a normal mRNA sequencing approach after amplification. The procedure is simple and resembles FACS sample preparation where antibodies bind on the cells, and then single cell technology is used to barcode individual cells, which are encapsulated in droplets and used to perform subsequent library construction steps. Dr. Stoeckius compared this technology with classical single cells RNAseq. He found that CITE-seq antibody tags are highly correlated to mRNAs and have no dropout. The technology can be nicely used with new antibodies to map them to the cell population to which they bind. CITEseq is comparable to cytometry, but can provide an additional comprehensive transcriptome profile for each cell. The technique can resolve cell types with subtle transcriptomic and post-transcriptomic differences. As an example, so far 3 to 5 T cell clusters were reported, but CITE-seq technology was recently able to identify 13 T cell clusters.Citation7 Similar to CITE-seq, Cell Hashing methodology uses DNA barcoding of antibodies to multiplex several single-cell sequencing experiments in one run. Ubiquitous protein markers are targeted with antibodies containing different DNA barcodes. Upon labeling, different samples can be merged into one and analyzed as such, much more efficiently sparing both time and cost.
SESSION III – PITCH SESSION: new approaches/technologies for mab target discovery & validation
The third session of the AIS2018 symposium was dedicated to short presentations describing original methods for target discovery and validation. Presentations about breakthrough technologies for early antibody development (selection and characterization) were also welcomed because this is an integral part of the target validation process. This session was opened by the talk from Dr. Astrid Musnier (MAbsilico), who presented their program concerning in silico methods for antibody characterization and development. Dr. Musnier presented the MAbtope docking-based epitope determination method that starts with the 3D structure of antigen (solved) and antibody (modeled). To perform the docking experiment, only the sequence of the antibody is necessary; the software gives as output the identification of peptides constituting the putative epitopes. Golimumab versus TNF-α was provided as an example. Use of the MAbtope docking algorithm allowed peptide (epitope) prediction, which was subsequently positively validated using homogeneous time-resolved fluorescence (HTRF) methodology. Further refinement of the epitope was achieved by mutating the individual residues in the peptide. Next, two additional algorithms, MAbsubstitute and MAbCross, were presented, both being based on the rationale that if two antibodies look alike, they will probably bind the same/similar target. The similarity is measured by looking at the linear sequence and the secondary structures. MAbsubstitute finds new antibodies that can replace a query one. MAbCross predicts the targets of the new antibodies as being off-targets of the query one and helps forecasting the cross-reactions. Collectively, MAbsilico presented a very complete suite of bioinformatics methods that can be used to refine the antibody choice and find more suitable antibodies, even for very difficult targets.
Dr. Bruno Tiller (Synthelis) presented a new strategy to use proteoliposomes as a tool to select active VHH binders by phage display technology. The advantage of the technology was put in perspective by the example of the NanoLOx project. The project aims to block the dialog between the leukemic cells and bone marrow microenvironment to prevent chemoresistance and relapse. In this project, Synthelis was in charge of the CXCR4 target production using its cell-free technology. Thus, the transcription and translation machinery was extracted from a bacterial cell culture and used outside of the cells to express the G protein-coupled receptor (GPCR) target. To obtain the active form of CXCR4, specific liposomes were added to the cell extract and proteoliposomes were collected after overnight reactions. The expression yield of the protein target reached 200 µg/mL, which is high for a GPCR. After validation of the proper receptor folding with different methods (surface plasmon resonance, bioluminescence imaging, and atomic force microscopy), the proteoliposomes were successfully used to select six different nanobody binders by phage display. A series of bioassays allowed the selection of one nanobody able to bind to CXCR4 with high affinity and to antagonize its activity.
Dr. Elsa Kress (Antineo) discussed the development of original in vivo tumor models for secondary resistance in immune-oncology. Dr. Kress noted that 60% of patients will not respond to immune checkpoint treatment, and that, among the responders, 25% will develop resistance. There is thus a genuine need for new targets and new preclinical models to study immuno-oncology. While syngeneic preclinical tumors are characterized for their sensitivity/primary resistance to immune checkpoint inhibitors, there is still a lack of models of secondary resistance. Antineo’s scientists selected parental tumors that are sensitive to immune checkpoint inhibitors, and then made them resistant. The parental and resistant models were characterized by immunophenotyping and RNAseq data to better understand the mechanisms of resistance. Several resistant models have been created for anti-PD-(L)1 therapies and new models are ongoing for anti-CTLA4. Those models are available for preclinical services in the framework of Antineo’s activities.
Mrs. Eva Sivado (Centre Léon Bérard & Covalab) discussed CovIsoLinkTM technology, which uses a new bacterial transglutaminase Q-tag substrate for the development of site-specific ADCs. Mrs. Sivado pointed that the main advantage of ADCs is their ability to extend the therapeutic window. The best are the 3rd generation of ADCs that are homogenous, have site-specific conjugation and improved stability. Covalab improved microbial transglutaminase-mediated conjugation by introducing a novel glutamine donor peptide substrate into the C-terminal part of various antibody fragments. They verified that the conjugation does not alter the antigen binding ability of the antibody, and showed a higher in vivo stability of their site-specific conjugate compared to non-specific counterpart.
Dr. Guilhem Richard (EpiVax) outlined an interesting bioinformatics approach to immune-engineering of better candidates. EpiVax is an immunoinformatics company that performs immunogenicity risk assessment of biologics and vaccine antigens, and can re-engineer protein sequences, such as mAb or vaccine antigens, to modulate their immunogenicity (either increasing or decreasing it depending on the application). T-cell epitopes can activate either effector or regulatory T cells. While most T-cell epitope prediction algorithms cannot differentiate between the two types of T-cell epitopes, EpiVax has designed specialized tools that evaluate which T cell population (effector or regulatory) is likely to recognize the predicted epitopes. The immunogenic potential of sequences is evaluated with EpiMatrix, while their tolerance potential is evaluated with JanusMatrix. Both immunogenicity and tolerance potentials can be ranked on a scale and compared to benchmark sequences. The immunogenic and tolerance potentials of the same sequence can change depending on the species/strain that is being considered (e.g., human, Bl/6 mice, Balb/c mice).
Dr. Julien Nourikyan (ALTRA Bio) presented a gating automation method. ALTRA Bio has expertise in the biostatistical and mathematical analysis of high-throughput data. They developed a machine-learning-based approach able to reproduce and standardize the application of predefined gating strategies to a high number of new flow and mass cytometry samples. This approach handles biological and technical variability, and has already proven its effectiveness on deep and complex gating strategies for the immunophenotyping of mouse and human samples.
Dr. Michael Schwenkert (Bio-Rad Laboratories) presented the discovery and validation of a novel chimeric mAb targeting MASP2. Human Combinatorial Antibody Library (HuCAL®) technology was used to isolate a chimeric antibody specific to mannose-binding lectin-associated serine protease 2 (MASP2) that inhibits the lectin pathway of the complement system in mice. The generation of mAbs against a murine protein through traditional methods can result in difficulties eliciting a sufficiently strong immune response against a self-antigen. HuCAL technology overcomes tolerance mechanisms and eliminates the need for animals. In this example, a human anti-rodent MASP2 antibody was selected, and then further engineered into a human/mouse IgG2a chimeric molecule for in vivo studies in mice. This enabled scientists to carry out target validation studies in a multitude of disease models, including a mouse model of myocardial infarction, demonstrating the therapeutic potential of inhibiting MASP2.Citation8 This validation of MASP2 as a feasible, druggable target provided important proof-of-concept data supporting the development of OMS721, a fully human mAb against MASP2 generated by the pharmaceutical company Omeros independently of Bio-Rad. Omeros holds worldwide exclusive licenses to rights related to MASP2, inhibitors targeting MASP2, and the therapeutic applications for these inhibitors.
Dr. Pierre Martineau (IMAb) presented a new generation of synthetic human antibody libraries displayed on filamentous phages. These libraries use a single hyperstable framework to reduce expression level and stability variability between the isolated clones. In addition, they presented a new display format that allows, with a single plasmid, to obtain antigen-binding fragment (Fab) display on phage particles and expression of full-length IgG in mammalian cells that can be directly used in functional screening experiments.
Prof. Pongrama Ramasoota (Mahidol University, CEAR) has shown a new neutralizing human mAb against dengue virus. Dengue is a big health problem around the world, and there is no drug for dengue fever. Prof. Ramasoota and his colleagues produced a fully human antibody using PBMC from infected patients fused with SPYMEG cells to create hybridomas producing mAbs. They produced 20 anti-Env mAbs with 95–100% neutralizing activity. The antibodies were successfully tested in mice that were rescued from virus for more than several months, while the controls died in 2 weeks. Next, they moved to experiments on monkeys, where the antibody reduced the viral load in a very significant manner. In order to move to human clinical trials, they have generated Chinese hamster ovary (CHO) cells expressing their antibody clones. Activity testing showed no alteration in activity compared to the original clones. Clinical studies are planned in the near future.
Dr. Jean-Jacques Mention (Axenis) presented a humanized mouse system for proof-of-concept in preclinical research. Axenis is a French contract research organization generating, exploiting and developing new immuno-deficient BRGS (BALB/c Rag2tm1Fwa IL-2Rγctm1Cgn SIRPαNOD) mouse model and the derivative immuno-deficient BRGSF (BALB/c Rag2tm1Fwa IL-2Rγctm1Cgn SIRPαNOD Flk2tm1Irl) mouse model, both being particularly permissive to the long-term establishment of a human immune system from umbilical cord blood-derived hematopoietic human stem cells. These optimized humanized mouse models represent a unique tool to evaluate human immune cell reactivity and introduce a variety of new solutions for academia, biotech companies and biopharmaceutical industries that seek innovative in vivo models for immuno-oncology, infectious diseases, inflammatory diseases and vaccination.
SESSION IV – PRE-CLINICAL VALIDATION: method, models & companion tests
The Keynote Lecture from Prof. Guus Van Dongen (VUmc, Amsterdam) opened the pre-clinical validation session. In his lecture, Prof. Van Dongen addressed the current status of immuno-positron-emission tomography (PET) with zirconium 89-labeled antibodies. He notably highlighted that we still do not understand the actual outcome of drugs in the body after administration to patients. Imaging is an excellent tool for better understanding drugs, better designing drug and optimizing them, and it can help in a more personalized application of drugs. In the clinic, imaging is important to diagnose the disease, to confirm that the therapy selectively hits the disease, and finally to demonstrate efficacy.
Two techniques are suitable for molecular imaging due to their sensitivity, nuclear and optical imaging. An example of anti-CD44v6 imaging using single photon emission computed tomography was shown that was done 10–15 years ago. Although revolutionary at that time, the resolution was very poor. Nevertheless, the images were very informative. Today, we have dramatic improvements in sensitivity, resolution, and mapping. An example is PET-CT/MRI, where anatomical features are combined with the imaging of metabolism (18F-FDG). The same principles can be used for visualizing, tracking and quantifying therapeutics in the human body. New antibody-based drugs are very potent and their in vivo behavior mostly unpredictable (ADCs, immune checkpoint inhibitors, bi- and multi-specific antibodies). It is thus crucial to understand what these drugs are actually targeting. A few appealing examples where imaging of the antibody might be very informative were presented. An anti-CEA mAb fused to IL-2 was developed to improve immune competence inside the tumor. However, IL-2 binds to a subset of CD25-positive cells in the blood and thus may induce toxicity, while tumor accumulation of the construct might be hampered. To overcome the latter, IL-2 has to be mutated and the selective tumor targeting can be then confirmed with imaging. Probodies cannot bind the target because the antigen-binding site is capped until this cap is cleaved in the tumor, activating the probody into an antibody. This is attractive because a tumor-specific target is thus not necessarily need since this probody is only active inside the tumor. Only imaging can validate such smart approaches, and should steer the selection of the most appropriate compounds.
In antibody radiolabeling, the labeling should not change the property of the drug (inert labeling), and the process should be GMP compliant and acceptable for both the pharma industry and regulatory agencies. For PET imaging of slow kinetic ligands such as antibodies, PET emitters with a long half-life such as zirconium-89 [89Zr] are required. Prof. Van Dongen presented generic procedures to label antibodies/proteins/cells with [89Zr]. Moreover, he introduced the novel DFO* chelate that is fully coordinating [89Zr], and as such prevents the release of the isotope during conjugate storage and after its administration. This reduces the unspecific signals in bones, joints and other structures. Prof. Van Dongen further showed the example of Kadcyla® (T-DM1, trastuzumab-DM1 ADC), where a scouting imaging with [89Zr]-labeled trastuzumab identified patients whose tumors are binding trastuzumab, and thus will benefit from the ADC therapy. Another example that Prof. Van Dongen brought up was the value of radiochemistry and PET imaging in the development of novel platinum II-based ADC linker technology (LinXis Pharmaceuticals, The Netherlands) that can easily couple a drug to an antibody (“Plug and Play”). They have used PET imaging to evaluate the in vivo stability of these ADCs with this linker, and proved full stability. To this end, each of the ADC components was labeled with a radionuclide, respective PET emitter, [89Zr] was used for the antibody, [195mPt] was used for the linker and a third radionuclide was used for the drug. In this way, biodistribution of each component was evaluated. Sijbrandi et al.Citation9 compared the therapeutic efficacy of Kadcyla® in tumor-bearing mice with that of trastuzumab-auristatin F using either the platinum linker or the Seattle Genetics linker technology, and demonstrated that the ADC with the platinum linker outperformed the two other ADCs. A potential explanation for the enhanced efficacy could be the fact that the platinum linker is a charged molecule, and, when coupled to a drug, it cannot enter a normal cell, resulting therefore in decreased toxicity. Alternatively, once a tumor cell internalizes the ADC and the antibody becomes catabolized inside the endosome, the released drug-linker complex is less prone to be a substrate for multi-drug resistance-mediated drug expulsion and remains in the tumor cell. So, it might well be that the therapeutic window of the ADC is broadened by this new linker.
Other appealing clinical examples were provided to demonstrate the value of [89Zr]-immuno-PET. One of the most challenging brain tumors in children is diffuse intrinsic pontine glioma (DIPG), a lethal childhood malignancy of the brainstem comprising 10% of all pediatric central nervous system tumors. DIPG tumors are resistant to all kinds of systemic therapies, including targeted agents, and hardly any patient survives beyond 2 years from diagnosis. One hypothesis for therapy failure is that drugs actually do not reach the tumors. Indeed, immuno-PET studies with [89Zr]-bevacizumab revealed that this antibody did not enter most of the tumors, and hence was ineffective. Alternative drug delivery options should be explored for these children. Immune checkpoint inhibitors are another example where we do not know what the ideal distribution would be, which patients will benefit and how efficacy can be improved. First-in-human imaging studies with a [18F]-labeled PD-L1 ligand and [89Zr]-labeled nivolumab (anti-PD1 to block interaction with PD-L1) demonstrated strong tumor distributions in responder patients. At the end of the lecture, Prof. Van Dongen stressed that immuno-PET should be used to determine the right patient, right disease, right drug, right dose, right moment and right outcome.
Next speaker was Dr. François Romagné (MI-MAbs) who gave a talk about knock-in mice as preclinical models for the evaluation of mAb candidates. He outlined the interest in genetically modified mice for target validation. Indeed, most therapeutic mAbs do not cross-react with mouse homologous targets. Most efficacy models are, however, based on mice. One has thus to use syngeneic mouse models and generate surrogate mAbs that do not completely reflect characteristics of the future drug candidate, whose efficacy can only be evaluated in xeno or xeno/humanized model (containing human immune cells). In the latter, the problem is that the target is only expressed on grafted tumors and do not reflect the global tissue distribution. These murine models are usually not adapted to find the therapeutic window and to evaluate tolerance. As an alternative, knock-in mice can be used. In this model the murine gene is replaced by its human ortholog, thus enabling testing of the actual drug candidate in physiological conditions. If such replacement alters the physiology of the target by impairing the target interaction with its interactant, Crispr/Cas9 technology expedites the replacement of several mouse genes with the human ones (i.e., the target and its interactants), and allows a more physiologically relevant model to be obtained.
As an example, Dr. Romagné discussed the replacement of mouse CD3ε gene by its human counterpart. In such mice, therapeutic activity could be demonstrated in a cancer mouse model using blinatumomab as a prototype of a T-cell engager, recognizing human CD3ε on one side and human CD19 on the other. To demonstrate activity, a BL/6 cell line transfected with human CD19 was used. The system could be further improved by crossing CD3ε knock-in mice with CD19 knock-in mice. Such a system may greatly facilitate the early testing of bispecific mAbs that must otherwise be evaluated in non-human primate to demonstrate safety and in parallel with Xeno/humanized mice to demonstrate efficacy. Alternatively, genetically modified mice can be used to obtain cross-reactive mAbs (mice/human) by immunizing knock-out mice with the target of interest. Dr. Romagné provided the example of anti-CD73 antibody, a target where the level of homology between mice and human is 88% at the protein level. They used a cell line-based screening system where three receptors (human, cynomolgus macaque, and mice) are expressed, which expedites characterization of cross-reactions in the primary screening of hybridomas. Immunization of wild-type mice gave no antibody cross-reactive with mice, whereas immunization of knock-out mice provided 20 cross-reactive antibodies between mice and human. Obtaining the cross-reactive antibodies greatly facilitated preclinical characterization of the lead candidates.
Dr. Eric Trinquet (CisBio) delivered the next talk on innovative fluorescent technologies to identify and characterize biologics. He highlighted the HTRF technology based on FRET and proprietary fluorescent donors that have long lifetime in fluorescence and time-resolved detections to remove the background. The tag-lite platform combines HTRF and SNAP-tag labeling technology. SNAP-tag involves site-specific covalent labeling of receptors at the surface of living cells. Using this technology, they have developed a wide range of binding assays.Citation10 This technology can be used to screen for binders from phage display libraries of mAbs.
Dr. Jacques Dumas (Sanofi) spoke on developability assessment of antibodies, which needs to be taken into account because scaling up and storage steps are necessary for the novel molecule before it is given to the patients and because stability/aggregation issues can be serious hurdles in drug development. Very stable molecules are needed, and this requires a strong interface between Research and Development teams. Developability is assessed during the entire process of mAb selection during the research stages, i.e., hits (in silico design, sequence optimization), leads (yield, purity, integrity), lead optimization (physical and chemical stabilities, stress tests, effects on functional activity), pre-candidate selection and final candidate.
Dr. Dumas presented a case study of humanization and amino-acid liabilities-derisking of an antibody where the same paratope was kept as the original mAb, but some residues were mutated in the Fc. Nano-differential scanning fluorimetry technology was used to stress the molecule with the temperature and to assess folding. Thermally stressed antibodies in different pHs were also analyzed by peptide mapping in order to assess the amino-acid liabilities. Antibody clones were then ranked based on the onset temperature and other physical and chemical criteria that characterize loss of stability. The aggregation status was evaluated using dynamic light scattering. Next, the impact of thermal stability on the affinity was assessed using ELISA and flow-cytometry. Immunogenicity risk was evaluated by measuring anti-drug antibody generation. Epivax analysis showed no T cell response. This interesting insight in the development process of the lead-candidate antibody was important because authorities are increasingly paying attention to the aggregation questions; for example, the US Food and Drug Administration specifies maximal aggregation parameters.
Dr. Alain Beck (Pierre Fabre) gave a presentation about strategies and challenges for mAbs and ADCs target selection in oncology, with a focus on JAM-A, CXCR4, IGF-1R, and cMet.
JAM-A. To identify new potential targets, functional approaches were developed using tumor cells as immunogens, to select mAbs targeting membrane receptors involved in cell proliferation. Cancer cells were injected into mice and the resulting hybridomas were screened for their ability to inhibit cell proliferation in vitro. Based on this functional approach coupled to proteomic analysis, a mAb specifically recognizing the human junctional adhesion molecule-A (JAM-A) was identified. JAM-A is mainly overexpressed on breast, lung, and kidney tumor tissues. In vivo experiments demonstrated that injections of anti-JAM-A mAb resulted in a significant tumor growth inhibition of xenografted human tumors. Treatment with Hz6F4 induced a decrease in Ki67 expression and downregulated JAM-A levels. The results also demonstrate that a functional approach coupled to a robust proteomic analysis can be successful to identify new antibody target molecules in oncology.Citation11
CXCR4. Amongst mechanisms favoring resistance to drugs in relapsed/refractory multiple myeloma, the homing of tumor cells to the bone marrow microenvironment allows them to abnormally survive, proliferate and resist to therapy. The CXCR4/SDF-1 axis has been found to play a major role in the bone marrow location of hematopoietic stem cells (HSC). F50067 (hz515H7-1), a hzIgG1 that targets CXCR4 demonstrated preclinical anti-tumor activity in MM. A clinical Phase 1 dose escalation study was performed to determine the maximum tolerated dose (MTD) for F50067 in monotherapy and in combination with lenalidomide. Nine patients were evaluable for response. However, because of significant hematological toxicity, this study had to be discontinued.Citation12
IGF-1R. Insulin-like growth factor receptor (IGF-1R) has been recognized for decades for its role in tumorigenesis and growth. In addition, overexpression of IGF-1R has been largely documented in numerous tumor tissues such as squamous NSCLC, squamous head and neck cancer, ER+ breast cancer, prostate cancer and some types of sarcomas. However, so far, therapeutic approaches based on naked mAbs and tyrosine kinase inhibitors failed to show clinical benefit in patients. W0101 is an ADC consisting in a hzIgG1 antibody specific for IGF-1R conjugated to a microtubule-targeting agent F554443 (DAR4). Pre-clinical pharmacology data suggest that the mAb component of W0101 effectively delivers the cytotoxic drug to tumor cells and that the sensitivity correlates with dose and expression level. A companion diagnostic test was developed, and a first-in-human trial of W0101 was initiated to address clinical safety and to find the MTD (NCT03316638).Citation13
cMet. The anti-tumor activity of c-Met inhibitors is generally limited to tumors that are MET-activated and driven predominately by c-Met signaling. Telisotuzumab vedotin (ABBV-399), a c-Met targeting ADC, represents a novel therapeutic delivering a potent payload to c-Met–overexpressing tumor cells, enabling cell killing regardless of reliance on MET signaling. ABBV-399 treatment, alone and in combination with standard-of-care chemotherapy, induces significant tumor growth inhibition and regressions in tumor cell lines and patient-derived xenograft models with overexpressed c-Met or amplified MET, including tumors refractory to other c-Met inhibitors. ABBV-399 killing requires a threshold level of c-Met, expressed by sensitive tumor but not normal cells. ABBV-399 may be an effective broad-acting c-Met–targeting therapeutic that can overcome limitations associated with other c-Met inhibitors. ABBV-399 has progressed to a Phase 1 study where it has been well tolerated and has produced objective responses in c-Met–expressing NSCLC patients,Citation14 and it is currently being investigated in a Phase 2 trial (NCT03539536).
Dr. Pierre Vidal (Histalim) closed this fourth session with his talk on how histopathology helps at all steps of antibody development. Histalim’s TISSUE SCREEN® (TS) validates novel antibody targets in order to consolidate industrial property strategies. TS is an approach based on frozen tissue analysis that can be combined with genomics and proteomics tools (next-generation sequencing (NGS), mass spectrometry). HISTOSELECT® is a new method for high-throughput screening of antibodies candidates by immunohistochemistry on tissue microarray (TMA), particularly for tissue cross-reactivity of the antibody. Finally, histology can serve as a companion to identify new appropriate indications by using TMA. Novel technologies based on fluorescence-based multiplexed IHC with up to six biomarkers per slides can serve as a companion for validation of immune mechanisms complementary to other OMICS methods such as mass spectrometry and NGS, as well-targeted approaches such as FACS and PCR.
SESSION V – EMERGING TARGETS: immune checkpoints & microenvironment
The tumor microenvironment, and more precisely the immune cells, currently represent the most exciting area for the discovery of novel targets and development of novel mAbs. This last session was opened by the keynote lecture of Dr. Christina Claus (Roche Innovation Center) who gave a talk about FAP-4-1BBL, a next generation, co-stimulatory agonist for cancer immunotherapy. FAP-4-1BBL, the first bispecific immune co-stimulator from Roche, is currently in clinical trials. In terms of T-cell infiltration status, tumors can be characterized as immune desert, immune excluded or inflamed. The goal of Roche is to move immune-excluded tumors towards inflamed tumors by targeting different membrane modulators on tumor-infiltrating immune cells. Immune modulators can be divided into activating and inhibitory receptors, which can be engaged or inhibited, respectively, to improve anti-tumoral immune responses. CD137 (4-1BB) receptor engagement enhances strength and durability of T cell responses. 4-1BB agonism leads to cytokine secretion (e.g., interferon (IFN)-γ, GnzB, IL-2, TNF-α, GM-CSF), immune cell proliferation, survival, and activation. There are agonistic antibodies against 4-1BB like urelumab and utomilumab that are still in Phase 2 trials. Utomilumab shows very limited functionality, whereas urelumab is more potent, but showed liver toxicity at higher doses. Lower doses show a better safety profile, but reduced functionality. The toxicity of urelumab is probably linked to the expression of FcγRs in the liver. Indeed, activation of CD8 T cells in the liver of normal mice by 4-1BB agonistic antibodies fully depends on their interaction with FcγRs. Based on these findings, the idea was to reduce the toxicity by eliminating FcγR binding and to deliver the 4-1BB agonist directly to the tumor by introducing a fibroblast activation protein alpha (FAP)-binding site leading to a FAP-targeted bispecific 4-1BB agonist. FAP is a good target because it is highly expressed on cancer-associated fibroblasts, and to a lower extent in lymph nodes on fibroblastic reticular cells, whereas expression in normal tissues is very low. FAP-4-1BBL binds FAP expressed on fibroblasts and 4-1BB expressed on activated human CD8 T cells. Due to this target-mediated cross-linking, FAP-4-1BBL has a different MOA compared to existing 4-1BB agonistic antibodies, which depend on FcγR-crosslinking. They showed this by using CHO cells expressing different FcγR and comparing FcγR-mediated crosslinking of the FAP-4-1BBL fusion protein with urelumab (human IgG4) and utomilumab (human IgG2) while measuring 4-1BB activation in luciferase assay using a 4-1BB-expressing reporter cell line.
In the case of TCR or CD3 stimulation in human T-cell activation assays, FAP-4-1BBL strictly mediates FAP-dependent 4-1BB agonism, leading to increased T-cell proliferation, activation, and cytokine secretion. FAP-4-1BBL enhances efficacy of a CEACAM5-targeted CD3 agonistic T cell bispecific antibody (CEA-TCB) in MKN45-3T3 fibroblast model in HSC-NSG mice. MKN45 is a CEACAM5+ human gastric cancer cell line. NIH/3T3 is a mouse fibroblast cell line. Both cell lines are co-injected in a 1:1 ratio (1x10^6 each) into the flank of the mice. The co-injection of MKN45 and NIH/3T3 is necessary to have a well fibroblast-infiltrated tumor as is usually found in humans. Mice are randomized at an average volume of 200 mm3 and treated with FAP-4-1BBL and CEA-TCB. CEA-TCB is a fusion antibody consisting of 1) IgG1 scaffold containing a P329G LALA mutation (impeding FcγR-binding), 2) two anti-human CEACAM5 Fabs, targeting the molecule to CEACAM5+ tumors, and 3) one agonistic anti-human CD3 Fab inducing signal 1 to tumor-infiltrating T cells. A strong activation of immunity, consisting of increased intratumoral CD8 T-cell infiltration and tumor rejection, was observed in this mouse model if CEA-TCB and FAP-4-1BBL were combined. Furthermore, FAP-4-1BBL has a synergistic effect if combined in vitro with atezolizumab, a checkpoint inhibitor of PD-L1. For this experiment, MKN45-huPD-L1 transfected cell line was mixed with NIH/3T3-huFAP fibroblasts and co-treated with CEA-TCB, FAP-4-1BBL, and PD-L1. The readout was cytokine release (IFN-γ, GM-CSF, and TNF) into the supernatant.
The next talk was given by Dr. Jean-Francois Prost (GamaMAbs Pharma), who focused on the anti-Mullerian receptor type II (AMHR2) target in solid tumors. AMHR2 is an embryonic morphogenic receptor re-expressed in cancer. In normal tissues, it is fully repressed all over the body and only expressed in Sertoli and Leydig cells in testis, and in granulosa cells in ovaries. Membranous expression of AMHR2 has been confirmed by IHC on average in 65% of gynecological cancers, including epithelial ovarian cancer and granulosa cell tumor (GCT). GamaMAbs Pharma has discovered the expression of this receptor in other solid tumors using FACS on fresh human samples. In CRC, 73% of samples were positive and 87000 copies of the receptor were present on the cell surface on average. The ovarian cancer microenvironment is enriched in M2 tissue-associated macrophages (TAM), while in CRC there is a mix of M1/M2 TAM. Glycoengineered GM102 displays both high affinity towards AMHRII (subnanomolar) and high affinity towards CD16 (nanomolar), resulting in a strong ADCC potential. In vitro, GM102 has been shown to induce M2-dependent antibody-dependent cell-mediated phagocytosis (ADCP), followed by T cell proliferation. Macrophage activation by GM102 is accompanied by substantial cytokine and chemokine release, which is involved in macrophages and T-cell recruitment. In vivo, GM102 delays tumor growth by CD16 engagement with a superiority to non-glycoengineered antibody, and appears to synergize with chemotherapy. GM102 is currently in early clinical trials in gynecological cancers and CRCs. The Phase 1 expansion cohorts in gynecological cancers are ongoing and early results show excellent tolerability with optimal dose of 7 mg/kg per week. Two GCT patients out of five exposed at active doses achieved a partial response according to the RECIST criteria with GM102 as a single agent. Overall, a reduction of tumor growth rate was achieved in half of the patients. Preliminary analysis on the patient-paired biopsies show a dramatic increase in CD16 + cells, in agreement with the results from pre-clinical studies.
Dr. Johanna Ruehmann (Glycotope) gave a talk concerning tumor-specific carbohydrate antigens as targets for novel bispecific antibody constructs. Malignant transformation is accompanied by changes in the cellular glycosylation machinery, which leads to the expression of altered carbohydrate structures on cancer cells. These glycans are interesting targets for antibody-based cancer therapies. Two GlycoTargets were introduced: TA-MUC1 (recognized by PankoMab) and the Thomsen-Friedenreich antigen (TFa) (recognized by KaroMab). Different case studies using these targets for bispecific approaches, i.e., T-cell engagement or dual targeting, were presented. TA-MUC1 is a tumor-specific epitope, has high therapeutic potential and is virtually absent from normal cells. PankoMab was designed to bind to a novel carbohydrate/protein mixed conformational epitope on the tumor marker MUC1. The main target indications are ovarian, lung and breast cancers having the highest TA-MUC1 expression. The antibody does not recognize the majority of normal human tissues. In the few positive cases, staining is confined to immune-privileged sites, which are not accessible by systemically administered antibody in vivo. Furthermore, it was confirmed that the target is not only expressed on primary cancers, but also on metastases and cancer stem cells. The first case study was a TA-MUC1 targeting T-cell engaging bispecific antibody. During development, 25 different constructs were generated by varying several parameters, such as valence of TA-MUC1 and CD3 binding, half-life extension, combination of NK cell and T cell MoA, as well as glyco-optimization. Binding valence was the most important factor, as monovalent binding constructs displayed the weakest effector functions. Fc constructs had preferable chemistry, manufacturing and controls characteristics compared to non-Fc constructs. Importantly, the selected lead construct induced T-cell activation and proliferation, but only in the presence of cancer cells, which is crucial for safety in patients. Effective T-cell infiltration was furthermore demonstrated in vitro using a 3D tumor spheroid model.
As second case study, a trifunctional checkpoint inhibitor was presented. Based on the complex molecule design consisting of a TA-MUC1 binding domain, a PD-L1 binding domain and a glyco-optimized Fc, different MOA are proposed to improve current PD-1/PD-L1 therapy: PD1/PD-L1 interaction blockade, which is accumulated directly to the tumor combined with double-targeted ADCC and ADCP. These MOAs are furthermore enhanced by glyco-optimization, notably not only classical NK cell ADCC, but also T-cell re-activation. This discriminates the antibody from current PD-L1 inhibitors, which either lack FcγR binding or are not glyco-optimized to enhance the immune response. As far as KaroMab is concerned, TFa (also known as CD176) is a pan-tumor marker that is usually covered by additional carbohydrates and thus hidden in normal cells. In cancer, this target becomes accessible due to incomplete glycosylation. Due to its unique tumor specificity, TFa is a predestined target for a T-cell engaging bispecific. Very good binding to both tumor and CD3 + T cells eventually leading to T-cell mediated cytotoxicity was demonstrated.
Dr. Cyril Rivat (The Institute for Neurosciences of Montpellier) presented the last talk, which concerned the FLT3-tyrosine kinase receptor as a new target for chronic pain management. Neuropathic pain is due to lesions of somatosensory nervous system caused by surgery, chemotherapy, alcohol, human immunodeficiency virus, and diabetes. It affects 7–10% of the population and has no specific treatment. There is thus a need for identification of new targets to develop new and innovative therapeutics. Maintenance of pain is caused by several factors responsible for inducing chronic changes in the peripheral and central nervous systems. Recently, the speaker demonstrated the critical role of FLT3 and its ligand FL in the development of neuropathic pain.Citation15 FLT3 is a class III receptor tyrosine kinase that, upon activation, induces Mek, Jak2 and PI3K signaling to produce proliferation and survival in hematopoietic cells. They demonstrated that FLT3 is expressed in sensory neurons, and showed that this receptor is involved in pain. In mice, FL produces pain hypersensitivity associated with molecular changes in sensory neurons and the dorsal horn of the spinal cord. FLT3 extinction prevents nerve injury-induced pain hypersensitivity. Thus, they tried to develop a specific antibody against the FLT3 receptor. They selected several clones able to block the FL binding to the FLT3 receptor. The antibodies cross-reacted with the murine protein, with an affinity in the low nM range in both cases. In vivo, the intra-peritoneally injected mAb was able to reverse pain hypersensitivity after nerve injury. The response was maintained if the antibody was regularly injected. Finally, they have also tested with success the ability of the mAb to prevent the development of exaggerated pain hypersensitivity in an animal model of post-operative chronic pain.
Conclusion
This successful 6th edition of the AIS confirmed the importance of the target choice for the development of efficient antibodies. There is no magic recipe, but the integration of several parameters from an in-depth knowledge of the biology of the potential target up to the developability of the newly isolated antibodies are certainly some of the most relevant points to consider. This outlines the necessity of collaborations between academic research teams, who are experts in biology/medicine, with industrial teams trained in developability of biopharmaceuticals. New high throughput methods, such as CITE-seq and F.I.R.S.TTM, that were presented during the meeting will certainly speed up this process. As often occurs in science, innovation can come from the unplanned meeting of different domains (as illustrated during the meeting by the anti-MISRII mAb mixing development and oncology, and the anti-FLT3 receptor mAb developed for peripheral neuropathic pain treatment mixing immunology and neurology). More exciting advances will be discussed at the next AIS meeting, “Harnessing the immune system with therapeutic antibodies”, which will be held in Tours on June 24–25, 2019.
Abbreviations
| 4-1BB | = | tumor necrosis factor receptor superfamily member 9 |
| ADC | = | antibody-drug conjugate |
| ADCC | = | antibody-dependentcell-mediated cytotoxicity |
| ADCP | = | antibody-dependentcell-mediated phagocytosis |
| AMHR2 | = | anti-Mullerian receptor type II |
| BRAF | = | serine/threonine-protein kinase B-raf |
| CD3 | = | T-cell surface glycoprotein CD3 |
| CD8 | = | T-cell surface glycoprotein CD8 |
| CD16 | = | low affinity immunoglobulin gamma Fc region receptor III-A |
| CD19 | = | B-lymphocyte antigen CD19 |
| CD20 | = | B-lymphocyte antigen CD20 |
| CD25 | = | interleukin-2 receptor subunit alpha |
| CD33 | = | myeloid cell surface antigen CD33 |
| CD39 | = | ectonucleoside triphosphate diphosphohydrolase 1 |
| CD44v6 | = | CD44 antigen variant 6 |
| CD73 | = | ecto-5‘-nucleotidase |
| CD94 | = | killer cell lectin-like receptor subfamily D, member 1 |
| CD137 | = | tumor necrosis factor receptor superfamily member 9 |
| CITE-seq | = | cellular indexing of transcriptomes and epitopes by sequencing |
| CRC | = | colorectal cancer |
| CTLA4 | = | cytotoxic T-lymphocyte-associated protein 4 (also known as CD152) |
| CXCR4 | = | C-X-C chemokine receptor type 4 |
| DC | = | dendritic cells |
| DIPG | = | diffuse intrinsic pontine glioma |
| Env | = | envelope |
| ER+ | = | estrogen receptor positive |
| FAP | = | fibroblast activated protein |
| FLT3 | = | cluster of differentiation antigen 135 |
| FRET | = | fluorescence resonance energy transfer |
| GCT | = | granulosa cell tumor |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| GPCR | = | G protein-coupled receptors |
| HERV-W | = | human endogenous retrovirus-w |
| HTRF | = | homogeneous time-resolved fluorescence |
| IHC | = | immunohistochemistry |
| IL-2 | = | interleukin 2 |
| IL-7 | = | interleukin 7 |
| IL-7R - interleukin-7receptor | = |
|
| IFN | = | interferon |
| MASP2 | = | mannose-binding protein-associated serine protease 2 |
| MEK | = | mitogen-activated protein kinase |
| MMR | = | mismatch repair |
| MRI | = | magnetic resonance imaging |
| MS | = | multiple sclerosis |
| MSI | = | microsatellite instability |
| MSRV | = | multiple sclerosis associated retrovirus |
| NOTCH | = | neurogenic locus notch homolog protein |
| NKG2A | = | NKG2-A/NKG2-B type II integral membrane protein |
| NSCLC | = | non-small-cell lung cancer |
| OX40 | = | tumor necrosis factor receptor superfamily, member 4 |
| PET-CT | = | positron emission tomography–computed tomography |
| PD1 | = | programmed cell death protein 1 |
| PD-L1 | = | programmed cell death ligand 1 |
| PD-L2 | = | programmed cell death ligand 2 |
| PBMC | = | peripheral blood mononuclear cells |
| STAT5 | = | signal transducer and activator of transcription 5 |
| TA-MUC1 | = | tumor associated mucin 1 |
| TAM | = | tissue-associated macrophages |
| TLR4 – toll-like receptor 4 | = |
|
| TNFα | = | tumor necrosis factor alpha |
| TMA | = | tissue micro array |
| Treg | = | regulatory T cells |
Disclosure of potential conflicts of interests
Authors have no potential conflicts of interest to disclose.
Acknowledgments
This work has been funded with the support from the French Higher Education and Research Ministry under the program “Investissements d‘avenir” grant agreement: LabEx MAbImprove ANR-10-LABX-53–01. The authors wish to thank all the speakers who participated in the congress and who reviewed their respective contributions outlined in this work. The authors are thankful to the scientific committee and to the organization committee as well as to Ana Sofia Maceira Antunes and Nicolas Groux from MabDesign. The authors are particularly grateful to all the sponsors for their help in organizing the congress.
Additional information
Funding
Related Research Data
References
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. mAbs. 2015;7:9–14. doi:10.4161/19420862.2015.989042.
- Kaplon H, Reichert JM. Antibodies to watch in 2018. mAbs. 2018;10:183–203. doi:10.1080/19420862.2018.1415671.
- Küry P, Nath A, Créange A, Dolei A, Marche P, Gold J, Giovannoni G, Hartung H-P, Perron H. Human endogenous retroviruses in neurological diseases. Trends Mol Med. 2018;24:379–94. doi:10.1016/j.molmed.2018.02.007.
- Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2017;13:25–36. doi:10.1038/nrneurol.2016.187.
- Segal NH, Naidoo J, Curigliano G, Patel S, Sahebjam S, Papadopoulos KP, Gordon MS, Wang D, Gómez Rueda A, Song X, et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J Clin Oncol. 2018;36:3540–3540. doi:10.1200/JCO.2018.36.15_suppl.3540.
- Cohen R, Fayette J, Posner M, Lefebvre G, Bauman J, Salas S, Even C, Seiwert T, Colevas D, Jimeno A, et al. Abstract CT158: phase II study of monalizumab, a first-in-class NKG2A monoclonal antibody, in combination with cetuximab in previously treated recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): preliminary assessment of safety and efficacy. Cancer Res. 2018;78:CT158–CT158.
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–68. doi:10.1038/nmeth.4380.
- Clark JE, Dudler T, Marber MS, Schwaeble W. Cardioprotection by an anti-MASP-2 antibody in a murine model of myocardial infarction. Open Heart. 2018;5:e000652. doi:10.1136/openhrt-2017-000652.
- Sijbrandi NJ, Merkul E, Muns JA, Waalboer DCJ, Adamzek K, Bolijn M, Montserrat V, Somsen GW, Haselberg R, Steverink PJGM, et al. A Novel Platinum(II)-based bifunctional ADC 89Zr-desferal and auristatin F-Conjugated trastuzumab. Cancer Res. 2017;77:257–67. doi:10.1158/0008-5472.CAN-16-1900.
- Maurel D, Comps-Agrar L, Brock C, Rives M-L, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prézeau L, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–67. doi:10.1038/nmeth.1213.
- Terral G, Champion T, Debaene F, Colas O, Bourguet M, Wagner-Rousset E, Corvaia N, Beck A, Cianferani S. Epitope characterization of anti-JAM-A antibodies using orthogonal mass spectrometry and surface plasmon resonance approaches. mAbs. 2017;9:1317–26. doi:10.1080/19420862.2017.1380762.
- Fouquet G, Guidez S, Richez V, Stoppa A-M, Le Tourneau C, Macro M, Gruchet C, Bobin A, Moya N, Syshenko T, et al. Phase I dose-escalation study of F50067, a humanized anti-CXCR4 monoclonal antibody alone and in combination with lenalidomide and low-dose dexamethasone, in relapsed or refractory multiple myeloma. Oncotarget. 2018;9:23890–99. doi:10.18632/oncotarget.v9i35.
- Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. doi:10.1038/nrd.2016.268.
- Strickland LA, Ross J, Williams S, Ross S, Romero M, Spencer S, Erickson R, Sutcliffe J, Verbeke C, Polakis P, et al. Preclinical evaluation of carcinoembryonic cell adhesion molecule (CEACAM) 6 as potential therapy target for pancreatic adenocarcinoma. J Pathol [Internet]. 2009; [cited 2009 Apr 3]. Available fromhttp://www.ncbi.nlm.nih.gov.gate2.inist.fr/pubmed/19334050.
- Rivat C, Sar C, Mechaly I, Leyris J-P, Diouloufet L, Sonrier C, Philipson Y, Lucas O, Mallié S, Jouvenel A, et al. Inhibition of neuronal FLT3 receptor tyrosine kinase alleviates peripheral neuropathic pain in mice. Nat Commun. 2018;9:1042. doi:10.1038/s41467-018-03496-2.