
ABSTRACT
Redirection of immune cells to efficiently eliminate tumor cells holds great promise. Natural killer cells (NK), macrophages, or T cells are specifically engaged with target cells expressing markers after infection or neoplastic transformation, resulting in their activation and subsequent killing of those targets. Multiple strategies to redirect immunity have been developed in the past two decades, but they have technical hurdles or cause undesirable side-effects, as exemplified by the T cell-based chimeric antigen receptor approaches (CAR-T therapies) or bispecific T cell engager platforms. Our first-in-class bispecific antibody redirecting innate immune cells to tumors (AFM13, a CD30/CD16A-specific innate immune cell engager) has shown signs of clinical efficacy in CD30-positive lymphomas and the potential to be safely administered, indicating a wider therapeutic window compared to T cell engaging therapies. AFM13 is the most advanced candidate from our fit-for-purpose redirected optimized cell killing (ROCK®) antibody platform, which comprises a plethora of CD16A-binding innate immune cell engagers with unique properties. Here, we discuss aspects of this modular platform, including the advantages of innate immune cell engagement over classical monoclonal antibodies and other engager concepts. We also present details on its potential to engineer a fit-for-purpose innate immune cell engager format that can be equipped with unique CD16A domains, modules that influence pharmacokinetic properties and molecular architectures that influence the activation of immune effectors, as well as tumor targeting. The ROCK® platform is aimed at the activation of innate immunity for the effective lysis of tumor cells and holds the promise of overcoming limitations of other approaches that redirect immune cells by widening the therapeutic window.
Introduction
Cancer immunotherapy is an innovative field in oncology that has yielded highly promising new cancer treatments harnessing the ability of the body’s innate immune system to fight cancer. Different immunotherapies are in development, including tumor-targeting monoclonal antibodies (mAbs), checkpoint modulators (CPMs), immune-activating cytokines, cancer vaccines, and adoptive cell transfer. Examples of mAb therapeutics targeting specific antigens overexpressed on cancer cells and activating the immune system through antibody-dependent cell-mediated cytotoxicity (ADCC) are rituximab, trastuzumab, and cetuximab, targeting CD20, human epidermal growth factor receptor 2 and epidermal growth factor receptor (EGFR), respectively. In contrast, mAbs targeting immune checkpoints directly activate immune cells, thus enabling them to recognize and eliminate cancer cells more efficiently. Examples of CPMs approved for cancer therapy are mAbs targeting programmed cell death 1 (PD-1), such as nivolumab or pembrolizumab, cytotoxic T-lymphocyte-associated protein 4, such as ipilimumab, or PD-L1, such as atezolizumab.
A different approach combines tumor targeting and immune cell engagement via bispecific antibodies.Citation1 The field currently focuses on cytotoxic T-cell engaging antibodies, with blinatumomab, a CD19/CD3-bispecific T-cell engager for acute lymphoblastic leukemia (ALL), being the only approved therapy in this class. Numerous T-cell engaging antibodies, as well as bispecific antibodies, targeting different immune effector cells such as natural killer (NK) cells are in early development. The most advanced bispecific NK cell engager is the tetravalent bispecific CD30/CD16A tandem diabody AFM13.Citation2,Citation3 AFM13 is in clinical development in CD30-positive lymphomas as monotherapy and in combination with the anti-PD-1 antibody pembrolizumab, and has shown signs of clinical efficacy with a good safety profile in both approaches. Other NK cell engagers targeting CD33, EGFR, or B-cell maturation antigen (BCMA) are currently in preclinical development.Citation1
Adoptive cell transfer is a highly personalized cancer immunotherapy in which patients receive immune cells exerting anticancer activity. These can typically be tumor-infiltrating lymphocytes collected from the patient, expanded and stimulated in vitro before reinfusion, or genetically engineered to express chimeric antigen receptors (CARs), thereby enhancing tumor cell targeting and killing. As with bispecific antibodies, adoptive cell therapy approaches have mainly focused on engaging cytotoxic T cells with antigen-expressing target cells, resulting in T cell activation and subsequent killing of cancer cells expressing those targets. First approvals of CAR T cell (CAR-T) therapies were the CD19-targeting CAR-T axicabtagene ciloleucel (Yescarta®) in large B-cell lymphoma and tisagenlecleucel (Kymriah®) in non-Hodgkin lymphoma and B cell ALL in 2017.
While these T-cell engaging approaches are efficacious, they are also associated with severe treatment-related toxicities like cytokine release syndrome (CRS) or tumor lysis syndrome, which cause very high fevers and dangerously low blood pressure. Other serious side effects observed with cytotoxic T cell engaging therapies include neurotoxicities such as, confusion, seizures and severe headaches. Despite the initial enthusiasm, most bispecific T-cell engaging antibodies have had difficulties establishing a therapeutic index that provides clinical benefit with acceptable side effects. In contrast, alternative immune cell engaging approaches such as bispecific NK cell engagers or adoptive NK cellular transfer have not only shown signs of efficacy,Citation4 but were also well tolerated in lymphoma,Citation3 acute myeloid leukemia (AML)Citation5,Citation6 or other hematological malignancies such as multiple myeloma.Citation7 NK cell-engaging bispecifc antibodies and NK cell-based cellular therapies might therefore be potentially safer alternatives to T-cell engagers and CAR-T therapies.
NK cells are cytotoxic lymphocytes of innate immunity and are essential for immunosurveillance of otherwise life-threatening infections and cancer.Citation8 NK cells, macrophages, neutrophils, and eosinophils can kill target cells opsonized with antibodies by ADCC after recognition of the Fc portion of human immunoglobulins with specific Fc receptors (FcR). ADCC has evolved as a defense mechanism against viral, fungal and parasitic infections, which are characterized by high expression levels of target antigens, allowing for activation of FcR-expressing immune effector cells upon engagement of a large number of opsonizing antibodies with rather low individual binding affinity of their Fc for the corresponding FcRs. By contrast, many tumors are characterized by heterogeneity in expression levels of tumor antigens or their downregulation as a common principle of tumor immune evasion; thus, the degree of avidity and power of ADCC is limited through lower and variable opsonization of tumor target cells with immunoglobulins.Citation9
Evidence from numerous studies supports the rationale of employing efficient NK cell engagement to achieve therapeutic benefits: In vivo, mice that have been NK cell-depleted or harbor NK cell deficiencies have a several-fold increased incidence of spontaneous or carcinogen-induced tumors.Citation10–Citation12 These findings are reflected in humans where abnormal cytolytic function of NK cells is associated with a severely increased risk to develop several types of malignancies, especially hematologic cancers.Citation13–Citation16 Moreover, tumor infiltration of NK cells is associated with a positive prognosis in patients suffering from various malignancies.Citation17–Citation21 A number of in vitro and in vivo studies have demonstrated that autologous and allogeneic cytokine-activated NK cells are capable of eliminating quiescent/non-proliferating target cells such as cancer stem cells.Citation22 Lastly, the finding that higher NK cell-binding affinity of mAbs correlates with enhanced ADCC has led to the development of a number of antibody modifications, including Fc-engineeringCitation23 or glycoengineering,Citation24–Citation26 which also translated into improved outcome in a number of clinical studies.Citation27–Citation32 Despite great promise, only a few classes of bispecific antibody formats specifically recruiting NK cells via antibody binding sites, the tandem diabodies, BiKEs and TriKEs, targeting tumor cells with their second specificity such as CD30 or CD33 have been developed so far.Citation2,Citation33 Those bispecific formats are solely composed of variable binding domains connected to each other via flexible linkers, and have much shorter serum half-lives than classical Fc-containing antibodies. NK cell recruitment by these bispecific antibodies is based on different CD16 (FcγRIII)-binding antibody domains differing in the isotype reactivity, recognition of CD16A (FcγRIIIA) and CD16B (FcγRIIIB), versus exclusive recognition of CD16A, affinity to different allotypes, and ability to compete with Fc binding or other CD16-binding antibodies, such as clone 3G8 that binds both CD16A and CD16B.Citation2,Citation34–Citation36
The CD30/CD16A tetravalent bispecific antibody AFM13 is the most advanced innate immune cell engager derived from the fit-for-purpose ROCK® platform, which includes a plethora of knownCitation37-Citation39 and newly designed bi- or multispecific antibody formats. The platform is equipped with unique CD16A-specific antibody variable domains optimized for affinity and avidity aimed at broad and sustainable activation of innate immunity, including NK cells and macrophages.Citation40 ROCK®-mediated innate cell engagement is independent of a patient’s CD16A allotype, binds to a specific epitope on CD16A distinct from the Fc binding site, is highly efficacious and virtually not inhibited by serum IgG. Despite bivalent CD16A engagement, ROCK® antibodies are engineered to prevent NK cell fratricide, a phenomenon described for mAbs such as daratumumab.Citation41 Based on the modularity of the ROCK® platform, therapeutic antibodies can be designed with different pharmacokinetic (PK) properties, allowing tailoring of formats to specific disease settings. The ROCK® antibody formats engaging innate immunity were designed to overcome limitations of T cell-based approaches and to be complementary to therapeutic agents stimulating adaptive immunity such as CPMs, as well as adoptive NK cellular therapy, thereby potentially enlarging the patient population benefiting from such immunotherapy.
Results
Rationale for generation of optimized anti-CD16A antibody domains
Most classical therapeutic antibodies containing an Fc portion show low intrinsic CD16A (FcγRIIIA) binding affinity, and immuno-engagers that employ the recognition site on CD16A also bound by the Fc proportion of IgG antibodiesCitation34,Citation42 have to compete with serum IgGs for CD16A-binding, thereby limiting CD16A occupancy and increasing the dose of therapeutic antibody required for efficacy. Competition with plasma IgGs might be even more pronounced in disease conditions that are characterized by high levels of plasma IgGs, such as multiple myeloma.Citation43 In addition, CD16A polymorphisms in humans,Citation44 with the most prominent variants CD16A-158V and CD16A-158F differing in binding affinities to IgG and hence FcR functions like ADCC, result in different levels of efficacy of classical mAb therapy depending on the patient’s individual CD16A genotype.Citation45,Citation46 Leveraging our ROCK® platform, we created an anti-CD16A domain that recognizes a unique epitope on CD16A,Citation2 and that is virtually unaffected by plasma IgG competition and CD16A polymorphism. In addition, we aimed for high affinity binding to CD16A mediating highly potent ADCC, and thus effective immuno-surveillance.
To fulfill the above-described criteria of antibody moieties with high affinity and selectivity for CD16A, a library of single-chain variable fragments (scFv) derived from human peripheral blood mononuclear cells (PBMCs) was screened. Using human CD16B variants for negative selection, an antibody with initially low affinity, but high selectivity for CD16A was identified (Ab16lo). Using different approaches for affinity maturation (e.g., light chain shuffling, rational mutagenesis in the complementarity-determining regions of heavy chain), the affinity toward CD16A could be increased by at least a factor of 100, up to a KD of 2 nM (Ab16hi, ). CD16A-binding properties of low affinity (Ab16lo), medium affinity (Ab16mid), and high-affinity scFv (Ab16hi) antibodies were characterized by surface plasmon resonance (SPR) and compared with the previously described and well characterized CD16 pan-specific antibody 3G8,Citation47 as well as IgG1 Fc or a genetically modified Fc variant with CD16A-binding- and Fc-mediated effector function enhancing mutations S239D/I332ECitation23 or silencing mutations L234F/L235E/D265A.Citation48–Citation50 Both Ab16mid and Ab16hi confirmed superior binding and retention on CD16A (). While CD16A-binding kinetics of wildtype IgG1 Fc, enhanced IgG1 Fc and of 3G8 depended on CD16A polymorphism, Ab16hi, as well as Ab16mid and Ab16lo, showed comparable binding kinetics regardless of the allelic forms, CD16A-158V and CD16A-158F (, and Supplementary Figure S3). Ab16mid and Ab16hi also showed good cross-reactivity with cynomolgus CD16 (, Supplementary Table S1). Despite the affinity improvements achieved by affinity maturation, the specificity to CD16A (and not binding to CD16B) and the cross-reactivity to cynomolgus CD16 was maintained (, Supplementary Table S1). Furthermore, the characterization of the anti-CD16A binders identified from the different affinity maturation strategies confirmed another unique characteristic: Engagement of CD16A uniformly and regardless of CD16A genotypic variation (, and Supplementary Figure S3).
Table 1. SPR binding kinetics of different CD16A binding scFvs or Fc portions. Mean binding constants and standard errors are shown (association rate constant ka, dissociation rate constant, Kd, and equilibrium dissociation constant KD). Sensorgrams are given in supplementary Figure S3.
Figure 1. SPR analysis of different anti-CD16 scFv. SPR sensorgrams of normalized relative binding signals of anti-CD16 binding scFv antibodies (Ab16hi, Ab16mid, 3G8, Ab16lo) compared to wildtype IgG1, and engineered human IgG1 Fc (S239D/I332E), enhancing binding to human CD16A-158V (a) and human CD16A-158F (b). Analytes were measured at a single concentration of 312.5 nM.
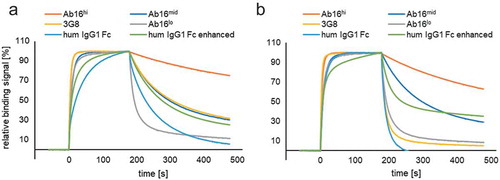
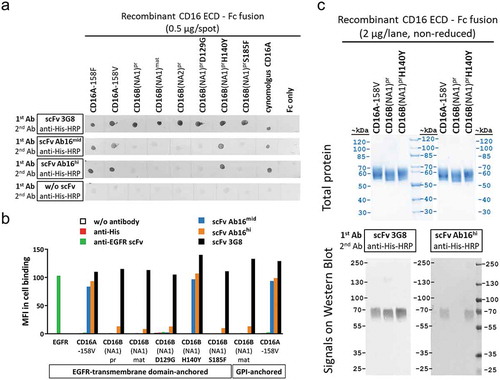
Figure 2. Tyrosine (Y) in position 140 is crucial for formation of a conformational epitope and CD16A-specific reactivity of scFv antibodies. Different recombinant CD16 variants expressed as fusion proteins of ECD sequences with monomeric Fc or membrane-anchor were analyzed. (a) Protein spots containing defined CD16 antigen variants on nitrocellulose membranes were tested for their reactivity with the indicated CD16 binding scFvs. (b) Reactivity of different anti-CD16 scFv, control scFv, or mAbs with CD16 antigen variants or EGFR antigen expressed on CHO cells anchored via fusion to the EGFR transmembrane domain or GPI, was analyzed by antibody staining and flow cytometry. (c) Binding of different anti-CD16 antibodies to CD16 antigen variants after separation by SDS-PAGE and Western blotting.
CD16A-specific binding to a distinct epitope and in the presence of serum IgG
In addition to the high degree of polymorphism shown by CD16A, allotypic variants of human CD16B (FcγRIIIB) differ in five amino acids in the extracellular domain (ECD) (Supplementary Figure S1) and incorporate additional glycosylation, resulting in three alloantigenic variants of the glycosylphosphatidylinositol (GPI)-anchored CD16B granulocyte antigens, NA1, NA2 and SH. Notably, only two amino acid positions consistently differ in the mature ECD of CD16A and B (Supplementary Figure S1). Additional variation resides in distinct modes of membrane-anchorage of CD16A and CD16B. A carboxyterminal propeptide sequence of CD16B cleaved off in the mature GPI-anchored form has high homology to the sequence connecting CD16A ECD to its transmembrane domain (Supplementary Figure S1). We generated and characterized a set of recombinant CD16 antigen mutants to investigate which of the CD16A/CD16B sequence variations determines the binding site for our strictly CD16A-selective antibodies: CD16B(NA1) ECD was either expressed as the maturated form (lacking the propeptide) (CD16Bmat), or as the precursor containing the propeptide sequence (CD16Bpr). In addition, CD16Bpr ECD mutants in which CD16B-specific amino acids were replaced with the homologous residues of CD16A were generated and tested in binding assays: CD16Bpr,D129G, CD16Bpr,H140Y, CD16Bpr,S185F. All tested CD16A and CD16B antigen variants were recognized by the CD16 pan-specific scFv antibody 3G8, confirming their molecular integrity (, Supplementary Table S1). scFv antibodies Ab16mid or the affinity-improved Ab16hi recognized the CD16A antigen, but not the mature or propeptide-containing ECD fusion antigens of CD16B. If, however, CD16B ECD Histidine (H) at position 140 was mutated to tyrosine (Y) – the amino acid present at the corresponding position in CD16A – this mutant form of CD16B (CD16Bpr,H140Y) was well recognized by the affinity-optimized scFv antibodies Ab16mid and Ab16hi (, Supplementary Table S1).
Both anti-CD16 scFv maintained very good cross-reactivity with cynomolgus CD16 (, Supplementary Table S1), despite the fact that cynomolgus CD16 ECD differs in a total of 16 amino acids from human CD16A (see alignment in Supplementary Figure S1). Binding selectivity and importance of tyrosine (Y) at position 140 in the ECD of CD16 for CD16A-specific recognition by Ab16mid and Ab16hi antibodies was confirmed using recombinant Chinese hamster ovary (CHO) cells with membrane-anchored expression of different CD16 antigen variants (). CD16A or CD16Bpr,H140Y in contrast to CD16Bpr or CD16Bmat antigen variants, were also recognized after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on Western blot using these scFv antibodies ( and data not shown). No signals were obtained on Western blots of reduced samples (data not shown), suggesting a conformational epitope, which, in the case of recognition by Ab16mid and Ab16hi, is likely determined by tyrosine in position 140 of CD16.
The IgG binding site on CD16A is a discontinuous binding area on the membrane proximal ECD of the receptor. Crucial amino acids for the low-affinity interaction of CD16A with the Fc part of IgG were previously identified and described.Citation51 Antibody 3G8 was reported to bind CD16A and CD16B and block IgG binding.Citation47 No blocking of binding of our antibodies to CD16A by IgGs was observed. While binding of 3G8 mAb to CD16A is outcompeted with increasing concentrations of a scFv antibody containing the corresponding variable domains of 3G8, the anti-CD16 scFv antibodies Ab16mid and Ab16hi do not block or displace 3G8 mAb on CD16A and bind independently (Supplementary Figure S2) and with similar affinity in the absence or presence of competing 3G8 mAb (Supplementary Table S2). Since occupation of the 3G8 binding site on CD16A is known to block IgG binding, we conclude that the binding site of our anti-CD16 antibodies on CD16A is distinct from the IgG binding site. Accordingly, NK cell-binding KD values of scFv antibodies Ab16hi and Ab16mid were only mildly affected by addition of 10 mg/mL human IgG, whereas binding affinity of CD16-binding scFv “3G8” was more than 20-fold reduced in the presence of physiological IgG concentrations (). These results suggest that our CD16A-binding antibodies Ab16hi and Ab16mid offer the advantage of high affinity binding to a CD16A-specific epitope that is not or only minimally impaired by competing IgG levels (e.g., in human blood), and we next attempted to exploit their power in multivalent anti-tumor NK cell-engaging ROCK® antibodies.
Table 2. Apparent affinities of anti-CD16 scFv to primary human NK cells.
Table 3C. Overview control antibodies used in functional assays.
The ROCK® platform
In order to demonstrate the versatility of the anti-CD16A domains in different antibody formats and to exploit its full potential for harnessing innate immunity, we created a wide spectrum of ROCK® architectures based on known antibody formatsCitation38 and newly designed variants thereof.
The ROCK® formats are multivalent and multispecific with at least one target-binding site and two CD16A-binding moieties enabling bivalent binding. Depending on certain design criteria, such as valency, multispecificity, PK, and pharmacodynamic properties, the engager constructs are modularly assembled using variable binding domains in combination with or without a constant region. The ROCK® antibody formats can be grouped into four different families comprising: (1) Fc-less ROCK®, (2) Fc fusion ROCK®, (3) IgG-like ROCK®, and (4) antibody binding fragment (Fab) fusion ROCK®. The target and effector cell-binding moieties of family members 2, 3, and 4 can be either formatted as scFv, single-chain diabody (scDb), diabody (Db), or as Fab, enabling a range of functionalities. The binding domains are either fused to the N- and C-terminus of CH2/CH3 constant domains via a flexible linker or, alternatively, to the N-terminus via a hinge or shorter middle hinge.Citation52 For family 1, the binding domains are connected by peptide linkers and assembled as TandAbCitation53 or aTriFlex molecules.Citation54 To ensure powerful engagement and activation of NK cells, the anti-CD16A moiety is designed for bivalent binding in all families. The tumor target-binding domain in ROCK® engagers can be either monovalent or bivalent, depending on desired binding strength and/or target biology. Modularity of the platform furthermore facilitates construction of multivalent and multispecific engagers for the cotargeting of more than one tumor antigen. shows all four ROCK® families with their associated formats. In order to show the versatility of the ROCK® formats, different target-binding moieties such as BCMA, CD19, CD20, EGFR, human serum albumin (HSA), or RSV were chosen. Representative examples of ROCK® constructs or control antibodies characterized in more detail in functional assays target the solid tumor target EGFR or BCMA, a target in multiple myeloma (). All ROCK® constructs are composed of human antibody domains, and have an approximate molecular weight of 100–250 kDa. In several cases tags (C-tag and/or 6xHis tag) were added at the C-terminus to enable selective purification of heterodimeric antibodies via affinity chromatography, analysis of the two different polypeptide chains in asymmetric products, and, finally, ensure high purity (> 90%) of the desired product species.
Table 3A. Overview of ROCK® examples containing CD16-binding moieties (depicted in blue) in Fab or scFv-fusion arrangement used in functional assays.
Figure 3. ROCK® platform overview.
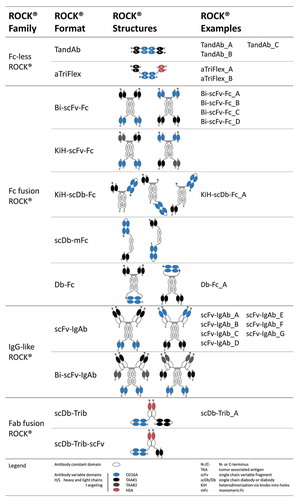
Fc-less ROCK® family 1: This group of Fc-less constructs comprises the well-known TandAb format, of which several products are already in clinical development,Citation2,Citation55–Citation57 and the aTriFlexCitation54 antibodies. The latter can be engineered as a trispecific format, employing two different scFv specificities to address two different tumor-associated antigens or target epitopes or be used for PK-extension. For that purpose, we used HSA-binding antibody domains to extend serum half-life of the otherwise short-lived Fc-less antibody format. Both formats within this family assemble as dimers by interaction of corresponding heavy and light chain variable domains on two separate polypeptide chains as head-to-tail homodimers (TandAb format) or as heterodimers in a head-to-tail or head-to-head fashion (aTriFlex). Members of this group are the smallest ROCK® bispecific constructs in size, with an approximate mass of 105 kDa of the functional dimer, and lack both an Fc and glycosylation.
Fc fusion ROCK® family 2: This family of Fc-containing antibody formats comprises several Fc variantsCitation48–Citation50 with the aim of avoiding Fcγ receptor binding, but still maintaining neonatal FcR (FcRn) binding for half-life extension. Thus, Fcγ receptor binding is exclusively mediated by the NK cell-binding domain and restricted to CD16A. In addition, the Fc included in this family can be either a IgG1-based homodimer (Bi-scFv-Fc, Db-Fc), a heterodimer (KiH-scFv-Fc, KiH-scDb-Fc) or a monomeric Fc (scDb-mFc). To facilitate heterodimerization, the “knobs-into-holes” (KiH) technology using classical Y407T (“holes”) and T366Y (“knobs”) mutations have been used.Citation58 The monomeric Fc (mFc) part has been engineered as previously publishedCitation59 using flexible connectors for fusion with target and NK cell-binding moieties. The anti-CD16A scDb is either located opposite to the target-binding domain at the N- or C-terminus or juxtaposed to it at the N-terminus as exemplified for KiH-scDb-Fc constructs (). A scDb format for the CD16A-binding domain was applied in KiH-scDb-Fc and scDb-mFc constructs. The Db-Fc format is a homodimer that exhibits a diabody module. This Db-Fc format assembles from two VL/VH domains that are fused to the constant regions via a flexible linker at the C-terminus or an extended hinge region at the N-terminus.
IgG-like ROCK® family 3: This bispecific antibody group encompasses the scFv-IgAbs and Bi-scFv-IgAbs, which are in principle “extended human IgG1” moleculesCitation60 with two CD16A antigen-binding moieties fused to the C-terminus or located at the N-terminus as variable fragments (Fv) within both Fab arms. The same holds true for the tumor target-binding domains that are located at the corresponding opposite ends. Bi-scFv-IgAb molecules exhibit a hexavalent trispecific IgG-like architecture with two additional pairs of antigen-binding scFvs fused to the C-terminus of a kappa or lambda constant region and CH3 via flexible linkers.
Fab fusion ROCK® family 4: Members of this group are “extended Fab” molecules and can be subdivided in the scDb-Trib and the scDb-Trib-scFv formats. Both contain CD16A-binding moieties as a bivalent scDb fused to the C-terminus of a kappa or lambda constant region in scDb-Trib-scFv or, in the case of scDb-Trib, to the C-terminal end of CH1 (). For PK-extension, the variable domain at the N-terminus of the Fab fragment provides HSA-binding. Tumor targeting is achieved either by monovalent (scDb-Trib-scFv) or bivalent (scDb-Trib) binding moieties (scFv vs. scDb) fused to the respective free C-terminus.
ROCK® engagers exploit avidity to maximize NK cell engagement via CD16A
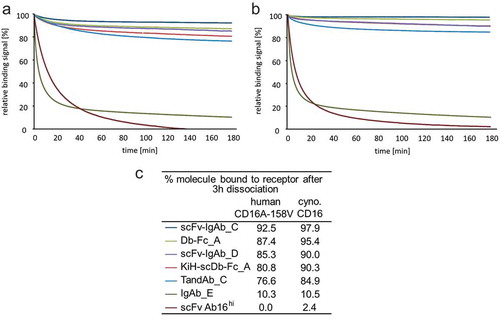
CD16A-binding retention of diverse ROCK® engager formats was investigated in SPR and compared to the monovalent binding of scFv Ab16hi and that of engineered human IgG1 Fc (S239D/I332E), enhancing binding to human CD16A, by comparing dissociation from human CD16A-158V or cynomolgus CD16. Relative binding signals of the tested antibodies during a 3 h dissociation showed clear superiority in receptor retention of all tested ROCK® engagers on both, human CD16A-158V (76.6% – 92.5%) and cynomolgus CD16 (84.9% – 97.9%) compared to the much faster dissociating IgG Fc (S239D/I332E) (10%) or monovalently binding scFv Ab16hi (0% – 2.4%) (). Consistent with the results of the dissociation measurements by SPR, ROCK® engagers, such as the TandAb containing Ab16hi Fv domains, exhibited substantial longer retention on the surface of NK cells than classical IgG with a wildtype Fc domain or enhanced IgG with S239D/I332E mutations in the Fc (Supplementary Figure S4).
Figure 4. SPR sensorgrams of normalized relative binding dissociation phases of diverse CD16A bivalently binding ROCK® engagers. Bivalent anti-CD16A domain containing scFv-IgAb_C, Db-Fc_A, scFv-IgAb_D, KiH-scDb-Fc_A, TandAb_C were compared to IgAb_E (Fc-enhanced; S239D/I332E) and monovalent anti-CD16A fragment scFv Ab16hi on immobilized (a) human CD16A-158V and (b) cynomolgus CD16. Legend and percentage of antibodies remaining on the receptor after 3 h of dissociation are shown in (c). Analytes were measured at a single concentration of 50 nM.
Apparent CD16-binding strengths of different ROCK® NK cell engagers, all containing two binding sites for CD16A, furthermore depended on the anti-CD16 variable domain identity (high-affinity Ab16hi, or medium affinity Ab16mid), antibody format and position of anti-CD16 moieties in the multispecific (bi- or trispecific) and multivalent (tetra or hexavalent) ROCK® NK cell engagers. Similar to the monovalent binding properties, bivalent CD16A-binding by Ab16hi-containing ROCK® antibodies exhibited higher apparent affinities in enzyme-linked immunosorbent assays (ELISA) than corresponding ROCK® engagers containing Ab16mid domains. Additionally, different positioning of anti-CD16A domains in ROCK® engagers enabled gradual affinity tuning: ROCK® architectures containing CD16A-binding moieties at the N-terminus of Fc domains mediated higher apparent binding affinity for CD16A than antibodies containing a fusion of anti-CD16A domains to the C-terminus of Fc (). Fine-tuning of binding affinities could be accomplished by using either Fab or diabody-based binding modules at the N-terminus of the Fc, which enhanced binding strength as compared to the fusion of two CD16-binding scFvs (). Further adjustment of CD16A-binding strength in ROCK® engagers containing anti-CD16A scFvs fused to the C-terminus of the Fc could be achieved by varying lengths of the connectors, whereby a longer connector facilitated higher apparent affinity CD16A-binding than a shorter connector (). Likely, avidity, but also sterical aspects, influence apparent binding affinities in these setups, whereas the impact of different target-binding specificities on CD16A-binding was negligible.
Figure 5. CD16A apparent affinity in ROCK® formats is tunable by variable domains, positioning and connector lengths. Binding of soluble CD16 antigen to ROCK® antibodies containing different CD16 binding Fvs (Ab16mid (open blue dots), Ab16hi (filled blue dots)) in different positions (N: anti-CD16 Fvs N-terminal of Fc, C: anti-CD16 Fvs C-terminal of Fc), antibody formats, and different domain orders was analyzed in ELISA. Representative pictograms with CD16-binding Fvs depicted in blue are shown below each group. Summary of CD16A apparent affinity by (a) Fab or scFv-based CD16 engagement, (b) diabody (Db)-based CD16 engagement, or (c) C-terminal scFv-based CD16 engagement comprising different connector lengths (10aa or 30aa) and domain orders of anti-CD16 Fv (HL: scFv domain order VH-VL, LH: scFv domain order VL-VH). All analyzed antibodies contain silenced Fc or lack Fc in the case of fusion to C-terminus of Fab. Binding specificities depicted in black or dark gray comprised antibody domains targeting BCMA, CD19, CD20, EGFR, HSA or RSV.
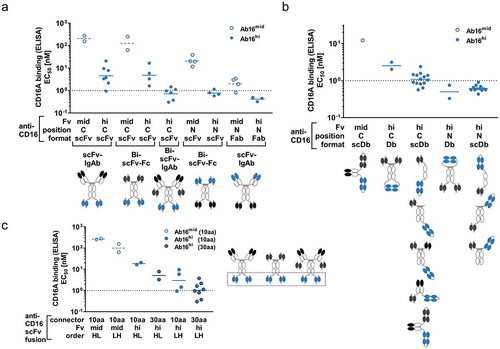
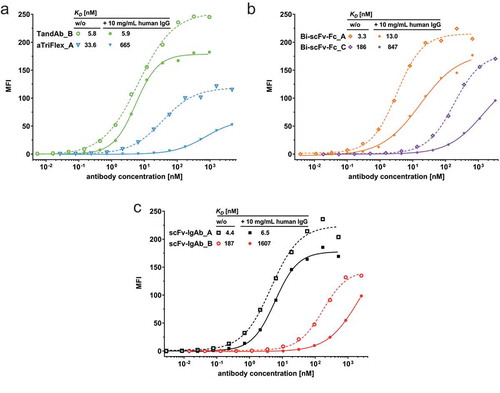
To assess whether the CD16A-binding properties measured by ELISA correlate with binding to NK cells, a selected set of ROCK® engagers containing the Ab16mid Fv domains was titrated on primary human NK cells in the absence or presence of polyclonal human IgG mimicking the physiological situation in human serum and the apparent affinities were determined by nonlinear regression (). Again, constructs with the anti-CD16A domains in a N-terminal Fab or scFv (Bi-scFv-Fc_A and scFv-IgAb_A) exhibited a substantially higher affinity to NK cells relative to the corresponding constructs with the anti-CD16A domains in a C-terminal scFv (Bi-scFv-Fc_C and scFv-IgAb_B). Interestingly, the TandAb with a central anti-CD16A diabody motif () showed similar high affinity to NK cells as constructs with N-terminal Fab or scFv, whereas the aTriFlex displayed the lowest affinity to NK cells despite a similar anti-CD16A diabody motif in the core of the construct but also an asymmetric variable domain order. High IgG concentrations had no substantial effect on the apparent NK cell-binding affinity of TandAb and scFv-IgAb constructs with anti-CD16A domains in the N-terminal Fab, moderate impact (factors 4–9) on binding of constructs containing anti-CD16A scFv independent whether N- or C-terminally fused to the construct, and the strongest inhibitory effect for the anyhow poorly binding aTriflex.
Table 3B. Overview of ROCK® examples containing CD16-binding moieties (depicted in blue) in diabody (Db)-like arrangement used in functional assays.
Figure 6. Binding of ROCK® antibodies to primary human NK cells in the presence or absence of human IgG. Primary human NK cells were stained with increasing concentrations of the indicated (a) Fc-less ROCK®, (b) Fc fusion ROCK®, (c) IgG-like ROCK® constructs in the presence or absence of 10 mg/mL polyclonal human IgG at 37°C. Cell bound antibodies were detected by flow cytometry, and median fluorescence intensities (MFI) were used for calculation of apparent affinities (KD) by non-linear regression.
NK cell fratricide and design of ROCK® formats
Multivalent high affinity binding of antibodies to CD16 harbors the risk of cross-linking the receptor on multiple NK cells, resulting in CD16A-mediated killing of cross-linked cells and thus depletion of the NK cell population.Citation61 This NK cell fratricide has already been shown for other mAbs such as daratumumab, which targets CD38 on the plasma membrane of NK cells.Citation41 Diminishing the main effector cell population presumably hampers the efficacy of an antibody-mediated antitumoral response. For that reason, an in vitro assay to monitor the degree of NK cell fratricide induced by various ROCK® formats was established. In this assay, primary human NK cells from the same donor were used as effector and target cells, thus allowing the determination of the potency of NK cell lysis as described above. As a positive control, daratumumab was included in the assay setup. We have never observed NK-NK fratricide with antibodies containing only a single binding site for CD16A, which suggests that bivalency of CD16A is a prerequisite for NK cell crosslinking and subsequent fratricide. Intriguingly, depending on the ROCK® antibody format or the anti-CD16A variable domain order, the occurrence of NK cell fratricide, as well as the potency of NK cell killing, could be modulated ().
Figure 7. Comparative analysis of various ROCK® antibody formats regarding NK fratricide in vitro. In vitro calcein-release cytotoxicity assays of enriched primary human NK cells in the presence of different ROCK® antibody formats toward autologous NK cells after 4 h co-incubation at an E:T of 1:1. Mean EC50 values of several independent experiments are summarized and plotted as individual dots. The ROCK® antibody formats are categorized based on the position of the anti-CD16 domain and the format/domain order. Only constructs containing the high affinity anti-CD16 domain and silenced Fc are included in the graph. HL: scFv domain order VH-VL, LH: scFv domain order VL-VH. N-term. = N-terminal, C-term. = C-terminal.
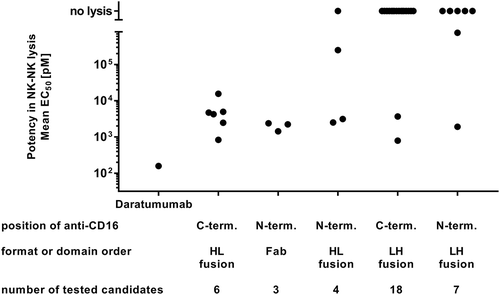
In general, all analyzed ROCK® antibody formats showed an ameliorated NK cell fratricide profile compared to daratumumab, which led to NK cell-directed killing with a mean potency of 158 pM. All tested ROCK® antibody formats either induced a less pronounced or no NK cell killing at all.
CD16A engagement via C-terminally fused anti-CD16 moieties of the ROCK® antibody appeared to avoid NK cell fratricide more robustly compared to CD16A engagement at the N-terminus. However, focusing on N-terminal CD16 engagement in detail revealed a more pronounced NK cell depletion using Fab-based CD16A engagement with mean potency values between 1439 and 2389 pM. Intriguingly, by analyzing the domain order of the CD16 Fv, a substantially lower number of constructs induced NK cell fratricide when the domain order was VL-VH in contrast to constructs containing the VH-VL domain order, which all induced NK cell lysis.
In summary, the proportion of NK cell fratricide-inducing antibodies as well as the potency of NK cell killing could be efficiently ameliorated by avoiding Fab-based CD16A engagement and focusing on the beneficial VL-VH order of the CD16 binding Fv domain.
Modulation of PK
Another important parameter for therapeutic antibodies is their serum half-life. For highly cytotoxic antibodies, a short half-life might be preferred because it enables rapid clearance. In contrast, for antibodies with good safety profiles, longer half-lives are likely to be favored to increase patient convenience by avoiding frequent dosing or the need for continuous infusion.
Basic PK parameters of a selected set of ROCK® formats were analyzed in CD-1 SWISS mice. For the IgG-like family of ROCK® engagers, two representative scFv-IgAb antibodies (scFv-IgAb_A and scFv-IgAb_D) either containing anti-CD16 moieties in N-terminal Fab or as scFvs fused to the C-terminus of CH3 were chosen. For the Fc fusion family of ROCK® engagers, a symmetric Bi-scFv-Fc_A and an asymmetric, scDb-containing KiH-scDb-Fc_A antibody were analyzed. Finally, for the Fab fusion and Fc-less families, the TandAb_B, aTriFlex_A, aTriFlex_B, and scDb-Trib_A antibodies were selected for PK analysis. The latter two employ a HSA-binding moiety with low affinity compared with a high affinity HSA-binding domain in aTriFlex_A for PK-extension. The tumor-targeting domains in the selected ROCK® engagers were either specific to human EGFR or BCMA. As both target- and CD16A-binding moieties are not cross-reactive to the corresponding mouse analogs (data not shown), no target-related pharmacodynamic effects could be postulated.
Blood serum samples from animals that received a single intravenous administration of 0.3 mg (~10 mg/kg body weight) of the different test items were collected at different time points for a period of 1 or 3 weeks and analyzed with established assays based on ELISA or MSD technology. Basic PK parameters were determined by noncompartmental PK analysis (). Following intravenous administration of the different ROCK® formats, serum concentrations declined in a biexponential manner ().
Table 4. ROCK® formats analyzed in mouse PK studies.
Figure 8. Comparison of the pharmacokinetics of different ROCK® engager formats: concentration of antibodies over time (1 or 3 weeks observation period) after single intravenous administration of 300 µg test item.
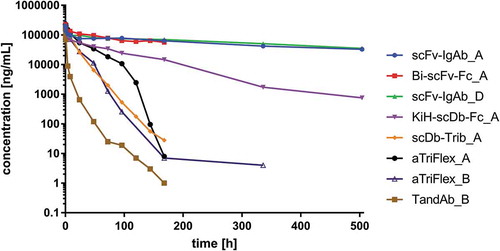
Half-lives of both IgG-like formats were shown to be in the same range of 329.2 and 364.3 h, respectively, and are comparable to standard IgG molecules.Citation62 Shorter half-lives of 95.8 or 74.3 h, respectively, were measured for the Fc fusion formats Bi-scFv-Fc_A and KiH-scDb-Fc_A. Serum half-lives of those were between the IgG-like and the Fc-less and Fab fusion ROCK® formats. However, the observation period for Bi-scFv-Fc_A was only 1 week compared to 3 weeks for both of the scFv-IgAb formats. Therefore, this value may be underestimated. The shortest half-lives were measured for the Fc-less and the Fab fusion ROCK® formats. Mean apparent terminal half-lives were 37.4 h for aTriFlex_A containing a high affinity anti-HSA moiety, and 18.0 h for the TandAb. The low affinity anti-HSA domain in scDb-Trib_A could not prolong the half-life of this construct (14.0 h). A similar ranking could be performed based on the mean residence times (MRT), which defines the unchanged drug in the circulation. The highest MRTs were shown for the IgG-like and Fc fusion test items with 528.5 to 110.6 h, followed by relatively short MRT values from 61.6 to 28.5 h for the Fc-less and Fab fusion formats represented by the TandAb, aTriFlex, and scDb-Trib, respectively. This ranking also holds true for the clearance (C) ranging between 0.006 and 1.228 mL/h with the highest value obtained for the TandAb construct containing neither Fc nor an anti-HSA domain.
An important parameter is also determined by the area under the curve (AUC), which defines the maximal amount or exposure of a molecule in the organism over time. As expected highest AUC values were achieved with the Fc-containing formats. Nearly 10-fold lower values were measured for the smaller Fc-less formats. Again, the TandAb demonstrated the lowest exposure with a 100-fold difference in AUC compared to the long-lived test items.
The steady-state distribution volume (Vss) that defines the apparent volume needed to account for the total amount of drug in the body if the drug was evenly distributed throughout the body was highest for the TandAb (4.8 mL), followed by both IgG-like (3.3 mL and 3.1 mL, respectively), the Fc fusion (2.2 mL), the aTriFlex (2.2 mL) and the scDb Tribody (1.5 mL). The lower Vss value of the scDb Tribody implies that the majority of the molecule remains in the blood compartment (blood).
In conclusion, different ROCK® formats showed distinct PK profiles in CD-1 mice, with half-lives ranging from 14 to 364 h.
Superior in vitro cytotoxicity of ROCK® engagers
In vitro cytotoxicity of various ROCK® engagers targeting EGFR or BCMA was assessed in calcein-release assays (). BCMA-targeting engagers with Ab16hi Fv domains (TandAb_A, scFv-IgAb_D, Bi-scFv-Fc_B, and KiH-scDb-Fc_A) were compared with the Ab16mid Fv-containing engager scFv-IgAb_E and Fc-enhanced anti-BCMA IgG1 with S239D/I332E mutations in the Fc (IgAb_C) on target cells expressing high (NCI-H929), mid (MM.1S), or low (MC/CAR) BCMA levels on the cell surface (). The exemplary cytotoxicity results reveal only minor differences in potency and efficacy among the different ROCK® engager formats on BCMAhigh NCI-H929 target cells (EC50 range: 9.5–52.7 pM), similar efficacy but stronger differences in potency (EC50 range: 1.8–29.1 pM) on BCMAmid MM.1S target cells, and strong differences in efficacy and potency (EC50 range: 59–859 pM) on BCMAlow MC/CAR target cells. Lower EC50 values were most pronounced for the BCMA-targeting TandAb_A, the KiH-scDb-Fc_A, and the scFv-IgAb_D. These results suggest that two valencies for CD16A and high apparent affinities for NK cells are advantageous for triggering NK cell-mediated lysis of less susceptible target cells and/or target cells expressing lower numbers of cell surface target antigens. Of note, bivalency for CD16A alone was not sufficient to mediate NK cell activation, as antibody-induced expression of NK cell activation marker CD69 and cytokine release in human PBMC cultures were strictly dependent on the presence of target cells (Supplementary Figure S5). Furthermore, inflammatory cytokine levels released in human PBMC cultures by NK cell engaging ROCK® antibodies were several orders of magnitude lower than those released by a BCMA-directed T cell engager (Supplementary Figure S6).
Figure 9. In vitro cytotoxicity of enriched primary human NK cells in the presence of several ROCK® antibodies toward cell lines expressing their corresponding tumor target. (a) Representative sigmoidal dose-response curves of the indicated BCMA-targeting ROCK® antibodies in 4 h calcein-release cytotoxicity assays with NK cells and target cell lines expressing differential levels of BCMA as indicated by the SABC values (mean of ≥3 assays) at an E:T ratio of 5:1. (b) Representative sigmoidal dose-response curves of the indicated EGFR-targeting ROCK® antibodies, comparators, or monovalently binding controls in 4 h calcein-release cytotoxicity assays with NK cells and SW-982 target cells at an E:T ratio of 5:1. (c) Correlation of ROCK® antibodies regarding NK cell-binding affinity and cytotoxic potency toward tumor target cells in vitro. Apparent affinities (KD) of ROCK® antibody formats on primary human NK cells are shown. EC50 values of the same ROCK® antibodies were determined in 3 h calcein-release cytotoxicity measurements with NK cells and BCMA-expressing RPMI-8226 target cells at an E:T ratio of 2:1 (left) or in 4 h calcein-release cytotoxicity assays with NK cells and BCMA-expressing NCI-H929 target cells at an E:T ratio of 5:1 (right). SABC, specific antibody-binding capacity.
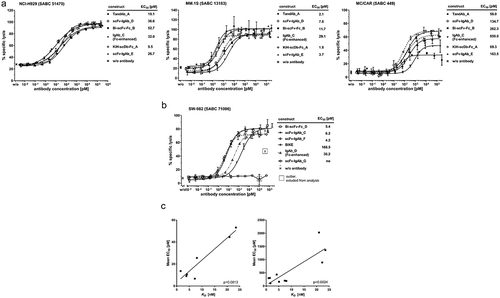
Similar to the results obtained with BCMA-targeting ROCK® engagers, EGFR-targeting ROCK® engagers showed superior potency in killing assays on SW-982 and A-431 target cells, reaching single digit pM EC50 values, compared to Fc-enhanced anti-EGFR IgG1 with S239D/I332E mutations in the Fc (IgAb_D), classical EGFR-targeting IgG1 (IgAb_B) or monovalently binding bispecific engager (BiKE) comprising the identical effector- and target-binding domains (, and Supplementary Figure S7). The EGFR/CD16A aTriFlex_A, despite substantial lower apparent affinity to human NK cells than other ROCK® engagers (), had similar potency as the Fc-enhanced anti-EGFR IgG1 (IgAb_D) (Supplementary Figure S7). Nonspecific killing can be excluded since the BCMA-targeting scFv-IgAb_E showed no killing activity on the EGFR positive, BCMA negative target cell line A-431 (Supplementary Figure S7).
The analysis of the apparent affinities of various BCMA/CD16A engagers to NK cells and the respective potency in in vitro cytotoxicity assays shown in suggests a correlation of the apparent affinity for CD16A on the NK cells and potency in cytotoxicity assays. The correlation was observed in 3 h cytotoxicity assays on RPMI-8226 target cells at an E:T of 2:1 as well as in 4 h assays on NCI-H929 target cells at an E:T ratio of 5:1, supporting the theory that high affinity binding to CD16A on NK cells contributes to superior cytotoxic potency. However, different binding affinities to BCMA+ target cells due to different architectures and orientation of the anti-BCMA Fv domains might also contribute to the potency of the engager constructs.
Superiority of a ROCK® CD30/CD16A-targeting TandAb over conventional and Fc-engineered anti-CD30 antibodies was even more pronounced when in vitro cytotoxicity assays were performed in the presence of physiological levels of competing IgG. Supplementation with 10 mg/mL polyclonal IgG clearly increased background lysis in all cultures (Supplementary Figure S8B), most likely due to serum IgG-induced degranulation by NK cells.Citation63 However, competition with monoclonal IgG (Supplementary Figure S8C) or Fc-engineered IgG (Supplementary Figure S8D) completely blocked activity or clearly impaired potency and efficacy of the conventional antibodies. The ROCK® CD30/CD16A TandAb antibody showed maximum cytotoxic potency and efficacy under all tested conditions, even in the presence of physiological concentrations of competing IgG (Supplementary Figure S8, Supplementary Table S3).
Discussion
One of the most promising therapeutic approaches in oncology is the redirection of immune effector cells, such as NK cells, macrophages, and T cells, to efficiently eliminate tumor cells. A number of technologies, including mono- and bispecific antibodies, and genetically engineered immune cells with artificial tumor-selective receptors such as CARs, have been approved or are in clinical development for the treatment of various malignancies. Even though many of these approaches have demonstrated efficacy, they all have limitations such as: (1) unfavorable benefit/risk ratio or narrow therapeutic window, including antibody-drug conjugates or T cell-based agents such as rovalpituzumab tesirineCitation64 and CD19/CD3-bispecific T cell engager duvortuxizumab,Citation65 respectively; and (2) lack of efficacy.Citation66,Citation67 Using the ROCK® platform, we have developed an innovative portfolio of multivalent, multispecific immune cell engagers with unique properties to overcome such limitations and to maximize the potential of innate immune effectors such as NK cells and macrophages. Although the ROCK® platform is not exclusive to a particular immune cell type, we focused this study on CD16A-specific immune cell engagers that mediate targeting of malignant cells by NK cells and other CD16A+ immune effector cells.
Our NK cell engagers have demonstrated potency in vitro and significantly lower levels of cytokine release than T cell engagers (i.e., BiTEs). This is an important safety consideration and supports the clinical experience with NK cell-based approaches: both NK cell engagement and adoptive NK cell therapy have shown a favorable safety profile and are thus differentiated from T cell-based therapy, which have often shown a narrow therapeutic window due to CRS, tumor lysis syndrome and neurotoxicity.Citation3,Citation68 In addition to toxicities, limited infiltration of effector cells in solid tumors is one of the major factors hindering development of T cell-based therapies in such cancers. Studies across several tumor indications suggest that prevalence of NK cells is associated with beneficial outcomes,Citation19,Citation69–Citation71 and we were able to show that a CD16A-based engager is able to restore dysregulated NK cell activity in patients.Citation72 In the case of adoptive NK cell therapy, good safety profiles and the ability to induce remissions in patients with AML have been shown. However, limitations of this approach are the short persistence of NK cells in vivo and the lack of redirection to tumor cells. A combination of engagers with adoptive NK cell therapy may overcome these hurdles.
CD16A, which is also known as FcγRIIIA receptor,Citation73 mediates ADCC by activation of NK cells and subsequent exocytosis of cytotoxic granules, death receptor signaling, and release of pro-inflammatory cytokines.Citation8 For most classical mAbs, ADCC is rather limited through: (1) low expression levels of antigens on tumor cells compared to, for example, virus-infected cells, and thus limited opsonization of target cells, (2) low affinities of the Fc region to CD16A, (3) polymorphisms in CD16A influencing efficacy,Citation74,Citation75 (4) competition with serum IgG for CD16A-binding, and (5) NK cell-mediated NK cell killing (NK cell fratricide).Citation41 Although higher affinities to FcγRIII have been achieved by engineering Fc portions of therapeutic antibodies, better clinical outcomes have only been achieved in some indications for anti-CD20 antibodiesCitation27-Citation76,Citation32 and the above limitations still need to be overcome. For our ROCK® platform, we initially combined unique CD16A-specific domains that have already been successfully applied in AFM13.Citation2,Citation3 This anti-CD16 domainCitation2 (Ab16mid) demonstrates high specificity for CD16A, virtually no competition with serum IgG, robust binding and engagement of NK cells and very good potency in tumor cell killing, and thus served as a bona fide effector domain that could be further engineered and applied as a backbone in the ROCK® platform.
In a first attempt, we performed an affinity maturation to enhance efficacy. This maturation step led to the second generation of anti-CD16A domains, which maintained the characteristics of the parental domain in terms of specificity and serum IgG competition. After maturation, the domains reached affinities up to 2 nM as measured in kinetic experiments via SPR. Thereby, these antibody domains enable substantially higher affinity binding to both prominent allelic variants CD16A-158V and CD16A-158F compared with wildtype IgG1 Fc portions, which bind to CD16A in the high nM to µM KD range and have a clear preference for the CD16A-158V allele that is maintained even in the context of antibody scaffolds with affinity enhanced Fc (e.g., S239D/I332E) as reported by us and others.Citation77 Due to recognition of an epitope on CD16A that is distinct from the Fc binding site, binding of our ROCK® engagers to CD16A is virtually not impaired in the presence of physiological or even pathophysiological levels of competing IgGs, allowing for efficacious redirection of NK cells independent of patient CD16A allotype. Notably the specific binding of our CD16A domain is different from clone 3G8,Citation34–Citation36 which binds both CD16A (FcγRIIIA) and CD16B (FcγRIIIB), thus avoiding a potential sink on polymorphonuclear granulocytes that express CD16B. In addition, the contribution of neutrophils, the most abundant type of granulocytes, and the role of CD16B in tumor control is not fully understood.Citation78,Citation79 Therefore, it is suggested that also those patients with a low affinity CD16A genotype (158F, roughly 85% of the Caucasian population)Citation74,Citation75 will respond better to immune cell engagers based on the ROCK® anti-CD16A domains. Moreover, the lack of binding to Fc receptors other than CD16A could substantially reduce sink-effects compared to conventional mAbs.
In a second step, we used the first (Ab16mid) and second generation of affinity-optimized anti-CD16A domains (Ab16hi) in different engager formats. Based on internal data (unpublished results), the Ab16mid binding moiety is fully functional in different setups, such as scFv, scDb, Db, or Fab that could be universally used as building blocks in different engager formats. Activity and functionality in a wide spectrum of different engager formats is crucial because the tumor target domain or epitope may be less flexible or accessible only with certain antibody formats or architectures. A plethora of bispecific or multispecific antibody formats based on scaffolds such as IgG, Fc, or Fab has previously been described and validated by othersCitation37,Citation38,Citation80 for immune cell engagement. Typically, binding domains in these architectures are arranged in Fab armsCitation81,Citation82 or as scFvs fused to the asymmetric or symmetric scaffolds.Citation60 Other bispecific formats are exclusively based on binding domains connected via flexible linkers as represented by the TandAb and other formats without a constant domain.Citation83–Citation87 In order to make further use of the unique anti-CD16A domains and profit from their unique features, we added both previously known and newly engineered multivalent and multispecific antibody formats to the ROCK® platform. All formats exhibit two CD16A-binding moieties in order to increase the binding strength through avidity effects. To avoid additional and less selective binding to other Fcγ receptors, Fc portions of Fc-containing ROCK® engagers comprise a combination of silencing mutations,Citation48-Citation50 but still maintain FcRn-binding for extended half-life. Fab fusionCitation81 and Fc-less engager constructs such as aTriFlexCitation54 complement and extend the versatility of the ROCK® platform. Finally, the platform can be used to generate molecules in four different families: (1) Fc-less ROCK®, (2) Fc fusion ROCK®, (3) IgG-like ROCK®, and (4) Fab fusion ROCK®, which have been further optimized for innate immune cell engagement.
In all formats, we were able to show that our highly CD16A-selective Fv domains, with its bivalent effector-binding characteristics in ROCK® engagers, fully leverage the avidity effect to maximize receptor retention, which is a strong differentiating feature of the platform compared to Fc-enhanced IgGs or monovalent recruiting antibodies. Apparent CD16A-binding strength was superior to wildtype IgG1 and Fc-enhanced IgGs for all tested ROCK® engagers, but still tunable with the use of different variable domains, N- or C-terminal positioning and connector lengths. Most importantly, fine-tuning of binding affinities could be accomplished by using either Fab- or Diabody-based binding modules that enhanced binding strength as compared to the fusion of two CD16-binding scFvs. Intriguingly, the Db-format is one of the smallest bivalent monospecific or monovalent bispecific antibody formats with a rigid antibody structure. Its short linkers between VL and VH impose major constraints to the association of the two chains. There are models that postulate a very compact structure with the size of a Fab,Citation88,Citation89 with each specificity pointing to opposite directions.Citation90,Citation91 Most likely, this structure may constitute a key element for an optimal innate immune cell engaging domain via CD16A. A similar rigid and compact structure with comparable architecture can also be postulated for the tandem diabody. A molecule of this type is already being tested clinically, i.e., AFM13,Citation2,Citation3 the most advanced NK cell engager format within the ROCK® platform. However, it remains to be shown if the Db, scFv or scDb format is the best fit-for-purpose NK cell engaging domain, and whether this principle can be transferred to other activation molecules, such as NKp46 or NKG2D.
Most notably, the ROCK® platform enabled the generation of NK cell engagers comprising two high affinity anti-CD16A domains that facilitate bivalent binding to NK cells but exclude crosslinking of neighboring NK cells. This is an important safety feature since NK-NK crosslinking would not only lead to off-target NK cell activation followed by secondary pharmacodynamic effects such as unintended cytokine release, but would also induce NK-NK cell fratricide and subsequently deplete the effector cell population. This effect was not only observed in vitro but also in vivo as shown for daratumumab.Citation41 Although daratumumab-induced NK cell depletion is caused by classical ADCC after binding to CD38 on the target cell population and to CD16A on the effector cell,Citation92 studies with daratumumab revealed that NK cell fratricide can be induced by antibodies, and that NK cells are not protected from killing by autologous NK cells. Employing the modularity of the ROCK® platform, the NK cell fratricide could be avoided by switching the CD16A antigen-binding moieties to VL-VH orientation, its positioning within the molecule (preferably at the C-terminus) and by modulation of linker- and connector-lengths.Citation93,Citation94
Adjustment of optimal PK is yet another important factor for antibody treatment in clinical development. Short-lived formats have the advantage of good tunability and rapid clearance from the patient’s body in case of toxic side effects, but lack patient convenience through frequent dosing or continuous infusions as has been seen for blinatumomab. A vice versa situation is given for a long-lived antibody molecule. The ROCK® platform accommodates both short and long-lived formats and those that can be modulated in between. Judging from a mouse PK study, the long-lived formats reach half-lives expected from canonical IgGs, whereas other formats exhibit half-life measurable in hours and are comparable to blinatumomab, BiKE and DART® molecules.Citation62
In vitro cytotoxicity assays with two independent sets of ROCK® engagers targeting either BCMA or EGFR demonstrated higher cytotoxic potency for engagers possessing high apparent affinity for NK cells and lower potency for constructs with low apparent affinity or only monovalent interaction with NK cells, such as classical IgG or engineered IgG with Fc-enhanced effector function. High affinity binding of ROCK® engagers to NK cells is not only beneficial for high cytotoxic potency on target cells expressing low cell surface levels, such as BCMA on MC/CAR cells, but also for high potency on target cells with medium or high copy numbers of the target antigen like BCMA on MM.1S or EGFR on A-431. In this context, the anti-CD16A diabody motif represents a remarkable structure that leads to high apparent affinities on NK cells and subsequent high potency in cytotoxicity assays in the bispecific TandAb, Db-Fc, KiH-scDb-Fc format, but surprisingly not in the asymmetric aTriFlex format. Since the anti-CD16A diabody without the distal tumor-targeting scFv, which are present in the aTriFlex format, exhibit high apparent affinities to NK cells (data not shown), it is tempting to speculate that the diabody fusion with the scFv itself changes the correct folding of the diabody or that the fused scFv lead to a sterical hindrance that reduces binding to CD16A on NK cells. In the present evaluation of the ROCK® engagers concerning NK cell-binding and cytotoxic activity, the apparent affinities of the engagers for the respective target antigens on cells was not considered. Although most of the ROCK® engagers are bivalent for the target antigen, their different apparent affinities may affect cytotoxic activity.
In summary, we presented a highly differentiated and novel fit-for-purpose technology platform allowing the engineering of innate immune cell engagers to overcome the therapeutic limitations of T cell-based approaches, monoclonal and Fc-enhanced antibodies. Key features of the ROCK® platform engagers are the unique CD16A domains, lack of serum IgG competition, independence of CD16A polymorphisms, and their multivalent architecture. In addition, the combination of CD16A antibody engineering in the context of a broad range of scaffolds allows for customized properties such as PK, high affinity/avidity and potency, and avoidance of NK cell fratricide. The ROCK® platform thus holds great promise for generating novel product candidates for tailored immunotherapy of cancer.
Materials and methods
Generation of ROCK® recombinant antibodies, controls, and antigen variants
Gene sequences for different recombinant proteins (antigens, antibodies, controls) were synthesized by GeneART Gene Synthesis (Thermo Fisher Scientific, Regensburg, Germany) or derived by PCR. Expression vectors, encoding these recombinant proteins, were generated by cloning of the respective sequence elements into the mammalian expression vector pcDNA5/FRT (Life Technologies) or a modified version thereof using standard molecular biology techniques. Soluble recombinant antigen variants were constructed as fusion proteins of the ECD sequences to monomeric Fc.Citation59 For the expression of cell surface-anchored antigen variants, ECD sequences were either fused to the transmembrane domain of human EGFRCitation95 or anchored via GPI using the human CD16B endogenous sequence with the full-length propeptide sequence for post-translational processing and lipidation for GPI-anchorage. In some expression constructs, the original vector was modified to contain two cytomegalovirus-promotor controlled expression cassettes for coexpression of two antibody chains. A further modification of the mammalian expression vector was the replacement of the hygromycin resistance with a puromycin resistance gene. Expression constructs were furthermore designed to contain coding sequences for N-terminal signal peptides to facilitate secretion. For recombinant fusion constructs (e.g., antigens with IgG1 Fc portion) sequences encoding the fusion partners were PCR amplified using elongated primers to construct corresponding linker or connector sequences for gene fusion or restriction enzyme digestion. The resulting overlapping DNA-fragments were inserted into the coexpression vector at the relevant position using Gibson Assembly® Master Mix (New England Biolabs, cat.: E2611) to yield the final construct. Sequences of all constructs were confirmed by Sanger sequencing performed at GATC (Köln, Germany) using custom-made primers.
Soluble antibodies or antigens or cell surface-anchored antigens were expressed in CHO as previously described.Citation57 Target proteins were purified from cell culture supernatant using one or two standard affinity chromatographic methods (Protein A, Protein L, Capture Select C-tag or IMAC) depending on monomeric or multimeric (homodimeric, heterodimeric, or tetrameric) product form and on fused affinity tags, presence of κ-light chain or Fc part. All protein preparations were further polished via size-exclusion chromatography (SEC) and analyzed using SEC-multi-angle light scattering, SDS-PAGE and UV-Vis spectroscopy.
Dotblot, SDS-PAGE, and western blot
Purified recombinant CD16 antigen variants were spotted on nitrocellulose membranes, or mixed with sample buffer and separated on 4–20% Criterion TGX Precast Gels (Bio-Rad). Total protein was imaged using the Bio-Rad Molecular Imager Gel Documentation system. Protein was transferred by Western blotting to PVDF Midi Membranes using the Trans-Blot Turbo system (Bio-Rad). Membranes were blocked for 30 min with 3% (w/v) skimmed milk powder (Merck) dissolved in tris-buffered saline (TBS) and incubated with 2–4 µg/mL of anti-CD16 scFv antibodies for 1 h at room temperature followed by washing with TBST (TBS containing 0.1% (v/v) Tween 20) and two times with TBS. Membranes were incubated with anti-Penta-His-HRP (Qiagen, cat.: 34460) 1:3,000 diluted as secondary detection conjugate for 1 h at room temperature. After washing, colorimetric development was started by addition of a freshly prepared mixture of 0.66 mg/mL DAB, 0.02% CoCl2, 0.015% H2O2 in TBS and stopped by washing membranes in water. Membranes were dried and photographed.
Analysis of CD16A antigen binding by ELISA
96-well ELISA plates (Immuno MaxiSorp; Nunc) were coated overnight at 4°C with recombinant antigens or different NK cell engager antibody formats in 100 mM carbonate-bicarbonate buffer. Depending on the molecular mass, 0.5–3 µg/mL of antigen or 2–5 µg/mL of antibody formats were coated, equalizing molarities to approximately 30 nM for coated antigens or 10–20 nM for coated antibodies. After blocking with 3% (w/v) skimmed milk powder (Merck) dissolved in phosphate-buffered saline (PBS), serial dilutions of His-tagged scFv antibodies or of a biotinylated CD16A-158V ECD-monomeric Fc fusion antigen in PBS containing 0.3% (w/v) skim milk powder were incubated on the plates coated with antigen or antibodies, respectively, for 1.5 h at room temperature. To assess binding competition, scFvs were titrated and co-incubated on the plates with 1 nM or 10 nM of 3G8 mAb. After washing three times with 300 µL per well of PBS containing 0.1% (v/v) Tween 20, plates were incubated with detection conjugates, Penta-His-HRP, at 1:3,000 dilution, Streptavidin-HRP (Roche, cat.: 1108915300) or for the detection of competitor 3G8, Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (Dianova, cat.: 115–035-003) at 1:10,000 dilution for 1 h at room temperature. After washing, plates were incubated with tetramethylbenzidine substrate (Seramun) for 1–2 min or until color development was clearly visible. Reaction was stopped by addition of 0.5 M H2SO4 (100 µL/well). Absorbance was measured at 450 nm (1 s) using a multiwell plate reader (Victor, Perkin Elmer). Absorbance values were plotted and EC50 values were determined by fitting a nonlinear regression model to sigmoidal dose-response curves (four parameters logistic fit) using GraphPad Prism version 6.07 (GraphPad Software, La Jolla California USA).
Cell lines and cell culture
NCI-H929 (DSMZ, cat.: ACC-163), MC/CAR (ATCC, cat.: CRL-8083), MM.1S (ATCC, cat.: CRL-2974), RPMI-8226 (DSMZ, cat.: ACC 402), KARPAS-299 (DSMZ, cat.: ACC 31), and SW-982 (ATCC, cat.: HTB-93) were purchased, and A-431 were provided by Dr. G. Moldenhauer (DKFZ Heidelberg). All cells were cultured under standard conditions in DMEM (cat.: 41965–039), IMDM (cat.: 12440–053), or RPMI 1640 (cat.: 21875–034) medium supplemented with: 10% heat-inactivated fetal calf serum (FCS) (cat.: 10270–106), 100 U/mL penicillin G/100µg/mL streptomycin (cat.: 1540–122), and 2 mM L-glutamine (cat.: 25030–024; all Life Technologies) referred to as complete RPMI 1640 medium at 37°C in a humidified 5% CO2 atmosphere.
Isolation of human NK cells
PBMC were isolated from healthy volunteers’ buffy coats (German Red Cross, Mannheim, Germany) by density gradient centrifugation using Lymphoprep (StemCell Technologies, cat.: 07861), as described before.Citation2,Citation96 PBMC were cultured overnight in complete RPMI 1640 medium at 37°C and 5% CO2 in a humidified atmosphere before NK cells were enriched by negative bead selection using EasySEP™ Negative NK Cell Enrichment Kit (StemCell Technologies, cat.: 17955) according to manufacturer’s instructions. The purity of NK cell isolation was determined by flow cytometry, and demonstrated usually >80% CD56+ cells (data not shown).
Cell binding assays and flow cytometric analysis
Aliquots of 0.2–1 × 106 enriched human NK cells were incubated with 100 µL of serial dilutions of the indicated constructs in FACS buffer (PBS, Invitrogen, cat.: 14190–169) containing 2% heat-inactivated FCS (Invitrogen, cat.: 10270–106), 0.1% sodium azide (Roth, cat.: A1430.0100) in the absence or, if indicated in the presence of 10 mg/mL polyclonal human IgG (Gammanorm, Octapharma) for 45 min at 37°C. After repeated washing with FACS buffer, cell-bound His-tagged scFv, TandAb, or aTriFlex antibodies were detected with 10 µg/mL anti-His mAb 13/45/31–2 (Dianova, cat.: DIA910-1MG) followed by 15 µg/mL FITC-conjugated goat anti-mouse IgG (Dianova, cat.: 115–095-062). Bispecific BCMA/CD16A antibody constructs were detected with soluble His-tagged BCMA followed by 10 µg/mL anti-His mAb 13/45/31–2 followed by 15 µg/mL FITC-conjugated goat anti-mouse IgG, and bispecific Fc-containing EGFR/CD16A constructs with 15 µg/mL FITC-conjugated goat anti-human IgG (Dianova, cat.: 109–095-098). After the last staining step, the cells were washed again and resuspended in 0.2 mL of FACS buffer containing 2 µg/mL propidium iodide (Sigma, cat.: P4170) in order to exclude dead cells. The fluorescence of 2–5 × 103 living cells was measured using a Millipore Guava EasyCyte flow cytometer (Merck Millipore, Schwalbach, Germany) or CytoFlex cytometer (Beckman Coulter, Krefeld, Germany), and median fluorescence intensities of the cell samples were determined. After subtracting the fluorescence intensity values of the cells stained with the secondary and/or tertiary reagents alone, the values were used for nonlinear regression analysis. Equilibrium dissociation constants (KD) were calculated using the one-site-binding (hyperbolic) fit and GraphPad Prism software V6 or V7 (GraphPad Software, La Jolla California USA).
Cytotoxicity assays
For the calcein-release assay, target cells were labeled with 10 mM calcein AM (Life Technologies, cat.: C3100MP) for 30 min in RPMI medium at 37°C, washed, and 1 × 104 tumor target cells were seeded, in individual wells of a 96-well micro plate, together with effector cells in a total volume of 200 µL at the indicated effector:target (E:T) ratios in the presence of increasing antibody concentrations. For the assessment of NK fratricide, 5 × 104 calcein-labeled primary human NK cells were coincubated with autologous NK cells at an E:T ratio of 1:1 in the presence of increasing antibody concentrations. After incubation at 37°C in a humidified 5% CO2 atmosphere for 4 h if not otherwise indicated, the fluorescence (F) of calcein released into the supernatant was measured by a plate reader at 520 nm (Victor 3 or EnSight, Perkin Elmer, Turku, Finland). The specific cell lysis was calculated as: [F(sample)–F(spontaneous)]/[F(maximum)–F(spontaneous)] x 100%. F(spontaneous) represents fluorescence released from target cells in the absence of effector cells and antibodies, and F(maximum)represents that released after total cell lysis induced by addition of Triton × 100 (Roth, cat.: 3051.2) to a final concentration of 1%. Mean values of specific target cell lysis (%) and standard deviations (SD) were plotted and in vitro potency (EC50) was determined by fitting the nonlinear regression model to sigmoidal dose-response curves (variable slope) using GraphPad Prism (v6 and v7; GraphPad Software, La Jolla California USA).
Surface plasmon resonance
Kinetic binding analyses of monovalent interactions to CD16A and qualitative dissociation phase comparison of monovalent and bivalent anti-CD16A-binding analytes were performed on a Biacore T200 Instrument (GE Healthcare) at 25°C using HBS-P+ (10 mM HEPES, 150 mM NaCl, 0.05% (w/v) polysorbate 20, pH 7.4) as running buffer and for dilutions. CAP sensor chips (Biotin Capture Kit, GE Healthcare) were preconditioned overnight using HBS-P+ buffer. In a precapturing step, chip surfaces were treated with Biotin CAPture reagent (GE Healthcare) at 5 µL/min for 100 s in flow cells 1–4.
For kinetic binding analyses of various anti-CD16A scFvs, human CD16A-158V, or human CD16A-158F ECDs fused to Avi-tagged and biotinylated monomeric Fc (silenced, L234F/L235E/D265ACitation48-Citation50) were prepared in HBS-P+ and captured (approx. 40 RU for measuring interaction with scFvs and approx. 10 RU for measuring interactions with IgG) in flow cells Fc 2 and Fc 4. IgGs and scFvs were prepared in HBS-P+ buffer at indicated concentration series and injected to reference surfaces (Fc 1, Fc 3) and receptor captured surface (Fc 2, Fc 4) at a flow rate of 40 µL/min (association time 180 s, dissociation time 300 s). Interactions were measured using Multi Cycle Kinetic mode (all antibodies were prepared in a fourfold dilution series: scFv-Ab16mid 150–0.586 nM; scFv-Ab16hi 50–0.195 nM, scFv-Ab16lo 500–1.953 nM, scFv-3G8, and human IgG1 Fc-enhanced 200–0.781 nM, human IgG1, and human IgG1 with silenced Fc (L234F/L235E/D265ACitation48-Citation50) 2000–7.813 nM) following a regeneration cycle with 6 M guanidine-HCl/250 mM NaOH at 10 mL/min for 120 s. Data were referenced by subtraction of signals from reference surfaces (Fc 2–1, Fc 4–3) and zero concentration (buffer control) signals. Kinetic evaluation was performed by fitting data to a 1:1 binding model (Rmax and RI locally fitted) using Biacore T200 Evaluation Software (v3.1). For qualitative comparison of sensorgrams, a single concentration (312.5 nM) of scFvs and IgGs was injected over reference and receptor captured surface at increased capture levels (human CD16A-158V, 180 RU and human CD16A-158F, 90 RU). Referenced curves were normalized to each curves maximum and overlaid. For investigation of receptor retention via qualitative dissociation phase comparison of monovalent and bivalent CD16A-binding antibodies or ligands, Avi-tagged and biotinylated monomeric Fc (silenced) receptor fusions were prepared in HBS-P+ and captured (human CD16A-158V, approx. 160 RU, cynomolgus CD16, approx. 320 RU) in flow cells Fc 2 and Fc 3. Antibodies of interest (scFv-IgAb, Db-Fc, KiH-scDb-Fc, TandAb) incorporating Ab16hi binding domain and comparators, enhanced human IgG1 Fc (S239D/I332E) and monovalent binding scFv-Ab16hi were diluted to a concentration of 50 nM in HBS-P+ buffer and are injected to reference surfaces (Fc 1) and receptor captured surface (Fc 2, Fc 3) at a flow rate of 30 µL/min (association time 180 s, dissociation time 3 h). Surface was regenerated with 6 M guanidine-HCl/250 mM NaOH at 10 mL/min for 120 s. Data were referenced by subtraction of signals from reference surfaces (Fc 2–1, Fc 4–3) and zero concentration (buffer control) signals. Referenced curves were normalized to each curves maximum and overlaid.
Pharmacokinetic studies of ROCK® engagers
Basic PK parameters of the different ROCK® platform formats were assessed in CD-1 SWISS mice. In life phases were conducted by Heidelberg Pharma GmbH. CD-1 SWISS female mice (30 g body weight) received a single intravenous administration of 0.3 mg (~10 mg/kg) of the test item. At different time points, blood was collected for a period of 1–3 weeks. A representative blood withdrawal schedule covered 13 timepoints, for example, predose day −7, 5 min, 30 min, 1, 4, 8 h, 1, 2, 3, 4, 7, 14, and 21 days posttreatment. Serum from the blood was taken and frozen for quantification. Different methods were developed using combinations of soluble antigens and/or mAbs as capture and/or detector for ROCK® engagers and ELISA or MSD technology. Assay development, qualification and determination of serum concentrations were executed by MicroMol GmbH using a Tristar LB941 instrument (Berthold instruments) based on enhanced chemoluminescence detection.
NK cell activation assay
5 x 105 human PBMCs were seeded in individual wells of a flat-bottom 96-well micro plate in the presence or absence of 1 × 104 RPMI-8226 cells in complete RPMI 1640 medium in the presence of the indicated antibody concentrations. Following 22 h incubation at 37°C with 5% CO2 in a humidified atmosphere, cells were harvested, washed, and resuspended in FACS/hIgG buffer (FACS buffer supplemented with 1 mg/mL polyclonal human IgG). Cells were then stained with CD56-PC7 and CD69-PC5 in FACS/hIgG buffer for 15 min on ice in the dark. After repeated washing with FACS buffer, 1 × 103–1 × 104 cells were analyzed by flow cytometry and percentage of CD69+ cells of CD56+ NK cells were quantified.
Cytokine release assay
Flat-bottom 96-well microtiter plates were blocked with RPMI-1640/5% FCS for 2–4 h at room temperature before seeding of 5 × 105 PBMC with or without 1 × 104 NCI-H929 tumor cells with or without 10 µg/mL of the indicated antibodies in a total volume of 200 µL/well. Plates were incubated for 24 h at 37°C and 5% CO2 in a humidified incubator. Cell culture supernatants were harvested and stored at −80°C until quantification of cytokines at Bioassay GmbH (Heidelberg, Germany) using BD™ Cytometric Bead Array (CBA) Human Th1/Th2 Cytokine Kit II (BD Bioscience).
Disclosure of potential conflicts of interest
KE, UR, IF, SW, TR, TM, US, TH, ER, JK, MTr, and MT are employees and hold stock options of Affimed.
Abbreviations
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| AML | = | acute myeloid leukemia |
| AUC | = | area under the curve |
| BCMA | = | B-cell maturation antigen |
| CAR | = | chimeric-antigen receptor |
| CAR-T | = | chimeric antigen receptor T cell |
| CD | = | Cluster of differentiation |
| CHO | = | Chinese hamster ovary |
| CPM | = | checkpoint modulator |
| CRS | = | cytokine release syndrome |
| Db | = | diabody |
| ECD | = | extracellular domain |
| EGFR | = | epidermal growth factor receptor |
| ELISA | = | enzyme-linked immunosorbent assays |
| Fab | = | antibody binding fragment |
| Fc | = | fragment, crystallizable |
| FcR | = | Fc receptors |
| FcRn | = | neonatal Fc receptor |
| Fv | = | variable fragment |
| GPI | = | glycosylphosphatidylinositol |
| HSA | = | human serum albumin |
| IMAC | = | immobilized-metal affinity chromatography |
| KiH | = | knobs-into-holes |
| mAb | = | monoclonal antibody |
| mFc | = | monomeric Fc |
| MRT | = | mean residence times |
| MSD | = | Meso Scale Discovery |
| NK | = | natural killer |
| PBMC | = | peripheral blood mononuclear cell |
| PBS | = | phosphate-buffered saline |
| PD-1 | = | programmed cell death 1 |
| PK | = | pharmacokinetic |
| ROCK | = | redirected optimized cell killing |
| RSV | = | respiratory syncytial virus |
| scDb | = | single-chain diabody |
| scFv | = | single-chain variable fragments |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEC | = | size-exclusion chromatography |
| SPR | = | surface plasmon resonance |
| TBS | = | tris-buffered saline |
Supplemental Material
Download MS Word (586.8 KB)Acknowledgments
We thank Wolfgang Fischer (Affimed N.V.), Adi Hoess (Affimed N.V.), Stefan Knackmuss (Affimed GmbH) and Holger Dulat (Affimed GmbH) for advice throughout all stages of this work, Anca Alexandru and Angela Sickmann for critical reading of the manuscript and Aline Reuter for literature support. The technical assistance of Claudia Linhard, Melanie Kuhse, Sonja Richt, Sarah Flößer, Philipp Fieger, Jessica Kientz, Jürgen Weik, Sonja Bastuck, Ineke Tellier, Susanne Eisel, Michael Damrat, Alexandra Ruf, Armin Beez, Stefanie Wolff, Raphael Bleiler, Vanessa Krenzer, and Sabrina Purr (all Affimed GmbH) was gratefully appreciated.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Related Research Data
References
- Dahlen E, Veitonmaki N, Norlen P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. 2018;6:3–17. doi:10.1177/2515135518763280.
- Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, Knackmuss SH, Kiprijanov S, Little M, Zhukovsky EA. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. 2014;6:728–39. doi:10.4161/mabs.28591.
- Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, Dietlein M, Kuhnert G, Kessler J, Buerkle C, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125:4024–31. doi:10.1182/blood-2014-12-614636.
- Berrien-Elliott MM, Romee R, Fehniger TA. Improving natural killer cell cancer immunotherapy. Curr Opin Organ Transplant. 2015;20:671–80. doi:10.1097/MOT.0000000000000243.
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Helman LJ, Kastan MB, Knapp DW, Levin WJ, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123. doi:10.1126/scitranslmed.aaf0746.
- Fehniger TA, Miller JS, Stuart RK, Cooley S, Salhotra A, Curtsinger J, Westervelt P, DiPersio JF, Hillman TM, Silver N, et al. A Phase 1 Trial of CNDO-109-Activated Natural Killer Cells in Patients with High-Risk Acute Myeloid Leukemia. Biol Blood Marrow Transplant. 2018;24:1581–89. doi:10.1016/j.bbmt.2018.03.019.
- Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, Muftuoglu M, Liu E, Orlowski RZ, Cooper L, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177:457–66. doi:10.1111/bjh.14570.
- Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi:10.1146/annurev.immunol.23.021704.115526.
- Koch J, Tesar M. Recombinant Antibodies to Arm Cytotoxic Lymphocytes in Cancer Immunotherapy. Transfus Med Hemother. 2017;44:337–50. doi:10.1159/000479981.
- Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–63.
- Haliotis T, Ball JK, Dexter D, Roder JC. Spontaneous and induced primary oncogenesis in natural killer (NK)-cell-deficient beige mutant mice. Int J Cancer. 1985;35:505–13. doi:10.1002/(ISSN)1097-0215.
- Talmadge JE, Meyers KM, Prieur DJ, Starkey JR. Role of NK cells in tumour growth and metastasis in beige mice. Nature. 1980;284:622–24. doi:10.1038/284622a0.
- Good RA. Relations between immunity and malignancy. Proc Natl Acad Sci U S A. 1972;69:1026–32. doi:10.1073/pnas.69.4.1026.
- Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vely F, Marabelle A, Papoular B, Piperoglou C, Ponzoni M, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med. 2015;7:283ra55. doi:10.1126/scitranslmed.aad3106.
- Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, Brunstein CG, Blazar BR, Wagner J, Diamond DJ, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia. 2016;30:456–63. doi:10.1038/leu.2015.260.
- Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356:1795–99. doi:10.1016/S0140-6736(00)03231-1.
- Xu B, Chen L, Li J, Zheng X, Shi L, Wu C,Jiang J. Prognostic value of tumor infiltrating NK cells and macrophages in stage II+III esophageal cancer patients. Oncotarget. 2016;7:74904–16. doi:10.18632/oncotarget.12484.
- Versluis MAC, Marchal S, Plat A, de Bock GH, van Hall T, de Bruyn M, Hollema H, Nijman HW. The prognostic benefit of tumour-infiltrating Natural Killer cells in endometrial cancer is dependent on concurrent overexpression of Human Leucocyte Antigen-E in the tumour microenvironment. Eur J Cancer. 2017;86:285–95. doi:10.1016/j.ejca.2017.09.008.
- Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. 2000;88:577–83.
- Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, Ferrelli F, Coppola A, Arriga R, Lauro D, et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology. 2014;3:e952197. doi:10.4161/21624011.2014.952197.
- Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, Patsouris ES, Peros G, Horcic M, Tornillo L, et al. Tumor infiltration by FcgammaRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer. 2011;128:2663–72. doi:10.1002/ijc.25609.
- Luna JI, Grossenbacher SK, Murphy WJ, Canter RJ. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin Biol Ther. 2017;17:313–24. doi:10.1080/14712598.2017.1271874.
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103:4005–10. doi:10.1073/pnas.0508123103.
- Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol. 1999;17:176–80. doi:10.1038/6179.
- Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SHA, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi:10.1074/jbc.M202069200.
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, Wakitani M, Niwa R, Sakurada M, Uchida K, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–22. doi:10.1002/bit.20151.
- Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–47. doi:10.1200/JCO.2003.05.013.
- Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med. 2006;203:743–53. doi:10.1084/jem.20052283.
- Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res. 2013;19:1126–38. doi:10.1158/1078-0432.CCR-12-0989.
- Byrd JC, Flynn JM, Kipps TJ, Boxer M, Kolibaba KS, Carlile DJ, Fingerle-Rowson G, Tyson N, Hirata J, Sharman JP. Randomized phase 2 study of obinutuzumab monotherapy in symptomatic, previously untreated chronic lymphocytic leukemia. Blood. 2016;127:79–86. doi:10.1182/blood-2015-03-634394.
- Temam S, Spicer J, Farzaneh F, Soria JC, Oppenheim D, McGurk M, Hollebecque A, Sarini J, Hussain K, Soehrman Brossard S, et al. An exploratory, open-label, randomized, multicenter study to investigate the pharmacodynamics of a glycoengineered antibody (imgatuzumab) and cetuximab in patients with operable head and neck squamous cell carcinoma. Ann Oncol. 2017;28:2827–35. doi:10.1093/annonc/mdx489.
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–58.
- Vallera DA, Felices M, McElmurry RT, McCullar V, Zhou X, Schmohl J, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33 + Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin Cancer Res. 2016;22:3440–50. doi:10.1158/1078-0432.CCR-15-2710.
- de Palazzo I, Gercel-Taylor C, Kitson J, Weiner LM. Potentiation of tumor lysis by a bispecific antibody that binds to CA19-9 antigen and the Fc gamma receptor expressed by human large granular lymphocytes. Cancer Res. 1990;50:7123–28.
- Arndt MA, Krauss J, Kipriyanov SM, Pfreundschuh M, Little M. A bispecific diabody that mediates natural killer cell cytotoxicity against xenotransplantated human Hodgkin’s tumors. Blood. 1999;94:2562–68.
- Li W, Yang H, Dimitrov DS. Identification of high-affinity anti-CD16A allotype-independent human antibody domains. Exp Mol Pathol. 2016;101:281–89. doi:10.1016/j.yexmp.2016.10.001.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212. doi:10.1080/19420862.2016.1268307.
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106. doi:10.1016/j.molimm.2015.01.003.
- Chen S, Li J, Li Q, Wang Z. Bispecific antibodies in cancer immunotherapy. Hum Vaccin Immunother. 2016;12:2491–500. doi:10.1080/21645515.2016.1187802.
- Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303–10. doi:10.1080/19420862.2015.1011450.
- Casneuf T, Xu XS, Adams HC, Axel AE, Chiu C, Khan I, Ahmadi T, Yan X, Lonial S, Plesner T, et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Advances. 2017;1:2105–14. doi:10.1182/bloodadvances.2017006866.
- de Palazzo IG, Holmes M, Gercel-Taylor C, Weiner LM. Antitumor effects of a bispecific antibody targeting CA19-9 antigen and CD16. Cancer Res. 1992;52:5713–19.
- Michallet M, Chapuis-Cellier C, Dejoie T, Lombard C, Caillon H, Sobh M, Moreau P, Attal M, Avet-Loiseau H. Heavy+light chain monitoring correlates with clinical outcome in multiple myeloma patients. Leukemia. 2018;32:376–382. doi:10.1038/leu.2017.209.
- Mahaweni NM, Olieslagers TI, Rivas IO, Molenbroeck SJJ, Groeneweg M, Bos GMJ, Tilanus MGJ, Voorter CEM, Wieten L. A comprehensive overview of FCGR3A gene variability by full-length gene sequencing including the identification of V158F polymorphism. Sci Rep. 2018;8:15983. doi:10.1038/s41598-018-34258-1.
- Bowles JA, Wang S-Y, Link BK, Allan B, Beuerlein G, Campbell M-A, Marquis D, Ondek B, Wooldridge JE, Smith BJ, et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood. 2006;108:2648–54. doi:10.1182/blood-2006-04-020057.
- Burchard PR, Malhotra S, Kaur P, Tsongalis GJ. Detection of the FCGR3a polymorphism using a real-time polymerase chain reaction assay. Cancer Genet. 2013;206:130–34. doi:10.1016/j.cancergen.2013.03.002.
- Tamm A, Schmidt RE. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for Ligand Binding. The Journal of Immunology. 1996;157:1576–81.
- Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi:10.3389/fimmu.2014.00520.
- Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S-I, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181:6664–69.
- Hezareh M, Hessell AJ, Jensen RC, van de Winkel JG, Parren PW. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J Virol. 2001;75:12161–68. doi:10.1128/JVI.75.24.12161-12168.2001.
- Ahmed AA, Keremane SR, Vielmetter J, Bjorkman PJ. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcgammaRIIIa. J Struct Biol. 2016;194:78–89. doi:10.1016/j.jsb.2016.02.001.
- Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, Huang A, Yang N-Y, Nishimura M, Greve J, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110:E2987–96. doi:10.1073/pnas.1302725110.
- McAleese F, Eser M. RECRUIT-TandAbs: harnessing the immune system to kill cancer cells. Future Oncol. 2012;8:687–95. doi:10.2217/fon.12.54.
- Gantke T, Weichel M, Herbrecht C, Reusch U, Ellwanger K, Fucek I, Eser M, Müller T, Griep R, Molkenthin V, et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng Des Sel. 2017;30:673–84. doi:10.1093/protein/gzx043.
- Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, Eser M, McAleese F, Molkenthin V, Gall FL, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs. 2015;7:584–604. doi:10.1080/19420862.2015.1029216.
- Ellwanger K, Reusch U, Fucek I, Knackmuss S, Weichel M, Gantke T, Molkenthin V, Zhukovsky EA, Tesar M, Treder M. Highly Specific and Effective Targeting of EGFRvIII-Positive Tumors with TandAb Antibodies. Front Oncol. 2017;7:100. doi:10.3389/fonc.2017.00100.
- Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, Knackmuss SHJ, Zhukovsky EA, Fox JA, Kunkel LA, et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin Cancer Res. 2016;22:5829–38. doi:10.1158/1078-0432.CCR-16-0350.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21.
- Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. J Biol Chem. 2012;287:19399–408. doi:10.1074/jbc.M112.368647.
- Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997;15:159–63. doi:10.1038/nbt0297-159.
- Choi EI, Wang R, Peterson L, Letvin NL, Reimann KA. Use of an anti-CD16 antibody for in vivo depletion of natural killer cells in rhesus macaques. Immunology. 2008;124:215–22. doi:10.1111/j.1365-2567.2007.02757.x.
- Unverdorben F, Richter F, Hutt M, Seifert O, Malinge P, Fischer N, Kontermann RE. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. MAbs. 2016;8:120–28. doi:10.1080/19420862.2015.1113360.
- Jacobi C, Claus M, Wildemann B, Wingert S, Korporal M, Römisch J, Meuer S, Watzl C, Giese T. Exposure of NK cells to intravenous immunoglobulin induces IFN gamma release and degranulation but inhibits their cytotoxic activity. Clin Immunol. 2009;133:393–401. doi:10.1016/j.clim.2009.09.006.
- Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, Byers LA, Johnson ML, Burris HA, Robert F, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18:42–51. doi:10.1016/S1470-2045(16)30565-4.
- Circosta P, Elia AR, Landra I, Machiorlatti R, Todaro M, Aliberti S, Hoadley KA, Print C, Knowlton N, Black MA, et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. Oncoimmunology. 2018;7:e1341032. doi:10.1080/2162402X.2018.1490854.
- Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195–208. doi:10.2147/DDDT.S151282.
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–87. doi:10.1038/nrc3236.
- Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8:1083.
- Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–28.
- Marechal R, De Schutter J, Nagy N, Demetter P, Lemmers A, Deviere J, Salmon I, Tejpar S, Van Laethem JL. Putative contribution of CD56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer. 2010;10:340. doi:10.1186/1471-2407-10-663.
- Villegas FR, Coca S, Villarrubia VG, Jiménez R, MaJ C, Jareño J, Zuil M, Callol L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. 2002;35:23–28.
- Reiners KS, Kessler J, Sauer M, Rothe A, Hansen HP, Reusch U, Hucke C, Köhl U, Dürkop H, Engert A, et al. Rescue of impaired NK cell activity in hodgkin lymphoma with bispecific antibodies in vitro and in patients. Mol Ther. 2013;21:895–903.
- Lanier LL, Ruitenberg JJ, Phillips JH. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J Immunol. 1988;141:3478–85.
- Koene HR, Kleijer M, Algra J, Roos D, Ae VDB, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90:1109–14.
- Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, Salmon JE, Kimberly RP. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–70. doi:10.1172/JCI119616.
- Pazina T, James AM, AWt M, Bezman NA, Henning KA, Bee C, Graziano RF, Robbins MD, Cohen AD, Campbell KS. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology. 2017;6:e1339853. doi:10.1080/2162402X.2017.1339853.
- Heider K-H, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, Baum A, Lamche H, Küpcü Z, Jacobi A, et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood. 2011;118:4159–68. doi:10.1182/blood-2011-04-351932.
- Treffers LW, Hiemstra IH, Kuijpers TW, TKvd B, Matlung HL. Neutrophils in cancer. Immunol Rev. 2016; 273:312–28.
- Treffers LW, van Houdt M, Bruggeman CW, Heineke MH, Zhao XW, van der Heijden J, Nagelkerke SQ, Verkuijlen P, Geissler J, Lissenberg-Thunnissen S, et al. FcgammaRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front Immunol. 2018;9:3124. doi:10.3389/fimmu.2018.03124.
- Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–47. doi:10.1016/j.drudis.2015.02.008.
- Schoonjans R, Willems A, Schoonooghe S, Fiers W, Grooten J, Mertens N. Fab Chains As an Efficient Heterodimerization Scaffold for the Production of Recombinant Bispecific and Trispecific Antibody Derivatives. J Immunol. 2000;165:7050–57.
- Schoonjans R, Willems A, Schoonooghe S, Leoen J, Grooten J, Mertens N. A new model for intermediate molecular weight recombinant bispecific and trispecific antibodies by efficient heterodimerization of single chain variable domains through fusion to a Fab-chain. Biomol Eng. 2001;17:193–202.
- Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J, Miller JS, Vallera DA. Engineering of Anti-CD133 Tri-Specific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC). Cancer Res Treat. 2017;49:1140–52. doi:10.4143/crt.2016.491.
- Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target Oncol. 2016;11:353–61. doi:10.1007/s11523-015-0391-8.
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–44. doi:10.1158/0008-5472.CAN-09-0547.
- Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, Riethmüller G, Dörken B, Bargou RC. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098–103.
- Rader C. DARTs take aim at BiTEs. Blood. 2011;117:4403–04. doi:10.1182/blood-2011-02-337691.
- Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993;90:6444–48. doi:10.1073/pnas.90.14.6444.
- Perisic O, Webb PA, Holliger P, Winter G, Williams RL. Crystal structure of a diabody, a bivalent antibody fragment. Structure. 1994;2:1217–26.
- Kim JH, Song DH, Youn S-J, Kim JW, Cho G, Kim SC, Lee H, Jin MS, Lee J-O. Crystal structures of mono- and bi-specific diabodies and reduction of their structural flexibility by introduction of disulfide bridges at the Fv interface. Sci Rep. 2016;6:34515. doi:10.1038/srep34515.
- Carmichael JA, Power BE, Garrett TPJ, Yazaki PJ, Shively JE, Raubischek AA,Wu AM, Hudson PJ. The Crystal Structure of an Anti-CEA scFv Diabody Assembled from T84.66 scFvs in VL-to-VH Orientation: implications for Diabody Flexibility. J Mol Biol. 2003;326:341–51.
- Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, Yu J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin Cancer Res. 2018;24:4006–17. doi:10.1158/1078-0432.CCR-17-3117.
- Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods. 2001;248:47–66.
- Le Gall F, Reusch U, Little M, Kipriyanov SM. Effect of linker sequences between the antibody variable domains on the formation, stability and biological activity of a bispecific tandem diabody. Protein Eng Des Sel. 2004;17:357–66. doi:10.1093/protein/gzh039.
- Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–63. doi:10.1182/blood-2011-02-337360.
- Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol. 2009;85:7.1. −7.1. 8.