
ABSTRACT
The success of therapeutic antibodies is largely attributed for their exquisite specificity, homogeneity, and functionality. There is, however, a need to engineer antibodies to extend and enhance their potency. One parameter is functional affinity augmentation, since antibodies matured in vivo have a natural affinity threshold. Generation of multivalent antibodies is one option capable of surpassing this affinity threshold through increased avidity. In this study, we present a novel platform consisting of an array of multivalent antibody formats, termed Quads, generated using the self-assembling tetramerization domain from p53. We demonstrate the versatility of this tetramerization domain by engineering anti-tumor necrosis factor (TNF) Quads that exhibit major increases in binding potency and in neutralizing TNF-mediated cytotoxicity compared to parental anti-TNF molecules. Further, Quads are amenable to fusion with different binding domains, allowing generation of novel multivalent monospecific and bispecific formats. Quads are thus a novel group of molecules that can be engineered to yield potential therapeutics with novel modalities and potencies.
Introduction
Monoclonal antibodies (mAbs) have become the dominant class of biological therapeutics since the licensing of the first monoclonal antibody, Orthoclone (OKT3), in 1986.Citation1 Concomitantly the evolution of antibody engineering has seen an even bigger transformation that has yielded a large diversity of antibody formats including bispecific antibodies.Citation2,Citation3 mAbs are generally developed as high-affinity antibodies using in vitro affinity maturation techniques as a means to overcome a barrier to in vivo affinity-maturated antibodies where they tend to have a natural affinity threshold.Citation4 In certain clinical settings where the antigen density is low or where cell-surface expressed antigen is down-regulated, the use of mAbs would potentially be limited. Clearly, the development of the next generation of antibody-based biologics would require improvement in their functional properties, such as enhancement in functional affinity and modality. As such, generation of multivalent biologicals is a promising conceptual innovation that has gained interest for their potential novel alternative modality.Citation5,Citation6 Multivalency would provide a simple approach to improve the functional affinity of antibodies through the combined binding strength of multiple binding domains known as avidity. Increased avidity is a key innovative feature of multivalent molecules that would allow for the generation of novel molecules that can surpass the antibody affinity limits without the need to engineer complex antibody libraries and perform intensive screening. Thus, multivalent molecules not only provide stronger binding to difficult or rare targets with high specificity, but they also have the potential to improve target selectivity, particularly in a cancer setting where nonspecific tumor-associated antigens are targeted.Citation7
While antibodies have been extensively engineered to generate a plethora of bispecific antibody formats,Citation3 the engineering of multivalent antibodies has been relatively slow. Despite this, a variety of engineered multivalent antibodies have been generated to date, with alteration to antibody size, shape, and valency yielding molecules with novel functionality.Citation5 The general strategy has been to generate multivalent antibodies by domain swapping, antibody domain fusion, and the use of self-assembling protein domains that structurally fall into two groups: IgG-like and non-IgG-like formats.Citation6 The use of self-assembly multimerization domains to generate multivalent antibodies using trimerization, tetramerization and pentamerization domains has been extensively studied.Citation8–Citation10 The self-assembling tetramerization domain (TD) from the tumor suppressor gene p53 allows two monomers to form a dimer via an antiparallel interaction, and two dimers interact with each other through hydrophobic and electrostatic contacts to form tetramers.Citation11,Citation12 Since the TD of p53 was defined,Citation13 the work to exploit this feature to generate multivalent antibodies has largely been restricted to the fusion of single-chain variable fragments (scFvs) to p53 TD, where the multivalent molecules were expressed in the periplasm of E. coli.Citation10,Citation14,Citation15
To extend the concept of multivalency and differentiate from previously reported multivalent antibody platforms, we report the establishment of a simple plug-and-play multivalent platform using the TD from p53 that allows self-assembly of multiple different antibody formats into tetramers with tetravalency or octavalency. These self-assembling antibody formats can be produced in high yields as soluble secreted proteins and with good purity using mammalian cell expression systems. We exemplify the simplicity and flexibility of our multivalent platform by generating an array of multivalent antibody formats, including multivalent bispecifics, termed Quads, against tumor necrosis factor (TNF, also known as TNFα). These multivalent anti-TNF Quads exhibit major improvements in binding potency and in neutralizing TNF-mediated cytotoxicity compared to the parental anti-TNF molecules. The Quad platform, therefore, provides an elegant strategy for improving preexisting mAbs and also facilitates the engineering of new Quads against different targets for a range of human clinical indications or veterinary applications.
Results
Modular design of Quads converted from anti-CD20 and anti-TNF mAbs
Previously, it was demonstrated that the TD of p53 could be used to multimerize scFv into tetramers.Citation10,Citation14,Citation15 To extend the concept of multimerization, we initially designed anti-CD20 scFvCitation16 -based Quads to compare the production of monovalent, tetravalent, and octavalent versions using the p53 TD. The simple modular design of Quads as a means to increase the binding domain valency can be seen in their molecular design and structural arrangement, as shown in Supplementary Figure S1A, B. Following expression in Expi293 cells and purification directly from culture supernatants, scFv protein purity was confirmed by SDS-PAGE (Figure S1 C). The anti-CD20 scFvs were analyzed for their ability to bind CD20 by indirect ELISA on CD20-coated plates. A dose-dependent binding to CD20 was observed where a stepwise increase in CD20 binding strength could also be seen with increasing binding domain valency. Of particular note, the octavalent anti-CD20 scFv Quad showed significantly higher binding strength to CD20 (>9 times) than the monovalent anti-CD20 scFv when half-maximal binding strengths are compared (Supplementary Figure S1D).
To further exemplify the modular design of Quads and to compare the potency of Quad proteins with parental IgG antibody, four groups of Quads were designed using different binding domains with specificity for anti-TNF (, Supplementary Figure S2). The first group of Quads was based on adalimumab (Humira®),Citation17 an anti-TNF IgG human monoclonal antibody. Two versions of Humira-based Quads were designed. The first version used the antigen-binding fragment (Fab) of Humira, with or without the Fc region, ensuring that the structural pairing of the variable heavy chain (HC) and variable light chain (LC) was retained in its native configuration. The TD was linked to the C-terminus of the CH1 domain in the Fab version without Fc (Humira Fab-TD) (illustrated in Supplementary Figure S2D). In the Fab version with Fc, the Fc region was devoid of the core hinge region effectively generating a monomeric Ig (mIg) where the TD was linked to the C-terminus of the Fc region (Humira Fab mIg-TD) (illustrated in Supplementary Figure S2E). The second version of Humira involved reformatting the variable HC and LC of Humira into an scFv (Humira scFv-TD) (illustrated in Supplementary Figure S2 F).
Figure 1. Schematic representation of plasmids used to transfect Expi293 cells for the production of benchmark and multivalent anti-TNF Quad proteins. Poly-histidine tag (6xHis) was included in each construct at the C-terminus except for where a second kappa light chain is used in conjunction with a first chain, which contained the his-tag. The * denotes Fc region lacking the core hinge region.
A second group of Quads was based on anti-TNF nanobody (referred to henceforth as dAb) from ozoralizumab. Two versions of anti-TNF dAbs were designed either with or without Fc region. Since dAbs are small in size (~13 kDa) and are highly versatile to engineering,Citation18 anti-TNF dAbs were designed to generate tetramers with either tetra- or octa-valent valences. The version without Fc region either had a single dAb linked to the N-terminus of the TD (TNF dAb-TD) or two dAbs linked at either end of the TD (TNF dAb-TD-dAb) yielding tetravalent and octavalent Quad molecules, respectively (Supplementary Figure S2 H, I). TNF dAb version with Fc was similarly designed to have either tetra- or octa-valency (Supplementary Figure. S2 J, K). In the tetravalent Fc version, TNF dAb was linked to Fc without core hinge where the TD was linked at the C-terminus of the Fc region (TNF dAb mIg-TD). The octavalent version included a second chain where TNF dAb was linked to kappa LC constant region (dAb-kappa). Co-expression of TNF dAb mIg-TD with TNF dAb-Kappa allowed octavalent dAb Quad expression (TNF dAb mIg-TD/dAb-Kappa).
A third group of Quads was designed to examine the expression of multivalent bispecific Quads in a unique 4 + 4 octavalent format. The binding domains of antibodies for TNF and interleukin 17a (IL17a), either as dAb or scFv, were used to produce three formats of multivalent bispecific Quads (Supplementary Figure S2 L-N). The first version was a dAb-TD-dAb version where the binding domain for both TNF and IL17a was dAbs (dAb sequences obtained from CN103547592A and WO2010/142551A2, respectively). In the second and third versions, the TNF binding arm was Humira scFv and the IL17a binding arm was an scFv that we reformatted from ixekizumab in the second version (scFv-TD-scFv) and a dAb in the third version (scFv-TD-dAb). In the fourth group of Quads, the extracellular portion of the TNF receptor-2 (TNFR2) was linked to Fc region lacking CH1 region, similar to etanercept, an inhibitor of TNF.Citation19 Etanercept mIg-TD was designed by linking the TD at the C-terminus of the Fc region.
Production of multivalent anti-TNF Quads
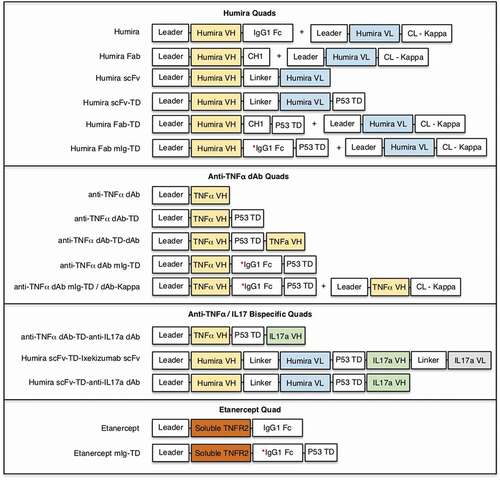
Soluble expression of the different anti-TNF multivalent Quad formats (Quad constructs illustrated in ) was examined following transfection in Expi293 cells. Secreted soluble Quad proteins were harvested directly from the culture supernatant and were purified by His-tag affinity chromatography in a single-step purification process. Expression and purity of the Quad proteins were confirmed by denaturing SDS-PAGE performed under non-reducing conditions (). Strikingly, all Quad proteins expressed as highly pure proteins and with the expected molecular weight, with the only exception being etanercept and Etanercept mIg-TD, which exhibited apparent molecular masses greater than the calculated molecular masses (approximately 104 kDa and 56 kDa, respectively). This increase in molecular mass is likely a result of O-linked glycosylation reported for etanercept.Citation20 The overall Quad expression yields were generally high and comparable to expression levels of IgG antibodies (Supplementary Table S1).
Figure 2. Characterization of secreted multivalent Quad proteins. Soluble expression and purity of the different groups of anti-TNF Quad proteins were demonstrated by SDS-PAGE analysis. Description and size of the protein monomers are indicated (a). The multimeric state and purity of four different Quad formats were further analyzed by size-exclusion chromatography, which included (b) Humira scFv-TD, (c) Humira Fab mIg-TD, (d) anti-TNF dAb-TD-dAb and (e) anti-TNF dAb-TD-IL17a dAb. Proteins eluted from the single main peaks were re-analyzed on SDS-PAGE under reduced conditions to further confirm the authenticity of the Quad proteins. Quad proteins are shown in blue lines and the monovalent control proteins in red.
Assessing the oligomeric state of Quad proteins
The native molecular mass and oligomeric state of the Quad proteins was analyzed by size-exclusion chromatography (SEC). All SEC profiles had a clear dominant peak, suggesting the protein samples were suitably homogenous and aggregates were absent (e). The Humira scFv and Humira scFv-TD proteins eluted in peaks corresponding to the expected molecular weights for monomers and tetramers, respectively (). The Humira Fab-mIg-TD protein, eluted at a volume consistent with a molecular weight of 315.8 kDa (). Gel filtration of TNF dAb and TNF dAb-TD-dAb also gave peaks that clearly defined monomeric and tetrameric species, respectively (). The TNF dAb-TD-IL17 dAb eluted at a volume appropriate for a protein of 122.8 kDa (). Protein eluted from each major peak was re-analyzed by SDS-PAGE under reducing conditions to confirm the authenticity of the Quad proteins. These data confirm the integrity of the Quad proteins as highly pure multimeric proteins.
Surface plasmon resonance and SAXS analysis of tetrameric Quads
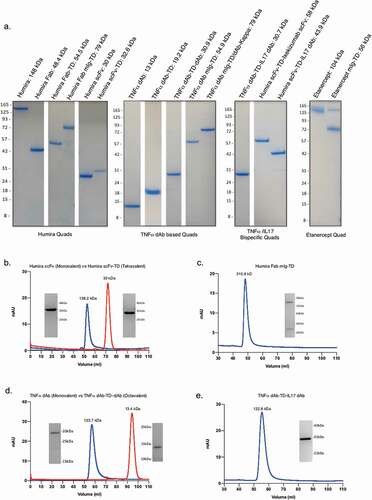
A surface plasmon resonance (SPR) assay was performed to confirm binding of our Humira scFv-TD to TNF, comparing it to Humira itself and an scFv engineered from Humira. The proteins were immobilized on the chip and recombinant TNF passed as the ligand (). TNF bound to all immobilized proteins with very high affinity. The very slow off rate was at the limit of accurate curve fitting for kinetic analysis. A reciprocal assay, in which TNF was immobilized as the ligand, was performed (). A summary of the Rmax values for Humira, Humira scFv, and Humira scFv-TD is shown in Supplementary Table 2.
Figure 3. SPR sensograms of Humira, Humira scFv-TD, and Humira scFv to TNF using immobilized Quad proteins (a) or immobilized recombinant TNF protein (b). SAXS analysis developed molecular envelopes and representative models of tetrameric and octameric proteins (Panels C and D). (c) Model of chimeric molecule CD7 scFv-TD (purple) fitted into the experimentally determined scattering envelope. The model suggests the chimeric molecule adopts a cruciform structure in good agreement with the envelope. (d) Model for construct CD20 scFv-TD-scFv (magenta) fitted to an experimentally determined scattering envelope, which again suggests a cruciform structure is adopted by the chimeric molecule.
The structures of Quad scFv-TD (tetravalent) and scFv-TD-scFv (octavalent) proteins were examined using small-angle X-ray scattering (SAXS);Citation21,Citation22 models of the two proteins are shown in ,d. Scattering curves of anti-CD7 scFv-TD and anti-CD20 scFv-TD-scFv were obtained and Geinier analysis of the data and demonstrated the proteins were pure and homogenous in solution. Further analysis (Kratky plots) revealed both samples to be globular with some degree of flexibility between domains. The molecular envelope calculated for the anti-CD7 scFv-TD construct (tetravalent) had a dumbbell shape, consisting of two large lobes that taper toward the center of the molecule (). A representative model, whereby scFv was fused to the C-terminus of the p53 TD has a cruciform structure and fits well into the molecular envelope with the scFv domains occupying the lobes and the smaller TD at the center of the molecule, coincident with the tapering of the envelope. The large size of the lobes relative to the model indicates the flexibility of the construct with the scFv domains free to rotate relative to the TD and each other. The envelope calculated for the anti-CD20 scFv-TD-scFv construct (octavalent) is again dumbbell shaped, but with a less pronounced tapering at the center, due to the increased steric bulk of placing scFv domains at both the N- and C-termini of the TD (). A molecular model generated for this construct fits well into the envelope, where again the increased size of the lobes relative to the model indicated the rotational freedom afforded each scFv domain.
Enhanced binding of multivalent anti-TNF Quads
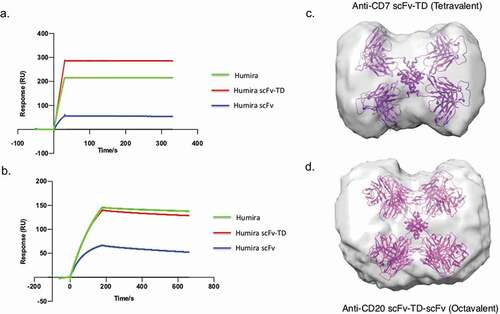
Anti-TNF Quads binding to TNF was analyzed by indirect ELISA to determine binding characteristics, but also to assess if antigen-binding potential was hampered, particularly when the valency goes beyond tetravalent. All anti-TNF Quads bound TNF in a dose-dependent manner confirming Quads were assembled as anticipated (). As expected, a stepwise increase in TNF binding strength with increasing binding domain valency was observed. For example, a clear increase in binding strength can be seen with Quads with increasing Humira Fab binding arms, as exemplified by Humira Fab (monovalent), Humira (bivalent) and Humira Fab-TD and Humira Fab mIg-TD (tetravalent) (). Interestingly, the TNF binding potential between the octavalent version of TNF dAb and its tetravalent version could not be resolved in these ELISA-based binding assays (). Similarly, only a small increase in TNF binding strength was observed for the octavalent version of TNF dAb mIg compared to tetravalent mIg version (). This may be because the tetramethylbenzidine-based colorimetric signal is rapidly saturated by these multivalent Quads, and thus the dynamic detection range for this ELISA binding assay is not adequate to differentiate the enhanced binding strength beyond a certain point.
Figure 4. Analysis of antigen-binding properties of anti-TNF Quads. Indirect binding ELISA was used to assess anti-TNF Quad binding to its cognate antigen (TNF). Anti-TNF Quads were analyzed in groups as follows (a) Humira-based Quads (b) anti-TNF dAb Quads without Fc (c) anti-TNF dAb Quads with Fc (d) bispecific anti-TNF/IL17a Quad formats (e). Monospecific anti-TNF dAb vs bispecific anti-TNF dAb and (f) Etanercept-based Quad (n = 2 ± SEM).
The three different bispecific Quad formats containing anti-TNF and anti-IL17a binding domains in either dAb/dAb, scFv/scFv or scFv/dAb configuration, respectively, can all bind TNF with similar binding strength to each other (). The anti-TNF dAb/dAb bispecific Quad version was used to analyze TNF binding compared to the monospecific anti-TNF dAb-TD version. The monospecific and bispecific anti-TNF dAb Quads were able to bind TNF with similar binding strength, confirming that the presence of a second binding arm in the bispecific Quad format does not hamper Quad binding to TNF (). Increase in binding strength to TNF with additional antigen-binding domain was also found to be true for Etanercept mIg-TD (tetravalent) compared to etanercept (bivalent) (). This increase in binding indirectly confirmed that the soluble TNFR2 binding domain in the Quad format multimerized into a stable multimer.
Engineered antibodies often use peptide linkers to provide flexibility and stability,Citation23 and thus the effect of including a peptide linker on antigen binding by Quads was also analyzed. The Quad molecules made in the presence or absence of a (G4S)4 peptide linker placed between the TD and the antibody binding domains of Humira scFv and Humira Fab Quads had similar binding properties when tested in ELISA binding assays (Supplementary Fig. S3A, B). These Quad molecules also had similar inhibitory effects in WEHI cell killing assays (see below, Supplementary Fig. S3 C, D). We conclude that adding a linker to our anti-TNF Quad proteins does not appear to improve their ability to bind TNF.
Multivalent Quads neutralize TNF-mediated cytotoxicity in WEHI cells
Due to the inherent drawbacks of ELISA to resolve the difference in avidity of the multivalent Quad molecules beyond tetravalency, we further analyzed Quad molecules in a cell-based cytotoxicity assay to determine more accurately the effect increased avidity has on potency to neutralize the effects of soluble TNF. The WEHI cell-based bioassayCitation24 was used to determine EC50 for neutralization of TNF-mediated cytotoxicity of the different anti-TNF Quads (). Overall, we observed a significant increase in potency when we employed Quad molecules containing increasing anti-TNF binding domains (EC50 values summarized in ). For example, although only a ~ 2 fold increase in potency was seen between Humira Fab (monovalent) and Humira IgG (bivalent), we saw a significant difference in TNF neutralization potency when compared to the tetravalent Humira Fab Quad formats. As an example, Humira Fab mIg-TD was found to be >15x more potent than Humira and >31x more potent than the monovalent Humira Fab control (). A difference in potency between the tetravalent Humira Fab Quad formats was seen, and this could be attributed to the Fc region in the mIg version, which spatially separates the binding domains from the multimerization domain allowing the Humira Fab mIg-TD version to bind TNF more efficiently. Although an increase in neutralization potency was observed with Humira scFv-TD when compared to the monovalent control (Humira-scFv) (), the effects were less pronounced than that seen with Quads containing the Fc region.
Table 1. A summary of the EC50 values of anti-TNF molecules to neutralize TNF-mediated cytotoxicity in WEHI 164 cells treated with 0.1 ng/ml of human TNF.
Figure 5. Functional characterization of anti-TNF Quad proteins. Anti-TNF Quad molecules were analyzed for their ability to neutralize TNF-mediated cytotoxicity in WEHI cells. The neutralization curves were plotted in different groups according to their format (n = 3 ± SEM). Benchmark antibodies and monovalent control were also included. The groups included Humira-based Quads (a), Humira-based scFv (b), anti-TNF dAb Quads without Fc (c) anti-TNF dAb Quads with Fc (d), bispecific anti-TNF/IL17a Quad formats (e), monospecific anti-TNF dAb vs bispecific anti-TNF dAb (f) and Etanercept-based Quad (g).
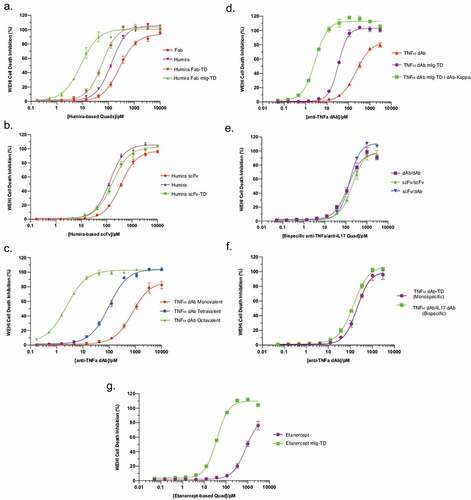
The TNF neutralization potency dramatically increased for TNF dAb Quads, with increasing TNF dAb binding domains (). TNF dAb-TD (tetravalent) and TNF dAb-TD-dAb (octavalent) Quads were approximately 8x and >317x more potent, respectively, than the monovalent TNF dAb control. A similar dramatic increase in potency was observed for the tetravalent and octavalent TNF dAb mIg Quad versions, with potencies >22x and >271x more potent than the monovalent TNF dAb control, respectively (). The octavalent TNF dAb-TD and TNF dAb mIg-TD versions were highly potent, with potencies approximately 55x and 47x more potent, respectively, than Humira benchmark antibody.
The bispecific Quads were all tetravalent for TNF, and as expected, no significant difference in their potencies could be seen in the WEHI cell bioassay (). Furthermore, no significant difference in potency could be seen between the dAb/dAb bispecific TNF and monospecific TNF Quad versions, confirming that the TNF binding arm in the bispecific Quad format was not hampered by the presence of a second antigen (IL17a) binding arm ().
The Quad monomeric-Ig form of etanercept showed a significant increase in TNF neutralization potency compared to etanercept. By increasing the binding valency from 2 to 4 in the Quad format, a 22-fold increase in potency was gained compared to etanercept (). These data support the postulate that an increase in avidity not only increases the overall affinity, but also significantly increases potency.
Discussion
One of the major rate-limiting steps in drug development is the validation of novel therapeutic targets. As well as novel target discovery, a parallel approach is required to develop more efficacious drugs against existing targets where current drugs fail to reach optimal therapeutic effect. In this study, we adapted the tetramerization domain of p53 to vastly extend the multimerization concept to allow generation of an array of novel multivalent antibody formats (). This was made possible through the demonstration of the flexibility and tolerability of p53 TD to N- and C-terminus fusion to different antigen binding domains such as dAbs, scFv, Fabs, and extracellular protein domains. Using the p53 TD, we demonstrated the generation of octavalent scFv and dAbs and also bispecific Quads in a unique 4 + 4 configuration. Through SEC analysis and more specifically SAXS structural analysis, the tetrameric structural integrity of different Quad formats was confirmed. These molecules can be made in high purity (>95%), suggesting a similar approach can be used to generate Quads in a binding domain agnostic manner and against any protein target of interest.
The Quad technology has several key attributes making it a platform for generating the next generation of multivalent antibody therapeutics. The simplicity and flexibility by which multivalent antibodies can be generated are key features. The initial plasmid construct generation requires only a single polypeptide monomer species containing the antibody binding domain(s) linked to the TD. Upon expression, Quads are generated through the self-assembly of individual monomers into stable tetramers as secreted soluble protein that can be isolated in high purity directly from culture supernatant (). Given that Quads are composed of fully human sequences where the TD is embedded in a central core, we do not anticipate Quads would be immunogenic, but further studies would be required to investigate this further. Furthermore, Quad expression in mammalian cells is scalable with protein yields similar to, or better than, those of mAbs (Supplementary Table 1). We show that the p53 TD is highly versatile, and this allows modular design of Quads into different antibody formats allowing simple plug-and-play design. The simple modular design to generate different Quads from monomeric building blocks can be seen in the example using anti-CD20 scFv (Supplementary Figure S1). Most importantly, antibodies where the variable heavy (VH) and variable light (VL) chains are naturally paired can also be converted into Quads in a manner in which the native VH and VL pairing is kept intact without effecting its antigen-binding properties.
As exemplification of the power of the tetramerization technology, we have extended the utility and potency of the clinically employed anti-TNF mAb Humira, by incorporating the VH and VL sequences of Humira into the Quad format. A modular plug-and-play strategy was used to generate multiple different Quad formats of anti-TNF either as Ig-like or non-Ig-like formats. The Ig-like Quad molecules were designed as monomeric Ig (mIg) by removing the core hinge region and we demonstrated that mIg-like Quads could be generated as stable tetramers with high purity (). These mIg formats provide the flexibility to limit the size of Quads without hampering the normal functions of Fc region to engage effector cells and undergo neonatal Fc receptor-mediated recycling.
The varied format options generated and analyzed in this study present their own unique attributes and characteristics, such as varying size, shape, affinities, valences, specificity, and effector function (Supplementary Figure S2). The flexibility to modulate Quads with these characteristics will provide substantial benefits to tailor Quads around a given target biology. Particularly in a cancer setting where one size does not fit all, having the choice from an array of multivalent Quad molecules with the flexibility to modulate format is an advantage in its own right. An example would be targeting the tumor microenvironment in solid cancers where molecules with large size would affect tissue penetration.Citation25
Multiple different anti-TNF Quad formats were generated, of which the Humira Fab and anti-TNF dAb-based Quads were of particular interest to demonstrate the superior potency of the Quad technology. As expected, the increase in binding domain valency increased Quad-binding strength to TNF when binding of Quads was compared to each other and to Humira in ELISAs with immobilized TNF (). Similarly, in the in vitro TNF cell-based neutralization assays, we saw significant improvements in efficacy between Quads and the parental anti-TNF molecules (), indicating the enhanced avidity improved TNF binding and neutralization potential of Quads. Humira Fab mIg-TD was strikingly potent (EC50 8.5 pM), making it >15x more potent than Humira. Interestingly, Humira Fab mIg-TD was almost 7x more potent than Humira Fab-TD even though both formats were tetravalent, containing four copies of Humira Fab. This suggests the structural configuration of the binding domain and the molecule size is both important features. The mIg-TD version also enhances molecule flexibility, which is equally, if not more, important than size.
A stepwise increase in TNF neutralization potency was evident between the tetra- and octa-valent anti-TNF dAb Quads both in the non-Ig-like and mIg-like formats (,d). Particularly the TNF neutralization potencies for the octavalent Quad versions (non-Ig-like and mIg-like formats) were extremely high, with EC50 of 2.4 pM and 2.8 pM, respectively, making them ~55x and ~47x more potent than Humira. It is noteworthy that in the WEHI in vitro bioassay, measurement of potencies was solely based on the neutralization of soluble TNF. However, in an in vivo setting, transmembrane bound TNF would also play an important role in cell cytotoxicity.Citation26 As such, determination of the true potencies of these multivalent anti-TNF Quad molecules would require further investigation in an in vivo setting.
The capacity to generate Quads with significantly enhanced functional affinity and potency surpassing that of the parental antibody presents several advantages that could be applied to the development of Quads as novel therapeutics. Targeting TNF is an example where Quads potentially can be used to repurpose this target in indications where mAbs have failed, such as in treating patients with sepsis.Citation27 The novel bispecific Quad formats with 4 + 4 binding configuration could further provide interesting opportunities in different clinical settings. For example, simultaneous targeting of two antigens with enhanced avidity and potency would be particularly beneficial in settings where antigen escapes through down-regulation is the common mechanism of escape. The potential clinical use of tetrameric or octameric Quads can be pursued in many diverse indications where potency is key. Two obvious areas are novel bispecific antibodies in immune-oncology, as well as super neutralizers of viruses. Further, given the substantial gain in potency, it could be envisioned that Quads can open novel treatment modalities for much smaller effective antibody doses and transform many current intravenous infusion medicines to future subcutaneous applications.
The work described here highlights some of the advantages the Quad technology can offer such as flexibility, modularity, and enhanced functionality. This sets the stage for further investigating the superior potency of the anti-TNF Quads developed in this study and further engineering Quads with novel modalities such as multispecific formats beyond bispecifics (e.g., tri- and tetra-specific Quads).
Materials and methods
Quad sequences and plasmid construction
All Quad constructs were designed using SnapGene viewer version 4.3.10 containing a 19 amino acid signal peptide of the Ig-heavy chain of a rat mAb against human CAMPATH-1Citation28 and a poly-histidine tag (6xHis) linked at the C-terminus. All sequences were synthesized and cloned into an expression vector by Twist Bioscience (California). Amino acid sequences of the mature peptide of the Quads used in this study can be found in Supplementary Figure S4. Large plasmid preps of Quad vectors were made using EndoFree plasmid Maxi kit (Qiagen).
Expression of Quad proteins in Expi293 F cells
Expi293 F™ cells (Thermo Fisher Scientific) were cultured in Expi293™ Expression Medium (Thermo Fisher Scientific) according to the manufacturer’s recommendations. The only exception was that 5% CO2 was added directly to the flasks when the cells were split and non-vented caps were used. Two methods involving different transfection reagents were utilized for protein expression. The methods for 30 ml cultures are described here, but the protocol was scaled up or down according to experimental requirements.
For PEI transfections the cells were counted 1 day prior to transfection using a NC-3000™ (ChemoMetec) and were diluted to 1.5 × 106 cells/ml using Expi293™ Expression Medium. The cells were cultured in 5% CO2 at 37°C, 125 rpm overnight. The following day the cells were counted, spun down for 5 min at 1000 rpm and resuspended at 2 × 106 cells/ml in 30 ml of fresh media. Thirty-three ug of plasmid DNA was added to 900 ul media and 90 ul of PEI Max (Polysciences Inc.) was added to 900 ul media. The DNA and transfection reagent samples were mixed and incubated at room temperature for 15 min. The DNA/transfection reagent mixture was added to the cells, which were cultured as before and incubated for a further 72 h.
For transfections with Expifectamine™ 293 Reagent (Thermo Fisher Scientific), the cells were also diluted to 1.5 × 106 cells/ml in Expi293™ Expression Medium 1 day prior to transfection. On the day of transfection, the cells were centrifuged and resuspended at 2.5 × 106 cells/ml in 30 ml of fresh media. Two tubes containing 1.5 ml of Gibco™ Opti-MEM™ (Thermo Fisher Scientific) were prepared. Thirty ug of plasmid DNA was added to one tube and 80 ul of Expifectamine was added to the other. The solutions were mixed and incubated at room temperature for 30 min. The DNA-transfection reagent complex was added to the cells, which were cultured in 5% CO2 at 37°C, 125 rpm. Following 16–18 hours incubation, transfection enhancers 1 and 2 were added to the cells according to the manufacturer’s protocol. The cells were incubated for a further 5–8 days.
Affinity protein purification
The cells were harvested by centrifugation for 10 min at 4000 rpm. The ~30 ml supernatant was filtered through a 0.22 μm filter and diluted to 50 ml with binding buffer (50 mM HEPES, pH 7,4, 250 mM NaCl, 20 mM imidazole) containing Complete™ EDTA-free protease inhibitors (Roche) to facilitate binding to the column. A 1 ml HisTrap™ HP column (GE Healthcare) was connected to an ÄKTA Start (GE Healthcare) and pre-equilibrated with binding buffer. The protein-containing media were loaded onto the column using a flow rate of 1 ml/min. The column was washed with >10x column volume of binding buffer before the protein was eluted using a 20–300 mM imidazole gradient over 12 ml. 0.5 ml fractions were collected and analyzed by SDS-PAGE. Protein containing fractions were pooled and concentrated using Amicon® Ultra centrifugal filter units (Millipore).
Following affinity chromatography the proteins were either snap frozen and stored at −80°C, dialyzed into an alternative buffer for a specific application or gel filtrated to assess the molecular weight of the various antibody formats. For the latter analyzes, protein samples were concentrated to 1.5–2 ml and subjected to gel filtration on a Superdex 75 16/600 column (GE Healthcare) using 10 mM HEPES, pH 7.4, 250 mM NaCl.
Protein analysis
Purified protein samples were separated on SDS-PAGE under non-reducing denaturing conditions. Typically, 1 μg samples of the purified proteins were loaded on to the gel alongside 5 μl of Prestained 10–245 kDa Protein Ladder. The gels were run in Tris-Glycine buffer containing 0.1% SDS. A constant voltage of 150 V was used and the gels were run for ~70 min until the dye front migrated fully.
Size-exclusion chromatography
The Quad proteins were concentrated for gel filtration analysis, typically to 1 mg/ml using Amicon® Ultra centrifugal filter units (Millipore). The proteins were purified using an ÄKTA Avant chromatography system (GE Healthcare), with a HiLoad 16/600 Superdex 75 pg prepacked column (GE Healthcare) at a flow rate of 1 ml/min. The size and molecular weight of the proteins were determined by calibration of the column with a gel filtration standard (Bio-Rad). Purification of Humira scFv and Humira scFv proteins were subjected to gel filtration using 10 mM HEPES, pH 7.4, 250 mM NaCl, while Humira Fab-mIg-TD, TNF dAb, TNF dAb-TD-dAb, and TNF dAb-TD-IL17 dAb used phosphate-buffered saline (PBS), pH 7.4. Protein containing fractions were analyzed by SDS-PAGE.
Surface plasmon resonance
Humira, Humira scFv-TD, and Humira scFv preparations were immobilized onto a CM5 sensor chip (GE Healthcare) using standard amine coupling on a Biacore T200 Instrument (GE Healthcare). The surfaces were activated for 5 min with 0.2 M N-ethyl-N-(dimethylaminopropyl) carbodiimide hydrochloride and 0.05 M N-hydroxysuccinimide at 20 μl/min, before the proteins were injected at 20 μg/ml in 10 mM sodium acetate, pH 4.5. The surfaces were blocked with a 5-min injection of 1 M ethanolamine, pH 8.5 at 20 µl/min. The immobilization levels of Humira, Humira scFv-TD, and Humira scFv were 16500, 8400, and 1100 RU, respectively. The running buffer HBS-EP (GE Healthcare), comprising 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20. Recombinant Human TNF (ab9649, Abcam) was diluted in running buffer to a concentration of 57 nM. For each analysis, three start up cycles were performed before a single-cycle kinetics high-performance run was implemented with a contact time of 30 sec and a dissociation time of 300 sec, at a flow rate of 30 µl/min. The sensorgrams were double referenced with an empty reference cell that had been through the same activation and deactivation cycles, and a buffer only injection. Binding data were collected at 37°C. The BIAevaluation 2.1 software (GE Healthcare) was used to analyze the data. Curve fitting to the association and dissociation phases was used to calculate the on and off rate and the Kd of the interaction was calculated from these values. The experiment was repeated with recombinant human lymphotoxin alpha1/beta2 (R&D Systems), at a concentration of 87 nM.
In an alternative experiment, to measure the avidity and stoichiometry of binding, recombinant human biotinylated TNF (ab167747, Abcam) or biotinylated KRAS (as a reference molecule) were immobilized on a SA chip (GE Healthcare). Briefly, biotinylated TNF, diluted to 0.20 µg/ml in HBS-EP, and biotinylated KRAS, diluted to 0.36 µg/ml in HBS-EP, were injected at a flow rate of 20 µl/min to a target response of 200 RU over the conditioned chip. Humira, Humira scFv-TD, and Humira scFv were diluted to 0.4 µg/ml and were passed over the chip for 180 s at 30 µl/min, with a dissociation time of 8 min, at 37°C. Ten mM glycine pH 1.7 was required for regeneration of the chip, but also denatured the ligand, so biotinylated TNF and biotinylated KRAS had to be re-immobilized prior to injection of each test molecule. Double-referenced sensorgrams were used for analysis on the BIAevaluation 2.1 software.
Small-angled X-ray scattering
All SAXS data were collected at beamline B21 (Diamond Light Source). Forty µl volumes of each sample, at concentrations of 1, 0.5, and 0.1 mg/ml in PBS buffer, were loaded into a quartz capillary cell using the sample-handling robot at the beam-line. For each sample 28 individual scattering curves were collected at a rate of 1 per second, under flow mode to minimize radiation damage, using an EIGER 4 M detector. Buffer curves for background correction were collected under identical conditions. All scattering data were collected at room temperature using x-rays with a wavelength of 1 Å. Manipulation and analysis of the scattering curves were performed using the SCÅTTER software suite (www.BIOISIS.net). Molecular envelopes were calculated using scattering data from samples at 0.5 mg ml−1 using DAMMIN/DAMMIFCitation29 with each envelope representing the average of 13 independent runs. Representative models for CD7 scFv-TD and CD20 scFv-TD-scFv were manually constructed within cootCitation30 using PDB files 1c26 (P53) and 5c6 w (IG HV1 domain) as building blocks. The resultant model was fitted to the molecular envelope using tools in Chimera.Citation31
Indirect ELISA
ELISAs were performed in duplicates to compare the binding affinities of the different antibody formats. Recombinant Human TNF (ab9649, Abcam) was diluted to 1 ug/ml in ELISA coating buffer (50 mM carbonate/bicarbonate). One hundred ul of 1 ug/ml TNF was added to each well of an ELISA plate and the plates were incubated overnight at 4°C. Replicate plates were set up containing coating buffer only (no TNF) as negative controls.
The plates were washed three times with PBS containing 0.05% Tween-20 before being blocked with 200 ul 5% bovine serum albumin in PBS for 4 h at room temperature. The plates were washed three times as before. Quad proteins were serially diluted in PBS containing 0.05% Tween-20. One hundred ul of sample was added to each well and the plates were incubated overnight at 4°C. The plates were washed four times with PBS containing 0.05% Tween-20. One hundred ul of detection antibody (anti-His-HRP, A7058, Sigma; or anti-Human-IgG HRP, 31410, Thermo Fisher Scientific; or Protein L HRP, M00098, Genscript) diluted in blocking buffer (according to the manufacturers’ recommendations) was added to each well and the plates were incubated at room temperature for 2 h. Following four plate washes, 25 ul of TMB substrate solution (Thermo Fisher Scientific) was added to each well. The reaction was terminated after ~15 min by the addition of 25 ul 3 M HCl. The absorbance at 450 nm was read using a CLARIOstar microplate reader (BMG Labtech).
WEHI functional bioassay
WEHI-13VAR cells (ATCC), which are highly-TNF sensitive, were employed to assess the bioactivity of the different antibody formats. We examined the neutralization activities of anti-TNF Quad proteins. WEHI-13VAR cells were seeded at 1 × 104 cells per well in a 96-well plate in RPMI-1640, 10% fetal bovine serum and incubated overnight at 37°C, 5% CO2. The media were aspirated from the cells and replaced with media containing 2 ug/ml actinomycin D, 0.1 ng/ml recombinant human TNF (ab9649, Abcam) and 0–2400 pM of Quad proteins. The samples were set up in triplicates with no TNF and no antibody controls. The cells were incubated under standard culture conditions for a further 20–22 hours.
To assess cell viability, ATP generated by metabolically active cells was quantified using the CellTiterGlo Luminescent Cell Viability Assay (Promega) according to the manufacturers’ instructions. Luminescent signals were measured using a CLARIOstar microplate reader (BMG Labtech). The luminescence signals obtained from the compound treated cells were normalized against the media only controls.
Disclosure of potential conflicts of interest
HA, AM and THR are shareholders in Quadrucept Bio Ltd. HA and THR are named inventors on patent applications covering multimerization technology.
Supplemental Material
Download Zip (1.5 MB)Acknowledgments
We are indebted to Dr. Chris Paluch for expert help with the SPR analysis and for discussion on the interpretation of the data. We would also like to thank Diamond Light Source for beam-time (proposal mx12346) and staff at beamlines B21 for assistance during x-ray data collection. AM was supported by Bloodwise Programme Grant (Grant No.12051).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Urquhart L. Top drugs and companies by sales in 2018. Nat Rev Drug Discov. 2019. doi:https://doi.org/10.1038/d41573-019-00049-0.
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–11. doi:https://doi.org/10.1016/j.molimm.2015.01.003.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212. doi:https://doi.org/10.1080/19420862.2016.1268307.
- Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: A threshold, a ceiling, and the importance of off-rate. Immunity. 1998;8:751–59. doi:https://doi.org/10.1016/S1074-7613(00)80580-4.
- Nuñez-Prado N, Compte M, Harwood S, Álvarez-Méndez A, Lykkemark S, Sanz L, Alvarez-Vallina L. The coming of age of engineered multivalent antibodies. Drug Discov Today. 2015;20:588–94.
- Cuesta ÁM, Sainz-Pastor N, Bonet J, Oliva B, Álvarez-Vallina L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010;28:355–62. doi:https://doi.org/10.1016/j.tibtech.2010.03.007.
- Slaga D, Ellerman D, Lombana TN, Vij R, Li J, Hristopoulos M, Clark R, Johnston J, Shelton A, Mai E, et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci Transl Med. 2018;10:eaat5775. doi:https://doi.org/10.1126/scitranslmed.aat5775.
- Fan C-Y, Huang -C-C, Chiu W-C, Lai -C-C, Liou -G-G, Li H-C, Chou M-Y. Production of multivalent protein binders using a self-trimerizing collagen-like peptide scaffold. Faseb J. 2008;22:3795–804. doi:https://doi.org/10.1096/fj.08-111484.
- Zhu X, Wang L, Liu R, Flutter B, Li S, Ding J, Tao H, Liu C, Sun M, Gao B, et al. COMBODY: one-domain antibody multimer with improved avidity. Immunol Cell Biol. 2010;88:667–75. doi:https://doi.org/10.1038/icb.2010.21.
- Liu M, Wang X, Yin C, Zhang Z, Lin Q, Zhen Y, Huang H. Targeting TNF-α with a tetravalent mini-antibody TNF-TeAb. Biochem J. 2007;406:237–46. doi:https://doi.org/10.1042/BJ20070149.
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–82. doi:https://doi.org/10.1146/annurev.biochem.77.060806.091238.
- Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 1995;267:1498–502. doi:https://doi.org/10.1126/science.7878469.
- Kraiss S, Quaiser A, Oren M, Montenarh M. Oligomerization of oncoprotein p53. J Virol. 1988;62:4737–44. doi:https://doi.org/10.1128/JVI.62.12.4737-4744.1988.
- Rheinnecker M, Hardt C, Ilag LL, Kufer P, Gruber R, Hoess A, Lupas A, Rottenberger C, Plückthun A, Pack P, et al. Multivalent antibody fragments with high functional affinity for a tumor-associated carbohydrate antigen. J Immunol. 1996;157:2989–97.
- Willuda J, Kubetzko S, Waibel R, Schubiger PA, Zangemeister-Wittke U, Plunckthun A. Tumor targeting of Mono-, Di-, and tetravalent anti-p185HER-2 miniantibodies multimerized by self-associating peptides. J Biol Chem. 2001;276:14385–92. doi:https://doi.org/10.1074/jbc.M011669200.
- Wu AM, Tan GJ, Sherman MA, Clarke P, Olafsen T, Forman SJ, Raubitschek AA. Multimerization of a chimeric anti-CD20 single-chain Fv-Fc fusion protein is mediated through variable domain exchange. Protein Eng. 2001;14:1025–33. doi:https://doi.org/10.1093/protein/14.12.1025.
- AbbVie. Humira (adalimumab) injection, for subcutaneous use; 2015. http://www.rxabbvie.com/pdf/humira.pdf.
- Beghein E, Gettemans J. Nanobody technology: A versatile toolkit for microscopic imaging, protein-protein interaction analysis, and protein function exploration. Front Immunol. 2017;8:1–14. doi:https://doi.org/10.3389/fimmu.2017.00771.
- Reddy SP, Shah VV, Lin EJ, Wu JJ. Etanercept. In: Wu J, Feldman S, Lebwohl M, editors. Therapy for severe psoriasis. Philadelphia (PA): Elsevier. 2016. p. 83–96.
- DiPaola M, Li J, Stephens E. Development of biosimilars: analysis of etanercept glycosylation as a case study. J Bioanal Biomed. 2013;5:180–86. doi:https://doi.org/10.4172/1948-593X.1000096.
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40(3):191–285. doi:https://doi.org/10.1017/S0033583507004635.
- Kikhney AG, Svergun DI. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015;589:2570–77. doi:https://doi.org/10.1016/j.febslet.2015.08.027.
- Chen X, Zaro JL, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–69. doi:https://doi.org/10.1016/j.addr.2012.09.039.
- Espevik T, Nissen-Meyer J. A highly sensitive cell line, WEHI 164 clone 13, for measuring cytotoxic factor/tumor necrosis factor from human monocytes. J Immunol Methods. 1986;95:99–105. doi:https://doi.org/10.1016/0022-1759(86)90322-4.
- Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer. 2007;109:170–79. doi:https://doi.org/10.1002/cncr.22402.
- Lim KJ, Lee SJ, Kim S, Lee SY, Lee MS, Park YA, Choi EJ, Lee EB, Jun HK, Cho JM, et al. Comparable immune function inhibition by the infliximab biosimilar CT-P13: implications for treatment of inflammatory bowel disease. J Crohns Colitis. 2017;11:593–602. doi:https://doi.org/10.1093/ecco-jcc/jjw183.
- Abraham E, Anzueto A, Gutierrez G, Tessler S, Pedro GS, Wunderink R, Nogare AD, Nasraway S, Berman S, Cooney R, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. Lancet. 1998;351:929–33. doi:https://doi.org/10.1016/S0140-6736(05)60602-2.
- Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332:323–27. doi:https://doi.org/10.1038/332323a0.
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–86. doi:https://doi.org/10.1016/S0006-3495(99)77443-6.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi:https://doi.org/10.1107/S0907444910007493.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi:https://doi.org/10.1002/jcc.20084.