
ABSTRACT
Complex cellular targets such as G protein-coupled receptors (GPCRs), ion channels, and other multi-transmembrane proteins represent a significant challenge for therapeutic antibody discovery, primarily because of poor stability of the target protein upon extraction from cell membranes. To assess whether a limited set of membrane-bound antigen formats could be exploited to identify functional antibodies directed against such targets, we selected a GPCR of therapeutic relevance (CCR1) and identified target binders using an in vitro yeast-based antibody discovery platform (AdimabTM) to expedite hit identification. Initially, we compared two different biotinylated antigen formats overexpressing human CCR1 in a ‘scouting’ approach using a subset of the antibody library. Binders were isolated using streptavidin-coated beads, expressed as yeast supernatants, and screened using a high-throughput binding assay and flow cytometry on appropriate cell lines. The most suitable antigen was then selected to isolate target binders using the full library diversity. This approach identified a combined total of 183 mAbs with diverse heavy chain sequences. A subset of clones exhibited high potencies in primary cell chemotaxis assays, with IC50 values in the low nM/high pM range. To assess the feasibility of any further affinity enhancement, full-length hCCR1 protein was purified, complementary-determining region diversified libraries were constructed from a high and lower affinity mAb, and improved binders were isolated by fluorescence-activated cell sorting selections. A significant affinity enhancement was observed for the lower affinity parental mAb, but not the high affinity mAb. These data exemplify a methodology to generate potent human mAbs for challenging targets rapidly using whole cells as antigen and define a route to the identification of affinity-matured variants if required.
Introduction
Monoclonal antibodies (mAbs) have emerged over the last three decades as a highly effective therapeutic modality for the treatment of a diverse range of diseases.Citation1,Citation2 The considerable effort that has been expended in developing mAbs and related molecular formats over this period is primarily due to the numerous benefits when compared with small molecules, including exquisite specificity, a lower risk of unanticipated safety issues and restricted central nervous system penetration, a longer duration of action due to neonatal Fc receptor-mediated recycling, and the ability to modulate effector functions via Fc engineering.Citation3,Citation4
A recurring technical hurdle in the discovery and development of large molecules, however, is the availability of sufficient quantities of target antigen in a clinically relevant conformation to support the identification of target-specific binders with desired functional properties. This is particularly evident in pursuit of high affinity mAbs directed against complex multi-transmembrane (TM) targets, including G protein-coupled receptors (GPCRs), ion channels, and other cell-surface targets, which often lack large extracellular domains that can be cloned and expressed recombinantly, enabling the delivery of soluble antigens to drive antibody discovery.Citation5-Citation7 Challenges in antigen availability for such targets include relatively low yields from recombinant cell lines, which creates issues in scaling protein production and limits the final quantity of purified antigen, and poor thermal stability upon extraction from the lipid membrane environment, hampering subsequent purification of antigen in a sufficiently stable, clinically relevant conformation. For GPCRs, these technical limitations hindered drug discovery and thwarted attempts to provide a more complete understanding of structure-function relationships within this target class until the first high resolution crystal structure emerged in 2000,Citation8 even though the first atomic model of a GPCR was reported in 1990.Citation9 Consistent with the challenging nature of purifying stable GPCR proteins, a further 7 years passed until the second GPCR crystal structure was reported publicly.Citation10,Citation11
A variety of solutions to this significant barrier to GPCR drug discovery have been exemplified, including screening for detergents to aid solubilization and stability,Citation12,Citation13 site-directed or high-throughput protein engineering,Citation14,Citation15 and directed evolution in microbial hosts.Citation16-Citation18 For a limited number of GPCRs, a stable, soluble, N-terminal extracellular domain construct can be expressed, secreted, and purified.Citation19-Citation21 For all other GPCRs, approaches that circumvent the need to purify the target protein can be applied, including the use of linear or constrained synthetic peptides representing exposed N-termini or extracellular loops,Citation22-Citation26 purification of recombinant virus-like particles (VLPs) formed by budding of replication-disabled viruses through cells transfected with the target of interest,Citation27 scaffold protein-mediated stabilization in lipid nanodiscs,Citation28-Citation30 or generating recombinant cell lines over-expressing the target of interest in mammalian or murine syngeneic/isogenic cell backgrounds.Citation3,Citation31-Citation33
DNA immunization represents a another approach that negates the need to develop antigen formats in vitro, where in vivo intradermal delivery of DNA encoding the target of interest under the control of an appropriate promoter results in transfection of host cells and subsequent target antigen presentation to the immune system.Citation34,Citation35 In addition to the ease of generating suitable DNA expression constructs, this approach has advantages in terms of displaying correctly folded target on cells that are regarded as ‘foreign’ by the immune system, albeit with the potential for murine post-translational modifications that may not be identical to the endogenously expressed human target. A key disadvantage of this technique is the relatively poor and slow immune response.Citation36 However, combining DNA immunization with other antigen formats can boost the target-specific immune response effectively.Citation6
Consistent with the challenging nature of delivering suitable quantities of GPCR in a clinically relevant conformation for the discovery of candidate-quality antibodies, only two anti-GPCR mAbs have been approved by the US Food and Drug Administration (FDA), specifically, mogamulizumab (POTELIGEO®), developed by Kyowa Hakko Kirin, an afucosylated (enhanced antibody-dependent cellular cytotoxicity) anti-CCR4 mAb for the treatment of cutaneous T-cell lymphoma,Citation37 and erenumab (Aimovig®), co-developed by Amgen and Novartis, an anti-CGRP receptor mAb for the treatment of chronic migraine.Citation38 Both mogamulizumab and erenumab were generated by immunization of mice with the relevant target N-termini; in the case of mogamulizumab, a linear synthetic peptide (aa 2–29) was used as antigen because of the short, relatively unstructured, N-terminus of CCR4,Citation38 whereas erenumab was generated using a purified recombinant soluble form of the large N-terminus of CGRP, which co-purified with the accessory protein RAMP1 using a proprietary process.Citation21 Other anti-GPCR mAbs are in late-stage clinical trials or pre-registration, including leronlimab (PRO-140), a humanized anti-CCR5 mAb undergoing regulatory review as a treatment for HIV infection, with additional clinical trials in progress or planned in acute GvHD (Phase 2) and metastatic triple-negative breast cancer (Phase 1b/2).Citation2,Citation5
We report here the identification of human antagonist mAbs directed against a GPCR of therapeutic relevance (CCR1) using a limited set of antigen formats combined with a yeast-based antibody discovery platform (AdimabTM). CCR1 and its chemokine ligands, primarily CCL3 (MIP-1α) and CCL5 (RANTES), are implicated in a variety of human disorders, including rheumatoid arthritis, multiple sclerosis, and osteolytic bone disease associated with multiple myeloma, and CCR1 has been the focus of intense interest in the discovery and development of small molecule antagonists, resulting in multiple Phase 2 human clinical trials.Citation39 Although peptides have been used successfully to generate FDA-approved anti-GPCR mAbs, we selected two ‘membrane-associated’ antigen formats over-expressing hCCR1 for these studies (recombinant Chinese hamster ovary (CHO) cells and VLPs, both biotinylated), on the basis that this may be more likely to deliver a diverse panel of lead antibodies directed against the full range of exposed epitopes across the entire extracellular surface of CCR1. We found that by applying this methodology high potencies could be achieved for a subset of hits without requiring further improvement via affinity maturation, resulting in a significant reduction in lead discovery timelines. To extend the potential utility of this approach for additional GPCR targets where affinity/potency goals may not be reached from naïve selections, we purified full-length CCR1 protein and constructed diversified complementarity-determining region (CDR) H1 and H2 libraries derived from the highest affinity lead mAb (single-digit nM EC50 on CHO-hCCR1 cells) and a lower affinity mAb (approximately 50–100 nM EC50 on CHO-hCCR1 cells). While this conferred a significant affinity enhancement for the lower affinity parental mAb, the high affinity mAb could not be improved upon further. These data exemplify a methodology to generate rapidly high affinity and high potency human mAbs for challenging targets using whole cells as antigen and define a route to the identification of affinity-matured variants where affinity/potency goals are not met from initial naïve library selections.
Results
Although in vivo immunization approaches have been exemplified for diverse GPCR targets,Citation3,Citation19,Citation31,Citation33,Citation40,Citation41 we wanted to assess the utility of an in vitro yeast-based discovery platform (AdimabTM), comprising large and diverse libraries of human mAbs, and magnetic-activated cell sorting (MACS®) selections to isolate target binders in order to reduce lead discovery timelines. We selected a GPCR of therapeutic relevance (CCR1, a class A chemokine receptor) for these investigations. With concerns over limited epitope diversity using linear peptide antigens based on the exposed CCR1 N-terminus, and without the availability of purified recombinant hCCR1 at the time of initiating these efforts, we instead compared two readily available ‘membrane-associated’ antigen formats over-expressing the target of interest, specifically, CHO-hCCR1 cells and hCCR1 VLPs.
Antigen quality control
Prior to initiating the antibody discovery campaign, we quantified the number of human CCR1 receptors expressed on the surface of CHO-hCCR1 cells using a R-phycoerythrin (PE)-labeled anti-human CCR1 mAb. Cells stained with this antibody were referenced to the signal from QuantiBRITETM PE-conjugated beads at the same flow cytometer settings. This gave a value of approximately 5.7 × 105 receptors/cell. At a concentration of 1 × 107 cells/ml this equates to a soluble hCCR1 concentration of approximately 9 nM. Similarly, we were able to estimate the level of hCCR1 presented on commercially sourced hCCR1 VLPs, provided at a concentration of 3.5 units/µl. According to the manufacturer’s specification, the suspension had an estimated hCCR1 receptor concentration of 82–123 pmol/ml, which we equated to a soluble hCCR1 concentration of approximately 100 nM.
Antibody selections from Naïve libraries
A scouting campaign was performed initially in order to determine the optimal hCCR1 antigen format to apply in subsequent selections exploiting the full diversity of the AdimabTM platform. As shown in , sequential selection rounds were performed with biotinylated CHO-hCCR1 cells or biotinylated hCCR1 VLPs in various combinations followed by MACS® or fluorescence-activated cell sorting (FACS). Control cells (CHO-hCCR5 and parental CHO cells), and VLPs without CCR1 (Null VLPs), were also applied as appropriate control reagents; in some rounds libraries were pre-cleared using the corresponding control VLPs or cell line and MACS® used to remove clones that were not specific for hCCR1.
Following four rounds of selection, a total of 1052 clones were expressed, and supernatants screened using fluorescent microvolume assay technology (FMAT), which is a homogenous binding assay. The FMAT assay was configured with a long (16 hr) incubation period of the mAb with target cells, allowing deselection of mAb clones that trigger internalization, which we considered to be an undesirable mechanism, and to improve the ability to identify weaker affinity hits and thereby maximize the diversity of the resulting panel of hits. A simple binding ratio (CHO-hCCR1:CHO-hCCR5), based on the FMAT signals achieved when individual mAb clones expressed in yeast supernatants were applied to the appropriate cell lines, was used to assess binding of each clone to hCCR1 and specificity for hCCR1 versus hCCR5, with an arbitrary cutoff of ≥5 applied to identify hits. As shown in , selections with CHO-hCCR1 cells as antigen gave the highest overall number of hits, and a greater proportion of hits with superior binding ratios indicating higher affinity for hCCR1. Specifically, selections that used CHO-hCCR1 cells yielded 87.9% of clones with the highest binding ratios, while selections using VLP yielded 22.1% of such clones. Selections incorporating both CHO-hCCR1 cells and hCCR1-VLPs gave a reduced number of hits with lower binding ratios compared with selections that applied CHO-hCCR1 cells as antigen only.
We therefore proceeded with a selection campaign using CHO-hCCR1 cells as antigen, utilizing the full diversity of the AdimabTM platform using the conditions identified during the initial scouting campaign (). Following the final selection round, 2816 individual clones were expressed, and yeast supernatants screened as before by FMAT. Specific hits were again identified using the cutoff as above. As shown in , a high number of hit mAb clones were identified and all clones that exhibited significant binding to CHO-hCCR1 showed minimal or no binding to CHO-hCCR5. A total of 183 unique clones based on V-gene sequences derived from either the scouting or full campaign were selected for further evaluation. We therefore calculate a hit rate of unique clones specific for hCCR1 from the combined scouting and full campaign of 4.7%.
Sequence analysis
The diversity and CDRH3 length of naïve selection outputs were determined. Sequences from these hit clones were derived from a total of 13 germlines representing four germline families (IGHV1-4), with high representation from IGHV4-39 and especially IGHV1-46. There was a diverse range of CDRH3 lengths across each of these VH gene-derived sequences (Supplementary Figure S1). Phylogenetic analysis showed that the CDRH3 loops associated with clones derived from particular VH genes were spread across the dendrogram, thus indicating good diversity at the sequence level (Supplementary Figure S2).
Characterization of Naïve selection outputs
Prior to comprehensive analyses of the binding characteristics and functional properties of the full panel of hits identified as above, we selected a subset of 10 mAbs for initial assessment in order to provide insights into the properties of the panel as a whole. Hits were ranked as ‘high’, ‘medium’, and ‘low’ based on the mean fold over background (FOB) CHO-hCCR1:CHO-hCCR5 binding ratios of >70, 40–70, or <40, respectively. A range of different clones were selected across each group, primarily focusing on ‘high’ and ‘medium’ binders. Heavy and light chain CDR sequences were reviewed to ensure the final panel was sufficiently diverse. One clone with a binding ratio of <1 was included as an internal negative control. Each mAb was expressed in yeast and purified in milligram quantities.
Initially, we assessed this mAb panel for hCCR1 apparent affinity (EC50), specificity for hCCR1 relative to a wider range of chemokine receptor paralogues (hCCR2, hCCR3, and hCCR5, on the basis of closest homology to hCCR1), and cross-reactivity to relevant preclinical orthologues. In each case, flow cytometry with relevant cell lines was used to determine binding. As expected, based on the specificity ranking, the selected clones had a range of apparent affinities, with 3 clones in the single-digit nM range, 3 clones in the 10–100 nM range, and 3 clones with EC50 values >100 nM (); the additional clone selected as an internal negative control did not bind to CHO-hCCR1 cells as anticipated. All clones within this panel showed no binding above background to recombinant CHO cell lines overexpressing the most closely related paralogues (hCCR2 and hCCR3, ); it was also confirmed in parallel that all clones bound CHO-hCCR1 cells, with no binding to CHO-hCCR5 cells as predicted based on the data presented in . The cross-reactivity profile was investigated using HEK or CHO cell lines overexpressing orthologues derived from relevant preclinical species. Intriguingly, although there was limited binding to rodent (mouse and rat) and non-human primate (cynomolgus macaque) CCR1 orthologues, we observed a number of clones binding to minipig CCR1 (), which offered a potential route forward for preclinical toxicology studies for any mAbs exhibiting the desired functional properties within this subset.
Table 1. Binding and functional properties of hits derived from the full naïve campaign
Next, we assessed functional properties, initially using a non-hydrolyzable and radioactive 35S derivative of guanosine triphosphate (GTP), GTPγS, to monitor antibody-mediated inhibition of G protein recruitment upon binding of the appropriate chemokine, a method we have described previously for other GPCR targets.Citation42 Using MIP-1α/CCL3 to drive signaling via hCCR1 on isolated cell membranes, we observed that 6 of the 9 hCCR1 binders within this panel showed some level of inhibition, with a range of mean IC50 values from 23 nM to 2.9 µM. There was a reasonable correlation between apparent affinity on CHO-hCCR1 cells and potency in the GTPγS assay, but, in some cases, high affinity clones such as 14Y029-130A06 did not inhibit CCR1 signaling driven by MIP-1α/CCL3.
Given the encouraging data described above, we proceeded to screen the full panel of hits derived from the naïve campaigns for functional activity in the GTPγS assay. The highest potency hits identified using this format were taken forward for assessment in a transwell chemotaxis assay using human donor monocytes, considered to be a more physiologically relevant assay than the GTPγS format. The most potent mAb tested was derived from the original panel of mAbs as described above (14Y029-133D02). As shown in , we obtained IC50 values as low as 371 pM using primary monocytes derived from specific donors in the chemotaxis assay format, with a mean IC50 of 2 nM across 8 independent donors.
Purification of CCR1 and affinity maturation
Although, as described above, we were able to achieve unexpectedly high affinities and potencies with clones isolated directly from the AdimabTM naïve library, we wanted to explore whether further improvements were achievable. Whole cells, however, would be expected to be problematic for affinity maturation using any in vitro selection system, due to the significant avidity effect that would be expected with a cell line expressing thousands of copies of the target, and complexities in visualizing/sorting individual higher affinity clones by FACS. Instead, we assessed the use of a detergent-solubilized and purified full-length form of hCCR1 that became available as an antigen after completion of the scouting and full naïve campaigns. His-tagged hCCR1 was expressed in a stable HEK293 cell line, solubilized with n-dodecyl-D-maltopyranoside (DDM) and cholesteryl hemisuccinate (CHS), and affinity-purified on TALON® immobilized metal affinity chromatography (IMAC) resin. The yield of CCR1 purified from adherent HEK293 cells was in excess of 1 mg/L (~2 x 109 total cells). Standard protein quality control (QC) methods (size-exclusion chromatography and SDS-PAGE, ), and AdimabTM yeast QC (data not shown) indicated the resulting purified protein was of surprisingly high quality relative to a typical heterologously expressed GPCR. Based on a crude analysis (Far Western blot using streptavidin-horseradish peroxidase), there were 4–5 different biotinylated species, indicating the material was not over-biotinylated. Our final criteria for validation of the purified CCR1 following biotinylation of the material was to demonstrate binding of conformation-sensitive anti-CCR1 mAbs, specifically a commercially available anti-CCR1 mAb as described in Materials and Methods (R&D Systems cat no. FAB145P), and anti-CCR1 hits including 14Y029-133D02 and 14Y029-142B03 identified from the naïve selections. In all cases we inferred the mAbs were binding to conformational epitopes by Western blotting (data not shown).
We selected two antibodies derived from the naïve library campaign with different affinities to assess whether improvements could be achieved using purified hCCR1. Libraries were generated by creating sequence diversity within CDRH1 and CDRH2 of the high affinity mAb (14Y029-133D02), and a second, lower affinity, antibody clone (14Y029-142B03). A simple selection strategy, as shown in , was applied. In parallel, a duplicate of the lower affinity library was spiked with the high affinity parent clone at a ratio of 100,000:1 to confirm that the maturation protocol would allow enrichment of high affinity clones in a background of lower affinity clones. The maturation libraries were subjected to one round of MACS® selection and 3 rounds of FACS selections (). At round 3, a negative or ‘null’ selection was performed using a polyspecificity reagent (PSR),Citation43 designed to remove clones that bind nonspecifically to the intended target. At round 4, the antigen concentration was reduced to enhance the identification of clones with improved affinity. Following this final round, individual clones were sequenced and evaluated for binding to CHO-hCCR1, as described below. Enrichment of the high affinity clone spiked into the lower affinity clone library was indeed confirmed in the maturation output by sequencing; > 6,000-fold enrichment of the high affinity clone over 3 rounds of positive selection on purified hCCR1 was observed (data not shown). As judged by the shift observed by flow cytometry, the binding population derived from the lower affinity parental mAb had improved significantly, in contrast to the binding population derived from the high affinity mAb.
Characterization of affinity maturation outputs
To assess improvements in binding in the outputs generated as above, we again used flow cytometry to determine apparent affinity (EC50) on CHO-hCCR1 cells with the expectation that any improvements in affinity would correlate with improved potency. 96 clones from each of the three libraries were selected based on observed affinity improvement by flow cytometry and sequenced to identify unique clones prior to purification. All parental and affinity-matured mAbs were expressed, purified, and screened for binding to CHO-hCCR1 cells in parallel to determine relative apparent affinities. No significant improvements were observed for outputs derived from the high affinity mAb clone (14Y029-133D02), indeed, the most improved clones showed very modest increases in maximal binding (up to 1.14-fold), with most clones showing no improvement. Similarly, only 1 clone was isolated with an improved EC50 (2.4-fold), with no improvement observed for all other clones derived from this output. In contrast with these data, significant affinity improvements were observed for the lower affinity parental mAb (14Y029-142B03), with some clones showing up to 18-fold improvement in EC50 (). Improved clones also showed a concomitant improvement in maximal binding, as judged by an increase in mean fluorescence intensity (MFI) of up to 8.9-fold. Although it was noted that the EC50 for the parental mAb 14Y029-142B03 was lower than that observed previously (), it is still valid to assess the relative apparent affinities between parental and affinity-matured mAbs expressed, purified, and screened in parallel, as noted above. There were no significant trends in the nature of the amino acid substitutions in affinity-matured variants.
As a measure of how highly mutated the most improved clones were, the total number of amino acid changes in CDRH1 and CDRH2 was analyzed. No changes were observed in CDRH3 since the affinity maturation libraries were H1/H2 diversified only, given that most of the diversity in the AdimabTM naïve libraries resides in CDRH3. A lower number of amino acid changes were observed for clones with the most improved EC50 values (4–5 substitutions) versus highest maximal binding (10–11 substitutions).
Discussion
We describe here an approach to isolate in vitro high affinity and high potency fully human mAbs for challenging multi-TM targets using live cells as antigen. When compared with well-established strategies for therapeutic antibody discovery for these target classes, such as in vivo immunization, the approach described provides a considerable advantage in terms of lead discovery timelines. In this study, which details our experience with applying the AdimabTM platform to isolate hits directed against human CCR1, a Class A chemokine receptor of therapeutic importance, candidate-quality mAbs were isolated without the need for affinity maturation. Hits were isolated from the AdimabTM libraries, expressed, purified, and characterized for binding to hCCR1 and selectivity over hCCR5, in less than 6 weeks, compared with several months to deliver characterized hits from traditional hybridoma approaches. Subsequent studies were undertaken to purify CCR1 in order to demonstrate that, for future multi-TM targets, naïve hits derived from this approach could be progressed toward preclinical studies even if affinity/potency goals had not been met. It was not considered viable to use whole cells as antigen for affinity maturation, given the significant avidity effect that would be expected with a cell line expressing thousands of copies of the target, and complexities in visualizing/sorting individual higher affinity clones by FACS.
The rapid timeline for naïve selections and characterization of the resulting output was achieved through the combination of several methodologies: 1) the inclusion of a prior campaign to select a superior antigen, 2) our ability to access the full diversity of the AdimabTM library in the subsequent campaign, 3) the application of MACS® selections to isolate target binders rapidly, and 4) the use of a homogenous high-throughput binding screen to determine binding to hCCR1, and specificity for hCCR1 versus hCCR5, in yeast supernatants. These results were unexpected since in vitro antibody discovery systems (like the AdimabTM platform) have traditionally proven more successful addressing soluble, as opposed to integral membrane, targets, with notable exceptions.Citation44
Since completing these investigations, we have applied this approach for an additional chemokine receptor target of therapeutic interest and achieved comparable results. In both cases, we calculated a hit rate of unique clones binding to the target of interest of approximately 5%. This compares very favorably with in vivo immunization approaches for the same targets, which were run in parallel (data not shown), in both cases yielding a hit rate of <1%. Recent trends toward direct B-cell cloning approaches to isolate target binders prior to subsequent characterization would be expected to improve this hit rate significantly.
The hit rate achieved with in vitro selection strategies using whole cells as antigen, while higher than the murine immunizations run in parallel, could be improved further by enhancing the effectiveness of ‘pre-clearing’ steps. In the case of CCR1, hit clones that exhibited significant binding to CHO-hCCR1 cells showed minimal or no binding to CHO-hCCR5 (). The overall hit rate of 4.7%, however, indicates that pre-clearing with CHO-hCCR5 cells was not effective in removing a large number of hCCR5 binders and/or clones that bind to endogenous CHO cell epitopes shared between the different cell lines. Increasing the stringency of these pre-clearing steps, for example by increasing the number or length of wash steps, may deliver some benefits, although at the risk of removing weaker target binders. Similarly, more stringent ‘null’ or negative selections using the PSRCitation43 may remove nonspecific binders more effectively, while potentially reducing the number and diversity of hits in the resulting output. These approaches would not deal with the inherent issue in combining the in vitro antibody discovery platform described with whole cells as antigen; specifically, the inability to visualize target binding events and sort binding populations by FACS, a key feature of the AdimabTM platform for soluble protein targets. Careful thought must be given to imaging/cell sorting techniques that can circumvent this issue to improve the positive selection of target binders using whole cells as an antigen format.
It was acknowledged during the CCR1 campaign that there was only a weak correlation between the specificity rank, as determined using a single-shot FMAT primary screen with individual mAb clones expressed in yeast supernatants, and the apparent affinity (EC50) on CHO-hCCR1 cells, as judged by flow cytometry with purified anti-CCR1 mAb hits. As shown in , two clones (14Y029-130A06 and 14Y029-142A07) ranked as ‘High’ gave relatively poor EC50 values (> 150 nM), while one clone ranked as ‘Low’ gave a relatively high EC50 value of 16.2 nM (14Y029-125A08). For future campaigns, therefore, specificity ranking should not be used as a reliable parameter to select clones for further evaluation. There are a number of possible reasons for the limited correlation, although the most likely explanation is that the assay format used to determine the specificity rank could be influenced heavily by the concentration of expressed antibody in yeast supernatants. This could be resolved by purifying individual clones and normalizing the concentration in a single-shot assay or running a full concentration-response curve, although at the expense of longer lead discovery timelines.
It was also clear that apparent affinity (EC50 on CHO-hCCR1 cells) and potency (IC50 for inhibition of MIP-1α in the GTPγS format) showed only a weak correlation. Indeed, clone 14Y029-131C10 exhibited the highest apparent affinity (6.2 nM), but was inactive in functional assays. Since we devised our functional assays to identify only antagonists, it is feasible that such clones may in fact stimulate rather than inhibit CCR1. For most target classes, it may not be surprising that individual mAb clones show no functional response, given the potential for a diverse range of epitopes that do not necessarily antagonize, or stimulate the intended target. For GPCRs, on the other hand, it might be expected that a high proportion of mAbs binding to such targets would inhibit their function either via simple steric hindrance, due to the size of the mAb versus the available epitopes as exemplified by crystal structures of antigen-binding fragments (Fabs) binding to GPCRs,Citation45 or by binding to an allosteric site. However, this theory is not supported by numerous GPCR antibody discovery campaigns that indicate that mAbs modulating the function of such targets are extremely rare.Citation3 Our ability to identify numerous antagonist mAbs as described in this report is therefore unexpected and highlights the potential utility of the approach for other GPCRs. An additional point of interest is that we did not observe an obvious connection in these investigations between increasing CDRH3 length and higher potency for CCR1 antagonist mAbs, as has been suggested for other GPCRs.Citation46 Indeed, there was a suggestion of an inverse correlation between CDRH3 length and potency, given that the highest potency clone identified (14Y029-133D02) possessed a relatively short CDRH3 (9 amino acids), and potency decreased as the CDRH3 length increased, indicating that this may not be a critical feature of therapeutic antibodies directed against all GPCR targets.
The superiority of a high-expressing hCCR1 recombinant CHO cell line over commercially sourced hCCR1 VLPs was evident in the scouting campaign that directly compared these two antigen formats, prior to a campaign accessing the full diversity of the AdimabTM library. Indeed, selections combining hCCR1-VLPs and CHO-hCCR1 cells appeared to reduce significantly the number of hits when compared with cells alone, and selections using VLPs alone reduced this still further (). It is unclear why in these investigations VLPs were not as effective as a recombinant cell line in isolating hits from naïve libraries. Although we attempted to normalize the concentration of membrane-associated hCCR1 used in selection strategies with either antigen based on quantification with QuantiBRITETM beads, it is possible that the concentration or density of the target expressed on a cell line is distinct from that expressed on VLPs; this may then reduce the potential for an avidity effect using VLPs during antibody selections. It is also feasible that the antigen as presented on the surface of the VLPs is distinct from that expressed on recombinant cell lines, such that clones binding to hCCR1 VLPs only would be missed during screening, although such mAbs would have little value from a drug discovery perspective.
As a result of the relatively limited homology between human, non-human primate, and rodent CCR1, the preclinical development path for therapeutic antibodies targeting CCR1 was identified as a risk at inception of the discovery campaign. For this reason, we included an additional higher species orthologue (minipig) in the cross-reactivity panel and considered inclusion of additional orthologues from non-human primate and higher species, although none appeared to provide any advantage in terms of amino acid similarity or identity in the exposed extracellular regions of CCR1. It was therefore unexpected to identify a number of clones that were cross-reactive with minipig CCR1; indeed, the highest potency clone identified (14Y029-133D02) bound human and minipig CCR1 on cell lines (), inhibited minipig CCR1 in the GTPγS assay format and exhibited concentration-dependent binding to primary human and minipig monocytes (data not shown). This indicates that one or more epitopes are conserved between human and minipig CCR1, that such epitope(s) are sufficiently exposed to enable binding of antibodies to this specific region, and that binding to this site can inhibit the functional responses elicited by human chemokine ligands. All 3 clones, including 14Y029-133D02, that were cross-reactive to only human and minipig CCR1 showed some inhibition of human CCR1 (); a further clone that bound human, minipig, and rat CCR1 was inactive in the GTPγS assay format, indicating engagement of a distinct, nonfunctional, epitope (). The remaining 5 clones within this panel that bound only human CCR1 also exhibited evidence of epitope diversity. Of these, 2 clones were completely inactive in the GTPγS assay format, and, of the clones exhibiting functional activity, 2 clones showed a direct correlation between affinity and potency (14Y029-14Y029-141A10, and -142B03) while 1 clone exhibited an inverse correlation ().
Finally, with respect to enabling further application of the approach for future GPCR targets, an important feature of the studies described here was defining a successful path to the identification of higher affinity variants. Although we isolated a diverse panel of mAbs with a range of different affinities for the specific GPCR target selected, we appreciated that affinity or potency goals may not always be reached for mAbs identified from naïve library selections for the vast majority of members of this target class. Access to high quality purified CCR1 in sufficient quantities enabled us to investigate affinity maturation of a high affinity (14Y029-133D02) and lower affinity mAb (14Y029-142B03). Following the use of a naïve library that incorporates most of the diversity in CDRH3, these further selections were performed using libraries with sequence diverse CDRH1 and CDRH2, and through this approach we were able to isolate significantly improved variants of the lower affinity mAb using purified hCCR1. In parallel, we recovered 14Y029-133D02 spiked into a duplicate library of the lower affinity mAb at a >6000-fold higher frequency over 3 rounds of positive selection, demonstrating enrichment of the high affinity clone against a background of lower affinity clones.
It is not clear why we were unable to isolate improved variants of the high affinity mAb. This is a typical observation during affinity maturation with mAb clones directed against any given target, i.e., improved variants are not always isolated for all parental mAb clones. Plausible explanations for these observations include the possibility that CDRH1 and H2 on 14Y029-133D02 are not orientated sufficiently to exert a positive effect on binding, or that the selective pressure used during affinity maturation did not allow sufficient enrichment of improved variants over the parent. Nevertheless, the data generated with diversified libraries derived from the low affinity mAb, and our ability to recover the high affinity mAb spiked into a duplicate library of the low affinity mAb, both demonstrate the feasibility of isolating higher affinity variants of anti-GPCR mAbs using the AdimabTM platform if a suitable reagent is available. Although we did not apply in silico methods to inform on the affinity maturation process, such methods have been applied successfully to affinity mature mAbs directed against a variety of target classes, and more recently guided the rational conversion of an antagonist GPCR domain antibody to a potent agonist.Citation47
Taken together, the results described here demonstrate an effective approach to isolate in vitro high affinity and high potency fully human mAbs directed against challenging multi-TM targets using live cells as antigen in a highly efficient and rapid manner. Additional optimization of the live cell selection processes described may yield further improvements in the identification of hit clones, and therefore enhance the diversity of antibody panels from which to select a final lead mAb. Given the success to date with two distinct chemokine receptor targets, we will continue to prosecute related GPCRs in this manner whilst assessing the further application in lead discovery campaigns directed against a wider array of multi-TM targets.
Materials and methods
Cells and reagents
CHO cell lines overexpressing human, cynomolgus, mouse, and rat CCR1, and human CCR2, CCR3, and CCR5, were generated at GlaxoSmithKline using standard techniques. VLPs were sourced from Integral Molecular (cat no. INT-1367A, lot no. RR0968A). RANTES/CCL5 was sourced from Peprotech (cat no. 300–06). Anti-human-CCR1, -CCR2, -CCR3, and -CCR5 and isotype control PE-conjugated antibodies were sourced from R&D Systems or Beckton Dickenson (cat no. FAB145P, FAB151P, BD558165, FAB182P, and IC002P, respectively).
Purification of full-length CCR1
Human full-length CCR1 was expressed in a stable HEK293 cell line and cells were disrupted by Dounce homogenization followed by cycles of centrifugation and resuspension in lysis buffer (10 mM HEPES pH 7.0, 10 mM MgCl2, 20 mM KCl) Purified membranes were solubilized by adding DDM (Anatrace) and CHS (Anatrace) to the membrane suspension at a final concentration of 1.0% (w/v) and 0.2% (w/v), respectively, in buffer of 20 mM HEPES, pH 7.0, and 500 mM NaCl, followed by continuous mixing at 4°C for 3 h. The supernatant was isolated by centrifugation, followed by overnight incubation in batch with TALON® immobilized metal affinity chromatography (IMAC) resin (Clontech) at 4°C. The resin was washed with 10 column volumes of wash buffer 1 (20 mM HEPES, pH 7.0, 500 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 20 mM imidazole and followed by five column volumes of wash buffer 2 (20 mM HEPES, pH 7.0, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS). Proteins were eluted in 5 column volumes of wash buffer 2 with 250 mM imidazole. Protein was concentrated to 2 mg/ml and monodispersity was assessed using analytical size-exclusion chromatography, while purity was estimated by SDS-PAGE followed by Coomassie dye staining.
Mammalian cell biotinylation
Human CCR1 over-expressing CHO cells (CHO-hCCR1) and control cells overexpressing human CCR5 (CHO-hCCR5) were biotinylated using EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific). Cells were resuspended in phosphate-buffered saline (PBS) at 2.5 × 107 cells/ml and incubated with 0.1 mg/ml biotinylation reagent for 15 minutes at 4°C. After washing, biotinylation was confirmed by staining non-biotinylated and biotinylated cells with a 1:50 dilution of ExtrAvidin-R-Phycoerythrin (Sigma Aldrich) and 2 µg/ml propidium iodide. Cells were frozen in 80% fetal calf serum (FCS) with 20% dimethyl sulfoxide. Human CCR1 positive cells were stained with anti-human-CCR1 PE-conjugated antibody before and after biotinylation, and before and after freezing, to confirm that CCR1 surface presentation was not disrupted.
Antibody selections
Antibody clones were selected from the AdimabTM platform libraries or newly generated libraries with re-diversified CDRs according to the protocols developed by AdimabTM. All antigens were biotinylated prior to use as described above. Magnetic bead selections were performed using streptavidin beads from Miltenyi (MACS®) in round 1 selections, or streptavidin M280 Dynal beads (Invitrogen) in subsequent rounds. FACS selection rounds were performed on a BD ARIA III, and yeast populations were sorted based on binding to hCCR1 positive antigens or cells, or lack of binding to a PSR.Citation43 In the ‘scouting’ experiment, the ratio of yeast to target cells was 400:1 in round 1, 500:1 in round 2, and 200:1 in round 3 and 4. Yeast cells were resuspended directly in the suspension containing hCCR1-VLPs. For these selections, the amount of hCCR1-VLPs used per 1 × 107 yeast cells was 2 units in round 1, 4 units in round 2 and 3, and 20 units in round 4. According to the manufacturer’s datasheet, hCCR1-VLPs were provided at 3.5 units/µl, and the suspension had an estimated hCCR1 receptor concentration of 82–123 pmol/ml which we equated to a soluble hCCR1 concentration of approximately 100 nM. In the naïve selections, the ratio of yeast cells to target cells was 200:1 in round 1 and 2, 40:1 in round 3, and 20:1 in round 4. Control cells were used for pre-clearing the libraries at yeast-to-control cell ratios of 200:1 in round 2, and 20:1 in round 3 and 4. The output yeast from the final round of selection were cultured on agar plates and 192 colonies were DNA sequenced from each output. For affinity maturation, new libraries re-diversified within the CDRH1 and CDRH2 regions were generated for selected clones using DNA supplied by Adimab (Lebanon, New Hampshire). Biotinylated full-length wild-type hCCR1 protein prepared as described above was used as antigen. The maturation libraries were subjected to one round of magnetic bead selection (MACS®) and 3 rounds of FACS selections.
Antibody expression and purification
Antibody clones were expressed as human IgG1 from a proprietary yeast strain (AdimabTM) and purified using Protein A affinity chromatography. Purified material in elution buffer was buffer exchanged into PBS using standard methods.
FMAT cell binding assay
Test antibodies were diluted in 384-well plates (4titude® VisionPlatesTM; Brooks Life Sciences) and CHO-CCR1 cells at 2 × 105 cells/ml added together with a 1:4000 dilution of a fluorescently labeled anti-human secondary antibody (Alexa Fluor® 647 goat anti-mouse Fcγ fragment; Jackson ImmunoResearch, West Grove, PA). Plates were then transferred to a 37°C/5% CO2 incubator for 16 hrs. Binding of test antibodies to CHO-CCR1 cells was measured using a FMAT 8200 Cellular Detection System. Data was collected as FL1, Cell Count, and FL1_Total; the latter parameter represents FL1 x Cell Count, therefore giving the well fluorescence combined with the number of cells labeled with a primary unlabeled mAb and the fluorescent secondary antibody. Samples were analyzed for EC50 using GraphPad Prism (GraphPad Software, San Diego, CA).
Apparent affinity, selectivity, and cross-reactivity binding assays
Stable cell lines or transiently transfected cells were harvested from tissue culture flasks and resuspended in assay buffer (PBS + 10% FCS). Dilution series of individual test mAbs, together with an irrelevant isotype control and relevant positive controls, or test mAbs and controls at a fixed concentration of 10 µg/ml, were prepared in a sterile polystyrene plate; appropriate cell lines or transiently transfected cells were added subsequently to a final density of 2 × 105 cells/ml. Samples were mixed gently, and the plates incubated at 4°C for 1 hr. Cells were washed in assay buffer twice and detection antibody (anti-human IgG, Southern Biotech cat no. 2040–09) then added at a 1:500 dilution and the plates incubated at 4°C for 30 mins. Cells were washed as before and finally resuspended in Cell Fix solution diluted 1:10 in dH20 (Becton, Dickinson & Company). Data was analyzed using FlowJo (Becton, Dickinson & Company).
GTPγS assay
The GTPγS assay format has been described previously for other chemokine receptor targets.Citation42 Briefly, CHO-CCR1 cell membranes (5 µg/ml) were mixed at a 1:1 ratio with 25 mg/ml WGA-coupled PS imaging (Leadseeker) beads (Perkin-Elmer) before being incubated for 1 h at 4°C. GDP was added to 384-well solid white plates (Nunc, FAC 4.4 μM) containing test compound. 35S-GTPγS (Perkin-Elmer) was diluted 1:1200 in assay buffer and 20 μl/well added to the plates before centrifugation at 1200 rpm for 30 s. After 3 h plates were read using Viewlux (Perkin-Elmer) with a 613/55(A09) emission filter. The raw data was analyzed using a 4-parameter logistic fit IC50 template (XLFit).
Human monocyte chemotaxis assay
Peripheral blood mononuclear cells (PBMCs) were prepared from human whole blood using a standard Ficoll density gradient centrifugation method (Sigma, St. Louis, MO). Blood was sourced ethically, and their research use was in accord with the terms of the informed consents. CD14+ monocytes were isolated from these PBMCs using MACS® MicroBead Technology and magnetic columns (Miltenyi Biotec, Germany). Cell number and viability were determined, and cells resuspended in an assay buffer to a concentration of 8 × 106 cells/ml.
Anti-CCR1 mAbs or irrelevant controls were pre-incubated with monocytes at RT for 1 hr. Chemotaxis assays were performed in 96-well plates (Neuro Probe, Inc. Gaithersburg, MD) according to the manufacturer’s instructions. Human RANTES/CCL5 (Peprotech, London, United Kingdom) at 5 ng/ml, or dilution buffer only, was added to the receiver chambers of the assay plate. Following re-incubation, monocytes were carefully deposited, in duplicate, onto the upper surface of the membrane and plates incubated at 37°C with 5% CO2. After 3 hrs the filter membrane was removed from the assay plate and the contents of each well of the receiver plate transferred to an opaque 96-well plate. An equivalent volume of CellTiter-Glo luminescent cell viability reagent (Promega, Madison, WI) was then added to each well and the plate incubated for a further 30 mins at 37°C. The plate was read on an EnVision 2103 Multilabel reader (Perkin Elmer, Waltham, MA). Raw data was analyzed, firstly by subtracting average minimum chemotaxis control values from both the maximum chemotaxis control values and the antibody treated samples, then by determining the percentage inhibition of chemotaxis for each treated sample by expressing values from each treated sample as a percentage of the average maximum chemotaxis control. IC50 values were calculated using GraphPad Prism.
Figure 1. (A). Scouting campaign selection strategy. In order to identify a single antigen format to take forward into a naïve campaign applying the full AdimabTM Library diversity, a subset of the library was subjected to multiple rounds of selection using biotinylated CHO-hCCR1 cells, hCCR1-VLPs, or combinations thereof. Some samples were additionally subjected to pre-clearing with control cells using MACS® prior to positive selection with the antigen as shown. Following selection round 4, clones from each output were generated as yeast supernatants and screened for hCCR1 binding, and specificity for hCCR1 vs hCCR5, on appropriate recombinant cell lines. (B). Characterization of scouting campaign output. Clones selected from the ‘scouting’ campaign, as outlined in Figure 1a, were screened using a homogenous cell binding assay (FMAT) to assess binding to CHO-hCCR1, and specificity for hCCR1 by comparing binding to CHO-hCCR1 and CHO-hCCR5 cells. Each full-length mAb clone was expressed in yeast supernatant; duplicate data points were averaged and the CHO-hCCR1:CHO-hCCR5 binding ratio calculated. A cutoff binding ratio of >2 was applied and the resulting dataset grouped depending on whether individual clones were derived from selections using CHO-hCCR1 cells only, hCCR1-VLPs only, or combinations of both antigens
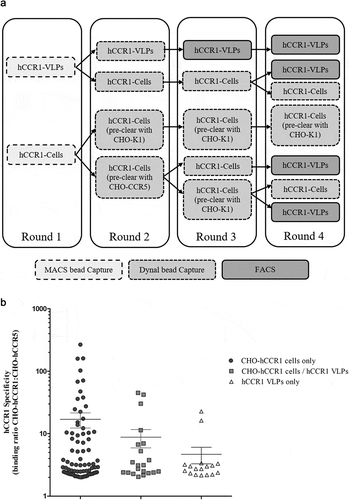
Figure 2. (A). Full naïve campaign selection strategy. The full AdimabTM library was subjected to four rounds of MACS® selections on biotinylated CHO-hCCR1 cells. At rounds 2 to 4, a preclear was performed using biotinylated CHO-hCCR5 or CHO-K1 (parental) cells. Following selection round 4, individual clones were screened as yeast supernatants using a homogenous cell binding assay (FMAT) to assess binding to CHO-hCCR1, and specificity for hCCR1 by comparing binding to CHO-hCCR1 and CHO-hCCR5 cells. (B). Full naïve campaign binding screen. Clones selected from the full naïve campaign, as outlined in (A), were assessed for binding to CHO-hCCR1 and -hCCR5 in a homogenous cell binding assay (FMAT). Each full-length mAb clone was tested in ‘single-shot’ over two independent assay runs. The data shown represents mean FMAT signal values ± SEM across the two runs for clones with a binding ratio (CHO-hCCR1:CHO-hCCR5) >20; these data were ordered from left to right (lowest to highest averaged FMAT signal for CHO-hCCR1)
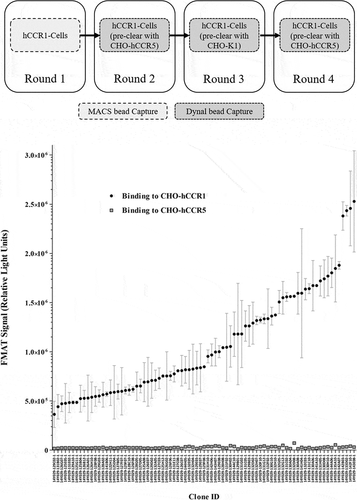
Figure 3. Inhibition of RANTES/CCL5-induced human donor monocyte chemotaxis. CD14+ monocytes were isolated from human PBMCs using MACS® technology, and chemotaxis of monocytes toward RANTES/CCL5 assessed in a transwell assay format. Monocytes were pre-incubated with mAb 14Y029-133D02 prior to addition of the chemokine. An IC50 value of 371 pM was calculated in this example dataset generated with monocytes derived from a specific human donor. The full dataset for 14Y029-133D02 using monocytes derived from multiple donors is shown in.
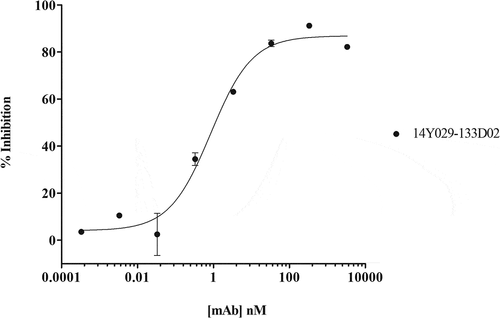
Figure 4. (A). QC of purified his-tagged hCCR1. His-tagged hCCR1 was expressed in a stable HEK293 cell line. Cells were disrupted and purified membranes solubilized with DDM/CHS; hCCR1-his was subsequently captured from the supernatant on TALON® immobilized metal affinity chromatography (IMAC) resin and eluted with 250 mM imidazole. Monodispersity was assessed using analytical size-exclusion chromatography coupled to tryptophan fluorescence detection, while purity was estimated by SDS-PAGE followed by Coomassie dye staining (inset). (B). Affinity maturation campaign selection strategy. Maturation libraries were subjected to one round of magnetic bead selection and 3 rounds of FACS selections. At round 3 a null selection was performed using a polyspecificity reagent (PSR)Citation41 designed to remove cross-reactive and sticky clones. At round 4 the antigen concentration was reduced to 1 µg/ml to aid identification of clones with improved affinity. Following the round 4 selection individual cones were evaluated for binding to CHO-hCCR1 cells. (C). Affinity-maturation of anti-CCR1 mAb clone 14Y029-142B03 using purified hCCR1 as antigen. Affinity-matured variants of parental mAb 14Y029-142B03, derived from the full naïve library selections, were isolated from diversified in CDRH1 and CDRH2 libraries using purified hCCR1 as antigen. Binding of test mAbs to CHO-hCCR1 cells was determined by flow cytometry via a fluorescently labeled detection antibody; a representative EC50 curve for the ‘most improved’ variant of parental mAb 14Y029-142B03 is shown
Table 2. Inhibition of RANTES/CCL5-induced human donor monocyte chemotaxis
Abbreviations
Supplemental Material
Download Zip (167.7 KB)Acknowledgments
We thank Christopher Plummer and Susannah Davis for critical review of the manuscript.
Disclosure statement
All authors hold stock in GSK.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Strohl W. Current progress in innovative engineered antibodies. Protein & Cell. 2018;9(1):86–13. doi:https://doi.org/10.1007/s13238-017-0457-8.
- Kaplon H, Reichert JM. Antibodies to watch in 2019. MABS. 2019;11(2):219–38. doi:https://doi.org/10.1080/19420862.2018.1556465.
- Dodd RB, Wilkinson T, Schofield DJ. Therapeutic monoclonal antibodies to complex membrane protein targets: antigen generation and antibody discovery strategies. BioDrugs. 2018;32(4):339–55. doi:https://doi.org/10.1007/s40259-018-0289-y.
- Heukers R, De Groof TWM, Smit MJ. Nanobodies detecting and modulating GPCRs outside in and inside out. Curr Opin Cell Biol. 2019;57:115–22. doi:https://doi.org/10.1016/j.ceb.2019.01.003.
- Jo E-K. Interplay between host and pathogen: immune defense and beyond. Exp Mol Med. 2019;51(12):1–9. doi:https://doi.org/10.1038/s12276-019-0281-8.
- Hutchings CJ, Koglin M, Olson WC, Marshall FH. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat Rev Drug Discov. 2017;16(11):787–810. doi:https://doi.org/10.1038/nrd.2017.91.
- Hutchings CJ, Colussia P, Clark TG. Ion channels as therapeutic antibody targets. MABS. 2019;11(2):265–96. doi:https://doi.org/10.1080/19420862.2018.1548232.
- Palczewski K. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–45. doi:https://doi.org/10.1126/science.289.5480.739.
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213(4):899–929. doi:https://doi.org/10.1016/S0022-2836(05)80271-2.
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi H-J, Yao X-J, Weis WI, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into 2-adrenergic receptor function. Science. 2007;318(5854):1266–73. doi:https://doi.org/10.1126/science.1150609.
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi H-J, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human 2-adrenergic g protein-coupled receptor. Science. 2007;318(5854):1258–65. doi:https://doi.org/10.1126/science.1150577.
- Lantez V, Nikolaidis I, Rechenmann M, Vernet T, Noirclerc-Savoye M. Rapid automated detergent screening for the solubilization and purification of membrane proteins and complexes. Eng Life Sci. 2015;15(1):39–50. doi:https://doi.org/10.1002/elsc.201400187.
- Mandon ED, Agez M, Pellegrin R, Igonet S, Jawhari A. Novel systematic detergent screening method for membrane proteins solubilization. Anal Biochem. 2017;517:40–49. doi:https://doi.org/10.1016/j.ab.2016.11.008.
- Tehan BG, Christopher JA. The use of conformationally thermostabilised GPCRs in drug discovery: application to fragment, structure and biophysical techniques. Curr Opin Pharmacol. 2016;30:8–13. doi:https://doi.org/10.1016/j.coph.2016.06.010.
- Soave M, Cseke G, Hutchings CJ, Brown AJH, Woolard J, Hill SJ. A monoclonal antibody raised against a thermo-stabilised β1-adrenoceptor interacts with extracellular loop 2 and acts as a negative allosteric modulator of a sub-set of β1-adrenoceptors expressed in stable cell lines. Biochem Pharmacol. 2018;147:38–54. doi:https://doi.org/10.1016/j.bcp.2017.10.015.
- Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, Pluckthun A. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci USA. 2008;105(39):14808–13. doi:https://doi.org/10.1073/pnas.0803103105.
- Dodevski I, Pluckthun A. Evolution of three human GPCRs for higher expression and stability. J Mol Biol. 2011;408(4):599–615. doi:https://doi.org/10.1016/j.jmb.2011.02.051.
- Mallipeddi S, Zvonok N, Makriyannis A. Expression, purification and characterization of the human cannabinoid 1 receptor. Sci Rep. 2018;8:2935.
- Mumaw MM, de la Fuente M, Arachiche A, Wahl JK 3rd, Nieman MT. Development and characterization of monoclonal antibodies against protease activated receptor 4 (PAR4). Thromb Res. 2015;135(6):1165–71. doi:https://doi.org/10.1016/j.thromres.2015.03.027.
- Ohta S, Sakaguchi S, Kobayashi Y, Mizuno N, Tago K, Itoh H. Agonistic antibodies reveal the function of GPR56 in human glioma U87-MG cells. Biol Pharm Bull. 2015;38(4):594–600. doi:https://doi.org/10.1248/bpb.b14-00752.
- Shi L, Lehto SG, Zhu DX, Sun H, Zhang J, Smith BP, Immke DC, Wild KD, Xu C. Pharmacologic characterization of AMG 334, a potent and selective human monoclonal antibody against the calcitonin gene-related peptide receptor. J Pharmacol Exp Ther. 2015;356(1):223–31. doi:https://doi.org/10.1124/jpet.115.227793.
- Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, Takeuchi G, Shimizu S, Ito M, Komatsu H, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Can Res. 2003;9:3625–34.
- Zhang Y, Pool C, Sadler K, Yan H-P, Edl J, Wang X, Boyd JG, Tam JP. Selection of active ScFv to G-protein-coupled receptor CCR5 using surface antigen-mimicking peptides†. Biochemistry. 2004;43(39):12575–84. doi:https://doi.org/10.1021/bi0492152.
- Harris GL, Creason MB, Brulte GB, Herr DR, Agoulnik I. In vitro and in vivo antagonism of a G protein-coupled receptor (S1P3) with a novel blocking monoclonal antibody. PLoS One. 2012;7(4):e35129. doi:https://doi.org/10.1371/journal.pone.0035129.
- Tohidkia MR, Asadi F, Barar J, Omidi Y. Selection of potential therapeutic human single-chain Fv antibodies against cholecystokinin-B/gastrin receptor by phage display technology. BioDrugs. 2013;27(1):55–67. doi:https://doi.org/10.1007/s40259-012-0007-0.
- Boshuizen RS, Marsden C, Turkstra J, Rossant CJ, Slootstra J, Copley C, Schwamborn K. A combination of in vitro techniques for efficient discovery of functional monoclonal antibodies against human CXC chemokine receptor-2 (CXCR2). MABS. 2014;6(6):1415–24. doi:https://doi.org/10.4161/mabs.36237.
- Ho TT, Nguyen JT, Liu J, Stanczak P, Thompson AA, Yan YG, Chen J, Allerston CK, Dillard CL, Xu H, et al. Method for rapid optimization of recombinant GPCR protein expression and stability using virus-like particles. Protein Expr Purif. 2017;133:41–49. doi:https://doi.org/10.1016/j.pep.2017.03.002.
- Denisov IG, Sligar SG. Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol. 2016;23(6):481–86. doi:https://doi.org/10.1038/nsmb.3195.
- Lindhoud S, Carvalho V, Pronk JW, Aubin-Tam M-E. SMA-SH: modified Styrene–Maleic acid copolymer for functionalization of lipid nanodiscs. Biomacromolecules. 2016;17(4):1516–22. doi:https://doi.org/10.1021/acs.biomac.6b00140.
- Frauenfeld J, Loving R, Armache J-P, Sonnen AF, Guettou F, Moberg P, Zhu L, Jegerschöld C, Flayhan A, Briggs J, et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat Methods. 2016;13(4):345–51. doi:https://doi.org/10.1038/nmeth.3801.
- Rossant CJ, Carroll D, Huang L, Elvin J, Neal F, Walker E, Benschop JJ, Kim EE, Barry ST, Vaughan TJ. Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MABS. 2014;6(6):1425–38. doi:https://doi.org/10.4161/mabs.34376.
- Koth CM, Murray JM, Mukund S, Madjidi A, Minn A, Clarke HJ, Wong T, Chiang V, Luis E, Estevez A, et al. Molecular basis for negative regulation of the glucagon receptor. Proc Natl Acad Sci USA. 2012;109(36):14393–98. doi:https://doi.org/10.1073/pnas.1206734109.
- Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, Cao F, Niekro W, Kempe T, Henning KA, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19(2):357–66. doi:https://doi.org/10.1158/1078-0432.CCR-12-2333.
- Peyrassol X, Laeremans T, Gouwy M, Lahura V, Debulpaep M, Van Damme J, Steyaert J, Parmentier M, Langer I. Development by genetic immunization of monovalent antibodies (nanobodies) behaving as antagonists of the human ChemR23 receptor. J Immunol. 2016;196(6):2893–901. doi:https://doi.org/10.4049/jimmunol.1500888.
- van der Woning B, De Boeck G, Blanchetot C, Bobkov V, Klarenbeek A, Saunders M, Waelbroeck M, Laeremans T, Steyaert J, Hultberg A, et al. DNA immunization combined with scFv phage display identifies antagonistic GCGR specific antibodies and reveals new epitopes on the small extracellular loops. MABS. 2016;8(6):1126–35. doi:https://doi.org/10.1080/19420862.2016.1189050.
- Saade F, Petrovsky N. Technologies for enhanced efficacy of DNA vaccines. Expert Rev Vaccines. 2012;11(2):189–209. doi:https://doi.org/10.1586/erv.11.188.
- Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, Matsushima K, Ueda R, Hanai N, Shitara K. Defucosylated chimeric anti-cc chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64(6):2127–33. doi:https://doi.org/10.1158/0008-5472.CAN-03-2068.
- Imai T, Nagira M, Takagi S, Kakizaki M, Nishimura M, Wang J, Gray PW, Matsushima K, Yoshie O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Immunol. 1999;11(1):81–88. doi:https://doi.org/10.1093/intimm/11.1.81.
- Zhang P, Dairaghi DJ, Jaen JC, Powers JP. Recent advances in the discovery and development of CCR1 antagonists. Ann Rep Med Chem. 2013;48:133–47.
- Hutchings CJ, Cseke G, Osborne G, Woolard J, Zhukov A, Koglin M, Jazayeri A, Pandya-Pathak J, Langmead CJ, Hill SJ, et al. Monoclonal anti-β1-adrenergic receptor antibodies activate G protein signaling in the absence of β-arrestin recruitment. MABS. 2014;6(1):246–61. doi:https://doi.org/10.4161/mabs.27226.
- Hennen S, Kodra JT, Soroka VX, Krogh BO, Wu X, Kaastrup P, Ørskov C, Rønn SG, Schluckebier G, Barbateskovic S, et al. Structural insight into antibody-mediated antagonism of the glucagon-like peptide-1 receptor. Sci Rep. 2016;6(1):26236. doi:https://doi.org/10.1038/srep26236.
- Procopiou PA, Barrett JW, Barton NP, Begg M, Clapham D, Copley RC, Ford AJ, Graves RH, Hall DA, Hancock AP, et al. Synthesis and structure–activity relationships of indazole arylsulfonamides as allosteric CC-chemokine receptor 4 (CCR4) antagonists. J Med Chem. 2013;56(5):1946–60. doi:https://doi.org/10.1021/jm301572h.
- Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Engineering Design and Selection. 2013;26(10):663–70. doi:https://doi.org/10.1093/protein/gzt047.
- Yang Z, Wan Y, Tao P, Qiang M, Dong X, Lin C-W, Yang G, Zheng T, Lerner RA. A cell–cell interaction format for selection of high-affinity antibodies to membrane proteins. PNAS. 2019;116(30):14971–78. doi:https://doi.org/10.1073/pnas.1908571116.
- Ishchenko A, Wacker D, Kapoor M, Zhang A, Han GW, Basu S, Patel N, Messerschmidt M, Weierstall U, Liu W, et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. PNAS. 2017;114(31):8223–28. doi:https://doi.org/10.1073/pnas.1700891114.
- Douthwaite JA, Sridharan S, Huntington C, Hammersley J, Marwood R, Hakulinen JK, Ek M, Sjögren T, Rider D, Privezentzev C, et al. Affinity maturation of a novel antagonistic human monoclonal antibody with a long VH CDR3 targeting the Class A GPCR formyl-peptide receptor 1. MABS. 2015;7(1):152–66. doi:https://doi.org/10.4161/19420862.2014.985158.
- Ma Y, Ding Y, Song X, Ma X, Li X, Zhang N, Song Y, Sun Y, Shen Y, Zhong W, et al. Structure-guided discovery of a single-domain antibody agonist against human apelin receptor. Sci Adv. 2020;6(3):eaax7379. doi:https://doi.org/10.1126/sciadv.aax7379.