
ABSTRACT
CXCR1 and CXCR2 signaling play a critical role in neutrophil migration, angiogenesis, and tumorigenesis and are therefore an attractive signaling axis to target in a variety of indications. In human, a total of seven chemokines signal through these receptors and comprise the ELR+CXC chemokine family, so named because of the conserved ELRCXC N-terminal motif. To fully antagonize CXCR1 and CXCR2 signaling, an effective therapeutic should block either both receptors or all seven ligands, yet neither approach has been fully realized clinically. In this work, we describe the generation and characterization of LY3041658, a humanized monoclonal antibody that binds and neutralizes all seven human and cynomolgus monkey ELR+CXC chemokines and three of five mouse and rat ELR+CXC chemokines with high affinity. LY3041658 is able to block ELR+CXC chemokine-induced Ca2+ mobilization, CXCR2 internalization, and chemotaxis in vitro as well as neutrophil mobilization in vivo without affecting other neutrophil functions. In addition to the in vitro and in vivo activity, we characterized the epitope and structural basis for binding in detail through alanine scanning, crystallography, and mutagenesis. Together, these data provide a robust preclinical characterization of LY3041658 for which the efficacy and safety is being evaluated in human clinical trials for neutrophilic skin diseases.
Introduction
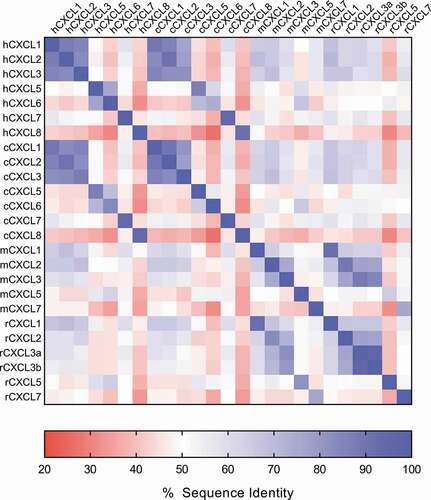
The CXC chemokine receptors 1 and 2 (CXCR1 and CXCR2) are G-protein coupled receptors (GPCR) expressed on granulocytes, monocytes, mast cells, and some natural killer cells that are capable of activating multiple downstream signaling pathways.Citation1,Citation2 Chemokines have been classified into 4 subfamilies, based on the presence of cysteines at the amino terminal: CXC, CC, CX3C and C.Citation3,Citation4 Among chemokine subfamilies, CXC chemokines can be further subclassified into Glu-Leu-Arg (ELR+) and ELR− CXC chemokines based on the presence or absence of a tripeptide ELR motif at the amine terminus. The ligands for CXCR1 and CXCR2 make up the ELR+CXC chemokine family. Despite the conserved structure and shared receptors, these chemokines share very little sequence homology outside of the ELRCXC motif (). In humans, CXCL6 and CXCL8 signal through CXCR1, while all seven chemokines (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8) signal through CXCR2.Citation5 Chemokine affinities to the receptors are in the single-digit nM range, and they typically have short turnover rates as they rapidly bind receptor, internalize, and are degraded.Citation6,Citation7 Characteristic of ELR+CXC chemokines is their ability to specifically recruit neutrophils into inflamed tissues.
Figure 1. ELR+CXC chemokine sequence diversity. The sequence identity matrix heat map shows the % amino acid sequence identity between the human, cynomolgus monkey, mouse, and rat ELR+CXC chemokines. % identity ranges from ~30-90% across the chemokine family both within a given species and across species
Since the discovery of CXCR1, CXCR2, and their ligands in the early 1990s, biological pathways that include these proteins have been targeted for drug development based on their relevance to neutrophil chemotaxis, angiogenesis, and tumorigenesis.Citation5,Citation7–11 Both small molecules and protein therapeutics have been investigated. The attenuation of neutrophil recruitment to sites of inflammation and reduction in inflammation-induced angiogenesis is believed to provide a key opportunity to influence the pathogenesis of a variety of both oncology and autoimmune/inflammatory conditions. Among these indications are neutrophilic dermatoses, which are diseases characterized by the accumulation of neutrophils in the skin and occasionally in internal organs, leading to inflammation and tissue damage.Citation12
Despite substantial investment by the biopharmaceutical industry, effective inhibition of both CXCR1 and CXCR2 has been elusive owing to the redundancy in the pathways. Several CXCR2-specific small molecule antagonists have entered clinical testing, e.g., AZD5069, AZD5122, danirixin (GSK-1325756), and SB-656933.Citation13–16 Several dual CXCR1/CXCR2 small molecule antagonists are also being tested in patients (navarixin/SCH527123/MK-7123, SX-682, and reparixin).Citation17–22 However, only CXCR2 is inhibited by navarixin at doses safely administered in the clinic, while reparixin is 400-fold less potent in inhibiting CXCR2 relative to CXCR1 in vitro. SX-682 shows promising in vitro and preclinical dual CXCR1/CXCR2 inhibition activity, but its clinical safety and efficacy remain to be seen. HuMab 10F8/HuMax IL-8/BMS-986253, a monoclonal antibody against CXCL8/IL-8, showed modest efficacy in early clinical trials.Citation23–26 Given the limitations of small molecules targeting the receptor(s) and antibodies targeting individual chemokines, as well as the likely substantial challenges of developing an antibody targeting two different high-turnover GPCRs present on a large population of cells, we chose to develop a pan-ELR+CXC chemokine neutralizing antibody.Citation27–30 Since the expression levels of the different chemokines varies across different disease states, an effective pan-ELR+CXC chemokine neutralizing antibody should neutralize all seven chemokines with high affinity.Citation31
LY3041658 is the first monoclonal antibody that effectively antagonizes both CXCR1 and CXCR2 signaling by neutralizing all seven of the ligands for these receptors. LY3041658 offers a novel approach to treating diseases where neutrophils play a pathogenic role, and it highlights the power of antibody engineering. As we enter the age of bispecific antibodies, LY3041658 is the first septa-specific monoclonal antibody, and a true first-in-class dual CXCR1/CXCR2 antagonist.
Results
Generation of a high affinity, pan-ELR+CXC chemokine neutralizing antibody
Chemokine signaling through either CXCR1 or CXCR2 mediates neutrophil recruitment to sites of inflammation. An effective therapeutic antagonizing both CXCR1 and CXCR2 signaling must neutralize all seven ELR+CXC chemokines with high affinity and have acceptable physiochemical and pharmacokinetic properties to enable its use in the clinic. To identify potential pan-ELR+CXC chemokine neutralizing antibodies, mice were immunized with human CXCL1 and subsequent hits were screened by ELISA for binding and FLIPR (Ca2+ flux) for neutralization of all seven human ELR+CXC chemokines. Mab1581 was identified with sub-nM affinity to CXCL1, 2, 3, and 5 and low-nM affinity to CXCL6, 7, and 8 (data not shown). Additionally, Mab1581 was able to neutralize the chemokine-induced intracellular Ca2+ mobilization in human CXCR2 transfected HMEC cells for all human ELR+CXC chemokines except for CXCL6 (data not shown). Mab1581 was humanized to an IgG4 antibody containing the hinge S228P mutation to prevent half-antibody formation, affinity matured, and optimized for developability properties resulting in LY3041658 (Figure S1). The IgG4 isotype was selected to minimize the potential for Fc-mediated effector function.Citation32
Affinity optimization was performed as described previously, with the notable addition of screening both single hit and combinatorial libraries against four antigens (hCXCL1, hCXCL5, hCXCL6, and hCXCL8) in parallel to increase the likelihood that selected hits maintained/improved binding across all ELR+CXC chemokines. Binding to all seven chemokines was then confirmed on select clones to identify the top combinatorial hit, which was designated as the Lead antibody. Lead optimization focused on improving sub-optimal biophysical and stability properties of the Lead antibody, including the hydrophobicity of the heavy chain (HC) complementarity-determining region 3 (CDR3). Saturation mutagenesis of CDR3 and screening against the same four chemokines as before revealed very few positions that would tolerate substitution while maintain broad specificity (data not shown). Select tolerated positions from this screen were scaled up, purified, and assessed for solubility, hydrophobicity, and stability. LY3041658 showed clear improvements compared to both Mab1581 and the Lead antibody (). Engineering of Mab1581 yielded a humanized antibody, LY3041658, with sub-nM affinity to all seven human ELR+CXC chemokines, and improved hydrophobicity, solubility, and stability properties.
Figure 2. Optimization of developability properties from the 1581 hit to LY3041658. (a) Solubility as a function of pH (vertical labels, pH = 3.5, 5.5, 6.5, and 7.4) and ammonium sulfate concentration (horizontal labels, [(NH4)2SO4] = 0.9, 1.1, 1.3, 1.5, 1.7, and 1.9 M). Solubility was scored based on a qualitative visual scale where 1 = light precipitation, 2 = medium precipitation, and 3 = heavy precipitation. Reduced precipitation was observed for LY3041658 compared to both 1581 and the Lead antibody, indicative of improved solubility. (b) aHIC analysis demonstrated the reduced hydrophobicity (earlier retention time) of LY3041658 compared to both 1581 and to the Lead antibody. (c) Relative to the Lead antibody, LY3041658 showed decreased growth in soluble aggregate as measured by aSEC after incubation at both 5°C and 40°C for 4 weeks at 25 mg/mL
![Figure 2. Optimization of developability properties from the 1581 hit to LY3041658. (a) Solubility as a function of pH (vertical labels, pH = 3.5, 5.5, 6.5, and 7.4) and ammonium sulfate concentration (horizontal labels, [(NH4)2SO4] = 0.9, 1.1, 1.3, 1.5, 1.7, and 1.9 M). Solubility was scored based on a qualitative visual scale where 1 = light precipitation, 2 = medium precipitation, and 3 = heavy precipitation. Reduced precipitation was observed for LY3041658 compared to both 1581 and the Lead antibody, indicative of improved solubility. (b) aHIC analysis demonstrated the reduced hydrophobicity (earlier retention time) of LY3041658 compared to both 1581 and to the Lead antibody. (c) Relative to the Lead antibody, LY3041658 showed decreased growth in soluble aggregate as measured by aSEC after incubation at both 5°C and 40°C for 4 weeks at 25 mg/mL](/cms/asset/c8219431-3e77-4a14-bdce-9a047fbce683/kmab_a_1831880_f0002_oc.jpg)
LY3041658 binding affinity, binding kinetics, and neutralization
The binding affinity and kinetics of LY3041658 to human, cynomolgus monkey, rat, and mouse ELR+CXC chemokines were measured by surface plasmon resonance (SPR) at 25°C (). The cynomolgus monkey, rat, and mouse chemokine affinities were measured to support preclinical work and to guide toxicology strategy. LY3041658 binds all human and cynomolgus monkey ELR+CXC chemokines with high affinity. Affinity for each ligand is comparable between human and cynomolgus monkey, and all but the cynomolgus monkey CXCL6 are bound with sub-nM dissociation constants (KD). LY3041658 also binds all five rat and mouse ELR+CXC chemokines, though binding to both CXCL5 and CXCL7 is considerably weaker, with KD values of ~30-900 nM compared to ~200 pM for CXCL1/2/3 (Supplemental Table 1). This high affinity binding across most family members from four different species, with rodent CXCL5 and CXCL7 being the exceptions, is remarkable given the low sequence identity across the chemokines ().
Table 1. In vitro binding kinetics of LY3041658 measured by surface plasmon resonance (SPR) at 25°C. Results reported as average ± standard deviation of 2 replicates
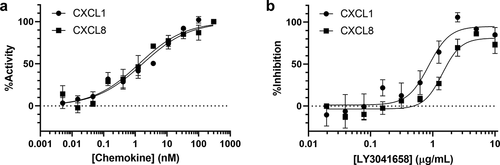
The ability of LY3041658 to neutralize ELR+CXC chemokine-induced intracellular Ca2+ mobilization via CXCR2 signaling was evaluated in vitro using HMEC-1 cells transfected with human CXCR2. CXCR2 was selected over CXCR1 because it binds all seven ELR+CXC chemokines. A dose response of human ELR+CXC chemokines was first established to determine the ability of the chemokines to induce Ca2+ mobilization (representative data in ) and S2a). All seven human chemokines were confirmed to induce Ca2+ influx, and LY3041658 was able to dose dependently inhibit this Ca2+ mobilization induced by 10 nM chemokine for all seven human chemokines (, ) and S2b). LY3041658 was also able to neutralize the tested cynomolgus monkey, mouse, and rat chemokines which were active in this assay (cCXCL5 and cCXCL7 were not active, cCXCL1 and cCXCL6 were not tested), but it did not neutralize rodent CXCL5 or CXCL7 (Table S2). Clearly, the ~30-900 nM affinity of LY3041658 for rodent CXCL5 and CXCL7 was too weak to achieve neutralization in this assay.
Table 2. In vitro neutralization of 10 nM human or cynomolgus monkey chemokine-induced intracellular Ca2+ mobilization in human CXCR2 transfected HMEC-1 cells measured by FLIPR. Results reported as average ± standard deviation of indicated number of replicates. No Ca2+ mobilization response was observed for cynomolgus monkey CXCL5 or CXCL7, therefore LY3041658 neutralization activity against these chemokines could not be measured
Figure 3. In vitro CXCR2 activity and neutralization in HMEC-CXCR2 FLIPR assay. (a) Representative dose response curves for CXCL1 or CXCL8 induction of Ca2+ flux. (b) Representative dose response curves for LY3041658 neutralization of Ca2+ flux induced by10 nM chemokine (EC70, approximately). Data points are the average ± SEM of 3 replicate points. Curve fits are four parameter logistic curve fits made using GraphPad Prism 8.3
LY3041658 is specific for ELR+CXC chemokines
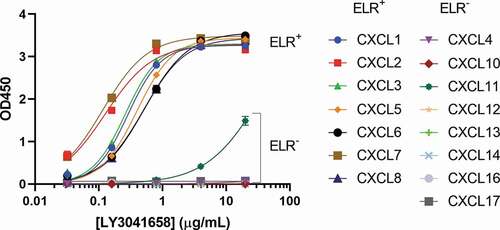
The ELR+CXC chemokines are a subset of the larger CXC chemokine family, which also includes the ELR−CXC chemokines CXCL4, CXCL9, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL16, and CXCL17. All CXC chemokines have a conserved structure and N-terminal CXC motif, but otherwise have very low sequence identity. To confirm specificity for ELR+CXC chemokines, binding of LY3041658 to all 16 human CXC chemokines was evaluated by ELISA, with the exception of CXCL9, which could not be used in the assay due to significant background binding (). Binding in this format to all seven ELR+CXC chemokines was confirmed. CXCL11 showed weak binding compared with the ELR+CXC chemokines, with a ~ 200-fold shift in EC50, and no binding was observed for any of the other ELR−CXC chemokines. CXCL9 and CXCL11 were further characterized by SPR. Due to high nonspecific, charge-mediated binding of the chemokines to the sensor chip surface, NaCl was added to the running buffer to a final concentration of 500 mM and maximum chemokine concentrations tested were 100 nM. ELR+CXC chemokine binding was not significantly affected by the addition of NaCl. Approximate affinities of ~100 nM were measured by equilibrium analysis for CXCL9 and CXCL11 (data not shown), consistent with the EC50 shift observed by ELISA for CXCL11. The >100-fold weaker apparent binding of CXCL9 and CXCL11 compared with the ELR+CXC chemokines is not expected to be pharmacologically relevant for LY3041658 in vivo.
Figure 4. Binding of LY3041658 is highly specific for ELR+CXC chemokines. Data shows strong binding of LY3041658 to the human ELR+CXC chemokines coated on an ELISA plate, but not to the ELR−CXC chemokines. CXCL11 is the only ELR−CXC chemokine which shows any binding, and it is significantly weaker compared to the ELR+CXC chemokines. CXCL9 could not be analyzed due to significant nonspecific background signal. Data represent the mean ± SEM blank subtracted OD values for duplicate wells within a single ELISA plate. Curve fits are four parameter logistic curve fits made using GraphPad Prism 8.3
Initial LY3041658/hCXCL8 epitope characterization
Given the low sequence identity outside of the conserved ELR+CXC motif and the potential for lack of specificity of a pan-ELR+CXC chemokine antibody, we sought to characterize the LY3041658 epitope in detail. Initial characterization by Western blot suggested a conformational epitope (Figure S3). The hCXCL8 epitope was characterized in more detail by alanine scanning in a yeast display system.
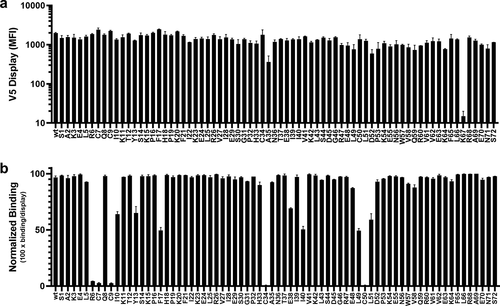
An alanine scan library of hCXCL8 fused to the anchor protein GPDL2 and N-terminally fused with a V5 tag was constructed and displayed on yeast. The library was analyzed by flow cytometry using anti-V5 staining to detect antigen display and anti-hIgG staining to detect LY3041658 binding (). With the exception of K67A, all mutants were displayed. Each of the four conserved cysteine residues (Cys7, Cys9, Cys34, and Cys50) were critical for binding, consistent with the Western blot results suggesting a conformational epitope dependent on the disulfide bonds. Aside from these positions, only mutation of the Arg6 of the ELR motif resulted in essentially no binding to the ligand. Interestingly, arginine is found at this position in several of the ELR−CXC chemokines: CXCL9, CXCL10, CXCL11, CXCL12, and CXCL13. While CXCL9 and CXCL11 are bound very weakly, CXCL10, CXCL12, and CXCL13 are not bound, which strongly suggests that the arginine at this position is necessary but not sufficient for high-affinity binding to LY3041658.
Figure 5. Alanine scanning of human CXCL8 by yeast display. (a) Expression/display of each mutant analyzed by V5+ staining. Only the K67 mutant showed poor display. (b) Relative binding was assessed by normalizing the V5+/hIgG+ population to total V5+ population. Substitution of any of the four conserved cysteines or the conserved arginine of the ELR motif effectively abolished LY3041658 binding. Several other positions with moderate loss of binding were identified: I10, Y13, F17, E38, I40, L49, and L51. In each plot, bars represent the mean ± SEM for two independent experiments
Mutation of the hydrophobic amino acids Ile10, Tyr13, Phe17, Ile40, Leu49, and Leu51 with alanine resulted in moderate loss of binding, as did mutation of Glu38, which is 100% conserved in human, cynomolgus monkey, mouse, and rat ELR+CXC chemokines. While it is not clear from these data if these amino acids are directly in the LY3041658 binding interface or are important for conformation of the chemokine, it is nevertheless striking that conservative substitutions of the aliphatic amino acids isoleucine and leucine with alanine resulted in a ~ 40–50% loss in binding.
High-resolution X-ray crystal structures of LY3041658/antigen complexes
In order to better define the LY3041658-ligand complex, a high-resolution X-ray crystal structure of the LY3041658 Fab in complex with a C-terminal truncated variant of human CXCL8 (1–66) was determined. The C-terminal helix truncation is reported to be monomeric at higher concentrations and was used given the potential for CXCL8 dimerization to negatively affect crystal formation.Citation33 As shown in ), the truncated CXCL8 still dimerized within the crystal with one Fab bound to each CXCL8 subunit distal to the dimer interface. After successfully obtaining this structure, we obtained three additional Fab/antigen structures to better understand how LY3041658 can selectively bind all human and cynomolgus monkey ELR+CXC chemokines with high affinity. Collection details and refinement statistics for all four structures are in Tables S3 and S4.
Figure 6. LY3041658/chemokine complex structure. (a) Two Fab/antigen complexes are observed in the asymmetric unit (human CXCL8 shown). The chemokines are present as homodimers with Fabs bound distal to the dimer interface. (b) The binding mode is conserved across cyno CXCL2 (blue), cyno CXCL3 (red), cyno CXCL7 (yellow), and human CXCL8 (salmon). The epitope surface is composed of the N-terminus (ELR motif and N-loop), the 30s-loop, and the β2/β3 strands
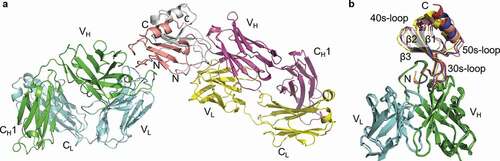
The binding mode is highly conserved in all four structures despite <30% overall antigen sequence identity ()). The buried surface area upon antigen binding ranges from ~850 to ~960 Å2, and shape complementarity of the binding interface (calculated using the shape correlation statistic Sc in CCP4) for each complex ranges from 0.76 to 0.80 on average (Table S5). This is indicative of a high degree of shape complementarity compared to average antibody-antigen complexes.Citation34 The chemokine N-loop sits in a groove between HC CDR2 and HC CDR3 that makes numerous interactions with Arg6 of the ELR motif, as well as Ile10 and Lys11 of hCXCL8. The conserved arginine sidechain is buried in an acidic pocket formed by Trp33, Glu99, Tyr102, and Tyr110 of the HC along with Asn91 and Trp94 of the LC (). The arginine sidechain is anchored by a salt bridge with Glu99 and by π-cation interactions with the Trp94 and Tyr110 aromatic sidechains. While the ELR leucine backbone carbonyl hydrogen bonds with the Trp94 LC backbone amide in all structures, the Leu sidechain is not specifically recognized (Figure S4). The ELR glutamic acid sidechain is at the edge of ordered structure and shows potential to interact with HC Arg108, with interactions observed in the cCXCL3 and cCXCL7 structures but not in cCXCL2 or hCXCL8 (Figure S4). This ambiguous interaction in the crystal structures is likely due to the conformational flexibility of both the glutamic acid and arginine sidechains as well as of the chemokine N-terminus, and this ambiguity is consistent with the yeast display data that did not show an effect on binding when the glutamic acid was substituted with alanine. Beyond recognition of the ELR motif, a hydrophobic pocket formed by Trp33, Ser52, Pro53, Asn54, and Tyr102 of the HC accommodates aliphatic residues in the chemokine: Leu12 in cCXCL2/3, Met8 in cCXCL7, and Ile10 in hCXCL8 ()). Finally, the HC Glu28, Thr30, and Ser31 can interact with charged/polar amino acids in the chemokine: Lys11 in hCXCL8, Gln13 in cCXCL2/3, and Lys9 in cCXCL7.
Figure 7. Detailed view of the LY3041658/hCXCL8 binding interface. (a) The Fab surface is colored by electrostatic potential using the APBS tool in PyMol 2.3 (Schrödinger, LLC) with red representing negative electrostatic potential and blue representing positive electrostatic potential (±5 kT/e scale).Citation35 A strong negative pocket shown in red binds the conserved arginine sidechain of the ELR motif, and the I10 sidechain sits in a hydrophobic pocket. (b) The R6 sidechain is anchored by a salt bridge to the HC E99 sidechain with several hydrogen bonds (dashed yellow lines) stabilizing the binding pocket. In addition to the salt bridge, cation-π interactions are observed with the R6 sidechain stacked between aromatic sidechains from both the HC and LC. (c) The paratope includes a hydrophobic pocket which accommodates I10 in hCXCL8 (shown), L12 in cCXCL2/3, and M8 in cCXCL7. (d) The HC CDR3 loop extends away from the Fab and forms a hydrophobic patch which interacts with hydrophobic residues in the N-terminus (Y13), β2 strand (I40), and β3 strand (L49) of hCXCL8. The hCXCL8 surface is colored based on residue hydrophobicity with purple representing hydrophobic residues and green representing hydrophilic residues. The color scale is normalized by consensus hydrophobicity.Citation36
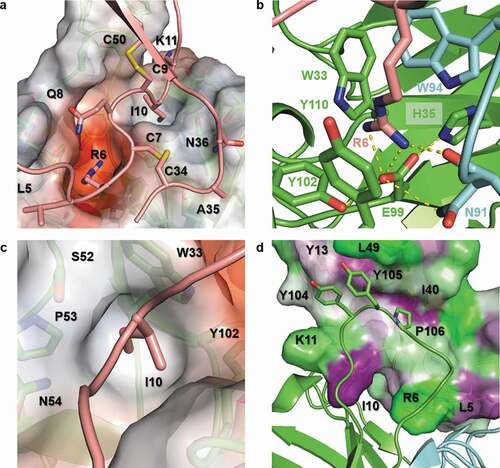
Aside from the ELR motif and the N-loop, several additional key contacts are observed. Both chemokine disulfides are contacted directly with a hydrogen bond between the HC Asn59 sidechain and the chemokine Cys7/Cys34 disulfide and multiple contacts <4.5 Å between both the HC Tyr104 backbone and the HC Pro106 and the chemokine Cys9/Cys50 disulfide (Figure S5). In all four structures, the HC Ser55 and/or HC Ser57 sidechain hydrogen bonds to a polar/charged amino acid in the same position in the chemokine: Gln37 in cCXCL2, Glu37 in cCXCL3, Gln33 in cCXCL7, and Asn36 in hCXCL8. A complete list of observed hydrogen bonding demonstrates several key conserved contacts, but also the overall plasticity of LY3041658 and its ability to recognize the diverse sequences in the ELR+CXC chemokine family (Table S6). The long HC CDR3 loop has several exposed hydrophobic residues (Tyr104, Tyr105, Pro106) that extend away from the antibody surface and interact with numerous conserved hydrophobic chemokine amino acids that were also identified by alanine scanning: Tyr104 with Leu15 in cCXCL2/3, Thr11 in cCXCL7, and Tyr13 in hCXCL8; Tyr105 with Ala50 in cCXCL2/3, Ile46 in cCXCL7, and Leu49 in cCXCL8; and Pro106 with Ile41 in cCXCL2/3, Ile37 in cCXCL7, Ile40 in hCXCL8 ()). The hCXCL8 Phe17, Glu38, and Leu51, which were identified by alanine scanning, are not observed in the LY3041658 binding interface, and therefore are instead likely important for conformation rather than part of the epitope. These positions are indicated in sequence alignment across the human, cynomolgus monkey, mouse, and rat chemokines in Figure S6, highlighting the diversity of sequences that LY3041658 is able to recognize.
To further evaluate the contribution of specific residues identified by both yeast display and analysis of the crystal structures, select human CXCL8 point mutants were made and tested by SPR to measure binding kinetics and affinity (). Alanine substitutions were made at Arg6, Ile10, Tyr13, Ile40, and Leu49. In addition, Ala35 was substituted with proline to probe the importance of this position. While some sequence diversity exists at this position in the ELR+CXC chemokines, proline is only observed in rat/mouse CXCL5 and was thought to be a potential contributor to the weak binding of LY3041658 to those chemokines (Figure S6). Unsurprisingly and consistent with both yeast display and crystallography results, the R6A mutation knocked out LY3041658 binding completely. The A35P substitution resulted in a faster on-rate, potentially due to decreased conformational flexibility of the chemokine, but a ~40-fold loss in affinity. Likely the proline at this same position in rodent CXCL5 is contributing to the weaker binding of these chemokines. The I10A and Y13A mutations all had modest effects, with ~2-3-fold loss in affinity, while the I40A and L49A mutations had more significant effects, with 70- and 10-fold losses in affinity, respectively. Given the relatively small contact surface area and the high conservation of the I40 and L49 positions, conformational changes may contribute to the loss in binding.
Table 3. LY3041658 SPR binding kinetics of select hCXCL8 mutants at 25°C. Results reported as average ± standard deviation of 2 replicates
Together, these data demonstrate that LY3041658 has a conformational epitope that broadly recognizes the CXC chemokine fold and relies on several key conserved contacts for specificity while accommodating diversity in other epitope positions. The arginine of the ELR motif is necessary, but not sufficient for binding.
LY3041658 neutralizes ELR+CXC chemokine-induced neutrophil signaling and neutrophil migration, but not neutrophil function in vitro
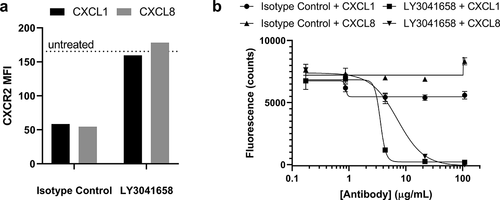
Functionally, ELR+CXC chemokines are chemoattractant ligands that direct neutrophils to the site of inflammation. Ligand binding to CXCR1 or CXCR2 causes rapid internalization of the receptor as the cells migrate with the chemokine gradient. In order to confirm biological activity of LY3041658 on cells naturally expressing both CXCR1 and CXCR2, the ability of LY3041658 to block both chemokine-induced CXCR2 internalization and neutrophil chemotaxis induced by either CXCL1 or CXCL8 was evaluated ex vivo using primary human neutrophils. LY3041658 was able to inhibit ligand-induced CXCR2 internalization, while the isotype control antibody demonstrated a significant reduction in cell surface CXCR2 expression due to hCXCL1- or hCXCL8-induced internalization. ()). In addition, LY3041658 dose dependently blocked human neutrophil migration induced by either hCXCL1 or hCXCL8 ()). In order to determine if LY3041658 inhibited neutrophil behaviors beyond those induced by ELR+CXC chemokines, an extensive panel of neutrophil functions was assessed. LY3041658 treatment did not, however, inhibit any critical neutrophil functions ex vivo, including fMLP activation, LTB4 activation, phagocytosis, or respiratory oxidative burst (Figure S7).
Figure 8. Neutralization of hCXCL1- or hCXCL8-induced CXCR2 internalization on primary human neutrophils or neutrophil migration by LY3041658 ex vivo. (a) 20 µg/mL LY3041658 was able to block hCXCL1 (28 nM) or hCXCL8 (8.8 nM) induced internalization of CXCR2. Data shown (n = 1) is cell surface CXCR2 measured using flow cytometry expressed as Mean Fluorescence Intensity (MFI). Chemokine doses reported were equipotent in inducing CXCR2 internalization (dose response data not shown). (b) LY3041658 dose dependently neutralizes neutrophil chemotaxis induced by either hCXCL1 (5.1 nM) or hCXCL8 (10 nM). Chemokine doses were selected to be equipotent in inducing chemotaxis as measured by fluorescence of the CellTracker Green dye. Fluorescence values (485 nm/535 nm ex/em) are plotted as the blank subtracted mean ± SEM of triplicate test wells. Curve fits are four parameter logistic fits made with GraphPad Prism 8.3
Together, these results demonstrate the ability of LY3041658 to neutralize CXCR1 and CXCR2 signaling and subsequent human neutrophil chemotaxis without inhibiting other neutrophil functions.
Pharmacodynamic studies
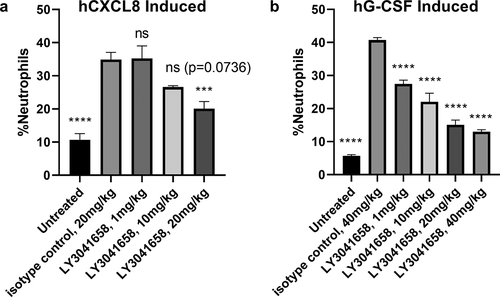
The pharmacodynamic (PD) effects of LY3041658 were evaluated in both a CXCL8 and a granulocyte-colony stimulating factor (G-CSF)-induced neutrophilia mouse model.Citation37 Human CXCL8 is able to signal through mouse CXCR2 directly, resulting in neutrophil mobilization into the blood, while G-CSF mediates thrombopoietin release triggering endogenous ELR+CXC chemokine-induced neutrophil mobilization from the bone marrow. LY3041658 dose dependently reduced hCXCL8-mediated neutrophil mobilization with statistical significance (p = .0009) at 20 mg/kg ()), and it dose dependently reduced hG-CSF mediated neutrophil mobilization with high statistical significance (p < .0001 compared with isotype control) at 1, 10, and 20, 40 mg/kg doses ()). While neutralization of both CXCL8- and G-CSF-induced neutrophilia was significant relative to isotype control, neutrophils were not reduced to the level of the untreated groups. This could be the result of upregulation of mouse CXCL5 or mouse CXCL7, neither of which are effectively neutralized by LY3041658, and thus would result in only partial inhibition of CXCR2 signaling. Together, these results demonstrate that LY3041685 is biologically active in in vivo PD models.
Figure 9. In vivo neutralizing activity of LY3041658. LY3041658 dose dependently reduced neutrophilia in mice induced by either hCXCL8 (a) or hG-CSF (b). The %neutrophils in blood was measured by flow cytometry as CD11b+/Ly6G+ cells normalized to total number of cells analyzed. Bars represent the mean ± SEM for n = 6 animals. Statistical analysis was done with GraphPad Prism 8 using one-way ANOVA with multiple comparisons to the isotype control (Dunnett’s test). Significant differences relative to isotype control are displayed: *** = 0.0001 ≤ p < .001, **** = p < .0001
Discussion
Here, we describe the generation and characterization of a humanized pan-ELR+CXC chemokine neutralizing antibody. The affinity maturation and humanization processes used to engineer this antibody included steps for optimizing its affinity for all seven of the human ELR+CXC chemokines, resulting in a novel septa-specific monoclonal antibody (mAb) that binds to and neutralizes this family of chemokines with sub-nM affinity. LY3041658 binds to all seven human and cynomolgus and three of five mouse and rat ELR+CXC chemokines with high affinity, but binds to mouse and rat CXCL5 and CXCL7 with relatively low affinity. LY3041658 is specific for ELR+CXC chemokines, and it blocks chemokine binding to its receptors CXCR1 and CXCR2, thereby neutralizing receptor signaling.
The ability of LY3041658 to block chemokine/receptor signaling was demonstrated in vitro, ex vivo, and in vivo. LY3041658 blocks ligand-induced Ca2+ mobilization in a CXCR2 transfected cell line, CXCR2 internalization in primary human neutrophils, chemotaxis of primary human neutrophils, and mobilization of neutrophils in mice. It is important to note that LY3041658 has these activities without blocking neutrophil function (activation, respiratory oxidative burst, or phagocytosis) and without significant cross-reactivity to ELR−CXC chemokines, which may provide a safe and efficacious treatment approach for neutrophilic diseases.
While Angelini and colleagues recently reported a directed evolution approach to develop broadly cross-reactive chemokine antibodies,Citation38 our work is the first to describe such an antibody derived from a traditional immunization/engineering approach. Moreover, LY3041658 binds all seven human ELR+CXC chemokines with sub-nM affinity compared to the ~10-100x weaker affinity and more limited cross-reactivity reported for the engineered single-chain variable fragments (scFvs).Citation38 Given the single digit nM potency of the chemokines to their receptors, the higher affinity of LY3041658 is likely to be pharmacologically relevant. LY3041658 does so while retaining high selectivity against ELR−CXC chemokines. This combination of high affinity and promiscuity within the ELR+CXC chemokines across multiple species yet selectivity against ELR−CXC chemokines is remarkable. Our approach of screening against multiple chemokines starting from initial hit screening and continuing throughout the entire humanization and optimization process contrasts with other previous reports of introducing additional specificity by engineering existing monospecific antibodies.Citation39,Citation40 This post-hoc introduction of multi-specificity, while successful for generating bispecifics, may well have limitations in adding further specificities and/or higher affinities.
Given the surprising and unique combination of high affinity and high selectivity for ELR+CXC chemokines, we sought to further characterize the epitope and structural basis of binding in detail. The epitope is conformational, dependent on the conserved disulfide bonds, and only highly selective for the conserved arginine residue of the ELR motif. The paratope features a negative electrostatic groove and an extended hydrophobic CDR3 loop, suggestive of a recognition mode similar to that shown for both CXCR2, vCKBPs, and the recently reported scFvs.Citation38,Citation41–43 Of note, the majority of mutations incorporated during affinity maturation (Figure S1) are found not in the paratope, but rather in the VH/VL interface or buried within the VH or VL. This highlights the power and necessity of the parallel library screening approach against multiple chemokines, as these mutants could not be rationally designed and do not have predictable effects on binding across the ELR+CXC chemokines.
While necessary for LY3041658 binding, the ELR arginine is not sufficient as evidenced by the selectivity against multiple ELR−CXC chemokines that retain the critical arginine. Based on the LY3041658/chemokine complex structures and sequence homology, we can speculate that LY3041658 has multiple mechanisms for selectivity against these ELR−CXC chemokines, such as unfavorable electrostatic interactions between HC Arg108 and positively charged residues corresponding to the E and X positions of the ELRCXC motif (CXCL9, CXCL11, CXCL12, and CXCL13) or steric hindrance between HC Asn59 and a proline at the position corresponding to Ala35 in hCXCL8 (CXCL10 and CXCL13). Perhaps not surprisingly, both of these differences are found in the rodent CXCL5 and CXCL7, which LY3041658 binds with significantly weaker affinity compared to all other ELR+CXC chemokines tested.
Prior efforts to develop therapeutic antibodies to chemokines (or their receptors) have been hampered by the redundant nature of the chemokine/receptor network in humans.Citation44 LY3041658 may have an advantage in this regard, given that it targets all seven ELR+CXC chemokines important in neutrophil chemotaxis via a common epitope, rather than targeting a single chemokine. Even so, a mAb therapy that disrupts neutrophil migration via blockade of a single chemokine, and therefore only partial blockade of CXCR1 and CXCR2, has shown measurable effects in vivo. Notably, HuMab IL-8/BMS-986253, an antibody to human CXCL8 (interleukin-8) that inhibits binding of CXCL8 to both CXCR1 and CXCR2 showed some effect on clinical indices in patients with palmoplantar pustulosis (PPP), a predominantly neutrophilic skin disease.Citation23 Results of this study also demonstrated the presence of CXCL1 in the washing fluid of blisters from PPP patients, suggesting that CXCL8 is not the only chemokine present in the blisters. These data support the hypothesis that inhibition of chemokine-mediated neutrophil migration is a potentially promising approach for pharmacotherapy in neutrophilic skin diseases, or other syndromes with prominent neutrophil-associated pathology, where approved treatment options are limited.
In conclusion, we report the generation and characterization of LY3041658, a high-affinity pan-ELR+CXC chemokine neutralizing antibody. A novel septa-specific antibody, LY3041658 binds and neutralizes all ELR+CXC chemokines, thereby blocking CXCR1 and CXCR2 mediated neutrophil migration without blocking neutrophil function and offering promising therapeutic potential in both neutrophilic inflammatory diseases and in oncology.
Materials and methods
All animal studies were performed in accordance with American Association for Laboratory Animal Care Institutional Guidelines. All Lilly-internal in vivo experimental protocols were approved by the Eli Lilly and Company Animal Care and Use Committee. Human blood used was donated by healthy volunteers according to the standards of the local ethical boards and the Research Biological Donation Program (RBDP) at Eli Lilly. All donors were kept anonymous and no identifying personal information was shared with the researchers who received these biological samples.
Immunizations and screening mouse antibodies
Immunization, isolation of antigen-binding clones, and screening of mouse antibodies was carried out as described previously.Citation45 Briefly, mice were immunized with recombinant human CXCL1. Isolated antigen-binding clones were screened for binding and neutralization of all seven human ELR+CXC chemokines. Based on its ability to bind to all seven and to neutralize six of the seven human ELR+CXC chemokines, clone 1581 was selected for humanization and affinity optimization.
Humanization and optimization
Humanization was performed using the same techniques as described previously.Citation45 In summary, clone 1581 CDRs were transferred onto human VH1-46 and VκL6 frameworks in an IgG4 isotype containing the S228P hinge stabilizing mutation and a kappa light chain, respectively.Citation46 Simultaneously, each position in both heavy chain CDR3 and light chain CDR3, as well as heavy chain CDR2 position Asn52, were randomized to all possible amino acids. These were subsequently screened for increased affinity to hCXCL1, hCXCL5, hCXCL6, and hCXCL8 by ELISA. Combinatorial libraries were made from the single hits and screened for binding and neutralization of all seven chemokines. The top combinatorial hit was further optimized for chemical and physical stability with particular focus on CDR3 of the heavy chain to reduce hydrophobicity and improve high concentration stability. Saturation mutagenesis of heavy chain CDR3 was performed with each mutant being screened for binding by ELISA to hCXCL1, hCXCL5, hCXCL6, and hCXCL8 as before. Permissive mutants were subsequently triaged using a high throughput, small scale solubility assay to rapidly identity variants with improved solubility.Citation47 Select mutants with improved solubility were scaled up and fully characterized for binding, neutralizing activity, and developability. From this work, the candidate mAb LY3041658 was identified.
Hydrophobic interaction chromatography
Hydrophobicity of antibodies was assessed by analytical hydrophobic interaction chromatography (aHIC) using a Source 15PHE PE 4.6 × 100 mm column (GE Healthcare) on an Agilent 1100 series HPLC. Mobile phase A was phosphate-buffered saline (PBS) pH 7.2, and mobile phase B was 60% acetonitrile/40% water. Samples were diluted to 0.5 mg/mL in PBS pH 7.2, and 0.05 mg was injected. Antibodies were eluted with a linear gradient from 0% to 100% B over 13 minutes at a flow rate of 1 mL/min, and detection was by UV absorption at 280 nm.
Stability analysis
Antibodies were dialyzed into 10 mM sodium citrate pH 6.5 and concentrated to 25 mg/mL using Amicon® Ultra-4 30 kDa NMWL centrifugal filters (Millipore Sigma). Samples were held at 5°C and 40°C. Percent soluble aggregate was measured at initial, 1, and 4-week timepoints by analytical size-exclusion chromatography (aSEC) using an Agilent 1100 series HPLC. aSEC was performed by injecting 1 µL of undiluted samples on a 7.8x300mm TSKgel G3000SWXL column (Tosoh Bioscience) in a 50 mM sodium phosphate pH 7.0 + 150 mM NaCl isocratic mobile phase at a flow rate of 0.5 mL/min with UV absorption detection at 280 nm. Chromatograms were integrated in ChemStation (Agilent) to determine the % of soluble aggregate.
Chemokine and antibody expression and purification
Cynomolgus monkey ligands were not commercially available and were therefore produced in-house. Briefly, cynomolgus monkey CXCL1, CXCL2, CXCL3, and CXCL6 were expressed in HEK293 EBNA cells. Chemokines were purified using SP sepharose and SEC. Cynomolgus monkey CXCL7 was expressed in E. coli cells as a His-sumo fusion protein. This protein was refolded, cleaved with yUlp1, and purified using IMAC, SP sepharose, and reverse phase chromatography. Cynomolgus monkey CXCL5 and CXCL8 were expressed and purified from a bacterial soluble protein expression system. Correct mass for all proteins was confirmed by MALDI-TOF, amino acid sequence was confirmed by N-terminal Edman sequencing, and in vitro activity was confirmed by a cell-based FLIPR assay. All other human, mouse, and rat ELR+CXC ligands were purchased from R&D Systems or PeproTech.
Human CXCL8 variants, both the 1–66 C-terminal truncated variant and the point mutants, were expressed, refolded, and purified using the same procedure used for cynomolgus monkey CXCL7.
The nomenclature, accession numbers, and relevant amino acid ranges for all chemokines as used in this study are provided in Tables S7 and S8. All chemokine amino acid numbering used in this manuscript is relative to these ranges.
Antibody expression and purification was carried out as described previously.Citation45
Binding measurements
Binding kinetic measurements were made at 25°C by surface plasmon resonance (SPR) using a Biacore 2000 instrument. A CM5 sensor chip with Protein A amine coupled to the chip surface and 1X HBS-EP+ running buffer were used. In each cycle, approximately 400–600 RU of LY3041658 was captured in flow cell 2 (Fc2) followed by a 250 second injection of chemokine at 100 µL/min, a 30-minute dissociation at 100 µL/min, and a 60 second injection of glycine pH 1.5 at 50 µL/min to regenerate the chip surface. Chemokine concentrations were 2-fold serially diluted down in running buffer from either 50 nM for high-affinity chemokines or 1 µM for low-affinity chemokines and injected in duplicate for each experiment. Reference subtracted data (Fc2-Fc1) was blank subtracted and fit to a 1:1 kinetic binding model using BIAEvaluation software.
Equilibrium binding measurements for the very weak binding human CXCL9 and CXCL11 were made using 1X HBS-EP+ with an additional 350 mM NaCl added and maximum ligand concentration of 100 nM due to significant nonspecific binding of the chemokines to the chip surface. Ligand injections were at 50 µL/min for 420 seconds to allow binding to reach equilibrium. Data were fit to the ‘One site – Specific binding’ saturation binding model in GraphPad Prism 8.3.0.
FLIPR neutralization assay
HMEC-huCXCR2 is an immortalized human mammary endothelial cell line (HMEC-1) transduced with retrovirus for expression of human CXCR2.Citation48 Cultures were maintained at sub-confluent densities (50% to 80% confluent). For each experiment, cells were harvested with TrypLE Express (Gibco), cell density was adjusted to 3 × 105 cells/mL in complete culture medium and 100 µL of the cell suspension were seeded into wells of black, clear-bottom assay plates (Costar 8671BC). Cell plates were incubated at room temperature for 30 minutes to allow cells to settle to the bottom of the wells before plates are incubated overnight at 37°C in 5% CO2. For each assay plate, the contents of one vial of Fluo-4NW reagent (Molecular Probes F36206) was suspended in 10 mL assay buffer and 100 µL probenecid from the kit to make 1x Fluo-4NW reagent. After incubation, culture medium was aspirated and 100 µL of the 1x Fluo-4NW solution was added to each well of the assay plate. Plates were incubated for 30 minutes at 37°C followed by an additional 30 minutes at room temperature, protected from light. LY3041658 was serially diluted at 3x the final assay concentration in assay buffer containing 0.2% bovine serum albumin (BSA; final concentrations ranging from 10 to 0.0195 µg/mL). Stock solutions of chemokines were prepared in Assay Buffer + 0.2% BSA at 30x the final assay concentration (final concentrations were 10 nM for all chemokines). Twenty microliters of chemokine were mixed with 180 µL of antibody in wells of v-bottom 96-well polypropylene plates and incubated at room temperature for 30 minutes. The cell plate and the ligand-antibody plate were loaded into a Fluorometric Imaging Plate Reader (FLIPR-3, Molecular Devices) programmed to transfer 50 µL of ligand-antibody solution to wells of the cell plate, and fluorescence was recorded every second for 90 seconds. The change in fluorescence (ΔRFU; Max-Min) was calculated from images 10 to 90. The ΔRFU versus log [antibody] was plotted, and IC50 values were determined by nonlinear regression using a four-parameter logistic model in GraphPad Prism.
Specificity ELISA
An ELISA plate was coated overnight with chemokines at 1 µg/mL in PBS. Coating solution was removed, the plate was blocked with 1% casein in PBS, and then LY3041658 was added at concentrations of 20, 4, 0.8, 0.16, 0.032, and 0 µg/mL in duplicate in blocking buffer. LY3041658 was detected with an anti-human Fc horseradish peroxidase secondary antibody (Southern Biotech, P/N 2014–05), developed with TMB, acid stopped, and OD at 450 nm was measured.
Yeast display
The hCXCL8 (1–72) gene was cloned into the yeast display plasmid pEMD3, which contains a tryptophan selection marker. The resulting construct contained a V5 tag at the N-terminus and a GPDL2 anchor protein at the C-terminus allowing display on the surface of yeast under the tryptophan selectable marker.
Single alanine mutations were introduced at every position, except for alanine positions at which glycine mutations were introduced. Mutations were made by Kunkel mutagenesis.Citation49 Briefly, uracil-containing single-strand DNA (ssDNA) of the pEMD3 vector was produced after transformation into CJ236 (New England Biolabs). A single colony of the transformation was grown overnight, the ssDNA was rescued following infection with M13K07 helper phage (New England Biolabs), and the ssDNA was purified using a QIAprep spin M13 kit. Oligonucleotides encoding mutations were annealed at a 20:1 molar ratio to the uracil template by denaturing at 85°C for 5 minutes, ramping to 55°C over 1 hour, holding at 55°C for 5 minutes, then chilling on ice. Second strand synthesis was then completed with T4 polymerase, T4 ligase and dNTPs (Invitrogen). The reaction was electroporated into Top10 E. coli (Invitrogen) and single colonies were picked, double-strand DNA prepared using the QIAprep miniprep kit (Qiagen), and mutations confirmed by sequencing. CXCL8 mutants were co-transformed into BJ5464 yeast (ATCC) and grown in complete minimal media without tryptophan and uracil.
Yeast were stained first with anti-V5 antibody (Invitrogen, P/N R960CUS) and LY3041658 and subsequently with a secondary goat anti-mouse IgG2a (Invitrogen, Alexa Fluor® 647, P/N A21241) to detect anti-V5 antibody (expression/display) and a goat anti-human kappa (Southern Biotech, PE, P/N 2060–09) to detect LY3041658. Yeast were analyzed by flow cytometry on a Becton Dickinson LSR11, where 50,000 events were collected based on gating cells by light scatter, V5/Alexa647, and LY3041658/PE staining. Relative binding was determined by normalizing the V5/LY3041658 double positives to total V5 positives.
Crystallization and structure determination
Purified antibody-ligand complexes at ~10 mg/mL were crystallized by the vapor diffusion method (sitting drop) at 21°C by mixing 0.3 μL of protein with 0.3 μL of well solution. Well solutions varied depending on the protein complex: Fab:cCXCL2 – 12% PEG 20 K, 100 mM MES pH 6.5; Fab:cCXCL3 – 25% PEG 4 K, 100 mM MES pH 6.5, 200 mM Magnesium Chloride; Fab:cCXCL7 – 25% PEG 3000, 100 mM HEPES pH 7.5; Fab:hCXCL8 – 20% PEG 3350, 200 mM Magnesium Chloride. Thin crystal prisms or plates, appeared within 7 days, were harvested and frozen in liquid nitrogen after quick passage in 80% well solution supplemented with 20% ethylene glycol as a cryoprotectant.
Synchrotron diffraction data was collected at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL), beamline 31-ID (LRL CAT) at 100 K. Data was processed using MOSFILM and scaled using CCP4 suite of programs.Citation50,Citation51 The structure was initially solved by molecular replacement using PHASER.Citation52 The model tracing was done in COOT and refined using BUSTER.Citation53,Citation54 Resolution of the structures ranged from 1.8 to 2.4 Å and belonged to P21 (cCXCL2 and cCXCL7 complexes) or to P212121 (cCXCL3 and hCXCL8) spacegroup. Data collection and refinement statistics are presented in Supplemental Table 3. Coordinates and structure factors for the antigen complexes with LY3041658 have been deposited in the RCSB Protein Data Bank with the accession codes 6WZJ, 6WZK, 6WZL, and 6WZM.
Neutralization of ligand-induced CXCR2 internalization
Human blood was collected in EDTA-containing Vacutainer tubes and held at room temperature. The hIgG4 isotype control antibody (20 µg/mL) or LY3041658 (20 µg/mL) was incubated with hCXCL1 (28 nM) or hCXCL8 (8.8 nM) at 37°C for 30 minutes.
After incubation, 10 µL of the antibody/chemokine samples was added to 90 µL of whole blood, vortexed for 5 seconds and incubated at 37°C for 30 minutes. The tubes were immediately transferred to ice following incubation. Surface CXCR2 was then measured by flow cytometry. Cells were stained with 5 µL of anti-CXCR2-PE (BD Biosciences, clone 6C6, P/N 555933) for 30 minutes on ice, transferred to a TQ-Prep workstation (Beckman Coulter) to lyse the red blood cells and fix the samples, then read on a FC500 (Beckman Coulter) flow cytometer. The neutrophil population was gated by forward/side scatter properties and the amount of CXCR2 present on the surface was analyzed by WinList Software and expressed as Mean Fluorescence Intensity (MFI).
Neutralization of human neutrophil chemotaxis
Peripheral blood from healthy volunteers was drawn into two 10 mL sodium heparin tubes. To isolate neutrophils, 5 mL of blood was layered over 5 mL of Polymorphprep in four 15 mL tubes. The tubes were centrifuged for 30 minutes at 470 x g, 18°C. The plasma and top cell band (mononuclear cells) were removed and discarded. The second band (neutrophils) was pooled from the 4 tubes and an equal volume of PBS was added. The tube was centrifuged for 10 minutes at 400 x g, 18°C. The pellet was washed with 12 mL of PBS, centrifuged as before, and the pellet was resuspended with 11 mL HBSS/BSA (7.5 mg/mL BSA, HBSS). Sixty million cells were suspended in 12 mL HBSS/BSA and 5 µM CMFDA CellTracker Green dye and incubated for 30 minutes at 37°C. Post-incubation, cells were pelleted by centrifugation, washed once with 12 mL HBSS/BSA, and then resuspended in 12 mL HBSS/BSA (5 x 106 cells/mL).
LY3041658 and isotype control were diluted to 216 µg/mL using HBSS/BSA and then serially diluted 1:5 with HBSS/BSA. hCXCL8 was diluted to 20 nM with HBSS/BSA, and hCXCL1 was diluted to 10.1 nM with HBSS/BSA. 70 µL of the test articles or HBSS/BSA were mixed with 70 µL of either the hCXCL8 or hCXCL1 and incubated at room temperature for approximately 30 minutes. 30 µL of the mixture was dispensed into the lower chamber wells of a ChemoTx plate in triplicate. Background signal was derived from wells containing only HBSS/BSA (no chemokine or antibody). The ChemoTx filter was placed over the lower chamber and 50 µL of cell suspension (250,000 cells) was dispensed above each well and the ChemoTx plate incubated for 3 hours at 37°C, 5% CO2. After the incubation, the cells were rinsed from the top surface with PBS and the ChemoTx filter removed. The fluorescence was read at 485 nm/535 nm ex/em using only the bottom detector (Wallac Victor3 1420 counter). The mean fluorescence of the background wells (HBSS/BSA only) was subtracted from the test well fluorescence.
Pharmacodynamic assays in mouse
The ability of LY3041658 to neutralize neutrophil mobilization induced via administration of either recombinant human CXCL8 or recombinant human G-CSF was evaluated. In the hCXCL8 study, six C57BL/6 mice per group were dosed intraperitoneally with 20, 10, or 1 mg/kg of LY3041658 or 20 mg/kg of isotype control 24 hours prior to CXCL8 stimulation. Recombinant hCXCL8 (Peprotech P/N 200–08 M) was administered intravenously at a dose of 5 µg. In the hG-CSF study, six C57BL/6 mice per group were dosed intraperitoneally with 40, 20, 10 or 1 mg/kg of LY3041658 or 40 mg/kg of isotype control 24 hours prior to G-CSF stimulation. Recombinant hG-CSF (R&D Systems P/N 214-CS) was administered intravenously at a dose of 2.5 µg. Both studies included an untreated control group to determine baseline neutrophil levels. Animals were euthanized 2 hours post G-CSF treatment or 50 minutes post CXCL8 treatment, and blood was collected by cardiac puncture into EDTA-containing syringes. Rat anti-mouse Ly-6 G antibody (BD Biosciences clone RB6-8C5 P/N 553126 or 1A8 P/N 551460 FITC conjugate) and rat anti-mouse CD11b antibody (BD Biosciences clone M1/70, PE conjugate, P/N 553311) were added to whole blood and incubated for 15–30 minutes. Stained samples were diluted ~20-fold in 1X Lyse/Fix buffer (BD Biosciences) and incubated for 10 minutes at room temperature. Cells were pelleted by centrifugation, washed twice with PBS, then resuspended in 1% paraformaldehyde. Cells were analyzed by flow cytometry and gated on CD11b+LY6G− (monocytes) and CD11b+LY6G+ (neutrophils) with neutrophils reported as % of total.
Disclosure of potential conflicts of interest
During the execution of this work, all authors were employees and stockholders of Eli Lilly and Company. No other conflicts of interest are disclosed.
Abbreviations
| BSA | = | bovine serum albumin |
| CDR | = | complementarity-determining region |
| EDTA | = | ethylenediaminetetraacetic acid |
| ELISA | = | enzyme-linked immunosorbent assay |
| FLIPR | = | fluorescence imaging plate reader |
| fMLP | = | N-formylmethionine-leucyl-phenylalanine |
| G-CSF | = | granulocyte colony-stimulating factor |
| HC | = | heavy chain |
| HEK | = | human embryonic kidney |
| HIC | = | hydrophobic interaction chromatography |
| HMEC | = | human mammary endothelial cells |
| HRP | = | horseradish peroxidase |
| IMAC | = | immobilized metal ion affinity chromatography |
| LC | = | light chain |
| LTB4 | = | leukotriene B4 |
| MAB | = | monoclonal antibody |
| MALDI-TOF | = | matrix assisted laser desorption/ionization time-of-flight |
| PBS | = | phosphate buffered saline |
| PD | = | pharmacodynamic |
| scFv | = | single chain variable fragment |
| SEC | = | size exclusion chromatography |
| SPR | = | surface plasmon resonance |
| ssDNA | = | single strand DNA |
| TMB | = | 3,3ʹ,5,5ʹ-tetramethylbenzidine |
Supplemental Material
Download Zip (3.3 MB)Acknowledgments
The authors would like to acknowledge Glenn Evans, Joseph Manetta, and Haiyan Long for their contributions to the in vitro and in vivo biological characterization; Karen Hill and Qing Chai for their contributions to the biophysical and developability characterizations; David Clawson for his contribution to the structure determination; Lisa Bafetti and Lawrence Dearth for the hCXCL8 yeast display assessment; and Kira Rubtsova for her review of the manuscript.
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Chuntharapai A, Lee J, Hébert CA, Kim KJ. Monoclonal antibodies detect different distribution patterns of IL-8 receptor A and IL-8 receptor B on human peripheral blood leukocytes. J Immunol. 1994;153:5682.
- Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491(7426):779–15. doi:https://doi.org/10.1038/nature11580.
- Gangur V, Birmingham NP, Thanesvorakul S. Chemokines in health and disease. Vet Immunol Immunopathol. 2002;86:127–36. doi:https://doi.org/10.1016/S0165-2427(02)00018-1.
- Murphy PM, Baggiolini M, Charo IF, Hébert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International Union of Pharmacology. XXII. Nomenclature for Chemokine Receptors. Pharmacol Rev. 2000;52:145.
- Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, et al. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer. 2009;125(5):1027–37. doi:https://doi.org/10.1002/ijc.24383.
- Ahuja SK, Murphy PM. The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J Biol Chem. 1996;271:20545–50. doi:https://doi.org/10.1074/jbc.271.34.20545.
- Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics. 2017;7:1543–88. doi:https://doi.org/10.7150/thno.15625.
- Acosta JC, Gil J. A role for CXCR2 in senescence, but what about in cancer? Cancer Res. 2009;69:2167–70. doi:https://doi.org/10.1158/0008-5472.CAN-08-3772.
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–21. doi:https://doi.org/10.1056/NEJMra052723.
- Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, Wu K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61–71. doi:https://doi.org/10.1016/j.cytogfr.2016.08.002.
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–41. doi:https://doi.org/10.1158/1078-0432.CCR-07-4843.
- Marzano AV, Borghi A, Wallach D, Cugno M. A comprehensive review of neutrophilic diseases. Clin Rev Allergy Immunol. 2018;54(1):114–30. doi:https://doi.org/10.1007/s12016-017-8621-8.
- Lazaar AL, Sweeney LE, MacDonald AJ, Alexis NE, Chen C, Tal-Singer R. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br J Clin Pharmacol. 2011;72(2):282–93. doi:https://doi.org/10.1111/j.1365-2125.2011.03968.x.
- Austin RP, Bennion C, Bonnert RV, Cheema L, Cook AR, Cox RJ, Ebden MR, Gaw A, Grime K, Meghani P. Discovery and evaluation of a novel monocyclic series of CXCR2 antagonists. Bioorg Med Chem Lett. 2015;25(7):1616–20. doi:https://doi.org/10.1016/j.bmcl.2015.01.067.
- Busch-Petersen J, Carpenter DC, Burman M, Foley J, Hunsberger GE, Kilian DJ, Salmon M, Mayer RJ, Yonchuk JG, Tal-Singer R, et al. Danirixin: a reversible and selective antagonist of the CXC Chemokine Receptor 2. J Pharmacol Exp Ther. 2017;362(2):338–46. doi:https://doi.org/10.1124/jpet.117.240705.
- Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A, Mårdh CK. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther. 2015;353(2):340–50. doi:https://doi.org/10.1124/jpet.114.221358.
- Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, Stryszak P, Gann L, Sadeh J, Chanez P. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy. 2012;42(7):1097–103. doi:https://doi.org/10.1111/j.1365-2222.2012.04014.x.
- Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, Horn LA, Palena C, Schlom J, Maeda DY, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-Cell immunotherapy in head and neck cancer models. Clin Cancer Res. 2020;26(6):1420–31. doi:https://doi.org/10.1158/1078-0432.CCR-19-2625.
- Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, Perez RP, Kato G, Wicha M. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. 2017;23:5358–65. doi:https://doi.org/10.1158/1078-0432.CCR-16-2748.
- Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V, Cesta MC, Galliera E. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci U S A. 2004;101(32):11791–96. doi:https://doi.org/10.1073/pnas.0402090101.
- Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, McCanna S, Butler B, Ruffini PA, Liu Y, et al. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. 2020;22(1):4. doi:https://doi.org/10.1186/s13058-019-1243-8.
- Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, Jiang S, Chang Q, Spring DJ, Sharma P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728–32. doi:https://doi.org/10.1038/nature21676.
- Skov L, Beurskens FJ, Zachariae CO, Reitamo S, Teeling J, Satijn D, Knudsen KM, Boot EPJ, Hudson D, Baadsgaard O, et al. IL-8 as antibody therapeutic target in inflammatory diseases: reduction of clinical activity in palmoplantar pustulosis. J Immunol (Baltimore, Md: 1950). 2008;181:669–79. doi:https://doi.org/10.4049/jimmunol.181.1.669.
- Yang XD, Corvalan JR, Wang P, Roy CM, Davis CG. Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol. 1999;66:401–10. doi:https://doi.org/10.1002/jlb.66.3.401.
- Bangsgaard N, Houtkamp M, Schuurhuis DH, Parren PW, Baadsgaard O, Niessen HW, Skov L. Neutralization of IL-8 prevents the induction of dermatologic adverse events associated with the inhibition of epidermal growth factor receptor. PLoS One. 2012;7(6):e39706. doi:https://doi.org/10.1371/journal.pone.0039706.
- Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA, Karzai F, Marté JL, Strauss J, Gatti-Mays ME. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019;7(1):240. doi:https://doi.org/10.1186/s40425-019-0706-x.
- Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol. 2019;40(7):584–97. doi:https://doi.org/10.1016/j.it.2019.04.013.
- Jo M, Jung ST. Engineering therapeutic antibodies targeting G-protein-coupled receptors. Exp Mol Med. 2016;48:e207. doi:https://doi.org/10.1038/emm.2015.105.
- Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11:355–63. doi:https://doi.org/10.1038/nri2972.
- Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12:5–13. doi:https://doi.org/10.1038/nrrheum.2015.157.
- Cheng Y, Ma X-L, Wei Y-Q, Wei X-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochimica et Biophysica Acta (BBA) - Rev Cancer. 2019;1871:289–312. doi:https://doi.org/10.1016/j.bbcan.2019.01.005.
- Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526:159–66. doi:https://doi.org/10.1016/j.abb.2012.03.021.
- Ravindran A, Joseph PR, Rajarathnam K. Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain: role of coupled interactions and dynamics. Biochemistry. 2009;48:8795–805. doi:https://doi.org/10.1021/bi901194p.
- Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi:https://doi.org/10.1006/jmbi.1993.1648.
- Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018;27:112–28.
- Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol. 1984;179:125–42. doi:https://doi.org/10.1016/0022-2836(84)90309-7.
- Kohler A, De Filippo K, Hasenberg M, van den Brandt C, Nye E, Hosking MP, Lane TE, Männ L, Ransohoff RM, Hauser AE, et al. G-CSF-mediated thrombopoietin release triggers neutrophil motility and mobilization from bone marrow via induction of Cxcr2 ligands. Blood. 2011;117:4349–57. doi:https://doi.org/10.1182/blood-2010-09-308387.
- Angelini A, Miyabe Y, Newsted D, Kwan BH, Miyabe C, Kelly RL, Jamy MN, Luster AD, Wittrup KD. Directed evolution of broadly crossreactive chemokine-blocking antibodies efficacious in arthritis. Nat Commun. 2018;9(1):1461. doi:https://doi.org/10.1038/s41467-018-03687-x.
- Bostrom J, Yu S-F, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323(5921):1610. doi:https://doi.org/10.1126/science.1165480.
- Lee CV, Koenig P, Fuh G. A two-in-one antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. mAbs. 2014;6(3):622–27. doi:https://doi.org/10.4161/mabs.28483.
- Felix J, Savvides SN. Mechanisms of immunomodulation by mammalian and viral decoy receptors: insights from structures. Nat Rev Immunol. 2017;17:112–29. doi:https://doi.org/10.1038/nri.2016.134.
- Prado GN, Suetomi K, Shumate D, Maxwell C, Ravindran A, Rajarathnam K, Navarro J. Chemokine signaling specificity: essential role for the N-terminal domain of chemokine receptors. Biochemistry. 2007;46(31):8961–68. doi:https://doi.org/10.1021/bi7004043.
- Proudfoot AE, Bonvin P, Power CA. Targeting chemokines: pathogens can, why can’t we? Cytokine. 2015;74:259–67. doi:https://doi.org/10.1016/j.cyto.2015.02.011.
- Comerford I, McColl SR. Mini-review series: focus on chemokines. Immunol Cell Biol. 2011;89:183–84. doi:https://doi.org/10.1038/icb.2010.164.
- Liu L, Lu J, Allan BW, Tang Y, Tetreault J, Chow C-K, Chow C-K, Barmettler B, Nelson J, Bina H. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J Inflamm Res. 2016;9:39–50. doi:https://doi.org/10.2147/JIR.S100940.
- Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27(8):767–71. doi:https://doi.org/10.1038/nbt.1553.
- Chai Q, Shih J, Weldon C, Phan S, Jones BE. Development of a high-throughput solubility screening assay for use in antibody discovery. MAbs. 2019;11:747–56. doi:https://doi.org/10.1080/19420862.2019.1589851.
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–90. doi:https://doi.org/10.1111/1523-1747.ep12613748.
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi:https://doi.org/10.1073/pnas.82.2.488.
- Leslie AGW, Powell HR. Processing diffraction data with mosflm. In: Read RJ, Sussman JL, editors. Evolving methods for macromolecular crystallography: the structural path to the understanding of the mechanism of action of CBRN agents. Dordrecht: Springer Netherlands; 2007. p. 41–51.
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi:https://doi.org/10.1107/S0907444910045749.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi:https://doi.org/10.1107/S0021889807021206.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi:https://doi.org/10.1107/S0907444904019158.
- Bricogne G, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.11.5. Cambridge (UK): Global Phasing Ltd; 2011.