
ABSTRACT
The increasing global occurrence of recalcitrant multi-drug resistant Klebsiella pneumoniae infections warrants the investigation of alternative therapy options, such as the use of monoclonal antibodies (mAbs). We used a target-agnostic phage display approach to K. pneumoniae bacteria lacking bulky, highly variable surface polysaccharides in order to isolate antibodies targeting conserved epitopes among clinically relevant strains. One antibody population contained a high proportion of unique carbohydrate binders, and biolayer interferometry revealed these antibodies bound to lipopolysaccharide (LPS). Antibodies that bound to O1 and O1/O2 LPS were identified. Antibodies were found to promote opsonophagocytic killing by human monocyte-derived macrophages and clearance of macrophage-associated bacteria when assessed using high-content imaging. One antibody, B39, was found to protect mice in a lethal model of K. pneumoniae pneumonia against both O1 and O2 strains when dosed therapeutically. High-content imaging, western blotting and fluorescence-activated cell sorting were used to determine binding to a collection of clinical K. pneumoniae O1 and O2 strains. The data suggests B39 binds to D-galactan-I and D-galactan-II of the LPS of O1 and O2 strains. Thus, we have discovered an mAb with novel binding and functional activity properties that is a promising candidate for development as a novel biotherapeutic for the treatment and prevention of K. pneumoniae infections.
Introduction
Klebsiella pneumoniae is a Gram-negative, encapsulated, rod-shaped bacterium within the Enterobacteriaceae family that was first identified as a causative agent of pneumonia by Carl Friedlander in 1882.Citation1 K. pneumoniae is found ubiquitously in nature, including in soil, surface water and sewage, and is also found in the healthcare environment on surfaces and on medical devices.Citation2,Citation3 Additionally, K. pneumoniae is a commensal organism in mammals, colonizing the gastrointestinal tract and nasopharynx in a benign manner.Citation2 From these sites, K. pneumoniae can disseminate to other tissues and cause severe infections of the urinary tract, lung, bloodstream and wound sites.Citation2 K. pneumoniae exists as both an opportunistic pathogen, causing infections in immunocompromised individuals in a nosocomial setting (classical K. pneumoniae), and as a virulent pathogen capable of causing community-acquired infections in otherwise healthy individuals (hypervirulent K. pneumoniae).Citation4
The increasing occurrence of antimicrobial resistance among classical K. pneumoniae isolates, and the more recent emergence of resistance among hypervirulent isolates,Citation5 has made K. pneumoniae infections particularly challenging to treat. The production of extended spectrum β-lactamases (ESBLs) and K. pneumoniae carbapenemases (KPCs) is particularly concerning, effectively disarming medical professionals of last-resort drugs. Clonal group (CG) 258, comprising sequence type (ST) 258, ST11 and ST512, and the recently expanding CG307, account for the vast majority of multi-drug resistant (MDR) K. pneumoniae isolates worldwide, whilst hypervirulent K. pneumoniae infections are dominated by ST23 clones.Citation6–8 A systematic review of K. pneumoniae infection mortality rates reported from 1999 to 2015 concluded that the mortality rate for carbapenem-susceptible K. pneumoniae infections was around 21%, whilst carbapenem-resistant infections were associated with a 42% mortality rate.Citation9 There is therefore an urgent need for the development of novel antimicrobial therapies for the treatment of MDR K. pneumoniae infections.
Monoclonal antibody (mAb) therapy offers an alternative to classical antimicrobials. Antimicrobial antibodies usually work in one of the two ways: firstly, by binding to and neutralizing bacterial virulence mechanisms including toxins, type III secretion systems, iron acquisition and adhesion; and secondly, by binding to bacteria and subsequently promoting the activation of the complement system and/or the recruitment of phagocytic cells mediated by Fc receptors.Citation10 In recent years, many antibody-discovery efforts have been directed against MDR bacteria. For example, a target-agnostic phage display campaign against Pseudomonas aeruginosa isolated single-chain variable fragments (scFv) toward Psl, a conserved extracellular polysaccharide involved with biofilm formation and cell attachment.Citation11 An scFv from this campaign is one component of the bispecific gremubamab,Citation12 for which Phase 2 clinical trials were completed in 2020.
Antibody-discovery campaigns targeting K. pneumoniae have also been described. Most were performed in a target-directed manner toward surface polysaccharides including lipopolysaccharide (LPS)Citation13–15 and capsular polysaccharide (CPS),Citation16–18 but a target-agnostic approach that included whole K. pneumoniae bacteria has also been used, and this led to mAbs targeting the major type III fimbrial subunit MrkA.Citation19 There remains an unmet and urgent need for the development of novel therapeutics for the prevention and treatment of K. pneumoniae infections. In this study, we aimed to use a target-agnostic approach to isolate therapeutic antibodies against K. pneumoniae.
Results
Target-agnostic phage display campaign against K. pneumoniae 43816 wild type and defined CPS and LPS mutants
We used an scFv phage display library to screen live K. pneumoniae with the ultimate aim of identifying high-affinity mAbs specific for surface antigens. A key characteristic of K. pneumoniae is its prominent CPS, and we hypothesized that CPS could present two major challenges during phage display: firstly, due to the abundance of CPS on the bacterial surface, we anticipated that a phage display campaign targeting wild-type (WT) K. pneumoniae could yield an antibody population dominated by CPS binders; and secondly, we hypothesized that CPS could mask surface antigens during phage display. Therefore, in our phage display campaign, we used both a WT strain, K. pneumoniae 43816, and a CPS-deficient strain, K. pneumoniae 43816 ΔcpsB. In addition, to enrich for surface antigens that could potentially be masked by O-antigen, a CPS and O-antigen double mutant, K. pneumoniae 43816 ΔcpsBwaaL, was also used.
Three rounds of enrichment were performed, and in some cases, the target strain was changed between rounds for example, performing two rounds of enrichment against K. pneumoniae 43816 ΔcpsBwaaL and a third round against K. pneumoniae 43816. In most cases, the third and final round of selection was performed against K. pneumoniae 43816, the most pathogenically relevant strain. After three rounds of enrichment, 11 antibody populations were generated. A summary of the selections performed and an overview of the discovery campaign is shown in .
Figure 1. Overview of the antibody discovery campaign. a) Flow diagram showing phage display selection campaign. The bacterial strain used at each round of selection is shown. WT (wild type) = K. pneumoniae 43816, M (mutant) = K. pneumoniae 43816 ΔcpsB, DM (double mutant) = K. pneumoniae 43816 ΔcpsBwaaL. b) Flow diagram summarizing the antibody discovery campaign. The number of antibodies triaged at each stage is indicated.
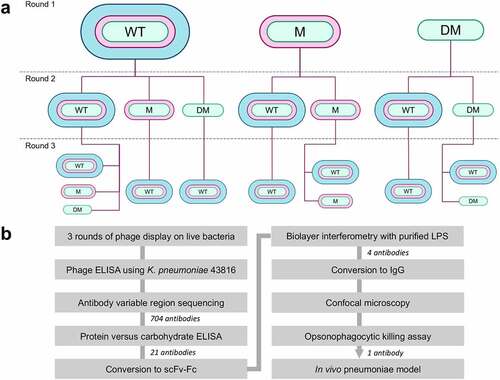
For each round three antibody population, 44 antibodies in scFv format were tested for binding to K. pneumoniae 43816 WT by phage ELISA to estimate the proportion of each population that bound to encapsulated bacteria. The 44 scFvs were also sequenced to estimate the VH and VL sequence diversity of each population. Lastly, the scFvs were tested for binding to bovine serum albumin (BSA) and E. coli by phage ELISA to determine the proportion of nonspecific binders in each population. Antibody populations with a low proportion of specific binders, a high proportion of nonspecific binders or a low complementarity-determining region 3 (CDR3) diversity (%) were eliminated at this stage, and eight antibody populations were selected for further characterization (Figure S1a).
A total of 88 scFv from the 8 antibody populations were next screened in a protein vs carbohydrate phage ELISA, using whole-cell lysates and proteinase K-digested whole-cell lysates. Of the 704 antibodies screened, 25 scFv with unique VH and VL CDR3 loop amino acid sequence were found to bind to carbohydrate antigens, and 21 of these originated from a single round three antibody population (Figure S1b). This population was enriched using K. pneumoniae 43816 ΔcpsB in the first and second rounds of selection and K. pneumoniae 43816 in the third round, suggesting that these antibodies bound to carbohydrate antigens not found in CPS that are accessible in encapsulated bacteria. The two major surface polysaccharides of K. pneumoniae are CPS and LPS, as such we hypothesized that these antibodies could bind to carbohydrate antigens found in the LPS. Targeting the O-antigen of the LPS has previously been shown to provide protection in vivo.Citation14,Citation20 Therefore, these 21 antibodies were converted to the dimeric scFv-Fc format for further characterization. At this stage, antibodies were assigned names. Antibodies binding to protein antigens were also isolated (data not shown).
In vitro characterization of carbohydrate-targeting scFv
To explore whether the antibodies bound to LPS, scFv-Fc were tested for binding to LPS purified from the O1 strain K. pneumoniae 43816 ΔcpsB. O1 O-antigen is composed of a polymer of D-galactan-I that is capped at the distal end with D-galactan-II. Since D-galactan-I is also the main component of O2 O-antigen, we also tested the scFv-Fc for binding to LPS purified from an O2 strain, K. pneumoniae 8570 ΔcpsB. All 21 scFv-Fc bound to O1 LPS by biolayer interferometry (BLI), exemplified by B02 and B09 (). Additionally, B14 and B39 bound to both O1 and O2 LPS (). No binding to LPS purified from O4 or O5 strains was observed (Figure S2), suggesting binding was specific to galactan-rich O-antigen polymers. B02, B09, B14 and B39 were converted to human IgG1 for further characterization. mAb characteristics are summarized in Table S1.
Figure 2. In vitro characterization of carbohydrate binding antibodies. a) Binding of scFv-Fc to LPS purified from K. pneumoniae 43816 ΔcpsB (O1, red) and K. pneumoniae 8570 ΔcpsB (O2, blue) strains using BLI. Association and dissociation steps were performed for 180 seconds. b) and c) Binding of IgGs to K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB. Fixed bacteria were treated with IgGs, then stained with nuclear stain DAPI (blue) and AF647 anti-human IgG (red). At high intensity, IgG binding signal appears white. Images were acquired using an Opera Phenix system (PerkinElmer) at 63x magnification and analyzed in Columbus (PerkinElmer). b. Representative images of IgGs binding to K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB. Scale bar represents 10 μm. c IgG binding intensity was calculated by dividing AF647 intensity sum by DAPI area.
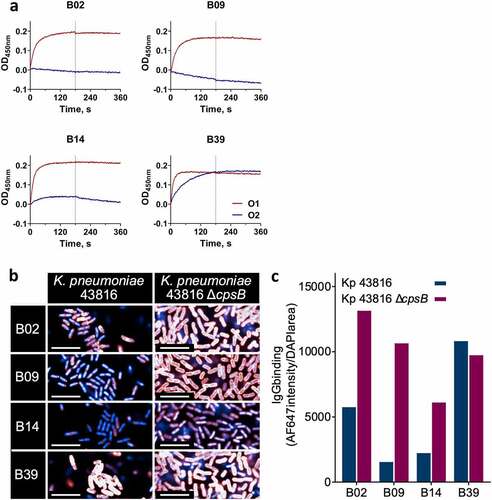
The mAbs were tested for binding to fixed whole K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB by high-content imaging (HCI) using immunofluorescence confocal microscopy to explore the surface accessibility of the antigens. All four mAbs bound with high intensity to K. pneumoniae 43816 ΔcpsB, which has an exposed O-antigen (). B02, B09 and B14 all showed a 50–80% reduction in binding to K. pneumoniae 43816, indicating some shielding of the O-antigen by CPS (). Contrastingly, B39 bound with roughly equal intensity to both strains ( and c), suggesting that CPS does not interfere with B39-antigen binding.
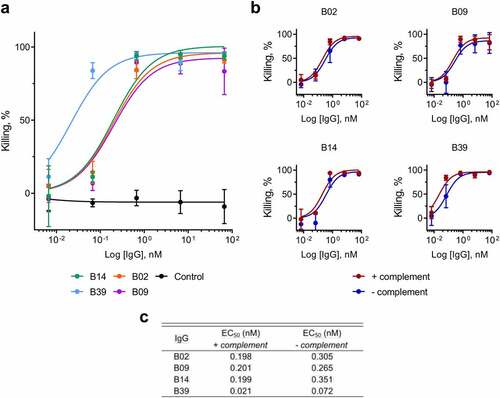
The mAbs were tested in an opsonophagocytic killing (OPK) assay using human monocyte-derived macrophages (MDMs). Due to the anti-phagocytic nature of CPS, both K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB strains were tested separately in this assay. These strains were genetically engineered to contain a plasmid encoding the luxABCDE operon, allowing a luminescence read-out as a measure of bacterial viability.Citation13,Citation14,Citation19 In line with previous reports,Citation19,Citation20 no activity against encapsulated bacteria was observed (Figure S3), but all four mAbs were capable of inducing the opsonization of K. pneumoniae 43816 ΔcpsB, promoting 90–95% killing by macrophages (). B39 was the most potent mAb in this assay, with a half maximal effective concentration (EC50) of 21 pM, which was ~ 10-fold lower than that of B02, B09 and B14 (). To understand more about how this activity was mediated, the OPK assay was repeated, except the addition of baby rabbit serum, serving as a source of complement, was omitted. We found little effect of complement on the OPK activity of the mAbs ( and c).
Figure 3. OPK of K. pneumoniae by macrophages in the presence of carbohydrate binding antibodies, measured by release of luciferase. a) Killing of K. pneumoniae 43816 ΔcpsB lux by primary human macrophages in the presence of IgGs. Bacteria, IgGs and complement were added to plates containing macrophages and incubated for 5 hours. Luminescence was measured using an Envision multilabel plate reader (PerkinElmer). Control = negative isotype control. Killing by test IgG or control IgG was calculated as a percentage of wells containing no IgG using the following calculation: (IgG treatment/no IgG)*100. Error bars represent 1 SD. N = 3 individual macrophage donors. b) Killing of K. pneumoniae 43816 ΔcpsB lux by primary human macrophages in the presence of IgGs, with (red) and without (blue) complement. c) EC50 values for IgG treatment in the presence and absence of complement.
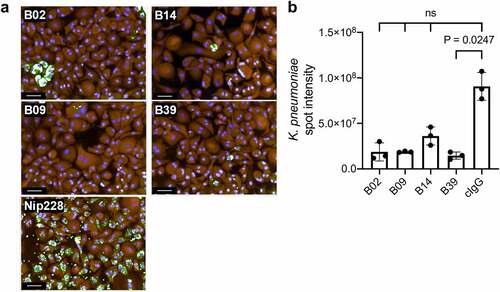
OPK activity was further explored using fluorescence microscopy, in which all four mAbs promoted macrophage-associated clearance of K. pneumoniae 43816 ΔcpsB (). B02, B09 and B39 led to a 6-fold reduction in the macrophage-associated K. pneumoniae intensity, whereas B14 led to a more modest 3-fold reduction in signal ().
Figure 4. OPK of K. pneumoniae by macrophages in the presence of carbohydrate binding antibodies by HCI. a) Representative images of bacterial clearance after a 7-hour incubation. Fixed and permeabilised cells were treated with rabbit polyclonal anti-K. pneumoniae 43816 and stained with the nuclear stain Hoechst (blue), macrophage stain cell mask Orange (Orange), and AF488 anti-rabbit IgG (green). Images from 15 fields were acquired using an Opera system (PerkinElmer) at 20 x magnification. Scale bar represents 50 μm. b) Quantification of AF488 intensity sum of K. pneumoniae spots in the macrophage cytoplasmic region. Kruskal-Wallis with Dunn’s correction for multiple comparisons test was used; Kruskal-Wallis statistic: 10.63. ns = not significant. cIgG = negative isotype control.
In vivo characterization of B39
Due to the potency of B39 in an OPK assay and the surface accessibility of the B39 target antigen, we aimed to test the efficacy of B39 in vivo. K. pneumoniae is the cause of severe pneumonia, therefore we used an acutely lethal murine model of pneumonia, which has a narrow window for successful therapeutic treatment.Citation13 As B39 was found to bind to both O1 and O2 K. pneumoniae LPS, it was tested in both O1 and O2 challenge models. The treatment of MDR isolates in the clinic is increasingly challenging, therefore we aimed to determine the effectiveness of B39 in vivo using two MDR isolates. We selected K. pneumoniae 1131115 (O1; SHV-1(b), CTX-M-15, IMP-4 positive), an O1 strain from the globally problematic clonal group ST25,Citation7 and K. pneumoniae 961842 (O2; SHV-12(e), TEM-OSBL(u), CTX-M-65, KPC-2 positive), an O2 ST258 strain. C57BL/6 WT mice were challenged with bacteria at an estimated dose of 6.0 × 107 (O1 model) and 2.0 × 108 (O2 model) cfu/mouse, 1 hour prior to administering B39 at various concentrations (as indicated in ). Actual inoculum size was determined post-infection by plating serial dilutions. The results from three independent challenges per strain are shown in .
Figure 5. Therapeutic activity of B39 antibody in a murine model of pneumonia. C57BL/6 mice were infected intranasally with the O1 strain K. pneumoniae 1131115 (a – c) or O2 strain K. pneumoniae 961842 (d – f) at an estimated inoculum size of 6.0 × 107 or 8.0 × 108 colony forming units (cfu) per mouse respectively. IgGs were administered one hour post challenge at various doses as indicated. Actual inoculum size, as indicated on each graph, was determined post challenge by plating serial dilutions. Data from each of three challenge experiments are shown separately. R347 = negative isotype control. A Mantel-Cox test was performed to compare each IgG treatment with R347. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method. P-values: * = P ≤ 0.05; ** = P ≤ 0.005. N = 8 mice per group.
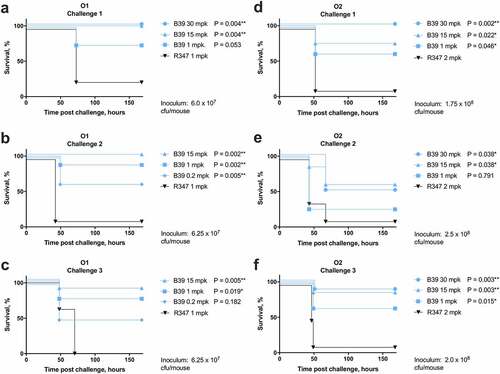
B39 at 1mpk (milligrams per kilograms) promoted 75–87.5% survival of mice challenged with the O1 strain after 168 hours (–c). This result was significant in comparison to the isotype control in both the second and third model (P = .002 and P = .019, respectively) ( and c), but not the first (). This could be due to the smaller inoculum size in the first challenge leading to a less severe infection, thereby enhancing survival of the mice. At a higher dose of 15 mpk, in two challenge models, B39 promoted 100% survival after 168 hours ( and b) (P ≤ 0.005).
In the O2 pneumonia challenge model, significant variation in B39 protectivity was observed between challenges 1 and 3 and challenge 2 (). B39 at 1 mpk promoted 62.5% survival in challenges 1 and 3 (P ≤ 0.05), compared to just 25% survival in challenge 2. This coincided with a larger inoculum used in challenge 2. We noted that this did not affect the survival of the group treated with the isotype control, suggesting the larger inoculum size affected the ability of B39 to clear the infection as opposed to increasing the lethality of the infection in all groups.
Binding of B39 to a panel of O1 and O2 strains
Several sub-serotypes exist within the O1 and O2 serotypes (Figure S4). Of marked importance are sub-serotypes that harbor the gmlABC locus, which encodes the conversion of D-galactan-I to D-galactan-III,Citation21 and which has been shown to be widely distributed among ST258 isolates,Citation22 leading to the proposal of D-galactan-III as an attractive therapeutic target.Citation22,Citation23 To investigate the potential scope of use of B39, we investigated whether B39 could bind to clinically relevant sub-serotypes within the O1 and O2 serotypes, namely O1 gmlABC− (O1−), O1 gmlABC+ (O1+), O2 gmlABC− (O2−), and O2 gmlABC+ (O2+). The O-antigen structures of these serotypes are shown in .
Figure 6. Characterization of binding to O1 and O2 strains with/without gmlABC locus by HCI and western blot. a) Structures of the O1 and O2 gmlABC ± O-antigens.Citation21 b) Representative images of B39 binding to each strain. Fixed bacteria were treated with B39, then stained with nuclear stain DAPI (blue, left panel) and AF647 anti-human IgG (red, right panel). 20 fields per well were acquired using an Opera system (PerkinElmer) at 60 x magnification and analyzed in Columbus. Scale bar represents 10 μm. N = 3. c) Quantification of IgG binding intensity. IgG binding intensity was calculated by dividing AF647 intensity sum by DAPI area. d) Quantification of % positive bacteria. % positive bacteria was calculated by dividing total AF647 positive area by DAPI area. e) Western blot analysis of B39 binding to a carbohydrate preparation from each strain. Bacterial whole cell lysates digested with proteinase K were immunoblotted and probed with B39 antibody. A strain with mannose-based O-antigen (O5) was included as a negative control. Strains used were as follows: O1 gmlABC – (O1−): K. pneumoniae 43816; O1 gmlABC + (O1+): K. pneumoniae 1131115; O2 gmlABC– (O2–): K. pneumoniae 845912; O2 gmlABC + (O2+): K. pneumoniae 961842; O5: K. pneumoniae 9181.
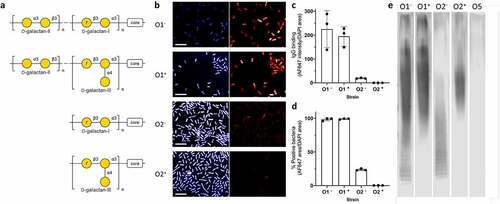
First, binding of B39 by HCI using fluorescence confocal microscopy was investigated. Uniform, high-intensity binding to bacterial cells was observed in O1− and O1+ strains as demonstrated by binding to K. pneumoniae 43816 and K. pneumoniae 1131115 (). Binding was observed to the O2− strain K. pneumoniae 845912, but binding appeared less intense than that observed in the O1 strains. Against the O2+ strain K. pneumoniae 961842, weak binding was only observed in a few bacteria per field, and most bacteria were not stained with B39. Quantification of mAb binding intensity revealed high-intensity binding to O1+ and O2- strains and low intensity to the O2− strain (). The binding intensity to the O2+ strain was negligible in this analysis. We also calculated the percentage of the bacterial population that was positive for B39 binding. Greater than 98% of O1− and O1+ bacteria were positive for binding, compared to 23% of O2− bacteria and 0.4% O2+ bacteria ().
B39 was further tested for binding to proteinase-K-digested bacterial whole-cell lysates isolated from the same strains used in the previous HCI experiment (). Against the O1− strain, binding of B39 was detected throughout the SDS-PAGE LPS profile, whereas only binding to the high molecular weight portion was observed in the O1+ strain. This binding pattern was also seen in the O2 strains, but the binding intensity appeared reduced in comparison to the O1 strains.
We next used fluorescence-activated cell sorting (FACS) to assess binding of B39 to K. pneumoniae O1 and O2 strains selected from a collection of 96 globally representative hospital isolates (Table S2). The 96 isolates were serotyped by FACS. A total of 29 O1 strains and 15 O2 strains were identified, representing 46% of the isolate panel (Figure S5). The genomes of the O1 and O2 strains were analyzed through the Kaptive web interface using the K. pneumoniae species complex LPS (O) locus database to identify the presence of gmlABC genes.Citation24 16/29 (55%) O1 strains and 8/15 (53%) O2 strains contained the gmlABC locus (Figure S6), which is within 10–15% of previous reports.Citation21,Citation22 Additionally, one O1 and one O2 strain contained gmlAC genes only. B39 was tested for binding to three strains from each group (O1+, O1−, O2+, and O2−) by FACS (). The ST and capsule (wzi) type for these strains is shown in Table S3. A number of globally problematic STs were tested,Citation7 as highlighted in Table S3. Additionally, an anti-O1 mAb () and anti-O2 mAb () were tested for comparison.
Figure 7. Characterization of binding to K. pneumoniae O1 and O2 strains with/without the gmlABC locus by FACS. a) Binding of B39 (blue) and an anti-O1 IgG (black) by FACS. b) Binding of B39 (blue) and an anti-O2 IgG (black) by FACS. The % positive-shift in binding for each antibody is labeled by color. The numbers in the top-center of each box are the K. pneumoniae strain name. c) Summary of binding to K. pneumoniae O1 (blue) and O2 (magenta) strains by FACS. O1− = K. pneumoniae O1 gmlABC −; O1+ = K. pneumoniae O1 gmlABC +; O2− = K. pneumoniae O2 gmlABC −; O2 + = K. pneumoniae O2 gmlABC +.
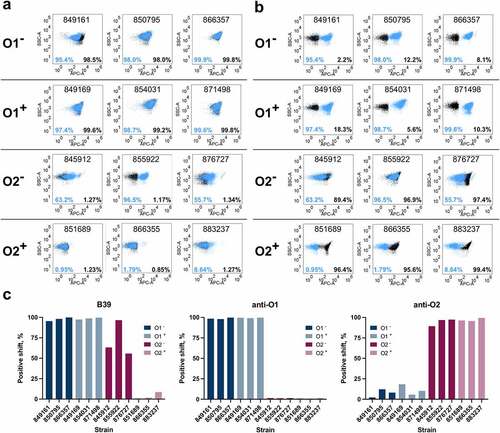
A greater than 95% positive shift for B39 was observed for all O1− and O1+ strains, a result that was consistent with the anti-O1 mAb binding (). Only a small shift for the anti-O2 mAb was observed in these strains. Against the O2 strains, an 89–99% positive shift for the anti-O2 mAb was observed irrespective of the presence or absence of gmlABC genes. Contrastingly, in the O2− group, a positive shift for B39 ranging from 55.7% to 96.5% was observed, but in the O2+ group, a positive shift for B39 was only observed for one strain, and this shift was small (8.64%) (). In both O2 groups, as expected, no positive shift for the anti-O1 mAb was observed (). B39 binding by FACS is summarized in .
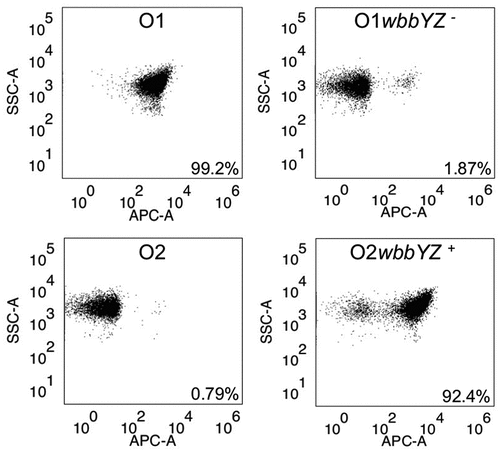
B39 was further tested for binding to two pairs of strains using FACS: K. pneumoniae 1131115 and the isogenic mutant K. pneumoniae 1131115 wbbYZ–, and K. pneumoniae 961842 and the isogenic mutant K. pneumoniae 961842 wbbYZ+ (). The wbbYZ locus encodes D-galactan-II, the O1 subunit that caps the O2 subunit, therefore deletion of these genes converts an O1 strain to O2, whilst incorporation of the genes converts an O2 strain to O1. Deletion of the wbbZY genes in K. pneumoniae 1131115 conferred a loss in binding of B39 in all but a small population (1.87%) of bacterial cells. Correspondingly, incorporation of the wbbYZ genes in the O2 strain led to a 92.4% positive shift for B39. It should be noted that the O1 and O2 isogenic strains were all gmlABC+.
Figure 8. Characterization of binding of B39 to K. pneumoniae O1/O2 isogenic mutants by FACS. Binding of B39 to K. pneumoniae 1131115 (O1), K. pneumoniae 961842 (O2), and the isogenic mutants K. pneumoniae 1131115 wbbYZ – (O1 wbbYZ –) and K. pneumoniae 961842 wbbYZ + (O2 wbbYZ +). The % positive-shift in binding for each strain is labeled.
Discussion
As the effectiveness of antibiotics continues to decline with the rise in antimicrobial resistance, there is an urgent need for alternative antimicrobial therapy options to treat MDR K. pneumoniae infections. In this work, we report the target-agnostic discovery of B39, a mAb that binds to the O-antigen of O1 and O2 K. pneumoniae. B39 was shown to promote the uptake and clearance of bacteria by primary human macrophages. B39 displayed novel in vivo activity in a lethal murine model of pneumonia, conferring therapeutic protection against both O1 and O2 K. pneumoniae strains. To the best of our knowledge, this activity represents the greatest LPS serotype coverage of a therapeutic mAb in the literature to date. O1 and O2 serotypes represent 45% of K. pneumoniae isolates, with this proportion rising to 73% among carbapenem-resistant isolates,Citation14 highlighting the wide potential scope of the development of B39 for use in the clinic to treat K. pneumoniae infections.
It should also be noted that the strains used in the in vivo challenges were both MDR strains from globally dominant clonal groups, highlighting the potential to use B39 against MDR infections. B39 could therefore be of use in situations where antibiotic treatment fails, or alternatively, as an adjunctive therapy alongside last-resort antibiotics. Indeed, synergy between anti-bacterial mAbs and antibiotics has been observed in preclinical models.Citation12,Citation14 Given the high levels of nephrotoxicity of last-resort drugs such as colistin, adjunctive therapies that could permit lower dosage of traditional antibiotics would be welcomed in the clinic.
B39 was shown to bind K. pneumoniae 871498, an ST23 strain with a type 1 capsule. ST23 is the dominant ST among hypervirulent K. pneumoniae isolates,Citation7 highlighting the potential to use B39 as a treatment for infections caused by hypervirulent K. pneumoniae. This is of particular importance due to the recent emergence of MDR hypervirulent K. pneumoniae isolates.Citation5
B39 was shown to mediate OPK by primary human macrophages. We observed that OPK activity was retained in the absence of complement, which could suggest that Fc-receptor recognition and subsequent complement-independent phagocytosis is important for B39 activity. Given that CPS can mediate resistance to complement-dependant opsonization,Citation25 this hypothesis is strengthened by the fact that B39 was active in vivo against two encapsulated strains. However, further work to explore Fc-receptor engagement and the role of complement in vivo is needed to confirm this. LPS-neutralization should also be explored using an endotoxemia model.
Anti-K. pneumoniae O-antigen mAbs cross-reactive to gut commensal bacteria have been reported, mediated by binding to mannose-based repeating units found in K. pneumoniae O3 and O5 O-antigens.Citation26 As yet, there have been no reports of cross-species reactivity observed in mAbs targeting the galactan-rich O-antigens of O1 and O2 strains. However, before the therapeutic or prophylactic use of B39 or derivatives can be considered, cross-species reactivity will need to be explored to assess any possible effects on the microbiota in different organs and locations in the body using a panel of commensal bacteria.
Efforts were also made to identify the target epitope of B39. Given that B39 bound to both O1 and O2 LPS, we hypothesized that B39 bound to the shared O1/O2 galactan repeat unit, D-galactan-I. However, FACS using isogenic mutants revealed that deletion of wbbYZ genes encoding D-galactan-II led to nearly 100% loss in positive shift for B39. Similarly, complementation of an O2 strain with wbbYZ led to a greater than 90% positive shift for B39. These data strongly suggests that B39 binds to D-galactan-II. However, we also reported BLI, HCI, western blotting, FACS, and in vivo data that all suggest some level of binding to some O2 strains. Since O2 strains lack wbbYZ genes, binding of B39 to D-galactan-II alone could not explain this data.
There was considerable variation in B39 binding to O2 strains, which correlated with gmlABC genotype. Negligible binding of B39 to the O2+ strain was observed by HCI, whereas comparatively high binding to the O2− strain was observed. A similar trend was seen when analyzed by FACS using a larger panel of strains. Given that the gmlABC locus encodes the conversion of D-galactan-I to D-galactan-III, these data suggest that B39 binds to D-galactan-I, and not D-galactan-III. Further to this, FACS revealed that one of the three O2+ strains showed a small (8.64%) positive shift for B39. It has been reported that in O2+ strains, 90% of D-galactan-I is converted to D-galactan-III,Citation21 so it is possible that this small positive shift represents binding to the unconverted D-galactan-I. However, given that this positive shift was only observed in one strain, the finding could also suggest that between O2+ strains, variability in the conversion of D-galactan-I to D-galactan-III exists. Another possible explanation for differences in binding by HCI and FACS could be differences in the expression of shielding CPS; indeed, wzi typing revealed 11 unique CPS types in the panel of 12 strains. However, it is noted that consistent binding of both B39 and the anti-O1 mAb to all 6 O1 strains was observed; likewise, binding of the anti-O2 mAb to all 6 O2 strains was also consistent. This consistent binding, despite differing wzi type, suggests that CPS type alone is unlikely to account for the variability in B39 binding observed within the O2+ and O2− groups.
Taken together, the data presented here suggests that B39 exhibits dual binding to D-galactan-I and D-galactan-II but does not bind to D-galactan-III. Given that the O1/O2 isogenic strains were both gmlABC+, both the loss of binding observed by deletion of wbbYZ in the O1 strain and the lack of binding to the O2 strain is therefore expected.
This dual binding is further supported by western blot analysis. Previous work has shown that mAbs targeting D-galactan-II only bind to high molecular weight O1 LPS, whilst binding of D-galactan-I mAbs is detected throughout the LPS size range.Citation27 B39 appears to bind to an antigen throughout the O1− LPS, which would suggest binding to D-galactan-I. In addition, B39 binding to O1+ LPS, in which D-galactan-I is largely absent, was only detected at high molecular weight, suggesting binding to D-galactan-II. This was echoed in the O2+ and O2- strains.
The in vivo activity of B39 against K. pneumoniae 961842, an O2+ strain, conflicts with the in vitro binding data, which could suggest that conversion of D-galactan-I → D-galactan-III may depend on environmental factors that differ between in vivo and in vitro conditions. Nonetheless, the high prevalence of the gmlABC locus in ST258 clones suggests the expression of D-galactan-III is important in a nosocomial setting. This work highlights the need for a wider phenotypical analysis of the O-antigens from a large collection of K. pneumoniae O1 and O2 strains. Moreover, given the discrepancies we observed here between in vitro binding data and in vivo mAb activity, future work should aim to explore the expression of D-galactan-I and D-galactan-III in vivo in different tissues, under different in vitro conditions, and in the environment.
In summary, we report the discovery of a mAb with novel binding and functional activity properties that is a promising candidate for development as a novel biotherapeutic for the treatment and prevention of K. pneumoniae infections.
Materials and methods
Bacterial strains and media
All bacteria were stored at −80°C as 25% glycerol stocks and cultured in Tryptone/Yeast extract (TY) broth. K. pneumoniae strains were purchased from the American Type Culture Collection (ATCC), National Collection of Type Cultures (NCTC) or International Health Management Associates (IHMA). A complete list of strains used in this study is shown in Table S4. K. pneumoniae cultures were grown overnight at 37°C with 280 revolutions per minute (rpm) shaking, then diluted to an optical density (OD) 600nm of 1.0 (~1 × 109 colony forming units (cfu)/ml) for use in phage display. The Escherichia coli strain TG1 was grown at 37°C with 300 rpm shaking to an OD600nm of 0.5 for use as the phagemid recipient in phage display. Where appropriate, antibiotics were used at the following concentrations: ampicillin (100 µg/ml), kanamycin (50 µg/ml) and gentamicin (10 µg/ml). Where necessary, broth was supplemented with 2% glucose to suppress recombinant scFv expression in E. coli.
Phage display on whole K. pneumoniae bacteria
Two naive human scFv libraries were combined and used in this work.Citation28,Citation29 Whole cell selections were performed as described previously,Citation19 using live K. pneumoniae 43816 (WT), K. pneumoniae ∆cpsB (mutant) and K. pneumoniae ∆cpsBwaaL (double mutant). For each selection, 1 × 109 cfu K. pneumoniae and 5 × 1010 phage particles were blocked in phosphate-buffered saline (PBS) supplemented with 3% nonfat dried milk and co-incubated. Unbound phage were removed by washing, and bound phage were eluted and used to infect mid-log phase E. coli TG1 cells for phage amplification in subsequent rounds of selection. scFv sequences were obtained by performing a colony PCR using primers to amplify the variable region, and sequences were analyzed using Blaze, an AstraZeneca in-house sequence analysis platform. Following three rounds of enrichment, scFv were assessed for binding to K. pneumoniae 43816 by phage ELISA. To identify nonspecific binding, scFv were tested for binding to BSA and E. coli TG1 by phage ELISA.
Antibody engineering
For scFv-Fc conversion, scFv sequences were ligated into a vector containing the antibody Fc and expressed in G22 Chinese hamster ovary (CHO) cells in 3 ml expression cultures. For IgG conversion, VH and VL DNA sequences were cloned into human IgG1 and human Lambda expression vectors, respectively. IgG were expressed in CHO cells in 20 ml expression cultures. scFv-Fc and IgG were purified by protein A affinity chromatography using ÄKTA systems.
Biolayer interferometry
Binding of scFv-Fc to various ligands was tested by BLI using the Octet RED384 system (Forte-Bio). The following ligands were used: O1, O2, O4, and O5 LPS, purified from K. pneumoniae 43816 ∆cpsB, 8570 ∆cpsB, 9135 K− and 9181 K−, respectively (10 µg/ml). Ligands and scFv-Fc were diluted in Octet buffer (PBS supplemented with 0.1% BSA (Sigma) and 0.05% Tween). scFv-Fc at a concentration of 100 nM were captured on anti-human Fc biosensors for 2 minutes. Coated biosensors were then incubated with the ligand for 3 minutes, followed by a dissociation step in which biosensors were incubated in Octet buffer for 3 minutes. A reference lane was also included, in which the capture well contained only Octet buffer and no scFv-Fc. For the analysis, the reference mean was subtracted from the data.
Opsonophagocytic killing assay
Luminescent K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB expressing a plasmid containing the luxABCDE operon have been described previously.Citation14,Citation19 Briefly, log-phase cultures were diluted to approximately 3.0 × 105 cells/ml in OPK buffer (RPMI 1640 medium without phenol red (Gibco) + 1% BSA (Sigma)). Baby rabbit serum (Cedarlane), as a source of complement, was diluted 1 in 10 in OPK buffer and incubated with K. pneumoniae for 1 hour to clear the complement of any preexisting antibodies against K. pneumoniae. Test antibodies were serially diluted in OPK buffer at 4x the test concentration. Human MDMs at 3.0 × 104 cells/well in 96-well white, clear bottom microplates (Corning) were suspended in 25 µl OPK buffer. 25 µl each of antibodies, bacteria, and complement were added to the 96-well plate containing macrophages, plates were sealed with Breathe-Easy sealing membranes (Merck) and incubated at 37°C for 5 hours with 5% CO2. Total luminescence units were read using an Envision multilabel plate reader (PerkinElmer). OPK activity of test IgG and isotype control IgG was calculated as a percentage of wells containing no IgG using the following calculation: (IgG treatment/no IgG)*100. In subsequent experiments, the OPK assay was performed without the addition of complement.
High-content imaging
For HCI binding characterization studies, ~5.0 × 105 K. pneumoniae bacteria were added to wells of a 96-well CellCarrier Ultra microplate (PerkinElmer) and incubated at 37°C for 2 hours. Bacteria were fixed in 4% paraformaldehyde (PFA) for 10 minutes, then treated with primary antibodies at 1 µg/ml for one hour. Bacteria were stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) (2 µg/ml) and AF647 anti-human IgG (Invitrogen #A-21445) (1 µg/ml). Images were acquired using an Opera Phenix system (PerkinElmer) at 63 x magnification or an Opera system (PerkinElmer) at 60 x magnification.
For HCI OPK studies, MDMs were seeded in tissue-culture treated 96-well optical-bottom plates (Nunc), and assays were prepared as described for the OPK assay, in the absence of complement. After 7 hours, cells were fixed in 4% PFA, then washed three times in PBS and stained with cell mask orange (1/25,000) (Invitrogen), Hoechst (1/10,000) (Thermo Scientific) and 0.3% Triton, prepared in HBSS supplemented with 5% donkey serum (Jackson Laboratory). Cells were washed three times in PBS and stained with rabbit polyclonal anti-K. pneumoniae 43816 antibody for 30 minutes, then washed once with PBS and stained with AF488 anti-rabbit IgG (Affinipure #111-545-003) for 30 minutes, before a final three washes in PBS. 15 fields per well were acquired using an Opera (PerkinElmer) at 20x magnification.
HCI analysis
HCI analysis was performed in Columbus (PerkinElmer). For quantification of mAb binding to K. pneumoniae 43816 and K. pneumoniae 43816 ΔcpsB, the total AF647 intensity per well was calculated using a cutoff of mean >5000 to eliminate nonspecific background signal. This was normalized to the density of bacteria per well by dividing the AF647 signal by DAPI area (px2). For quantification of mAb binding to K. pneumoniae O1 and O2 strains ± gmlABC, the same analysis pipeline was used, and a cutoff of mean >50 was used to eliminate background. For quantification of the proportion of bacteria that were positive for binding, the AF647 area sum (px2) was divided by DAPI area sum (px2). To eliminate background, a cutoff of mean >50 was used. For quantification of macrophage-associated K. pneumoniae spot intensity in the high-content OPK assay, the macrophage cytoplasm image area was first defined using cell mask orange (CMO) signal. K. pneumoniae spots within the macrophage cytoplasm image area were then defined using AF488 signal, and the total AF488 intensity sum was calculated, excluding spots with a mean AF488 intensity <5 to eliminate nonspecific background signal.
Murine model of pneumonia
C57BL/6 mice were procured from Jackson Laboratory and maintained in a pathogen-free facility. All animal experiments were conducted in accordance with the AstraZeneca Institutional Animal Care and Use Committee protocol and guidance. To prepare bacteria for infection, K. pneumoniae strains (1131115 or 961842) were grown overnight on TY agar. Bacterial scrapes were diluted in saline to the required concentration just prior to infection. To determine the inoculation titer, serial dilutions were plated onto TY agar plates. Mice were anesthetized with isoflurane, then 50 µl inoculum was administered intranasally; test and isotype control mAbs were administered intravascularly 1 hour post challenge (p.c.). Mouse survival was monitored daily for up to 7 days.
Klebsiella isolate sequencing and analysis
Clinical isolates were obtained from the International Health Management Associates (IHMA) as part of a collection from an international antibiotic resistance surveillance program. Basic demographic data (age, sex, hospital location, sample type, and length of stay) were provided for each isolate by the IHMA using a unique study number that was delinked from any patient identification. DNA was purified from bacterial cultures via bead beating followed by extraction using a PureLink Genomic DNA Mini Kit (ThermoFisher). Sequencing libraries were prepared by mechanical shearing of DNA (Covaris) followed by a NEBNext Ultra DNA Library Prep Kit for Illumina (New England BioLabs). Libraries were then run on an Illumina HiSeq, 2×150bp configuration. Sequence data was quality-trimmed, de-novo assembled and annotated using BugBuilder,Citation30 and multilocus sequence typing (MLST) was performed using SRST2.Citation31 In some cases, where whole genome sequences were unavailable, housekeeping genes used for MLST were PCR-amplified using methods described previously,Citation32 with primers listed in Table S5, and then analyzed via the BIGSdb online server (http://bigsdb.pasteur.fr/klebsiella/klebsiella.html).
SDS-PAGE and immunoblotting
Whole-cell lysates were prepared as described previouslyCitation33 with some modifications. Briefly, 1 ml OD600nm cells were pelleted by centrifugation at 21,100 xg, the supernatant was removed, and the bacterial pellet was re-suspended in 100 μl tris-glycine SDS sample buffer (Invitrogen) and lysed for 10 minutes at 95°C prior to proteinase K (Sigma) digestion. Samples were separated on Novex 12% tris-glycine gels (Invitrogen), then transferred onto nitrocellulose membranes using an iBlot device (Invitrogen). Membranes were blocked with Odyssey blocking buffer (Licor) and then probed with B39 at 10 μg/ml; binding was detected using IRDye 680RD Goat anti-Human IgG (Licor #926-68078) and visualized using the Odyssey CLx imaging system (Licor).
Flow cytometry
For flow cytometry experiments, all K. pneumoniae strains were cultured in TY broth. 1 μl overnight K. pneumoniae cultures were added to 200 μl FACS buffer (PBS + 3% fetal bovine serum, 0.1% Tween) and collected by centrifugation at 21,100 xg. This wash step was repeated once, then bacteria were incubated with shaking at 4°C for 1 hour in round-bottom plates with B39, anti-O1 mAb or anti-O2 mAb diluted to 5 μg/ml in FACS buffer. Bacteria were washed twice in FACS buffer, then stained with AF647 goat anti-human IgG (Invitrogen #A-21445) diluted 1/200 at 4°C for 30 minutes, followed by a final two washes. Bacterial cells were analyzed using a Symphony A5 system (BD Biosciences) and data was analyzed using the FlowJo program.
Ethics statement
For use in OPK assays, human peripheral blood mononuclear cells were isolated from leukocyte cones. Cones were supplied by NHS Blood and Transplant Service (UK) as anonymized samples from consenting donors. All animal studies were performed under the guidance and protocol approval of the AstraZeneca Institutional Animal Care and Use Committee. Additional oversight was also provided by Office of Research Protections, US Army Medical Research and Material Command, Animal Care and Use Review Office.
Statistical analysis
All statistical analyses were performed in GraphPad Prism, version 8. For in vivo murine pneumonia models, survival data were plotted as Kaplan Meier curves and analyzed with the logrank Mantel-Cox test; animals treated with B39 were compared with animals treated with RS347, the human isotype control antibody. P-values were corrected for multiple comparisons using the Benjamini-Hochberg methodCitation34 with a false discovery rate of 5%. P values of less than 0.05 were considered statistically significant.
Author contributions
SKB performed the phage display campaign, biolayer interferometry, OPK assay, high-content imaging and western blotting. CC performed in vivo experiments and FACS. SKB, LI and JBS participated in design and analysis of high-content imaging experiments. DT performed genome sequencing and analysis. SKB, AJG, SR, RM, PW, GC and GD participated in experimental design. SKB, AJG, SR and RM devised the study. SKB, AJG, SR and RM interpreted the data. SKB and AJG wrote the manuscript. All authors reviewed the manuscript.
Supplemental Material
Download Zip (1.6 MB)Acknowledgments
This work was funded by AstraZeneca and the Cambridge Biomedical Research Centre. SKB was funded by a Cambridge AstraZeneca PhD studentship. The authors are grateful for the assistance of AstraZeneca scientists John Ferguson, Augustin Harvey and Zachary Britton for the preparation of monocyte-derived macrophages and for antibody production. The authors thank University of Cambridge scientist Sally Forrest for her assistance with high-content imaging.
Disclosure statement
SR, RM, LI, CC, PW, and DT are current/former employees of the AstraZeneca Group and may have/had stock options in AstraZeneca.
Data availability statement
The K. pneumoniae genome sequencing datasets that support the findings of this study can be accessed using the following link https://doi.org/10.17863/CAM.78423 The rest of the data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Friedlaender C. Ueber die Schizomyceten bei der acuten fibrösen Pneumonie. Arch für Pathol Anat und Physiol und für Klin Med. 1882;87:319–13.
- Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11(4):589–603. doi:10.1128/CMR.11.4.589.
- Rock C, Thom KA, Masnick M, Johnson JK, Harris AD, Morgan DJ. Frequency of Klebsiella pneumoniae carbapenemase (KPC)–producing and non-KPC-producing Klebsiella species contamination of healthcare workers and the environment. Infect Control Hosp Epidemiol Off J Soc Hosp Epidemiol Am. 2014;35(4):426. doi:10.1086/675598.
- Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev [Internet]. 2019;32(3):e00001–19. doi:10.1128/CMR.00001-19.
- Lee C-R, Lee JH, Park KS, Jeon JH, Kim YB, Cha C-J, Jeong BC, Lee SH. Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front Cell Infect Microbiol. 2017;7:483. doi:10.3389/fcimb.2017.00483.
- Wyres KL, Hawkey J, Hetland MAK, Fostervold A, Wick RR, Judd LM, Hamidian M, Howden BP, Löhr IH, Holt KE. Emergence and rapid global dissemination of CTX-M-15-associated Klebsiella pneumoniae strain ST307. J Antimicrob Chemother [Internet]. 2019;74(3):577–81. doi:10.1093/jac/dky492.
- Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18(6):1–16. doi:10.1038/s41579-019-0315-1.
- Van Duin D, Arias CA, Komarow L, Chen L, Hanson BM, Weston G, Cober E, Garner OB, Jacob JT, Satlin MJ. Molecular and clinical epidemiology of carbapenem-resistant Enterobacterales in the USA (CRACKLE-2): a prospective cohort study. Lancet Infect Dis. 2020;20(6):731–41. doi:10.1016/S1473-3099(19)30755-8.
- Xu L, Sun X, Ma X. Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Ann Clin Microbiol Antimicrob. 2017;16(1):1–12. doi:10.1186/s12941-017-0191-3.
- Nagy E, Nagy G, Power CA, Badarau A, Szijártó V. Anti-bacterial monoclonal antibodies. Adv Exp Med Biol. 2017;1053:119–53.
- DiGiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, Minter R, Camara MM, Venkatraman V, MacGill RS, Lin J, et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J Exp Med. 2012;209(7):1273–87. doi:10.1084/jem.20120033.
- DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, Bonnell J, Fleming R, Bezabeh B, Dimasi N, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med. 2014;6(262):262ra155. doi:10.1126/scitranslmed.3009655.
- Cohen TS, Pelletier M, Cheng L, Pennini ME, Bonnell J, Cvitkovic R, Chang CS, Xiao X, Cameroni E, Corti D, et al. Anti-LPS antibodies protect against Klebsiella pneumoniae by empowering neutrophil-mediated clearance without neutralizing TLR4. JCI Insight. 2017;2(9):e92774. doi:10.1172/jci.insight.92774.
- Pennini ME, De Marco A, Pelletier M, Bonnell J, Cvitkovic R, Beltramello M, Cameroni E, Bianchi S, Zatta F, Zhao W, et al. Immune stealth-driven O2 serotype prevalence and potential for therapeutic antibodies against multidrug resistant Klebsiella pneumoniae. Nat Commun. 2017;8(1):1991. doi:10.1038/s41467-017-02223-7.
- Szijártó V, Guachalla LM, Hartl K, Varga C, Badarau A, Mirkina I, Visram ZC, Stulik L, Power CA, Nagy E, et al. Endotoxin neutralization by an O-antigen specific monoclonal antibody: a potential novel therapeutic approach against Klebsiella pneumoniae ST258. Virulence. 2017;8(7):1203–15. doi:10.1080/21505594.2017.1279778.
- Diago-Navarro E, Motley MP, Ruiz-Peréz G, Yu W, Austin J, Seco BMS, Xiao G, Chikhaly A, Seeberger PH, Fries BC. Novel, Broadly Reactive Anticapsular Antibodies against Carbapenem-Resistant Klebsiella pneumoniae Protect from Infection. MBio. 2018;9(2):e00091–18. doi:10.1128/mBio.00091-18.
- Diago-Navarro E, Calatayud-Baselg I, Sun D, Khairallah C, Mann I, Ulacia-Hernando A, Sheridan B, Shi M, Fries BC, Burns DL. Antibody-based immunotherapy to treat and prevent infection with hypervirulent Klebsiella pneumoniae. Clin Vaccine Immunol. 2017;24:e00456–16. doi:10.1128/CVI.00456-16.
- Kobayashi SD, Porter AR, Freedman B, Pandey R, Chen L, Kreiswirth BN, DeLeo FR, Keim P. Antibody-mediated killing of carbapenem-resistant ST258 Klebsiella pneumoniae by human neutrophils. MBio. 2018;9(2):e00297–18. doi:10.1128/mBio.00297-18.
- Wang Q, Chang CS, Pennini M, Pelletier M, Rajan S, Zha J, Chen Y, Cvitkovic R, Sadowska A, Thompson JH, et al. Target-agnostic identification of functional monoclonal antibodies against Klebsiella pneumoniae multimeric MrkA fimbrial subunit. J Infect Dis. 2016;213(11):1800–08. doi:10.1093/infdis/jiw021.
- Rukavina T, Tićac B, Susa M, Jendrike N, Jonjić S, Lučin P, Marre R, Dorić M, Trautmann M. Protective effect of antilipopolysaccharide monoclonal antibody in experimental Klebsiella infection. Infect Immun. 1997;65(5):1754–60. doi:10.1128/iai.65.5.1754-1760.1997.
- Clarke BR, Ovchinnikova OG, Kelly SD, Williamson ML, Butler JE, Liu B, Wang L, Gou X, Follador R, Lowary TL, et al. Molecular basis for the structural diversity in serogroup O2-antigen polysaccharides in Klebsiella pneumoniae. J Biol Chem. 2018;293(13):4666–79. doi:10.1074/jbc.RA117.000646.
- Szijártó V, Guachalla LM, Hartl K, Varga C, Banerjee P, Stojkovic K, Kaszowska M, Nagy E, Lukasiewicz J, Nagy G. Both clades of the epidemic KPC-producing Klebsiella pneumoniae clone ST258 share a modified galactan O-antigen type. Int J Med Microbiol. 2016;306(2):89–98. doi:10.1016/j.ijmm.2015.12.002.
- Stojkovic K, Szijártó V, Kaszowska M, Niedziela T, Hartl K, Nagy G, Lukasiewicz J. Identification of D-galactan-III as part of the lipopolysaccharide of Klebsiella pneumoniae serotype O1. Front Microbiol. 2017;8:684. doi:10.3389/fmicb.2017.00684.
- Wick RR, Heinz E, Holt KE, Wyres KL, Diekema DJ. Kaptive Web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J Clin Microbiol. 2018;56(6):e00197–18. doi:10.1128/JCM.00197-18.
- Domenico P, Salo RJ, Cross AS, Cunha BA. Polysaccharide capsule-mediated resistance to opsonophagocytosis in Klebsiella pneumoniae. Infect Immun. 1994;62(10):4495–99. doi:10.1128/iai.62.10.4495-4499.1994.
- Rollenske T, Szijarto V, Lukasiewicz J, Guachalla LM, Stojkovic K, Hartl K, Stulik L, Kocher S, Lasitschka F, Al-Saeedi M, et al. Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen. Nat Immunol [Internet]. 2018;19(6):617–24. doi:10.1038/s41590-018-0106-2.
- Whitfield C, Richards JC, Perry MB, Clarke BR, MacLean LL. Expression of two structurally distinct D-galactan O antigens in the lipopolysaccharide of Klebsiella pneumoniae serotype O1. J Bacteriol. 1991;173(4):1420–31. doi:10.1128/jb.173.4.1420-1431.1991.
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14(3):309–14. doi:10.1038/nbt0396-309.
- Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel. 2009;22:159–68. doi:10.1093/protein/gzn058.
- Abbott JC. BugBuilder - an automated microbial genome assembly and analysis pipeline. bioRxiv [Internet]. 2017:148783. Available from: http://biorxiv.org/content/early/2017/06/11/148783.abstract.
- Inouye M, Dashnow H, Raven L-A, Schultz MB, Pope BJ, Tomita T, Zobel J, Holt KE. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med [Internet]. 2014;6(11):90. doi:10.1186/s13073-014-0090-6.
- Diancourt L, Passet V, Verhoef J, Grimont PAD, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol [Internet]. 2005;43(8):4178–82. doi:10.1128/JCM.43.8.4178-4182.2005.
- Hitchcock PJ, Brown TM. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol. 1983;154(1):269–77. doi:10.1128/jb.154.1.269-277.1983.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B [Internet]. 1995;57:289–300. Available from: http://www.jstor.org/stable/2346101.