
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Monoclonal antibodies are susceptible to chemical and enzymatic modifications during manufacturing, storage, and shipping. Deamidation, isomerization, and oxidation can compromise the potency, efficacy, and safety of therapeutic antibodies. Recently, in silico tools have been used to identify liable residues and engineer antibodies with better chemical stability. Computational approaches for predicting deamidation, isomerization, oxidation, glycation, carbonylation, sulfation, and hydroxylation are reviewed here. Although liable motifs have been used to improve the chemical stability of antibodies, the accuracy of in silico predictions can be improved using machine learning and molecular dynamic simulations. In addition, there are opportunities to improve predictions for specific stress conditions, develop in silico prediction of novel modifications in antibodies, and predict the impact of modifications on physical stability and antigen-binding.
Introduction
Monoclonal antibodies (mAbs) have become one of the predominant classes of therapeutic proteins and they are used to treat various diseases, such as breast cancer, multiple sclerosis, and asthma.Citation1 Therapeutic antibodies can be engineered to have high specificity and affinity for their targets.Citation2 However, physical and chemical instabilities can have a negative impact on the manufacturability, safety, and efficacy of therapeutic antibodies.Citation3 Like all proteins, mAbs are susceptible to chemical degradation (e.g., oxidation)Citation4 and enzymatic modifications (e.g., sulfation) during cell culture.Citation5 Chemical and enzymatic modifications contribute to heterogeneity. For example, asparagine (Asn) deamidation can generate charge variants; tryptophan (Trp) oxidation can generate hydrophilic or hydrophobic variants.Citation6 In addition, chemical modifications can affect the physical stability and biological activity of antibodies. For example, isomerization within the Fab region can reduce conformational stability,Citation7 whereas deamidation within complementary-determining region (CDR) loops can reduce binding affinity.Citation8 Therefore, identifying liable sites for post-translational modifications (PTMs) has become a critical step in assessing the developability of therapeutic candidates.
Developability assessments aim to identify candidates with long-term stability, manufacturability, and low heterogeneity.Citation9 Forced degradation with thermal, pH, and light stress has been used to accelerate chemical degradation and identify liable residues.Citation10 Peptide mapping can identify the specific sites for chemical modifications after forced degradation. In contrast, chromatographic techniques such as cation exchange chromatography and hydrophobic interaction chromatography can monitor the overall change in charge and hydrophilic variants, respectively.Citation11 However, experimental approaches for identifying PTM liabilities are time-consuming and require high quantities of the purified protein.Citation12 The sample preparation and data analysis for peptide mapping is incredibly labor-intensive.Citation13 At earlier stages of drug development, the number of forced degradation conditions is limited by the low availability of the purified protein.Citation10 Computational tools are becoming more common during developability assessments due to the low cost, lack of sample consumption, and high speed. In the past decade, computational tools have been used to predict PTM liable sites and engineer antibodies with better chemical stability.Citation14
Computational approaches for predicting PTMs
Computational approaches for predicting PTMs can be divided into three categories: sequence-based, structure-based, and physics-based. Sequence-based approaches either flag individual residues prone to chemical degradation (e.g., methionine oxidation) or liable motifs (e.g., NG, NS, and NT for deamidation).Citation15 Liable motifs for deamidation and isomerization were identified by investigating the effects of protein sequence on deamidationCitation16 and isomerizationCitation17 rates for model peptides. Model peptides are suitable for assessing the effects of protein sequence on chemical degradation due to the generation of substantial chemical degradation in a short time.Citation17
Structure-based approaches predict PTM liabilities by using structural features correlated with enzymatic and chemical modifications.Citation18 Common structural features include, but are not limited to, secondary structure, water coordination number (WCN), and solvent-accessible surface area (SASA).Citation9 WCN represents the average number of water molecules within the radius of an atom;Citation19 SASA represents the surface area of the protein that interacts with the solvent.Citation20 Structural features such as SASA are typically extracted from crystalCitation21 or predicted structures for antibodies.Citation22 Alternatively, machine learning algorithms (e.g., NetSurfP) can also be used to predict structural features such as secondary structure, solvent exposure, and structural disorder for protein sequences.Citation23
Physics-based approaches are based on physical principles: for example, molecular dynamic (MD) simulations use Newtonian physics to predict the spatial position of atoms over time.Citation24 Physics-based approaches can predict the free energy barriers for chemical modificationsCitation25,Citation26 and probe protein dynamics.Citation27 MD simulations can be used to estimate averaged SASA, which captures changes in solvent exposure due to conformational changes. In addition, MD simulation can provide the root-mean-square fluctuations (RMSF) for Cα atoms, which captures structural flexibility.Citation28
Comparison of computational approaches
Sequence-based approaches are simple and easy to implement. Once the protein sequence is available, candidates with poor chemical stability can be eliminated by checking for liable motifs.Citation15 However, using liable motifs alone can overestimate the number of liable residues or miss potential degradation hotspots.Citation12 The rate of chemical degradation for model peptides does not represent chemical degradation in native proteins, where the tertiary structure plays a role in chemical and enzymatic modifications.Citation16 For example, high solvent exposure can increase the risk of chemical modifications due to the increased exposure to degrading agents such as water, peroxides, and light.Citation27
Structure-based approaches have better accuracy than sequence-based approaches because the in silico prediction includes the impact of secondary and tertiary structure on PTMs.Citation29 The accuracy of structure-based approaches is dependent on the quality of crystal or predicted structures and the selection of structural features. Knowledge of the chemical mechanisms for modifications (e.g., deamidation) can improve the accuracy of structure- and physics-based approaches. For example, some studies use the nucleophilic attack (Cγ – Nn+1) distance to predict the risk of deamidation due to the nucleophilic attack on the side chain by the backbone nitrogen during succinimide formation.Citation30,Citation31
Although physics-based approaches provide valuable insight into the dynamics and energetics of chemical degradation, physics-based approaches are more computationally demanding compared to sequence- and structure-based approaches. It is challenging and time-consuming to predict free energy barriers and conduct MD simulations for larger proteins like antibodies.Citation32 Improvements in the speed and accuracy of MD simulations and quantum chemical calculations are required for the broader adoption of physics-based approaches. Moreover, coarse-grained models can potentially improve the efficiency of MD simulations for larger proteins such as antibodies.Citation33
More recent studies combine these three computational approaches for in silico prediction of PTMs. Protein sequence and structural features have been incorporated into machine learning algorithms (e.g., random forest) to classify cold and hot spots for chemical modifications.Citation34 This review discusses current computational approaches for predicting isomerization, deamidation, oxidation, and lysine glycation in mAbs. In silico prediction of novel PTMs in mAbs, such as carbonylation, sulfation, and hydroxylation, is also considered.
Aspartic acid isomerization
Aspartic acid (Asp) residues in mAbs can undergo isomerization to form IsoAsp. During isomerization, there is a nucleophilic attack on the carbonyl group of Asp by the ionized amine group at the n + 1 residue. The Asp side chain requires a proton (H+) to form the succinimide intermediate and leaving water molecule, and thus isomerization is more favorable at low pH.Citation35 Finally, the succinimide intermediate is hydrolyzed at the n + 1 peptide bond to form isoAsp.Citation17 At pH 4–7, Asp isomerization forms succinimide, which can cause a change in net charge. In contrast, IsoAsp formation can cause conformational changes because of the methyl group added to the peptide backbone. These conformational changes can cause changes in surface charge distribution or surface hydrophobicity.Citation5 Depending on the location of IsoAsp formation, isomerization can generate basic,Citation8 acidic,Citation36 hydrophilic, or hydrophobic variants.Citation37 Aspartate isomerization at the CDR loops can decrease the antigen-binding affinity.Citation38
Asp isomerization is sensitive to temperature and the dielectric constant of the solvent. At neutral pH, a high dielectric constant for solvents increases the pKa of Asp, which increases the concentration of the carboxylic acid (COOH) form of the Asp side chain. The COOH form is more reactive and prone to isomerization than the carboxylate form (COO−).Citation39 High temperatures can also accelerate the rate of isomerization reactions. Moreover, flanking residues, ionization state, and higher-order structure also influences isomerization.Citation40 For example, the risk of isomerization is highest for Asp residues within random coils due to structural flexibility and higher solvent exposure.Citation41 The risk of isomerization also differs for different CDR loops. Asp isomerization at liable motifs is more likely with the CDR H3, H2, and L1 loops.Citation42
Sequence-based approaches
Sequence-based approaches for predicting Asp isomerization flag liable motifs for Asp isomerization such as DG, DS, DD, DT, and DH.Citation42 Flanking glycine (Gly) residues make succinimide formation more favorable due to the lower steric hindrance. Serine and threonine residues can act as proton donors during isomerization.Citation40 Positively charged residues at n + 1 or n-1 position can accelerate Asp isomerization due to electrostatic effects.Citation35 Moreover, flanking residues that act as proton donors and acceptors can make Asp residues more reactive and prone to isomerization. For example, histidine residues can act as proton donors (H+) to OH− ions at low pH during succinimide formation.Citation43
During the developability stage, protein engineering can be used to remove PTM liabilities and improve chemical stability.Citation14 The risk of isomerization can be reduced by substituting Asp with glutamic acid, which is less prone to isomerization. Patel et al. identified a liable DS motif within the CDR loop that was prone to isomerization: substituting Asp with glutamic acid led to a loss in bioactivity. However, engineering the n + 1 residue (i.e., serine) retained the bioactivity while reducing the risk of Asp isomerization.Citation44
Structure-based approaches
The higher-order structure of antibodies also plays a crucial role in the risk for Asp isomerization. Dihedral angles can influence the reactivity of Asp residues and affect the risk of isomerization.Citation26 Diepold and colleagues observed isomerization for liable motifs within the solvent-exposed and flexible CDR loops. However, isomerization did not occur at the DG motif in the conserved region of the antibody.Citation45 Asp isomerization is the least favorable within beta-sheets due to structural rigidity. Sreedhara and colleagues observed succinimide formation for Asp74, which was solvent-exposed and part of a loop, whereas the Asp73 within beta-helix was less prone to isomerization.Citation40 In addition, hydrogen bonding for peptide backbone and side chains can make succinimide formation less favorable.Citation46
Structure-based approaches for predicting Asp isomerization incorporate solvent exposure, secondary structure, nucleophilic attack (Cγ – Nn+1) distance, and hydrogen bonding.Citation46 Sydow and colleagues used the root mean square deviation of Asp Cα atoms, size of the n + 1 residue, and secondary structure to develop a decision tree for classifying liable Asp residues.Citation46 They reported that Asp residues with high conformational flexibility and small size for n + 1 residue were more prone to isomerization. Although, a smaller side chain leads to lower steric hindrance for forming succinimide;Citation26 using the size of n + 1 residue for predicting isomerization neglects potential electrostatic effects from flanking residues on isomerization.Citation35
Physics-based approaches
MD simulations can supplement structure-based approaches for predicting Asp isomerization. Sharma et al. developed a logistic regression model to predict Asp isomerization at liable motifs (i.e., DG, DS, DT, DD, DH). Citation29 The averaged SASA for Asp residues, RMSF for Cα atoms, and the averaged SASA for the n + 1 residue correlated well with Asp isomerization liability. Sharma and colleagues’ regression model successfully predicted five out of six liable Asp residues and all non-liable sites. Most of the parameters used in the study were derived from MD simulations. Thus, MD simulations can provide insight into how conformational flexibility plays a role in chemical degradation.
Recent studies have used MD simulations to predict chemical modifications; however, few studies attempt to predict the free energy barriers for chemical modifications. Plotnikov et al. used quantum mechanical/molecular mechanics (QM/MM) MD simulations to identify isomerization and deamidation hotspots. They estimated the free-energy barriers at different steps for isomerization and deamidation. The steric hindrance for succinimide formation is captured by the free energy barriers for adopting more reactive conformations to form succinimide.Citation26 In addition, Plotnikov et al. included long- and short-range electrostatic interactions in their QM/MM model. Compared to structure-based approaches, free energy predictions can incorporate electrostatic effects on Asp isomerization.Citation26
Therapeutic candidates with high and low risk for isomerization can be classified based on liable motifs and structural predictors such as SASA. In addition, knowledge of the antigen-binding sites, either from crystal structures or in silico predictions, can guide protein engineering to improve chemical stability. For example, if a liable Asp residue is involved in antigen binding, then the n + 1 residue can be substituted to improve chemical stability without affecting the bioactivity of the antibody.Citation44
Asparagine deamidation
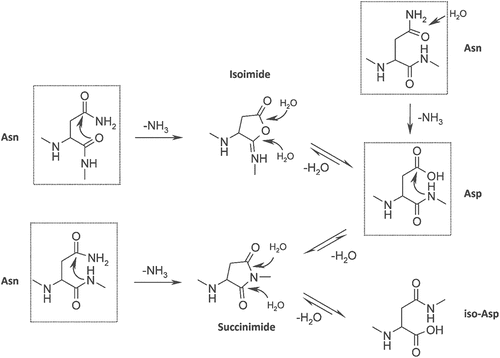
Asn residues can undergo deamidation to form Asp or IsoAsp. There are three possible pathways for Asn deamidation. A nucleophilic attack on the backbone nitrogen at the n + 1 residue forms succinimide, which is hydrolyzed to form Asp. Citation46 Alternatively, the nucleophilic attack by the backbone carbonyl group can form isoimide, which is hydrolyzed to IsoAsp and Asp (). At acidic pH (pH ≤ 4), direct hydrolysis of Asn can also form Asp ().Citation47 The direct hydrolysis pathway competes with succinimide-mediated deamidation.Citation48 Overall, deamidation is more favorable at pH ≥ 6. Forced degradation by thermal and high pH stress are used to confirm liable Asn residues.Citation10 Flanking residues, secondary and tertiary structure, solvent exposure, and structural flexibility can affect deamidation.Citation49
Figure 1. Asparagine deamidation and aspartic acid isomerisation pathway. Reproduced from Sydow JF et al. (2014).Citation46 (Creative Commons Attribution License).
Sequence-based approaches
Liable motifs for deamidation include NG, NS, NN, NT, and NH:Citation42 the NG motif has the highest deamidation rate, followed by NS, NT, and NH.Citation16 The high deamidation rate for the NG motif can be attributed to lower steric hindrance for forming succinimide. Alternatively, Gly residues at the n + 1 position can provide more conformational flexibility, allowing Asn residues to adopt more reactive conformations to form succinimide.Citation41 The n + 1 flanking residues can also play a role in deamidation. Chelius et al. reported the highest level of deamidation at SNG, ENN, LNG, and LNN.Citation50 Lu et al. investigated the deamidation for 131 clinical-stage antibodies and reported deamidation at liable motifs was more common at CDR-H2 and CDR-L1 loops.Citation42 Deamidation at NT, NF, and NY only occurred within the CDR-H3 loop,Citation14 which could be due to the length and flexibility of the CDR-H3 loop, allowing it to adopt more conformations.Citation51 Excluding the CDR-H3 loop, the remaining CDR loops are clustered into canonical structures, where a few critical amino acids at specific positions can determine the CDR conformation.Citation52 Non-H3 CDR loops are clustered based on structural similarity: dihedral angles and root mean square deviation have been used to compare and cluster structures for the same type of CDR loops (e.g., CDR-L1).Citation53 For example, the L1-11-A cluster refers to CDR-L1 loops with a length of 11 residues, which fall into the first cluster of CDR-L1 structures. Citation53 Future studies could investigate how differences in conformations for different CDR-L1 and H2 clusters affect the risk of deamidation.
Structure-based approaches
The higher-order structure also influences the rate of Asn deamidation.Citation45 The risk of deamidation is low for Asn within beta-sheets due to structural rigidity and an extensive network of hydrogen bonds. Structure-based approaches for predicting deamidation incorporate secondary structure, hydrogen bonding, SASA, dihedral angles, and nucleophilic attack (Cγ – N) distance.Citation30 Random forest and decision trees are the most common machine learning algorithms for classifying hot and cold spots (). Delmar et al. developed a random forest model to identify liable Asn residue and predict the deamidation rate.Citation30 The backbone and side-chain dihedral angles are used to assess if the Asn side chain and backbone alignment are favorable for forming succinimide. However, the backbone dihedral angles are more critical than side-chain dihedral for predicting deamidation hot spots.Citation30,Citation54
Table 1. Summary of parameters used for machine learning methods for predicting asparagine deamidation in mAbs. Abbreviations: RMSD, root mean square deviation; SASA, solvent-accessible surface area
There are contradicting results for the impact of Cγ – Nn+1 distance on Asn deamidation. Yan et al. reported that a Cγ – Nn+1 distance > 3.4 Å was unfavorable for Asn deamidation, Citation54 whereas Sydow et al. found no correlation between deamidation risk and Cγ – Nn+1 distance.Citation46 The discrepancy in results could be due to different stress conditions used to identify liable Asn residues for both studies. Yan et al. used high pH stress to identify liable residues, while Sydow et al. thermally stressed their samples at the formulation pH of 6.0. At basic pH, deamidation by forming succinimide is more favorable, and the Cγ – Nn+1 distance is a useful predictor for succinimide formation.Citation48 Therefore, the impact of structural features such as Cγ – Nn+1 distance on deamidation can differ between thermal stress at acidic and basic pH. Future studies could select different structural features to develop machine learning models to predict the risk of deamidation for different types of stress conditions.
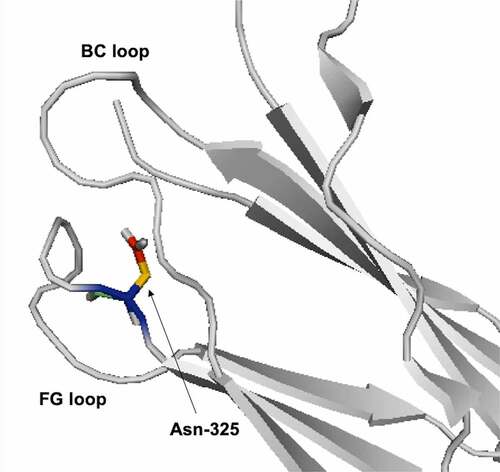
Antibodies can also change conformations at different pH, which can cause a change in the solvent exposure for Asn residues. At high pH, heavy chain (HC)-Asn325 at the FG loop of the CH2 domain is buried between the FG and BC loop (). There is a conformational change in the BC loop at low pH, which causes HC-Asn325 to become exposed and susceptible to deamidation.Citation54 The CDR-H3 loop can adopt multiple conformations due to its length and flexibility. Lan et al. observed different conformations for the CDR-H3 loop at different pH. At neutral pH, the CDR-H3 loop is extended and solvent-exposed. In contrast, at low pH, the CDR-H3 loop is bent. Citation55 Future studies could use MD simulations to identify more reactive conformations for deamidation at different pH.
Figure 2. Ribbon diagram representation for HC-Asn325 between BC and FG loop (PDB: 4BYH).
Oxidation
Oxidation of antibodies can occur due to peroxides within formulations, exposure to trace metals during manufacturing, and light exposure. Aromatic amino acids and the sulfur groups of methionine and cysteine residues are the most susceptible to oxidation.Citation56 Methionine (Met) oxidation generates sulfoxides and sulfone. Histidine (His) oxidation generates oxo-His. For tryptophan (Trp) oxidation, singlet oxygen reacts with the indole group to form kynurenine and N-formylkynurenine.Citation4,Citation49 Met is the most prone to oxidation due to the reactive sulfur atom.Citation57
Sequence-based approaches flag all Met and Trp residues in the variable domains as liable residues.Citation58 However, sequence-based approaches can overestimate the risk of oxidation due to the impact of higher-order structure on oxidation. Exposed Met, Trp, and His are more prone to oxidation. SASA is a common predictor for identifying liable Met and Trp residues.Citation9 More recent studies have successfully incorporated MD simulations and machine learning to predict Met and Trp oxidation.Citation29,Citation59However, specific factors that affect oxidation risk can vary depending on the type of liable residue and the type of oxidative stress.Citation60 The subsequent sections outline in silico prediction for Met, Trp, and His oxidation in more depth.
Methionine oxidation
Met is oxidized by peroxides, light stress, and trace metal ions. Oxidation by peroxides involves transferring oxygen from the peroxide to Met through nucleophilic substitution.Citation61 Water molecules stabilize the transition state near the sulfur atom.Citation62 Alternatively, Met can also react with singlet oxygen to form Met sulfoxide. Met oxidation in mAbs can reduce conformational stability,Citation63 generate hydrophilic variants,Citation5 cause structural changes,Citation64 and affect antigen binding.Citation65 Met residues within CDR loops are more prone to oxidation than Met residues within framework regions. For example, Yan et al. reported oxidation of Met-56 in the solvent-exposed CDR-H2 loops.Citation7
SASA and WCN are useful predictors when water molecules play a role in stabilizing the transition state during oxidation by H2O2.Citation62 WCN represents the average number of water molecules within the radius of an atom.Citation19 The accuracy of WCN as a predictor depends on the selected radius. If the radius is too small, the risk of Met oxidation is underestimated; if the radius is too big, the risk of Met oxidation is overestimated. A radius of 5 Å- 6 Å for WCN is sufficient for predicting Met oxidation. Citation19,Citation66 Recent studies use the averaged SASA values of Met residues from MD simulations. MD simulations capture changes in solvent exposure for Met residues due to the flexibility of flanking residues and conformational changes.Citation23 In addition, the SASA of the sulfur atom can be a better predictor than overall SASA for Met residues.Citation19
Machine learning models have been used to identify liable Met residues.Citation22,Citation59 Yang and colleagues used the SASA of Met side chains derived from a random forest model to identify liable Met residues for 121 antibodies.Citation67 They observed a strong correlation between side-chain solvent exposure and experimental Met oxidation, but there were some false negatives. The study used SASA values from static crystal structures, which fails to capture changes in solvent exposure of Met residues due to conformational changes.Citation67 Sankar et al. developed a random forest model incorporating SASA, WCN, and distance between sulfur and aromatic residues, to predict Met oxidation by 2,2ʹ-azobis(2-amidinopropane) dihydrochloride (AAPH). Most of the incorrect predictions for Met oxidation were for the CDR-H3 loop. Citation22 Predicting the structure of CDR-H3 is challenging due to the longer length and high structural variation of the CDR-H3 loops.Citation68 Thus, the accuracy of SASA values is limited by the quality of the crystal or predicted structures.
Although MD simulations have provided more accurate SASA values, most studies conduct MD simulations with water. Excipients and buffers impact the risk of Met oxidation and the hydrodynamic radius of antibodies.Citation69 Changes in hydrodynamic radius of IgG can lead to changes in solvent exposure for Met residues. Conducting MD simulations with the formulation buffer can further bridge the gap between in silico predictions and experimental data. In addition, in silico tools can be used to identify formulations with a lower risk of Met oxidation.Citation70
In silico prediction of Met oxidation by peroxides is very well established. More recent studies have explored in silico prediction for Met oxidation under light stress and forced oxidation by AAPH. Citation22,Citation59 The mechanism for Met oxidation by peroxides and photolysis is different. Hydrogen peroxide oxidizes Met residues through nucleophilic substitution, whereas light exposure oxidizes Met by generating free radicals.Citation4 In addition to using solvent exposure, Delmar and colleagues also used the closest atomic distance between phenylalanine and Met residues as a predictor for Met oxidation under light stress. Neighboring aromatic residues can prevent Met oxidation by acting as scavengers for free radicals.Citation59 Forced oxidation by H2O2 mimics exposure to trace peroxides from polysorbates in formulations, whereas photolysis is meant to mimic degradation due to light exposure during shipping and storage.Citation71 Therefore, different predictors for Met oxidation (e.g., WCN) should be selected for different types of forced oxidation (e.g., peroxides and photolysis).Citation19
Tryptophan oxidation
Trp oxidation can cause color changes due to kynurenine formation,Citation72 reduce physical stability,Citation73 and oxidation of Trp residues in CDR loops can reduce binding affinity.Citation65 Structural flexibility, solvent exposure, secondary structure, and side-chain and backbone conformations affect Trp oxidation.Citation59 Most aromatic residues get buried within the hydrophobic core during protein folding;Citation74 however, the CDR loops are enriched with tyrosine and Trp residues, which play a role in antigen binding.Citation75
Trp oxidation is more common in the solvent-exposed and flexible CDR loops:Citation9 Trp oxidation in CDR-H3,Citation76 CDR-L1,Citation77 CDR-L3,Citation78,Citation79 CDR-H2, and CDR-L2 loopsCitation60 has been reported. Neighboring aromatic residues and disulfide bonds can prevent photooxidation of Trp residues by acting as a light sink.Citation80 IgG1 antibodies have a conserved Cys-Cys-Trp triad, where the disulfide bond protects the neighboring Trp residue from photooxidation. Electron transfer from the Trp residue to the disulfide bond occurs as the disulfide bond dissipates the UV energy absorbed by Trp residues. Citation80 Solvent exposure is the predominant predictor for Trp oxidation. SASA from static structures does not capture fluctuations in the exposure of Trp residues due to conformational changes. An averaged SASA sampled from MD simulations can provide a more accurate prediction for Trp oxidation. Solvent exposure for Trp side chains is a good predictor because the oxidation of the indole ring is a critical step for forming kynurenine.Citation27 In addition, SASA for Trp side chains can be an accurate predictor for oxidation in cases where the Trp backbone is buried, but the side chain is exposed. Sharma et al. observed that a Trp side chain SASA of > 80 ÅCitation2 correlated well with Trp oxidation after incubation with AAPH.Citation29
Other structural factors also influence the oxidation rate and liability for different Trp residues (e.g., side chain and backbone dihedral angles) and the electrostatic environment.Citation60 Recently, Delmar and colleagues developed a random forest model to predict Trp oxidation under light stress, which incorporated secondary structure and psi and phi angles.Citation59 Future studies could investigate the role of other structural factors on Trp oxidation for a broader panel of antibodies.
The risk of Trp oxidation can also differ under long-term thermal stress compared to forced oxidation by AAPH and photolysis. For example, Jacobitz et al. reported that solvent exposure of Trp residues correlated better with Trp oxidation under long-term thermal stress than oxidation by AAPH and light stress.Citation60 Future studies can continue to tailor in silico prediction of Trp oxidation to specific stress conditions, such as thermal stress, photolysis, and forced oxidation by AAPH and peroxides combined with trace metals.
Histidine oxidation
Photo-oxidation and metal-catalyzed oxidation (MCO) are the primary mechanisms for His oxidation. During light stress or MCO, a singlet oxygen reacts with the imidazole ring of His to form peroxide intermediates that then yield oxo-His or cross-linked products.Citation4 Recently, His oxidation has been reported for mAbs: Luo et al. observed oxidation at His-304 and His-428 for IgG2 antibodies after incubation with Cu2+/ascorbate,Citation81 and Amano et al. reported His oxidation in the CH2 domain of an IgG1 antibody under light stress.Citation82 Studies have also reported cross-linking of mAbs caused by His oxidation, with cross-linking being more favorable at higher pH.Citation83 For IgG1 antibodies, oxidized His residues in the Fc and hinge region can cross-link with His, cysteine, and lysine residues after light stress.Citation84 Interestingly, His-His cross-linking has also occurred between the His side chain of an IgG4 antibody and the free His in the formulation.Citation85 Therefore, in silico tools can be used to select formulations for antibodies with a higher risk of His oxidation.
Solvent exposure,Citation85 conformational flexibility,Citation84 surrounding residues, and pKa influence His oxidation. Not all solvent-exposed His residues will get oxidized,Citation82 but solvent exposure is still a prerequisite for His oxidation. His residues within highly flexible and solvent-exposed regions are more prone to oxidation. For example, the oxidized HC-His440 and HC-His292 were located at the CH2-CH3 interface within loops,Citation84 whereas the oxidized His231Citation84 and His220Citation83 were within the hinge region. Surrounding residues can also enhance or inhibit His oxidation. Neighboring Trp residues can act as photosensitizers and increase the risk of His oxidation. Under light stress, Trp residues can generate singlet oxygen or superoxide anions, which can oxidize neighboring His residues.Citation86,Citation87
Studies on the impact of pKa on the risk of His oxidation have yielded inconsistent results. For example, Amano et al. reported no correlation between pKa of His residues and His oxidation,Citation82 whereas Miyahara et al. reported His residues with lower pKa values were more prone to photooxidation.Citation88 The discrepancy in the reported role of pKa on His oxidation could be due to the different stress conditions (i.e., MCO versus UVC light exposure) used in these studies and the different by-products formed after the stress condition (i.e., oxo-His versus Asn and Asp residues). Compared to Met and Trp oxidation, in silico prediction of His oxidation is understudied.Citation49 Future studies could investigate the impact of pKa on His oxidation for different types of oxidative stress (e.g., photooxidation versus MCO).
Lysine glycation
Glycation is a non-enzymatic modification, where amino groups at lysine (Lys) and arginine residues or the N-terminal are glycated by reducing sugars such as glucose.Citation89 The Schiff base is formed after a condensation reaction between the aldehydes of the reducing sugars and the amine groups of Lys residues ()). Schiff base formation is reversible; however, the multistep Amadori rearrangement can generate more stable ketoamines ()).Citation90 Glycated Lys residues can further degrade to form advanced glycation end products,Citation91 which can cause an immunogenic response. Lys glycation typically occurs during long-term storage in formulations, cell culture, and in vivo.Citation92 Disaccharides in formulations can degrade to form reducing sugars, leading to glycation during storage.Citation92 During cell culture, a high glucose concentration can accelerate Lys glycation and reduce protein yield.Citation89 The effects of Lys glycation on antigen binding varies from decreased binding affinityCitation93 to minimal change in binding affinity.Citation94 Lys glycation can increase protein aggregation by affecting the net surface charge, reducing the electrostatic repulsion between antibodies.Citation95 Forced glycation is used to identify liable Lys residues by incubating the IgG with high concentrations of reducing sugars (e.g., glucose) at high temperatures.Citation92 Forced glycation in citrate buffers correlates well with glycation during cell culture.Citation96
Figure 3. (a) Mechanism for Schiff base formation. (b) Mechanism for Amadori rearrangement on glycated lysine.
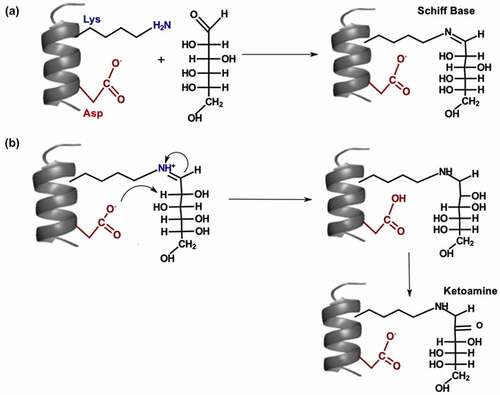
The use of SASA to identify liable residues is a popular approach.Citation27 High solvent exposure is a prerequisite for Lys glycation, but not all exposed Lys residues are glycated.Citation96 Lys glycation is also influenced by flanking residues (i.e., polar, Citation97 acidic, and basic residuesCitation92), three-dimensional structure, and pKa of Lys residues.Citation90 Neighboring Asp and His residues can slow Schiff base formation; however, flanking His and Asp residues can promote Amadori rearrangement by acting as proton donors and acceptors ()).Citation90,Citation97 Liable motifs for glycation include KD, KXD, KXK, and KXE.Citation96 Structure-based approaches can identify acidic residues that are not part of motifs, but are close enough to catalyze glycation.Citation98 For example, Zhang et al. reported glycation at light chain (LC)-Lys49, which was 11 Å apart from LC-Asp31.Citation97 They suggested that the flexibility of Lys and Asp side-chains promoted Amadori rearrangement by reducing the distance between LC-Lys49 and LC-Asp31.Citation97
Basic residues such as arginine can promote Schiff base formation by lowering the pKa of the Lys side chain.Citation90,Citation95 Lys side chains with lower pKa values are more likely to adopt the deprotonated form (NH2), which is more favorable for forming the Schiff base. The deprotonated form of the Lys side chain can perform a nucleophilic attack on the carbonyl carbon of the sugar to form the Schiff base ()). Miller et al. suggested that neighboring arginine and His residues increased reactivity of HC-Lys98 by influencing the pKa of HC-Lys98.Citation95 However, Zhang et al. found no correlation between Lys side chain pKa and glycation risk.Citation97 They inferred that Amadori rearrangement ()) was the rate-limiting step for the glycation of a recombinant humanized mAb. Thus, the impact of the Lys side chain pKa on glycation may depend on the rate-limiting step for Lys glycation. QM/MM simulations of free energy calculations could be used to help identify the rate-limiting step for the glycation of different antibodies. Moreover, future studies could investigate if different backbone and side-chain conformations are more favorable for glycation.
Carbonylation
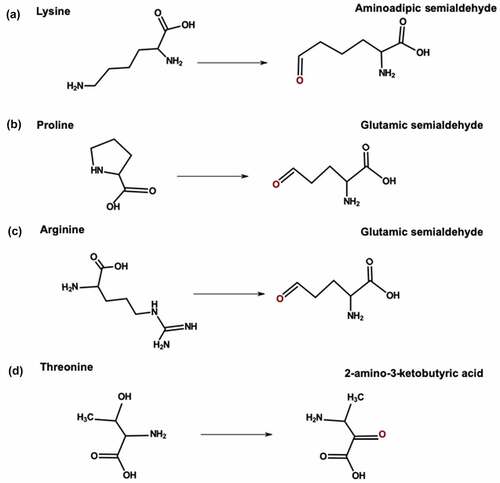
Metal ions can catalyze oxidative carbonylation of arginine, Lys, proline (Pro), and threonine residues. Transition metals such as iron and copper can convert oxygen (O2) to superoxide radical anions (O2–·).Citation99 During carbonylation, free radicals attack the side chain and add an amine or ketone group. Arginine and Pro are converted to glutamic semialdehyde; Lys is converted to aminoadipic semialdehyde, and threonine is converted to 2-amino-3-ketobutyric acid ().Citation100 Exposure to trace metals from stainless steel surfaces and glass vials can cause carbonylation of mAbs during manufacturing and storage.Citation101 Carbonylation of arginine and Lys residues leads to a loss in positive charge, which generates acidic variants. For example, Yang et al. reported increased acidic variants after forced oxidation with ferrous sulfate and hydrogen peroxide.Citation102 Oxidative carbonylation of mAbs can also increase protein aggregation.Citation103
Figure 4. Carbonylation pathway for (a) Lysine (b) Proline (c) Arginine and (d) Threonine.
Lys is most susceptible to carbonylation, followed by Pro as the next most susceptible residue, and arginine and threonine are the least susceptible residues. Pro residues tend to form kinks that make Pro residues more exposed and increases the risk of carbonylation.Citation104 Carbonylation sites are more likely to occur in regions enriched with arginine, Lys, Pro, and threonine residues. Regions at the N- and C-terminal of the carbonylation sites tend to be enriched with positively charged residues.Citation105,Citation106 Solvent exposure and structural flexibility also increase the risk of carbonylation.Citation105 Currently, there is no clear association between carbonylation sites and different types of secondary structures.Citation104,Citation107 Multiple carbonylation predictors such as CarSPred,Citation106 iCar-PseCp,Citation108 and iCarPS,Citation109 have been developed using random forest and support vector machine (SVM) algorithms (). The most common features of machine algorithms for predicting carbonylation include the position-specific propensity of amino acid, hydrophilicity, SASA, side-chain interaction parameter, and positive charge (). Citation106,Citation108,Citation109
Table 2. Comparison of online carbonylation predictors used for machine learning methods. Abbreviations: KNN, k-nearest neighbor; ROC, Receiver Operating Characteristic
More precise, sensitive, and site-specific analytical methods for detecting carbonylation in antibodies have been developed in the past decade.Citation5,Citation99,Citation100,Citation110 Joshi et al. identified 27 carbonylation sites within trastuzumab. Most carbonylation sites were within the framework region; they reported no carbonylation sites within the CH3 domain and CDR loops.Citation101 The abundance of carbonylation sites within the structurally rigid framework regionsCitation100,Citation101 is surprising because previous studies have suggested that carbonylation is more common in structurally flexible or disordered regions. Citation104,Citation107 However, residues within the framework region can have a high solvent exposure and undergo carbonylation. Citation100 Future studies could investigate the impact of the structure of antibodies on carbonylation. In addition, carbonylation sites from a wide panel of antibodies could be used to develop machine learning models for specifically predicting carbonylation sites for therapeutic antibodies.
Tyrosine sulfation
Tyrosine sulfation is an enzymatic modification that is catalyzed by tyrosylprotein sulfotransferases (TPSTs). During sulfation, a sulfate group is attached to the hydroxyl group of tyrosine residues.Citation111 Tyrosine sulfation has been reported for a few monoclonal and bispecific antibodies.Citation111–114 Sulfation of mAbs can occur in Chinese hamster ovary (CHO) cells during the cell culture. The degree of tyrosine sulfation in CHO cells varies due to differential expression of phosphoadenosine-5ʹ-phosphosulfate (PAPS) synthetase and TPST. PAPS synthetase converts ATP to PAPS; TPST transfers the sulfo group from PAPs to tyrosine residues.Citation115 Sulfation in mAbs generates acidic variants,Citation111 but the impact of tyrosine sulfation on the safety and efficacy of therapeutic antibodies has not yet been established.Citation5
Common predictors of tyrosine sulfation include secondary structure, flanking residues, structural flexibility,Citation101, and SASA.Citation116 Sulfation is more favorable for structurally flexible or disordered regions that can fit into the cleft of TPSTs. Tyrosine residues flanked by acidic residues are also more likely to get sulfated ()).Citation111 During sulfation, the flanking acidic residues within the IgG form electrotactic interactions with arginine and Lys residues of TPST.Citation117 The role of structural flexibility in sulfation suggests that sulfation is more likely to occur in the solvent-exposed and flexible CDR loops than in the framework regions.Citation51 Zhao et al. reported tyrosine sulfation in the CDR-L1 loop,Citation111 and Tyshchuk et al. reported sulfation in the CDR-L2 loop.Citation112
Figure 5. Sequence logos showing the most conserved amino acids around (a) sulfotyrosine (sTyr). (b) Sequence logos showing the most conserved amino acids around 4-hydroxyproline (4Hyp) residues. Reproduced from Tyshchuk, O. et al.Citation112 (Creative Commons Attribution License).
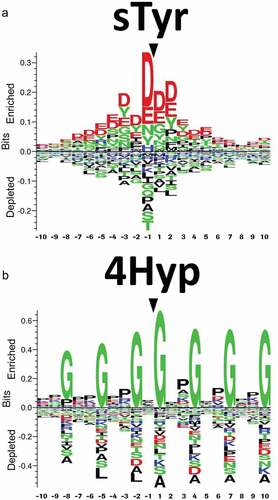
There are multiple online tools for predicting sulfation, such as Sulfinator,Citation118 PredSulSite,Citation119 and Sulfotyrosine.Citation120 However, the accuracy of online tools is limited by the training set used to develop machine learning algorithms to predict PTMs.Citation34 The training sets for most online predictors include experimentally identified tyrosine sulfation sites from databases like Swiss Prot. However, mAbs are underrepresented, and an overtrained machine learning algorithm may fail to identify sulfation sites in mAbs. Hence, sulfation predictors have to be used with caution for assessing the risk of tyrosine sulfation in mAbs. Tyrosine sulfation needs to be identified in a broader panel of antibodies to improve tyrosine sulfation prediction for mAbs. Future studies could also explore the impact of structural features such as dihedral angles, pKa of surrounding residues, and position within IgG (i.e., CDR or framework region) on tyrosine sulfation.
Hydroxylation
Hydroxylation is an enzymatic modification that is catalyzed by hydroxylases. Hydroxylation can occur at arginine, tyrosine, Trp, and phenylalanine, but it is more common for Pro and Lys.Citation121 During hydroxylation, a hydroxyl (OH) group is added to Pro or Lys residues. As a result, Pro is converted to 3-hydroxyPro or 4-hydroxyPro, whereas Lys is converted to 5-hydroxyLys.Citation122 Hydroxylation of Lys and Pro residues at consensus motifs (Xaa-Lys-Gly or Xaa-Pro-Gly) is common in collagen, where hydroxylation helps stabilize the collagen triple-helix.Citation123 Common parameters for designing hydroxylation predictors include solvent exposure, intrinsic disorder, hydrophilicity, and sequence.Citation122,Citation123 Hydroxylation sites tend to be disordered, exposed, and enriched with Pro and Gly residues.Citation124
There are multiple online predictors for hydroxylation sites such as PredHydroxy, HydPred, and ModPred.Citation123 Online hydroxylation predictors are suitable for collagen. However, most hydroxylation predictors have limited training sets, which leads to a high number of false positives and false negatives for non-collagen proteins and uncommon hydroxylation motifs. To date, two studies have reported hydroxylation in antibodies. Xie et al. reported Lys hydroxylation for an IgG1 antibody,Citation125 and Tyshchu et al. observed Pro hydroxylation for a bispecific antibody.Citation112 Using consensus motifs can lead to the overestimation of hydroxylation sites. For example, Xie et al. did not report Lys hydroxylation at all of the consensus motifs.Citation125 Most hydroxylation predictors are not specific to mammalian and human proteins, limiting their utility for therapeutic antibodies.Citation123 Tyshchuk and colleagues used hydroxylation sites for mammalian proteins to generate sequence logos ()) and develop a kNN model:Citation112 the in silico prediction correlated well with the experimental data for the bispecific antibody. Huang et al. used a similar approach to create HydLoc, which had a training set composed of human proteins and had better accuracy for predicting hydroxylation of human proteins.Citation124 Machine learning methods for predicting hydroxylation can be improved with more experimental data for uncommon hydroxylation sites, as well as hydroxylation sites for mammalian proteins, human proteins, and antibodies.Citation123
Perspectives and future directions
In the past decade, in silico prediction of PTMs has evolved from simple sequence-based approaches to more sophisticated approaches incorporating antibody modeling, MD, and machine learning.Citation12 Although the structure- and physics-based approaches have better accuracy than sequence-based approaches, there is still a gap between the in silico prediction and experimental data from forced degradation studies. External factors such as formulation and cell culture conditions can contribute to the gap between in silico predictions and forced degradation results.Citation3 The quality of predicted structures is a limiting factor for structure- and physics-based approaches. Homology modeling is suitable for modeling the conserved framework regions. Recent advances in loop modeling have provided more accurate predictions for the L1, L2, L3, H1, and H2 loops.Citation126 However, modeling the CDR-H3 loop is still challenging due to the length and structural variation.Citation127 Future advancements in antibody modeling will improve the accuracy in silico prediction of PTMs. In addition, MD simulations can provide more representative SASA values for predicted and crystal structures and refine the quality of predicted structures.
Machine learning algorithms such as random forest and decision trees have been used to predict the risk of deamidation, isomerization, and oxidation. However, machine learning models are limited by the training set and selection of parameters. Building a training set can require a lot of in-house experimental data. Experimentally confirmed liable sites for publicly available antibody sequences are essential for developing better machine learning models in the future. Selecting the best parameters is complicated by contradictory findings for the impact of structural features (e.g., pKa values) on PTMs. Studies investigating the effect of different structural features on PTMs for a broad panel of antibodies are crucial for improving in silico prediction of PTMs. Citation42,Citation67 Moreover, the selection parameters for machine learning models should account for different mechanisms for chemical modifications (e.g., oxidation) under different types of stress conditions, such as forced oxidation by peroxidesCitation66 and photolysis.
Therapeutic antibodies can also undergo in vivo chemical modifications (e.g., deamidation and oxidation) due to rapid temperature and pH changes after drug administration.Citation128 In vivo chemical modifications can impact the safety and efficacy of therapeutic antibodies. For example, in vivo deamidation can lead to a loss in activity and cause immunogenicity. Citation57 Few studies have investigated in vivo modifications by monitoring PTMs for IgG in serum or by using in vitro conditions to mimic in vivo degradation.Citation57 Monitoring in vivo modifications is difficult because the human serum is a mixture of endogenous proteins and the administered therapeutic antibodies. It is challenging to separate and distinguish therapeutic antibodies from endogenous proteins. Fluorescence labeling techniques or affinity purification with anti-Fc antibodies are required for mass spectrometry (MS) analysis of in vivo samples. The harsh purification conditions (e.g., low pH elution) can introduce artifacts in liquid chromatography-MS analysis.Citation129 Computational approaches for improving the stability of therapeutic antibodies focus on preventing chemical degradation under the types of stresses (e.g., thermal stress) encountered during manufacturing and storage.Citation27 In silico prediction of in vivo modifications remains relatively unexplored. Developing computational approaches to predict in vivo chemical modifications provides opportunities for improving in vivo stability of therapeutic candidates.
The impact of PTMs on heterogeneity, safety, and efficacy can vary depending on the location of the liable residue. A more holistic approach to engineering mAbs could identify PTM hotspots and predict the impact of PTMs on binding affinity and physical stability. Removing liable sites is not always feasible, especially if the amino acid residue plays a role in antigen binding. Paratope prediction and molecular docking of IgG-antigen complex can guide antibody engineering for developability.Citation130 The surface charge distribution and surface hydrophobicityCitation131 could be used to predict the impact of modifications of liable residues on colloidal stability.
Substantial strides have been made in silico prediction of deamidation, isomerization, and Met oxidation.Citation22,Citation30,Citation46 However, the in silico prediction of glycation, sulfation, hydroxylation, and carbonylation mAbs is underdeveloped compared to other PTMs. Another understudied area is in silico prediction of in vivo chemical modifications.Citation57 Advances in analytical characterization of novel modifications (e.g., sulfation) and in vivo modifications are crucial for developing computational approaches for predicting PTMs.
After two decades of establishing a solid foundation for in silico prediction of chemical modifications, future studies can focus on tailoring in silico predictions to more specific stress conditions and formulations.
Abbreviations
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–15. PMID: 19459844. doi:10.1111/j.1476-5381.2009.00190.x.
- Kapingidza AB, Kowal K, Chruszcz M. Antigen-antibody complexes. Subcell Biochem. 2020;94:465–97. PMID: 32189312. doi:10.1007/978-3-030-41769-7_19.
- Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. PMID: 30543482. doi:10.1080/19420862.2018.1553476.
- Grassi L, Cabrele C. Susceptibility of protein therapeutics to spontaneous chemical modifications by oxidation, cyclization, and elimination reactions. Amino Acids. 2019;51(10–12):1409–31. PMID: 31576455. doi:10.1007/s00726-019-02787-2.
- Beck A, Liu H. Macro- and micro-heterogeneity of natural and recombinant IgG antibodies. Antibodies. 2019;8(1):18. PMID: 31544824. doi:10.3390/antib8010018.
- Barton C, Spencer D, Levitskaya S, Feng J, Harris R, Schenerman MA. Heterogeneity of IgGs: role of production, processing, and storage on structure and function [Internet]. In: ACS Symposium Series. American Chemical Society, State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space; 2014. 69–98. doi:10.1021/bk-2014-1176.ch003
- Yan Y, Wei H, Fu Y, Jusuf S, Zeng M, Ludwig R, Krystek SR, Chen G, Tao L, Das TK. Isomerization and oxidation in the complementarity-determining regions of a monoclonal antibody: a study of the modification-structure-function correlations by hydrogen-deuterium exchange mass spectrometry. Anal Chem. 2016;88(4):2041–50. PMID: 26824491. doi:10.1021/acs.analchem.5b02800.
- Du Y, Walsh A, Ehrick R, Xu W, May K, Liu H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs. 2012;4(5):578–85. PMID: 22820257. doi:10.4161/mabs.21328.
- Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–98. PMID: 25821140. doi:10.1002/jps.24430.
- Nowak CK, Cheung JM, Dellatore S, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies: a practical guide. MAbs. 2017;9(8):1217–30. PMID: 28853987. doi:10.1080/19420862.2017.1368602.
- Michels DA, Ip AY, Dillon TM, Brorson K, Lute S, Chavez B, Prentice KM, Brady LJ, Miller KJ. Separation Methods and Orthogonal Techniques [Internet]. In: State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization Volume 2. Biopharmaceutical characterization: the NISTmAb case study. American Chemical Society; 2015, p. 237–84. DOI:10.1021/bk-2015-1201.ch005.
- Kuroda D, Tsumoto K. Engineering stability, viscosity, and immunogenicity of antibodies by computational design. J Pharm Sci. 2020;109(5):1631–51. PMID: 31958430. doi:10.1016/j.xphs.2020.01.011.
- Temel DB, Kinderman F, Eryilmaz E. Developability in biophysical characterization. In: Houde, Damian J and Berkowitz, Steven A, editors. Biophysical Characterization of proteins in developing biopharmaceuticals. Elsevier; 2019. 505–26. DOI:10.1016/B978-0-444-64173-1.00017-2.
- Zurdo J, Arnell A, Obrezanova O, Smith N, Gómez De La Cuesta R, Gallagher TRA, Michael R, Stallwood Y, Ekblad C, Abrahmsén L, et al. Early implementation of QbD in biopharmaceutical development: a practical example. Biomed Res Int. 2015;2015:1–19. PMID: 26075248. doi:10.1155/2015/605427.
- Strohl WR, Strohl LM. Development issues: antibody stability, developability, immunogenicity, and comparability [Internet. In: Therapeutic antibody engineering. Elsevier: Woodhead Publishing; 2012. 377–595. DOI:10.1533/9781908818096.377.
- Robinson NE, Robinson AB. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc Natl Acad Sci U S A. 2001;98(8):4367–72. PMID: 11296285. doi:10.1073/pnas.071066498.
- Wakankar AA, Borchardt RT. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J Pharm Sci. 2006;95(11):2321–36. PMID: 16960822. doi:10.1002/jps.20740.
- Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21(5):1549–67. PMID: 31626279. doi:10.1093/bib/bbz095.
- Agrawal NJ, Dykstra A, Yang J, Yue H, Nguyen X, Kolvenbach C, Angell N. Prediction of the hydrogen peroxide–induced methionine oxidation propensity in monoclonal antibodies. J Pharm Sci. 2018;107(5):1282–89. PMID: 29325924. doi:10.1016/j.xphs.2018.01.002.
- Ali S, Hassan M, Islam A, Ahmad F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr Protein Pept Sci. 2014;15(5):456–76. PMID: 24678666. doi:10.2174/1389203715666140327114232.
- Yang Y, Zhao J, Geng S, Hou C, Li X, Lang X, Qiao C, Li Y, Feng J, Lv M, et al. Improving Trastuzumab’s stability profile by removing the two degradation hotspots. J Pharm Sci. 2015;104(6):1960–70. PMID: 25820189. doi:10.1002/jps.24435.
- Sankar K, Hoi KH, Yin Y, Ramachandran P, Andersen N, Hilderbrand A, McDonald P, Spiess C, Zhang Q. Prediction of methionine oxidation risk in monoclonal antibodies using a machine learning method. MAbs. 2018;10(8):1281–90. PMID: 30252602. doi:10.1080/19420862.2018.1518887.
- Xu KN, Uversky V, Xue B. Local flexibility facilitates oxidization of buried methionine residues. Protein Pept Lett. 2012;19(6):688–97. PMID: 22519542. doi:10.2174/092986612800494084.
- Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129–43. PMID: 30236283. doi:10.1016/j.neuron.2018.08.011.
- Kaliman I, Nemukhin A, Varfolomeev S. Free energy barriers for the N-terminal asparagine to succinimide conversion: quantum molecular dynamics simulations for the fully solvated model. J Chem Theory Comput. 2010;6(1):184–89. PMID: 26614331. doi:10.1021/ct900398a.
- Plotnikov NV, Singh SK, Rouse JC, Kumar S. Quantifying the risks of asparagine deamidation and aspartate isomerization in biopharmaceuticals by computing reaction free-energy surfaces. J Phys Chem B. 2017;121(4):719–30. PMID: 28051868. doi:10.1021/acs.jpcb.6b11614.
- Kumar S, Plotnikov NV, Rouse JC, Singh SK. Biopharmaceutical informatics: supporting biologic drug development via molecular modelling and informatics. J Pharm Pharmacol. 2018;70(5):595–608. PMID: 28155992. doi:10.1111/jphp.12700.
- Salsbury FR. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010;10(6):738–44. PMID: 20971684. doi:10.1016/j.coph.2010.09.016.
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111(52):18601–06. PMID: 25512516. doi:10.1073/pnas.1421779112.
- Delmar JA, Wang J, Choi SW, Martins JA, Mikhail JP. Machine learning enables accurate prediction of asparagine deamidation probability and rate. Mol Ther Methods Clin Dev. 2019;15:264–74. PMID: 31890727. doi:10.1016/j.omtm.2019.09.008.
- Jia L, Sun Y, de Brevern AG. Protein asparagine deamidation prediction based on structures with machine learning methods. PLoS One. 2017;12(7):1–17. PMID: 28732052. doi:10.1371/journal.pone.0181347.
- Koehl P, Delarue M. Combined approaches from physics, statistics, and computer science for ab initio protein structure prediction: ex unitate vires (unity is strength)? F1000Research. 2018;7:11. PMID: 30079234. doi:10.12688/f1000research.14870.1.
- Lai PK, Swan JW, Trout BL. Calculation of therapeutic antibody viscosity with coarse-grained models, hydrodynamic calculations and machine learning-based parameters. MAbs. 2021;13(1). PMID: 33834944. doi:10.1080/19420862.2021.1907882.
- Tatjewski M, Kierczak M, Plewczynski D. Predicting post-translational modifications from local sequence fragments using machine learning algorithms: overview and best practices. Methods Mol Biol. 2017;1484:275–300. PMID: 27787833. doi:10.1007/978-1-4939-6406-2_19.
- Yi L, Beckley N, Gikanga B, Zhang J, Wang YJ, Chih HW, Sharma VK. Isomerization of Asp-Asp motif in model peptides and a monoclonal antibody fab fragment. J Pharm Sci. 2013;102(3):947–59. PMID: 23280575. doi:10.1002/jps.23423.
- Miao S, Xie P, Zou M, Fan L, Liu X, Zhou Y, Zhao L, Ding D, Wang H, Tan WS. Identification of multiple sources of the acidic charge variants in an IgG1 monoclonal antibody. Appl Microbiol Biotechnol. 2017;101(14):5627–38. PMID: 28439623. doi:10.1007/s00253-017-8301-x.
- Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. MAbs. 2014;6(4):852–58. PMID: 24751784. doi:10.4161/mabs.28693.
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752(2):233–45. PMID: 11270864. doi:10.1016/S0378-4347(00)00548-X.
- Wakankar AA, Liu J, Vandervelde D, Wang YJ, Shire SJ, Borchardt RT. The effect of cosolutes on the isomerization of aspartic acid residues and conformational stability in a monoclonal antibody. J Pharm Sci. 2007;96(7):1708–18. PMID: 17238195. doi:10.1002/jps.20823.
- Sreedhara A, Cordoba A, Zhu Q, Kwong J, Liu J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm Res. 2012;29(1):187–97. PMID: 21809161. doi:10.1007/s11095-011-0534-2.
- Radkiewicz JL, Zipse H, Clarke S, Houk KN. Neighboring side chain effects on asparaginyl and aspartyl degradation: an Ab initio study of the relationship between peptide conformation and backbone NH acidity. J Am Chem Soc. 2001;123(15):3499–506. PMID: 11472122. doi:10.1021/ja0026814.
- Lu X, Nobrega RP, Lynaugh H, Jain T, Barlow K, Boland T, Sivasubramanian A, Vásquez M, Xu Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs. 2019;11(1):45–57. PMID: 30526254. doi:10.1080/19420862.2018.1548233.
- Brennan TV, Clarke S. Effect of adjacent histidine and cysteine residues on the spontaneous degradation of asparaginyl- and aspartyl-containing peptides. Int J Pept Protein Res. 2009;45(6):547–53. PMID: 7558585. doi:10.1111/j.1399-3011.1995.tb01318.x.
- Patel CN, Bauer SP, Davies J, Durbin JD, Shiyanova TL, Zhang K, Tang JX. N+1 engineering of an aspartate isomerization hotspot in the complementarity-determining region of a monoclonal antibody. J Pharm Sci. 2016;105(2):512–18. PMID: 26869414. doi:10.1016/S0022-3549(15)00185-9.
- Diepold K, Bomans K, Wiedmann M, Zimmermann B, Petzold A, Schlothauer T, Mueller R, Moritz B, Stracke JO, Mølhøj M, et al. Simultaneous assessment of Asp isomerization and Asn deamidation in recombinant antibodies by LC-MS following incubation at elevated temperatures. PLoS One. 2012;7(1):1–11. PMID: 22272329. doi:10.1371/journal.pone.0030295.
- Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9(6):e100736. PMID: 24959685. doi:10.1371/journal.pone.0100736.
- Pace AL, Wong RL, Zhang YT, Kao YH, Wang YJ. Asparagine deamidation dependence on buffer type, pH, and temperature. J Pharm Sci. 2013;102(6):1712–23. PMID: 23568760. doi:10.1002/jps.23529.
- Catak S, Monard G, Aviyente V, Ruiz-López MF. Deamidation of asparagine residues: direct hydrolysis versus succinimide-mediated deamidation mechanisms. J Phys Chem A. 2009;113(6):1111–20. PMID: 19152321. doi:10.1021/jp808597v.
- Shire SJ. Stability of monoclonal antibodies (mAbs). In: Monoclonal antibodies. Elsevier: Woodhead Publishing; 2015, 45–92. DOI:10.1016/b978-0-08-100296-4.00003-8.
- Chelius D, Render DS, Bondarenko PV. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal Chem. 2005;77(18):6004–11. PMID: 16159134. doi:10.1021/ac050672d.
- Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies. 2019;8(4):55. PMID: 31816964. doi:10.3390/antib8040055.
- North B, Lehmann A, Dunbrack RL. A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406(2):228–56. PMID: 21035459. doi:10.1016/j.jmb.2010.10.030.
- Nowak J, Baker T, Georges G, Kelm S, Klostermann S, Shi J, Sridharan S, Deane CM. Length-independent structural similarities enrich the antibody CDR canonical class model. MAbs. 2016;8(4):751–60. PMID: 26963563. doi:10.1080/19420862.2016.1158370.
- Yan Q, Huang M, Lewis MJ, Hu P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 2018;10(6):901–12. PMID: 29958069. doi:10.1080/19420862.2018.1478646.
- Lan W, Valente JJ, Ilott A, Chennamsetty N, Liu Z, Rizzo JM, Yamniuk AP, Qiu D, Shackman HM, Bolgar MS. Investigation of anomalous charge variant profile reveals discrete pH-dependent conformations and conformation-dependent charge states within the CDR3 loop of a therapeutic mAb. MAbs. 2020;12(1):1763138. PMID: 32432964. doi:10.1080/19420862.2020.1763138.
- Le Basle Y, Chennell P, Tokhadze N, Astier A, Sautou V. Physicochemical stability of monoclonal antibodies: a review. J Pharm Sci. 2020;109(1):169–90. PMID: 31465737. doi:10.1016/j.xphs.2019.08.009.
- Liu H, Ponniah G, Zhang HM, Nowak C, Neill A, Gonzalez-Lopez N, Patel R, Cheng G, Kita AZ, Andrien B. In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs. 2014;6(5):1145–54. PMID: 25517300. doi:10.4161/mabs.29883.
- Xu A, Kim HS, Estee S, Viajar S, Galush WJ, Gill A, Hötzel I, Lazar GA, McDonald P, Andersen N, et al. Susceptibility of antibody CDR residues to chemical modifications can be revealed prior to antibody humanization and aid in the lead selection process. Mol Pharm. 2018;15(10):4529–37. PMID: 30118239. doi:10.1021/acs.molpharmaceut.8b00536.
- Delmar JA, Buehler E, Chetty AK, Das A, Quesada GM, Wang J, Chen X. Machine learning prediction of methionine and tryptophan photooxidation susceptibility. Mol Ther Methods Clin Dev. 2021;21:466–77. PMID: 33898635. doi:10.1016/j.omtm.2021.03.023.
- Jacobitz AW, Liu Q, Suravajjala S, Agrawal NJ. Tryptophan oxidation of a monoclonal antibody under diverse oxidative stress conditions: distinct oxidative pathways favor specific tryptophan residues. J Pharm Sci. 2021;110(2):719–26. PMID: 33198947. doi:10.1016/j.xphs.2020.10.039.
- Shah DD, Zhang J, Hsieh MC, Sundaram S, Maity H, Mallela KMG. Effect of peroxide- versus alkoxyl-induced chemical oxidation on the structure, stability, aggregation, and function of a therapeutic monoclonal antibody. J Pharm Sci. 2018;107(11):2789–803. PMID: 30075161. doi:10.1016/j.xphs.2018.07.024.
- Chu JW, Brooks BR, Trout BL. Oxidation of methionine residues in aqueous solutions: free methionine and methionine in granulocyte colony-stimulating factor. J Am Chem Soc. 2004;126(50):16601–07. PMID: 15600366. doi:10.1021/ja0467059.
- Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47(18):5088–100. PMID: 18407665. doi:10.1021/bi702238b.
- Tang L, Sundaram S, Zhang J, Carlson P, Matathia A, Parekh B, Zhou Q, Hsieh MC. Conformational characterization of the charge variants of a human IgG1 monoclonal antibody using H/D exchange mass spectrometry. MAbs. 2013;5(1):114–25. PMID: 23222183. doi:10.4161/mabs.22695.
- Torosantucci R, Schöneich C, Jiskoot W. Oxidation of therapeutic proteins and peptides: structural and biological consequences. Pharm Res. 2014;31(3):541–53. PMID: 24065593. doi:10.1007/s11095-013-1199-9.
- Chennamsetty N, Quan Y, Nashine V, Sadineni V, Lyngberg O, Krystek S. Modeling the oxidation of methionine residues by peroxides in proteins. J Pharm Sci. 2015;104(4):1246–55. PMID: 25641333. doi:10.1002/jps.24340.
- Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I, Sun T, Caffry I, Brown M, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 2017;9(4):646–53. PMID: 28281887. doi:10.1080/19420862.2017.1290753.
- Regep C, Georges G, Shi J, Popovic B, Deane CM. The H3 loop of antibodies shows unique structural characteristics. Proteins Struct Funct Bioinf. 2017;85(7):1311–18. PMID: 28342222. doi:10.1002/prot.25291.
- Baek Y, Singh N, Arunkumar A, Zydney AL. Effects of histidine and sucrose on the biophysical properties of a monoclonal antibody. Pharm Res. 2017;34(3):629–39. PMID: 28035628. doi:10.1007/s11095-016-2092-0.
- Razinkov VI, Treuheit MJ, Becker GW. Accelerated formulation development of monoclonal antibodies (MABS) and mab-based modalities: review of methods and tools. J Biomol Screen. 2015;20(4):468–83. PMID: 25576149. doi:10.1177/1087057114565593.
- Hawe A, Wiggenhorn M, van de Weert M, Garbe JHO, Mahler H, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci. 2012;101(3):895–913. PMID: 22083792. doi:10.1002/jps.22812.
- Li Y, Polozova A, Gruia F, Feng J. Characterization of the degradation products of a color-changed monoclonal antibody: tryptophan-derived chromophores. Anal Chem. 2014;86(14):6850–57. PMID: 24937252. doi:10.1021/ac404218t.
- Alam ME, Slaney TR, Wu L, Das TK, Kar S, Barnett GV, Leone A, Tessier PM. Unique impacts of methionine oxidation, tryptophan oxidation, and asparagine deamidation on antibody stability and aggregation. J Pharm Sci. 2020;109(1):656–69. PMID: 31678251. doi:10.1016/j.xphs.2019.10.051.
- Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem. 2010;35(4):139–48. PMID: 20022755. doi:10.1016/j.physbeh.2017.03.040.
- Dalkas GA, Teheux F, Kwasigroch JM, Rooman M. Cation–π, amino–π, π–π, and H‐bond interactions stabilize antigen–antibody interfaces. Proteins Struct Funct Bioinf. 2014;82(9):1734–46. PMID: 24488795. doi:10.1002/prot.24527.
- Wei Z, Feng J, Lin H-Y-Y, Mullapudi S, Bishop E, Tous GI, Casas-Finet J, Hakki F, Strouse R, Schenerman MA. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal Chem. 2007;79(7):2797–805. PMID: 17319649. doi:10.1021/ac062311j.
- Hensel M, Steurer R, Fichtl J, Elger C, Wedekind F, Petzold A, Schlothauer T, Molhoj M, Reusch D, Bulau P. Identification of potential sites for tryptophan oxidation in recombinant antibodies using tert-butylhydroperoxide and quantitative LC-MS. PLoS One. 2011;6(3):e17708. PMID: 21390239. doi:10.1371/journal.pone.0017708.
- Qi PP, Volkin DB, Zhao H, Nedved ML, Hughes R, Bass R, Yi SC, Panek ME, Wang D, Dalmonte P, et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J Pharm Sci. 2009;98(9):3117–30. PMID: 19009595. doi:10.1002/jps.21617.
- Dashivets T, Stracke J, Dengl S, Knaupp A, Pollmann J, Buchner J, Schlothauer T. Oxidation in the complementarity-determining regions differentially influences the properties of therapeutic antibodies. MAbs. 2016;8(8):1525–35. PMID: 27612038. doi:10.1080/19420862.2016.1231277.
- Wecksler AT, Yin J, Lee Tao P, Kabakoff B, Sreedhara A, Deperalta G. Photodisruption of the structurally conserved Cys-Cys-Trp triads leads to reduction-resistant scrambled intrachain disulfides in an IgG1 monoclonal antibody. Mol Pharm. 2018;15(4):1598–606. PMID: 29502420. doi:10.1021/acs.molpharmaceut.7b01128.
- Luo Q, Joubert MK, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem. 2011;286(28):25134–44. PMID: 21518762. doi:10.1074/jbc.M110.160440.
- Amano M, Kobayashi N, Yabuta M, Uchiyama S, Fukui K. Detection of histidine oxidation in a monoclonal immunoglobulin gamma (IgG) 1 antibody. Anal Chem. 2014;86(15):7536–43. PMID: 24940720. doi:10.1021/ac501300m.
- Liu M, Zhang Z, Cheetham J, Ren D, Zhou ZS. Discovery and characterization of a photo-oxidative histidine-histidine cross-link in IgG1 antibody utilizing 18O-labeling and mass spectrometry. Anal Chem. 2014;86(10):4940–48. PMID: 24738698. doi:10.1021/ac500334k.
- Xu CF, Chen Y, Yi L, Brantley T, Stanley B, Sosic Z, Zang L. Discovery and characterization of histidine oxidation initiated cross-links in an IgG1 monoclonal antibody. Anal Chem. 2017;89(15):7915–23. PMID: 28635253. doi:10.1021/acs.analchem.7b00860.
- Powell T, Knight MJ, O’Hara J, Burkitt W. Discovery of a photoinduced histidine-histidine cross-link in an IgG4 antibody. J Am Soc Mass Spectrom. 2020;31(6):1233–40. PMID: 32392057. doi:10.1021/jasms.0c00076.
- Sreedhara A, Lau K, Li C, Hosken B, MacChi F, Zhan D, Shen A, Steinmann D, Schöneich C, Lentz Y. Role of surface exposed tryptophan as substrate generators for the antibody catalyzed water oxidation pathway. Mol Pharm. 2013;10(1):278–88. PMID: 23136850. doi:10.1021/mp300418r.
- Bane J, Mozziconacci O, Yi L, Wang YJ, Sreedhara A, Schöneich C. Photo-oxidation of IgG1 and model peptides: detection and analysis of triply oxidized his and trp side chain cleavage products. Pharm Res. 2017;34(1):229–42. PMID: 27800571. doi:10.1007/s11095-016-2058-2.
- Miyahara Y, Shintani K, Hayashihara-Kakuhou K, Zukawa T, Morita Y, Nakazawa T, Yoshida T, Ohkubo T, Uchiyama S. Effect of UVC irradiation on the oxidation of histidine in monoclonal antibodies. Sci Rep. 2020;10(1). PMID: 32286391. doi:10.1038/s41598-020-63078-5.
- Quan C, Alcala E, Petkovska I, Matthews D, Canova-Davis E, Taticek R, Ma S. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: where it is, how it got there, and how it affects charge-based behavior. Anal Biochem. 2008;373(2):179–91. PMID: 18158144. doi:10.1016/j.ab.2007.09.027.
- Venkatraman J, Aggarwal K, Balaram P. Helical peptide models for protein glycation: proximity effects in catalysis of the Amadori rearrangement. Chem Biol. 2001;8(7):611–25. PMID: 11451663. doi:10.1016/S1074-5521(01)00036-9.
- Goetze AM, Liu YD, Arroll T, Chu L, Flynn GC. Rates and impact of human antibody glycation in vivo. Glycobiology. 2012;22(2):221–34. PMID: 21930650. doi:10.1093/glycob/cwr141.
- Wei B, Berning K, Quan C, Zhang YT. Glycation of antibodies: modification, methods and potential effects on biological functions. MAbs. 2017;9(4):586–94. PMID: 28272973. doi:10.1080/19420862.2017.1300214.
- Kennedy DM, Skillen AW, Self CH. Glycation of monoclonal antibodies impairs their ability to bind antigen. Clin Exp Immunol. 1994;98(2):245–51. PMID: 7955529. doi:10.1111/j.1365-2249.1994.tb06133.x.
- Vrdoljak A, Trescec A, Benko B, Hecimovic D, Simic M. In vitro glycation of human immunoglobulin G. Clin Chim Acta. 2004;345(1–2):105–11. PMID: 15193984. doi:10.1016/j.cccn.2004.03.026.
- Miller AK, Hambly DM, Kerwin BA, Treuheit MJ, Gadgil HS. Characterization of site-specific glycation during process development of a human therapeutic monoclonal antibody. J Pharm Sci. 2011;100(7):2543–50. PMID: 21287557. doi:10.1002/jps.22504.
- Jacobitz AW, Dykstra AB, Spahr C, Agrawal NJ. Effects of buffer composition on site-specific glycation of lysine residues in monoclonal antibodies. J Pharm Sci. 2020;109(1):293–300. PMID: 31150698. doi:10.1016/j.xphs.2019.05.025.
- Zhang B, Yang Y, Yuk I, Pai R, McKay P, Eigenbrot C, Dennis M, Katta V, Francissen KC. Unveiling a glycation hot spot in a recombinant humanized monoclonal antibody. Anal Chem. 2008;80(7):2379–90. PMID: 18307322. doi:10.1021/ac701810q.
- Saleem RA, Affholter BR, Deng S, Campbell PC, Matthies K, Eakin CM, Wallace A. A chemical and computational approach to comprehensive glycation characterization on antibodies. MAbs. 2015;7(4):719–31. PMID: 26030340. doi:10.1080/19420862.2015.1046663.
- Ros J, Ashraf G., Madian, Fred E., Regnier, Ao, Zeng. Protein carbonylation: principles, analysis, and biological implications. In Editor JoaquimRos. John Wiley & Sons, Inc; 2017. p. 24–47. doi:10.1002/9781119374947.
- Yang Y, Stella C, Wang W, Schöneich C, Gennaro L. Characterization of oxidative carbonylation on recombinant monoclonal antibodies. Anal Chem. 2014;86(10):4799–806. PMID: 24731230. doi:10.1021/ac4039866.
- Joshi S, Kumari S, Rathore AS. Identification and characterization of carbonylation sites in trastuzumab biosimilars. Int J Biol Macromol. 2021;169:95–102. PMID: 33338527. doi:10.1016/j.ijbiomac.2020.12.095.
- Yang Y, Mah A, Yuk IH, Grewal PS, Pynn A, Cole W, Gao D, Zhang F, Chen J, Gennaro L, et al. Investigation of metal-catalyzed antibody carbonylation with an improved protein carbonylation assay. J Pharm Sci. 2018;107(10):2570–80. PMID: 29935298. doi:10.1016/j.xphs.2018.06.015.
- Bee JS, Davis M, Freund E, Carpenter JF, Randolph TW. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol Bioeng. 2010;105(1):121–29. PMID: 19725039. doi:10.1002/bit.22525.
- Shyama Prasad Rao R, Møller IM. Pattern of occurrence and occupancy of carbonylation sites in proteins. Proteomics. 2011;11(21):4166–73. PMID: 21919202. doi:10.1002/pmic.201100223.
- Weng SL, Huang KY, Kaunang FJ, Huang CH, Kao HJ, Chang TH, Wang HY, Lu JJ, Lee TY. Investigation and identification of protein carbonylation sites based on position-specific amino acid composition and physicochemical features. BMC Bioinform. 2017;18(S3). PMID: 28361707. doi:10.1186/s12859-017-1472-8.
- Lv H, Han J, Liu J, Zheng J, Liu R, Zhong D, Lisacek F. CarSPred: a computational tool for predicting carbonylation sites of human proteins. PLoS One. 2014;9(10):1–8. PMID: 25347395. doi:10.1371/journal.pone.0111478.
- Maisonneuve E, Ducret A, Khoueiry P, Lignon S, Longhi S, Talla E, Dukan S, Hofmann A. Rules governing selective protein carbonylation. PLoS One. 2009;4(10):1–10. PMID: 19802390. doi:10.1371/journal.pone.0007269.
- Jia J, Liu Z, Xiao X, Liu B, Chou KC. ICar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016;7(23):34558–70. PMID: 27153555. doi:10.18632/oncotarget.9148.
- Zhang D, Xu ZC, Su W, Yang YH, Lv H, Yang H, Lin H, Xu J. ICarPS: a computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics. 2021;37(2):171–77. PMID: 32766811. doi:10.1093/bioinformatics/btaa702.
- Yi Y, Zang L. High-throughput carbonyl content method of therapeutic mAb using size-exclusion chromatography with ultraviolet and fluorescence detection. Anal Biochem. 2019;571:25–36. PMID: 30653944. doi:10.1016/j.ab.2018.12.025.
- Zhao J, Saunders J, Schussler SD, Rios S, Insaidoo FK, Fridman AL, Li H, Liu YH. Characterization of a novel modification of a CHO-produced mAb: evidence for the presence of tyrosine sulfation. MAbs. 2017;9(6):985–95. PMID: 28590151. doi:10.1080/19420862.2017.1332552.
- Tyshchuk O, Gstöttner C, Funk D, Nicolardi S, Frost S, Klostermann S, Becker T, Jolkver E, Schumacher F, Koller CF, et al. Characterization and prediction of positional 4-hydroxyproline and sulfotyrosine, two post-translational modifications that can occur at substantial levels in CHO cells-expressed biotherapeutics. MAbs. 2019;11(7):1219–32. PMID: 31339437. doi:10.1080/19420862.2019.1635865.
- Jiang H, Xu W, Liu R, Gupta B, Kilgore B, Du Z, Yang X. Characterization of bispecific antibody production in cell cultures by unique mixed mode size exclusion chromatography. Anal Chem. 2020;92(13):9312–21. PMID: 32497423. doi:10.1021/acs.analchem.0c01641.
- Gomez N, Lull J, Yang X, Wang Y, Zhang X, Wieczorek A, Harrahy J, Pritchard M, Cano DM, Shearer M, et al. Improving product quality and productivity of bispecific molecules through the application of continuous perfusion principles. Biotechnol Prog. 2020;36(4). PMID: 31991523. doi:10.1002/btpr.2973.
- Liu R, Zhang Y, Kumar A, Huhn S, Hullinger L, Du Z. Modulating tyrosine sulfation of recombinant antibodies in CHO cell culture by host selection and sodium chlorate supplementation. Biotechnol J. 2021;16(9):2100142. PMID: 34081410. doi:10.1002/biot.202100142.
- Chang WC, Lee TY, Shien DM, Hsu JBK, Horng JT, Hsu PC, Wang TY, Huang HD, Pan RL. Incorporating support vector machine for identifying protein tyrosine sulfation sites. J Comput Chem. 2009;30(15):2526–37. PMID: 19373826. doi:10.1002/jcc.21258.
- Teramoto T, Fujikawa Y, Kawaguchi Y, Kurogi K, Soejima M, Adachi R, Nakanishi Y, Mishiro-Sato E, Liu MC, Sakakibara Y, et al. Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction. Nat Commun. 2013;4(1). PMID: 23481380. doi:10.1038/ncomms2593.
- Monigatti F, Gasteiger E, Bairoch A, Jung E. The Sulfinator: predicting tyrosine sulfation sites in protein sequences. Bioinformatics. 2002;18(5):769–70. PMID: 12050077. doi:10.1093/bioinformatics/18.5.769.
- Huang SY, Shi SP, Qiu JD, Sun XY, Suo SB, Liang RP. PredSulSite: prediction of protein tyrosine sulfation sites with multiple features and analysis. Anal Biochem. 2012;428(1):16–23. PMID: 22691961. doi:10.1016/j.ab.2012.06.003.
- Yang ZR. Predicting sulfotyrosine sites using the random forest algorithm with significantly improved prediction accuracy. BMC Bioinform. 2009;10(1):361. PMID: 19874585. doi:10.1186/1471-2105-10-361.
- Raju TS. Hydroxylation of proteins. In: Co- and post-translational modifications of therapeutic antibodies and proteins. John Wiley & Sons Inc; 2019. p. 119–31. doi:10.1002/9781119053354.ch10.
- Hu LL, Niu S, Huang T, Wang K, Shi XH, Cai YD, Uversky VN. Prediction and analysis of protein hydroxyproline and hydroxylysine. PLoS One. 2010;5(12):1–8. PMID: 21209839. doi:10.1371/journal.pone.0015917.
- Piovesan D, Hatos A, Minervini G, Quaglia F, Monzon AM, Tosatto SCE, Iakoucheva LM. Assessing predictors for new post translational modification sites: a case study on hydroxylation. PLoS Comput Biol. 2020;16(6):1–15. PMID: 32569263. doi:10.1371/journal.pcbi.1007967.
- Huang Q, Chen X, Wang Y, Li J, Liu H, Xie Y, Dai Z, Zou X, Li Z. HydLoc: a tool for hydroxyproline and hydroxylysine sites prediction in the human proteome. Chemom Intell Lab Syst. 2020;202:104035. doi:10.1016/j.chemolab.2020.104035.
- Xie Q, Moore B, Beardsley RL. Discovery and characterization of hydroxylysine in recombinant monoclonal antibodies. MAbs. 2016;8(2):371–78. PMID: 26651858. doi:10.1080/19420862.2015.1122148.
- Choi Y, Deane CM. Predicting antibody complementarity determining region structures without classification. Mol Biosyst. 2011;7(12):3327–34. PMID: 22011953. doi:10.1039/c1mb05223c.
- Almagro JC, Teplyakov A, Luo J, Sweet RW, Kodangattil S, Hernandez-Guzman F, Gilliland GL. Second Antibody Modeling Assessment (AMA-II). Proteins Struct Funct Bioinforma. 2014;82(8):1553–62. PMID: 24668560. doi:10.1002/prot.24567.
- Schuster J, Koulov A, Mahler HC, Detampel P, Huwyler J, Singh S, Mathaes R. In vivo stability of therapeutic proteins. Pharm Res. 2020;37. PMID: 31900680. doi:10.1007/s11095-019-2689-1.
- Xu X, Huang Y, Pan H, Molden R, Qiu H, Daly TJ, Li N, Popoff MR. Quantitation and modeling of posttranslational modifications in a therapeutic monoclonal antibody from single- And multiple-dose monkey pharmacokinetic studies using mass spectrometry. PLoS One. 2019;14(10):e0223899. PMID: 31618250. doi:10.1371/journal.pone.0223899.
- Qiu H, Wei R, Jaworski J, Boudanova E, Hughes H, VanPatten S, Lund A, Day J, Zhou Y, McSherry T, et al. Engineering an anti-CD52 antibody for enhanced deamidation stability. MAbs. 2019;11(7):1266–75. PMID: 31199181. doi:10.1080/19420862.2019.1631117.
- Hebditch M, Warwicker J. Web-based display of protein surface and pH-dependent properties for assessing the developability of biotherapeutics. Sci Rep. 2019;9(1):1–9. PMID: 30760735. doi:10.1038/s41598-018-36950-8.