
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
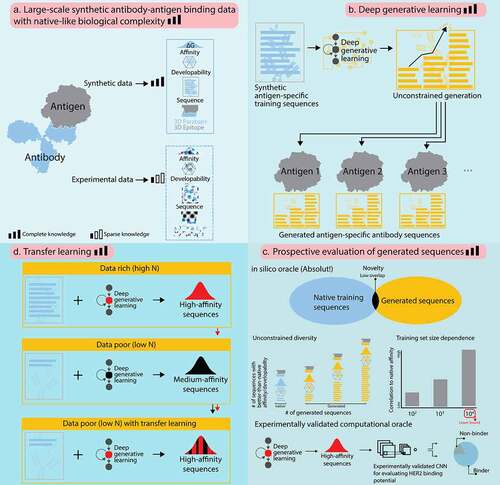
Generative machine learning (ML) has been postulated to become a major driver in the computational design of antigen-specific monoclonal antibodies (mAb). However, efforts to confirm this hypothesis have been hindered by the infeasibility of testing arbitrarily large numbers of antibody sequences for their most critical design parameters: paratope, epitope, affinity, and developability. To address this challenge, we leveraged a lattice-based antibody-antigen binding simulation framework, which incorporates a wide range of physiological antibody-binding parameters. The simulation framework enables the computation of synthetic antibody-antigen 3D-structures, and it functions as an oracle for unrestricted prospective evaluation and benchmarking of antibody design parameters of ML-generated antibody sequences. We found that a deep generative model, trained exclusively on antibody sequence (one dimensional: 1D) data can be used to design conformational (three dimensional: 3D) epitope-specific antibodies, matching, or exceeding the training dataset in affinity and developability parameter value variety. Furthermore, we established a lower threshold of sequence diversity necessary for high-accuracy generative antibody ML and demonstrated that this lower threshold also holds on experimental real-world data. Finally, we show that transfer learning enables the generation of high-affinity antibody sequences from low-N training data. Our work establishes a priori feasibility and the theoretical foundation of high-throughput ML-based mAb design.
Introduction
Monoclonal antibodies (mAbs) have proven incredibly successful as treatments for cancer and autoimmune disease (and recently, viral infections) with estimated market size of 300 billion USD by 2025.Citation1 Efforts to use mAbs for the neutralization of viral agents, such as human immunodeficiency virus (HIV), influenza and, more recently, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)Citation2–5 are ongoing as well. Excluding anti-SARS-CoV-2 antibodies, lead times to mAb discovery and design are typically >3 years.Citation6–9 The reason for this is that current mAb development pipelines mostly rely on a combination of large screening libraries and experimental heuristics with very little to no emphasis on rule-driven discovery.Citation10 Recently, it has been increasingly postulated that machine learning (ML) may be useful in accelerating antibody discovery, especially when applied to large-scale antigen-specific display library screening antibody sequencing data.Citation11 However, formal proof that ML can generate antibody sequences that are three-dimensionally (3D)-antigen-specific (affinity, paratope, epitope) if only provided with one-dimensional (1D)-sequence training data (the most abundant class of available antigen-specific antibody data) alone is missing.
Recent reports suggest that ML may be able to learn the rules of efficient antibody (protein) design.Citation7,Citation11–22 Specifically, Amimeur and colleaguesCitation23 trained generative adversarial networks (GANs)Citation24 on sequences obtained from the Observed Antibody Space (OAS) databaseCitation25 to demonstrate the capacity of deep generative networks to discover mAbs with certain developability parameters. Friedensohn and colleaguesCitation26 trained variational autoencoders (VAE)Citation27 on mouse B-cell receptor data for both identifying immunized-cohort-associated sequences and generating novel antigen-binding sequences. Widrich et al. and Davidsen et al. have used long short-term memory (LSTM) or VAE models to generate T-cell-receptor (TCR) β sequences with the aim to generate realistic immune repertoires.Citation28,Citation29 Saka et al. used RNN-LSTMs to examine the capacity of deep generative models to improve the affinity of kynurenine-binding antibodies.Citation30 Finally, Eguchi et al. built class-specific backbones using VAE to generate 3D coordinates of mAbs.Citation31 However, while several generative deep learning methods have been explored for the in silico generation of immune receptor sequences, these strategies did not allow the exhaustive examination of whether the generated sequences follow the same antigen-specificity distribution as the input training data. This is due to the absence of large-scale antigen-annotated antibody sequence training data and the lack of high-throughput techniques for validating antigen binding of ML-generated antibody sequences.
Here, we investigated whether generative deep learning can learn 3D-affinity and epitope information from 1D antibody sequence data. This was done by using two oracles (external validator functions). The first oracle is an in silico framework that enables unrestricted validation (prospective evaluation) of the biological activity (paratope, epitope, affinity) of generated antibody sequences. Specifically, we used an in silico antibody-antigen binding simulation framework (which respects the biological complexity of antibody-antigen binding to the largest extent possible), called Absolut!.Citation32 The framework can annotate large collections of antibody sequences with synthetic binding affinities (specificity) to a synthetic 3D-antigen, which allows the assembly of large-scale complete-knowledge training data.Citation33,Citation34 Due to its ability to annotate newly ML-generated antibody sequences with antigen-binding information, Absolut! resolves the current problems of large-scale validation of generated sequences.Citation35,Citation36 The second oracle is an experimentally validated deep learning classifier that was trained on binders and non-binders to human epidermal growth factor 2 (HER2).Citation37 Our work provides a complete-knowledge simulation-based foundation for the ML-driven design of fit-for-purpose antibodies with respect to binding affinity, epitope, and developability ().
Figure 1. In silico proof of principle of ML-based antibody design at unconstrained scale. We leveraged large synthetic ground-truth antibody sequence data with known paratope, epitope, and affinity to demonstrate in a proof-of-principle the (a,b) unconstrained deep generative learning-based generation of native-like antibody sequences. (c) An in silico oracle (Absolut!Citation32) enables the prospective evaluation of conformational (3D) affinity, paratope-epitope pairs, and developability of in silico generated antibody sequences. We also leveraged an experimentally validated oracleCitation37 to test antibody design conclusions gained based on the synthetic antibody sequence data. (d) Finally, we show that transfer learning increases generation quality of low-N-based ML models.
Results
Deep learning generates novel antigen-specific CDR-H3 sequences across a wide range of developability parameters
ML-based generation of new antibody sequences with desired biological properties requires large experimental datasets and a method to test the generated sequences for such properties. To address the absence of large experimental antigen-specific antibody-antigen datasets for training and testing deep antibody generative models, we leveraged Absolut!, which is a software suite that simulates the binding of antibody sequences to 3D antigens. Absolut! replicates and recaptures the biological properties and complexity of experimental antibody-antigen binding to a large extent.Citation32 We used our previously published dataset of seven million (7x106) murine native antibody (CDR-H3) amino acid sequencesCitation38 (see Methods) and (via Absolut!) computed their binding to 10 protein antigens (, ). Briefly, synthetic lattice-based antibody-antigen complexes were obtained by iterating over all possible binding positions between a sequence and an antigen to find the optimal binding position and by calculating the resulting binding affinity, paratope, epitope, and structural fold for each antibody CDR-H3 sequence. We note that the affinity, paratope, epitope, and structural fold were calculated according to Absolut!’s lattice representation (see Methods). Following affinity annotation, a set of six developability parameters () were calculated for each CDR-H3 sequence (). CDR-H3 amino acid sequences equipped with affinity, paratope, epitope, and developability information are henceforth termed antigen-annotated CDR-H3 sequences.
Table 1. List of 3D-antigens used in the deep-learning-based antibody generation pipeline
Table 2. Antibody developability parameters and their computational implementation
Figure 2. Computational workflow for ML-based antibody design and evaluation thereof. (a) Generation of in silico training datasets with binding paratope, epitope, and affinity annotation. Briefly, PDB (Protein Data Bank) 3D antigen structures were obtained from the Antibody DatabaseCitation39 and native antibody sequences (CDR-H3) were obtained from Greiff and colleagues.Citation38 CDR-H3 sequences were annotated with their corresponding affinity and epitope to each antigen using the Absolut! software suite.Citation32 In addition, six widely used developability parameters were calculated for each CDR-H3 sequence (see ). (b) Training a generative model on high-affinity CDR-H3 sequences to each antigen. Native linear 1D antigen-specific CDR-H3 sequences were used as input to train sequence-based RNN-LSTM generative models. Of note, the RNN-LSTM architecture did not receive any explicit 3D information on the paratope, epitope, affinity, nor the developability of a given sequence. (c) Large-scale in silico CDR-H3 sequence generation and binding validation. Following training, the deep models were used to generate new CDR-H3 sequences, which were then evaluated (prospectively tested) for their antigen-specificity (affinity, paratope, epitope) using Absolut! (simulation) and annotated with developability-associated parameters. (d) Comparison of training and generated affinities. The affinity of training antigen-specific CDR-H3 sequences (nseq = 70,000, blue) to 10 different 3D antigens obtained from PDB (see ). The affinity of the 70,000 generated CDR-H3 sequences from the 10 RNN-LSTM models is shown in yellow. (e) Comparison of training and generated sequences for paratope-epitope recognition. Absolut! was used to compute the affinity and paratope fold/epitope of the training data (see Methods: Generation of lattice-based antibody-antigen binding structures using Absolut!). For readability, paratope and epitope statistics in the training (native) and generated datasets are visualized at larger proportions for the antigen 1OB1. (f) Pearson correlation (range: 0.864–0.907) of CDR-H3 sequence composition between training (“native”) and generated datasets quantifying the preservation of long-range dependencies. CDR-H3 sequence composition was measured using gapped k-mers where the size of the k-mer was 1 and the size of the maximum gap varied between 1 and 5. (g) CDR-H3 sequence similarity (Levenshtein distance, LD) distribution determined among training (native) and generated CDR-H3 sequence datasets (see Supplementary Fig. S4 for the LD distribution of CDR-H3 sequences with the native and generated set, respectively). (h) CDR-H3 sequence novelty (overlap) defined as CDR-H3antigen_x∩CDR-H3antigen_y/70,000, where x and y are the 10 antigens listed in ) of CDR-H3 sequences (median overlap <0.5% → novelty: >99.5%) between both “native and generated” and “generated and generated” datasets across all antigen combinations. (i) Developability parameter distribution between training and generated CDR-H3 sequences overlaps substantially (see for a description of the developability parameters used).
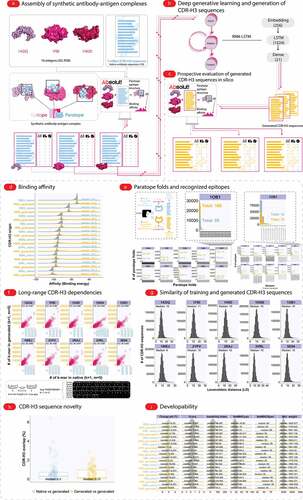
We examined the capacity of a deep (autoregressive) generative model (recurrent neural networks with long short-term memory (RNN-LSTM) to generate (design) novel antigen-specific sequences as follows. We first trained the RNN-LSTM model on antigen-specific CDR-H3 sequences (top 1% affinity sorted sequences, nseq = 70,000, also called “high-affinity” in the following) (). Importantly, we did not provide explicitly the affinity or paratope/epitope information in the training process. Subsequently, we used the trained model to generate new CDR-H3 sequences (nseq = 70,000) (), which we then evaluated in terms of antigen specificity (using Absolut!, ), sequence novelty, and developability (–i).
The binding affinity (), as well as the paratope fold and epitopes of generated CDR-H3 sequences (), mirrored very closely those of the native (training) CDR-H3 sequences. Novel paratope folds and epitopes were also discovered as observed by the paratope fold and epitope diversityCitation41 of generated CDR-H3 sequences that were higher than those of native (training set) sequences (). The within-sequence similarity, as measured by the distribution of Levenshtein distance (LD) between CDR-H3 sequences within the set of native or generated CDR-H3 sequence datasets was preserved (, Supplementary Fig. S4) as were long-range sequence dependencies (gapped k-mer decomposition, Pearson correlation 0.864–0.907, ). To exclude the possibility that generated CDR-H3 sequences showed high affinity merely by virtue of their similarity to the training input, we validated that the generated CDR-H3 sequences were novel (<1% overlap between generated and native antigen-specific sequences, ) both measured by exact sequence identity or based on sequence similarity (median Levenshtein distance between generated and native CDR-H3 sequences: ≈9–10 amino acids, ). Thus, deep generative learning explores non-trivial novel sequence spaces. We excluded the possibility that the chosen RNN-LSTM-architecture would be biased to generate high-affinity CDR-H3 sequences by showing that training on the following two CDR-H3 sequence sets did not lead to the generation of high-affinity CDR-H3 sequences: 1) exclusively low-affinity CDR-H3 sequences [generating exclusively low-affinity CDR-H3 sequences] (Supplementary Fig. S3A), and 2) CDR-H3 sequences spanning the entire affinity spectrum (generating CDR-H3 spanning the entire affinity spectrum) (Supplementary Fig. S3A). As an additional control, we analyzed CDR-H3 sequences that follow the positional amino acid distribution (position-specific weight matrix (PWM)) of the high-affinity training data and showed that these sequences span the entire affinity spectrum (Supplementary Fig. S3B). Finally, the distribution of developability parameters of generated CDR-H3 sequences largely mirrored but also expanded the range of parameters of native antigen-specific sequences ().
On-demand generation of large amounts of CDR-H3 sequences with broad developability and affinity that match or exceed the training sequences
Following the observation that deep generative models were capable of generating novel CDR-H3 sequences that mirror very closely the binding and developability properties of native CDR-H3 sequences (), we hypothesized that such models are useful for generating large quantities of CDR-H3 sequences with similar or better affinities than those of the native ones. To assess this hypothesis, we first grouped the native antigen-specific CDR-H3 sequences (nseq,training = 70,000, top 1%) into four affinity categories based on their binding energy (low energy → high affinity): 1) ultimate binder (max native–⅓), 2) penultimate binder (⅓–⅔), 3) binder (⅔–min native), and 4) hyperbinder (affinity>native max, i.e., higher affinity than found in the training data CDR-H3 sequences, see schematic in ), and then we generated, for each antigen, 7 × 105 unique antigen-specific CDR-H3 sequences (i.e., 10 times larger than the training dataset), and evaluated the generated CDR-H3 sequences with respect to the four categories. Broadly, we found that the number of binders in all four categories increased as the generated sequences increased (). Specifically, when the number of generated CDR-H3 sequences equaled that of the training data (nseq,generated = 70,000), we found binders in the same order of magnitude in all categories of binders (binder–ultimate binder) compared to the native (training) dataset (blue lines), except for hyperbinders (as the native populations have, per definitionem, no hyperbinders). At nseq,generated = 7x105, the quantities of discovered binders far eclipsed those of the native binders in all four categories by ~4-fold () suggesting that generative learning may be used for a highly exhaustive discovery of novel binders. Importantly, the discovery of CDR-H3 sequences with superior predicted binding affinity compared to the native sequences (hyperbinder) further illustrates the importance of deep generative models in the design and discovery of high-affinity CDR-H3 sequences.Citation12,Citation23,Citation26,Citation42 Hyperbinders showed affinity improvements over native CDR-H3 sequences in the range of 0.4–4.4% (percentages were calculated against the maximum affinity [lowest energy] of each antigen’s training dataset) and median LD (against native binders) of 10 to 14. To summarize, our RNN-LSTM models were able to generate large quantities of non-redundant CDR-H3 sequences that match or exceed the affinity of the training sequences.
Figure 3. Exhaustive generation reveals better antibodies than are present in the training dataset. (a) To examine the ability of the RNN-LSTM model to generate CDR-H3 sequences beyond the native realms (in terms of quantity and affinity), we first binned the native high-affinity antigen-specific training CDR-H3 sequences into four affinity classes: hyperbinder (affinity >max native), ultimate binder (max native>–1/3), penultimate binder (1/3–2/3), and binder (2/3–min native). Following binning, we used deep generative models to generate 700 K new sequences, devised 10 cutoffs in the increment of 70 K (70 K[native sized], 140 K … 700 K[large]), subsampled 10 times (from the 700 K generated sequences) and counted the number of novel sequences in each cutoff. Native (training) and generated sequences are shown in blue and yellow; error bars are shown for subsampled sequences. We found that, for all affinity classes, the number of unique sequences in each class increases as a function of the total number of generated sequences. In addition, we found sequences that possess a higher affinity than the native-training sequences (called hyperbinders) with affinity improvements over native CDR-H3 sequences ranging between 0.04–4.4% [depending on the antigen, percentages were calculated relative to the minimum affinity per antigen]. (b) To examine the diversity and preferences of developability combinations, we annotated each CDR-H3 sequence with a binary developability encoding. Briefly, we binned each developability parameter in two bins (low = min–median and high = median–max) and annotated each sequence with a composite binary encoding from all six developability parameters (i.e., 0_0_0_0_0_1 indicates that the sequence has a low charge, low molecular weight, low gravy index, low instability index, low affinity to MHCII and high affinity to MHC). We found that the generated CDR-H3 sequences yielded larger ranges of developability combinations in native-sized generation (nseq = 70,000) and large generation (nseq = 7x105). Error bars indicate the standard deviation for the subsampling.
![Figure 3. Exhaustive generation reveals better antibodies than are present in the training dataset. (a) To examine the ability of the RNN-LSTM model to generate CDR-H3 sequences beyond the native realms (in terms of quantity and affinity), we first binned the native high-affinity antigen-specific training CDR-H3 sequences into four affinity classes: hyperbinder (affinity >max native), ultimate binder (max native>–1/3), penultimate binder (1/3–2/3), and binder (2/3–min native). Following binning, we used deep generative models to generate 700 K new sequences, devised 10 cutoffs in the increment of 70 K (70 K[native sized], 140 K … 700 K[large]), subsampled 10 times (from the 700 K generated sequences) and counted the number of novel sequences in each cutoff. Native (training) and generated sequences are shown in blue and yellow; error bars are shown for subsampled sequences. We found that, for all affinity classes, the number of unique sequences in each class increases as a function of the total number of generated sequences. In addition, we found sequences that possess a higher affinity than the native-training sequences (called hyperbinders) with affinity improvements over native CDR-H3 sequences ranging between 0.04–4.4% [depending on the antigen, percentages were calculated relative to the minimum affinity per antigen]. (b) To examine the diversity and preferences of developability combinations, we annotated each CDR-H3 sequence with a binary developability encoding. Briefly, we binned each developability parameter in two bins (low = min–median and high = median–max) and annotated each sequence with a composite binary encoding from all six developability parameters (i.e., 0_0_0_0_0_1 indicates that the sequence has a low charge, low molecular weight, low gravy index, low instability index, low affinity to MHCII and high affinity to MHC). We found that the generated CDR-H3 sequences yielded larger ranges of developability combinations in native-sized generation (nseq = 70,000) and large generation (nseq = 7x105). Error bars indicate the standard deviation for the subsampling.](/cms/asset/641ac1e1-eb98-44c9-9f61-96e5be78bf13/kmab_a_2031482_f0003_oc.jpg)
In the same vein, we hypothesized that deep generative models would prove useful for generating CDR-H3 sequences with similar or richer developability profiles to native CDR-H3 sequences (higher number of combinations or constraints on developability parameter values). To this end, we devised a binary developability encoding wherein each developability parameter () is grouped into two categories: low (parameter values that range between the min and median of the distribution of the parameter) and high (parameter values that range between the median and max of the distribution of the parameter) and annotated each CDR-H3 sequence with a composite developability encoding combining all six developability parameters here examined (). For instance, the encoding 0_0_0_0_0_1 indicates that the thus annotated CDR-H3 sequence has a low charge (0), low molecular weight (0), low gravy index (0), low instability index (0), and low affinity to MHCII (0), but a high affinity for MHCI (1). Subsequently, we compared the total number of developability parameter combinations populated by the generated sequences (against native sequences) in two conditions: native-sized wherein the number of generated sequences matches the number of sequences in the native (training) dataset (nseq,generated = 70,000) and large where the number of generated sequences is an order of magnitude larger than the native training sequences (nseq,generated = 7x105). We observed a larger number of developability parameter combinations in the generated populations (). Specifically, native-sized generation yielded 29–39 developability parameter combinations (45–61% of all possible combinations), large generation yielded 33–44 (52–69% of all possible combinations) as compared to native sequences that yielded 21–37 combinations (33–58% of all possible combinations). Pearson correlation between the counts of developability parameter combinations in native and generated sequences was high (Pearson cor: 0.74–0.99, ). In other words, deep generative models can be leveraged to generate antibody sequences that are equally or more diverse than native (training) ones in terms of developability profile even when restricted to the constraint of generating high-affinity CDR-H3 sequences.
The quality of ML-based antibody sequence generation is a function of the size of the training data
The absence of large antigen-annotated antibody sequences and structural datasets remains a major challenge in developing robust machine learning methods for antibody-antigen binding prediction as well as antigen-specific generation of mAbs.Citation12,Citation43 Furthermore, the precise amount of antibody sequence data necessary to recover native-like antibody affinity, epitope, and developability is a subject of ongoing investigations.Citation11–13 Therefore, within the framework of our simulation suite Absolut!, we examined how the number of training CDR-H3 sequences affects the resulting binding affinity of the generated CDR-H3 sequences. To this end, from the top 1% antigen-specific CDR-H3 sequences (nseq = 70,000), we created smaller datasets of antigen-specific CDR-H3 sequences (nseq,subsample = 700; 7,000; 10,000; 20,000; 30,000; 40,000; 50,000; 60,000, and nreplicates = 5); trained deep generative models on these subsets; and compared the resulting binding affinity against native CDR-H3 sequences and CDR-H3 sequences generated by models trained on the top 1% (nseq = 70,000) antigen-specific CDR-H3 sequences (called “data-rich” model). We found that the correspondence between the binding affinity and epitope recognition of native and generated CDR-H3 sequences increased as a function of the number of training CDR-H3 sequences (, Supplementary Fig. S5). Specifically, our models recovered very closely the native affinity (as measured by median energy) when we used 20,000 or more training CDR-H3 sequences (Supplementary Fig. S5). Similarly, the agreement of epitope occupancy between generated CDR-H3 sequences increases as a function of the sequence size of the training set (Supplementary Fig. S8, Supplementary Fig. S11). Of note, we found that the agreement of epitope occupancy between native and generated CDR-H3 sequence was already reasonable at a small training dataset (ntrain = 700) for antigens with fewer epitopes (e.g., 3VRL, see ) (Supplementary Fig. S8, Supplementary Fig. S11). In contrast, antigens with more epitopes (e.g., 1H0D, ) required larger training datasets for reaching a high concordance with the epitope occupancy observed in the training dataset (Supplementary Fig. S8, Supplementary Fig. S11).
Figure 4. Generation quality of antibody sequences depends on the size of the training dataset and transfer learning enables the generation of higher-affinity CDR-H3 sequences from lower-sized training datasets. (a) To examine the impact of sample size on the resulting binding affinity and epitope (see Supplementary Fig. S8) of generated CDR-H3 sequences, we created smaller training datasets (nseq,subsample = 700; 7,000; 10,000; 20,000; 30 000; 40,000; 50,000; 60,000, and nreplicates = 5) from the full antigen-specific CDR-H3 sequences (nseq,training = 70,000), trained deep generative models on the subsets and compared the binding affinity and epitope against affinity and epitope from models trained on the full data and the native affinity and epitope (see Supplementary Fig. S8 for correlations of CDR-H3 epitope occupancy). We found that models trained on the larger dataset sizes (>2x104), but not the smaller subsets (in the order of 103 or 102), sufficiently replicate the distribution of binding affinity and epitope CDR-H3 sequences. (b) To investigate whether transfer learning may be used to improve the affinity and epitope (see also Supplementary Fig. S9–Supplementary Fig. S13) binding of CDR-H3 sequences generated by models trained on smaller-sized datasets, we constructed a transfer architecture wherein embedding and RNN-LSTM layers from a “data-rich” model (high N, nseq, training = 70,000) were stacked atop of a fresh dense layer and training the resulting ‘transfer’ model on lower-sized datasets (data-poor; low N, nseq, training = 700/7,000). Two types of transfer experiments were performed: a within-antigen transfer experiment (e.g., between a data-rich model of an antigen V and data-poor models of the same antigen V) and a between-antigens (across antigens) transfer experiment (e.g., between data-rich model of an antigen V and data-poor model of antigen G). We used Kolmogorov–Smirnov distance (KSD, range: 0 for identical distribution, increasing value for increasing dissimilarity between distributions) to quantify the similarity between affinity distributions of CDR-H3 sequences generated by the models with transfer learning (+T) and without transfer learning (-T). Smaller KSD values indicate that the compared affinity distributions are similar and a larger value signifies dissimilarity of affinity distributions. For within transfer experiments, we found marked reductions of KSD values (against the native population) in all antigens signifying the transferability of general antibody-antigen binding features within antigens. For across-antigens transfer experiments, 7 out of 10 antigens showed reductions in KSD values in at least one transfer scenario (nseq, training = 700 or 7,000, Figure 4b) suggesting the transferability of antibody-antigen binding features across antigens.
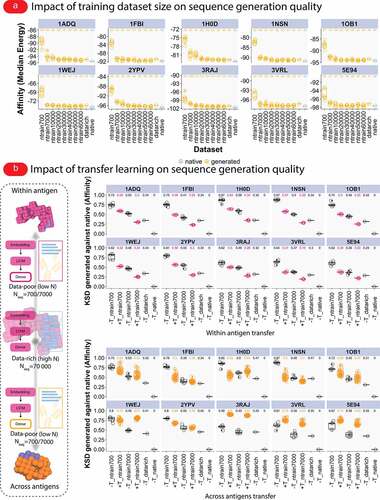
In summary, 20 000 CDR-H3 sequences were sufficient to train models that reproduce native-like affinity. We note that our simulation framework Absolut! does not operate at atomistic resolution,Citation32 thus, nseq,training in the order of 20,000 should only be regarded as a lower bound of the number of training CDR-H3 sequences necessary to train a robust deep generative model, in comparison with a higher dimensionality of binding modes in experimental datasets.
Transfer learning enables the generation of high-affinity CDR-H3 sequences from lower-sized (low-N) training datasets
Based upon the observation that lower-sized training datasets failed to produce CDR-H3 sequences with native-like binding affinity and epitope binding, we asked whether the generation quality of models trained on lower-sized datasets (data-poor, “low-N,Citation44”, nseq,training = 700 and 7,000) may be improved by transferring learned features from models trained on larger training datasets, which were found to be sufficient for achieving a native-like affinity (data-rich, nseq,training = 70,000, ). We examined this question by constructing a transfer learning architecture wherein pre-trained embedding and RNN-LSTM layers from a data-rich model were stacked atop of a new fully connected layer with the resulting “transfer” model subsequently being trained on lower-sized datasets (). We performed two different transfer learning experiments, termed within antigen and across antigens transfer. Within antigen transfer describes a transfer experiment involving the same antigen (this transfer setting serves as a positive control for the functioning of the transfer architecture). That is, pre-trained embedding and LSTM layers from a data-rich model based on CDR-H3 sequences specific for an antigen V were stacked atop of a new dense layer; the resulting architecture was trained on lower-sized datasets (nseq,training = 700 and 7,000) of antigen V. In contrast, across antigens transfer identifies a transfer experiment involving different antigens, e.g., a data-rich model of an antigen V and data-poor (lower-sized datasets nseq,training = 700 and 7,000) models of antigen G (see Methods and ). Following training, for each antigen, we generated a total of 100,000 CDR-H3 sequences (10,000 sequences, 10 replicates) and measured the generation quality with respect to affinity and epitope. We used the Kolgomorov–Smirnov distance (KSD) to quantify the similarity between the generated binding affinity distributions and the native affinity distribution. A small KSD indicates that the compared affinity distribution is similar and increasing KSD indicates increased dissimilarity. We observed marked reductions of KSD values (against the affinity distribution of the native population) for the within antigen transfer in all models (, upper panel and Supplementary Fig. S7), which signifies the availability, learnability, and transferability of general antibody-antigen binding features within an antigen. For the across antigens transfer experiments, 7 out of 10 antigens showed reductions in KSD values in at least one transfer scenario (nseq,training = 700 or 7,000, , lower panel) suggesting the transferability of antibody-antigen binding features across antigens and the multi-faceted nature of the signal per antigen learned (nota bene, the medians of binding affinities in the across antigens transfer scenario were closer to the native and data-rich affinities in all 10 antigens, Supplementary Fig. S6). For epitope similarity, we used Pearson correlation (Supplementary Fig. S9, Supplementary Fig. S10) and overlap (Supplementary Fig. S12, Supplementary Fig. S13) to quantify the concordance between epitopes recognized by native and generated CDR-H3 sequences. Similar to affinity, we found better concordance both for the within and across antigens transfer (increasing Pearson correlation values, Supplementary Fig. S9 and Supplementary Fig. S10). Interestingly, the number of recognized epitopes jumped in across-antigens transfer (Supplementary Fig. S13) whereas in the within-antigen transfer (Supplementary Fig. S12) the number of recognized epitopes dropped, hinting at the utility of across-antigens transfer in generating epitope diversity. In summary, our in silico experiments suggest that transfer learning may represent a suitable method for generating high-affinity CDR-H3 sequences from lower-sized training datasets.
Antibody-design conclusions gained from simulated antibody-antigen binding data on required sequence diversity hold on experimental antibody-antigen data
Experimental validation at the scale of the number of antibody sequences that can potentially be ML-generated () remains an unresolved technological problem. One potential solution to this challenge is the development of experimentally validated ML-classifiers (also called oracles) that can screen the potential sequence space for binders. One such classifier for HER2 binders was previously developed by Mason et al.Citation37 Briefly, this convolutional neural network (CNN)-based classifier (oracle) discriminates CDR-H3 amino acid sequences for their potential to bind HER2; all CDR-H3 sequences annotated with a binding probability of p > 0.5 are considered binders (we also investigated a probability threshold of p > 0.7). Mason et al. validated the CNN-based classifiers experimentally by the expression and testing for binding of predicted HER-2 binders (experimentally validated computational oracle). Given that the experimental system by Mason et al. is similar to the one simulated in this work, i.e., testing of binding of CDR-H3 sequences, we concluded that the CNN-classifier can be used to evaluate the experimental HER-2 binding potential of the output of our RNN sequence generator ().
We used the CNN-classifier to investigate whether the lower threshold of sequence diversity necessary for high-accuracy generative antibody ML determined using simulations () also holds on experimental real-world data. To this end, we performed the following experiment: we trained separate RNN-LSTMs on the experimentally verified 11,300 HER2 binders (“RNN-LSTM binder model”) and the 27,539 HER2 non-binders (“RNN-LSTM non-binder model”) of the Mason et al. dataset and used the RNN-LSTM models to generate 7 × 105 sequences in each case. To examine the impact of dataset size on the percentage of generated binders, we created smaller training datasets (700 and 7,000) by subsampling five times from the original binders and non-binders datasets (). Importantly, while the CNN-based classifier was trained using both HER2 binder and non-HER2-binder data, our RNN-LSTM models were trained on binder and non-binder data separately. That is, the RNN-LSTM trained on binders did not have access to non-binder data and vice-versa. These generated CDR-H3 sequences were assessed for their HER2-binding potential using the experimentally verified CNN-classifier (). We found that 68% (HER2-binding probability cutoff [Pbind]>0.5; 63% for Pbind>0.7) of the generated CDR-H3 sequences from the RNN-LSTM binder model trained on all 11,300 HER2 binders were scored as binders and we ascertained that the generated sequences follow the positional amino acid dependencies (position weight matrix [PWM]) of the experimentally verified training data (Supplementary Fig. S15; PWM-generated sequences yielded a markedly lower percentage of binders at 43% (Pbind>0.5), Supplementary Fig. S17B; MSE values are shown in Supplementary Fig. S17A). To verify that binders are enriched in the generated sequences, we computed as a baseline the percentage of CNN-predicted binders on an unrelated human IgM naive B cell dataset (1,307,472 sequences, Supplementary Fig. S16; see Methods) finding a substantially reduced number of CDR-H3 sequences that were classified as binders (10.5% for the cutoff value >0.5; 7.6% for the cutoff value >0.7).
Figure 5. RNN-LSTM model trained on experimentally validated binders (not synthetic sequences) generated native-experimental-like binders. (a) To validate that our RNN-LSTM model cannot only reproduce properties of native-like synthetic sequences of binders but also experimentally determined binders, we trained the model with varying numbers (700–Max; max for binders ~11 K and max for non-binders ~27 K) of binders and non-binders obtained from recently published experimental data against human epidermal growth factor 2 (HER2),Citation37 generated 7 × 104 sequences and scored the sequences with the Mason et al. CNN classifier (the CNN classifier outputs a HER2 binding probability value between 0–1). Subsequently we used the CNN as an experimentally validated oracle to create datasets of binders (Pbind>0.7 or Pbind>0.5) and non-binders (Pbind≤0.3 or Pbind≤0.5) for our 7 × 106 mouse sequences, trained our model with the oracled datasets (700–Max; max for binders and non-binders is 7 × 104). We subsampled five times for the lower sized datasets (700 and 7,000). Finally we compared the proportion of predicted HER2 binders and non-binders across models trained on experimental data and models trained on oracled data. (b) We found good correspondence between the experimental and oracled datasets in terms of fraction of correctly predicted sequence (binders, non-binders). For binders, RNN-LSTM models trained on the smallest training datasets yielded the least fraction of correct prediction (Exp.: 0.25; Orac. 0.16) and models trained on the largest training datasets yielded the maximum fraction of correct prediction (Exp.: 0.68; Orac.: 0.71). For non-binders, we found that already at the smallest datasets the models were able to yield non-binders both for experimental and oracled datasets. Specifically, the fraction of correct prediction for the smallest non-binder datasets were 0.95, 0.88, and 0.91 for the experimental and oracled (Pbind≤0.3 and Pbind≤0.5) category respectively; and at the largest non-binder datasets, the fractions of correct prediction were 0.84, 0.92, and 0.94 for the experimental and oracled category respectively. Distributions of amino acids per position are summarized in Supplementary Fig. S15 and distributions of predicted binding probability of the here shown data are in Supplementary Fig. S14. Baseline HER2 binding probability distributions of human and mouse CDR-H3 sequences are shown in Supplementary Fig. S16.
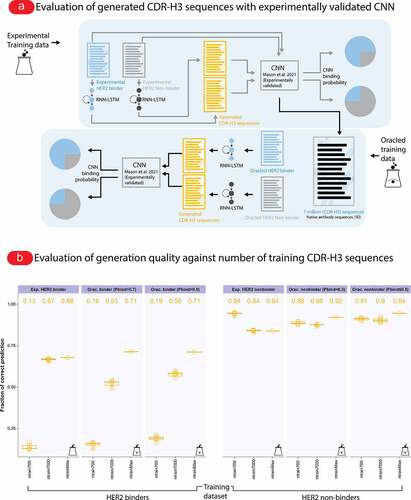
When CNN-scoring the CDR-H3 sequences from the RNN-LSTM non-binder model, only 16% of the generated CDR-H3 sequences were scored as binders (84% of the non-binder CDR-H3 sequences were correctly classified as non-binders; the percentages are similar for the smaller datasets 94% and 84% for 700 and 7000, respectively, which is in line with detection rates (89.5% for the cutoff value ≤0.5; 86% for the cutoff value ≤0.3) of non-binders baseline data (unrelated human IgM naive B cell data as mentioned above) indicating that the RNN-LSTM model does not learn to generate non-binder better than a random dataset.
To summarize, we showed using the experimentally validated CNN that the RNN-LSTM trained on experimentally determined HER2-binding sequences, successfully generated sequences classified as HER2-binders and that a training dataset in the order of 1–2x104 sequences (as also observed with our synthetic data, ) is sufficient to generate CDR-H3 sequences that bind the target antigen.
Subsequently, to repeat the above results on a different set of experimental antibody sequences, we also used the CNN to create binder and non-binder datasets from our native sequences (7 million murine CDR-H3) and trained separately RNN-LSTMs on these ‘oracled’ datasets as described above (). We found good correspondence between the experimental and oracled datasets with respect to percentages of generated binders vs non-binders (, for a baseline HER2-binding probability distribution, see Supplementary Fig. S16 and Supplementary Fig. S17B) as a function of dataset size suggesting that ML conclusions gained on Absolut!-selected data are transferable to datasets selected by experimentally validated oracles. Of note, these results also suggest that de novo antibody design is feasible using only binding sequences (positive data) for ML model training even when binder and non-binder data are relatively similar.
Discussion
We have here provided the in silico proof-of-principle that deep learning can learn the non-linear rules of 3D-antibody-antigen interaction from 1D antibody sequence data alone by showing (in a 3D-lattice space) that novel antibody variants with high affinity and specific epitope binding can be generated based on sufficiently large training data (). Among the generated antibodies, for all tested antigens (10 out of 10), we detected novel antibody sequences that exceeded in affinity those found in the training dataset (). ML-based sequence generation also allowed for the discovery of novel developability parameter combinations (). For the ML model used, we determined the number of training CDR-H3 sequences necessary (>2x104) for generating high-affinity CDR-H3s and demonstrated that these numbers may be reduced by transfer learning (). Finally, we experimentally validated the antibody-design conclusions drawn from ML training on simulated antibody-antigen binding data (). More broadly, while our primary objective was the proof-of-principle study of antibody generative learning, the secondary objective was to leverage large-scale synthetic antibody-antigen binding data that replicates many complexities of biological antibody-antigen binding and develop a set of analytical approaches that may help in the study of the quality of generated antibody sequences in future studies with a similar aim.Citation45
In this work, we chose an RNN-LSTM-based language modeling approach because it represents a competitive baseline to the state-of-the-art transformer-based architecture.Citation46 Recently, VAEs, as well as other deep generative approaches, have also been used for generating T- and B-cell receptor sequences.Citation23,Citation26,Citation29,Citation47 However, both in the area of natural language processing as well as in the area of generative models for small molecules, GANs and VAEs remain less competitive.Citation35,Citation48 Although we decided to use an RNN-LSTM as a generative model, we hypothesize that any accurate language model, e.g., transformer architectures,Citation32 would lead to similar results and conclusions. Further benchmarking is needed in the area of generative protein design.
A common problem with deep learning-generated sequence data is that methods may reproduce the training data with minimal changes, which has been termed the “copy problem” by Renz and colleagues.Citation49 The copy problem is especially prevalent when the capacity for high-throughput testing of molecular properties (in our case, antigen binding and developability) is unavailable. The absence of prospective testing capacity precludes the functional (e.g., antigen binding) evaluation of the generated dataset, which renders addressing the copy problem somewhat unfeasible (merely testing sequence diversity on sequences of which the binding mode is unknown does not elucidate the extent of diversity for a given binding mode for example). In this work, we were able to address and exclude the copy problem by evaluating all generated sequences for both binding as well as for sequence diversity due to the capacity of unrestricted prospective sequence evaluation afforded by the Absolut! platform ().Citation32
The transfer learning experiments demonstrated the capacity of deep learning models trained on large collections of CDR-H3 sequences to augment weaker datasets (smaller datasets that fail to reproduce faithfully the affinity and epitope of native sequences) for both within and across antigens scenarios (). Although transfer learning improved (smaller KSD values against native) the generation quality of weaker models in all 10 antigens for the within antigen transfer scenario, three antigens (3RAJ, 3VRL, and 5E94) did not show improvements (larger KSD values against native) for the across antigens transfer scenario (although closer examination of the generated affinity distributions revealed that the median affinity values of across antigens transfer learning were closer to the median affinity values of native CDR-H3 sequences, Supplementary Fig. S6). Furthermore, the number of recognized epitopes in any transfer learning was notably larger than the number of recognized epitopes in sequence generation without transfer learning and in native CDR-H3 sequences (Supplementary Fig. S13) independent of the KSD values against native CDR-H3 sequences. This illustrates the key challenges remaining in the prospective testing of many orthogonal variables wherein several parameters must be captured and justly reflected in order to communicate faithfully the underlying trends in the data. Indeed the success of cross tasks transfer has been shown to be heavily influenced by the compatibility of the source and target task types.Citation50 Nevertheless, our cross-antigens transfer learning experiments show that, at least in the case of our antibody sequence datasets, neural network models can extrapolate 3D non-linear dependencies to CDR-H3 sequences outside the training distribution.Citation50–52
One may argue that the Absolut! antibody-antigen binding simulation framework generates sequences that are binders within the lattice framework but would not be binders if tested in vitro/vivo. That said, we ensured that the Absolut! framework is state-of-the-art surpassing all currently available large-scale antibody-antigen binding simulation frameworksCitation34 (e.g., the inclusion of discretized Protein Data Bank (PDB)-stored antigens, 3D-binding [albeit on a 90°-grid], experimentally determined physiologically relevant amino-acid interaction potentialsCitation32). The inbuilt physiological relevance of our antibody-antigen simulation model affords a more precise understanding of how the accuracy of computational models increases with the number of available antibody sequences for training, which will help in planning experimental validation studies. We also avoided the possibility that the generative model learns to exploit the affinity models by refraining from a full reinforcement learning setting, in which the affinity model would be used as a reward function.Citation49 Specifically, the major challenge of predicting antigen reactivity of an antibody sequence lies in recapitulating the residue interactions between the antibody and antigen structures in 3D space. Even our simplified computational model of antibody structure includes physical antibody-antigen interactions in 3D space entailing non-linearities and positional dependencies reminiscent of the biological complexity.Citation32 Consequently, one may argue that our simulation framework and investigations are suitable for establishing an informative lower bound of the complexities encountered in machine and deep-learning-based biological sequence design. Indeed, a recent study by Mason et al.Citation37 that leverages experimental deep mutational scanning data showed that a training dataset size in the order of 104 (as also shown in this study for generative models; ) was sufficient to train ML models that discriminate binders and non-binders. Furthermore, the study also highlights that a large proportion of dissimilar sequences (LD>2) bind to the target antigen (as also shown in this study in and in Robert et al.Citation32). These parallels (with results from experimental data) reiterate the utility and relevance of simulated custom-designed synthetic datasets in advancing the development of computational approaches for antibody design and discovery.
For future investigations, we cannot emphasize enough the need for experimental validations to compliment the herein in silico results. Recently, Saka and colleagues showed that RNN-based generated antibody sequences bind the desired target providing experimental proof of principle of our computational framework.Citation30 Here, we validated the conclusions gained profiling the RNN-LSTM framework () by scoring the generated CDR-H3 sequences using an experimentally validated oracle (CNN-classifier)Citation37 (). Furthermore, our RNN-LSTM models were trained separately on binders (positive data) and non-binders (negative data) suggesting that the design of native-like CDR-H3 sequences is possible without the need for negative examples and accuracy is likely to be further improved with more training examples (the 68%-HER2 generation rate by the RNN is fairly close to the CNN prediction accuracy of ≈80%,Citation37 and substantially different from the percentage of HER2 binders on an unrelated baseline dataset, Supplementary Fig. S16). This could potentially reduce the cost to generate training datasets given that the HER2 generation rate of the RNN was remarkably high despite only being trained on positive data. Indeed earlier studies have shown that performance improvements/reductions by including more or less negative data vary across models and application domains.Citation53,Citation54 This highlights the potential applicability of our framework in real-life settings beyond the synthetic simulated setting earlier described. We strongly believe that the synergistic combination of simulation and experimental strategies is necessary for the time- and cost-efficient discovery of antibody therapeutics. Furthermore, we would like to stress that it is currently virtually unfeasible to exhaustively generate experimental data for validation of ML methods at a scale and breadth of conditions corresponding to the simulation-based analyses reported in this work, as antibodies would have had to be expressed for each simulation condition. If we had also benchmarked different ML architectures, the predictions of each of those would also have had to be validated separately, leading to an endless number of conditions for which to express antibodies. This Gedankenexperiment underlines the immense importance of both simulation frameworks for benchmarking biology-focused ML applications,Citation45,Citation55 as well as the availability of experimentally validated in silico oracles for (multiparameter) scoring of ML-generated protein sequences.Citation56 Naturally, future refinements to the Absolut! simulation framework would further improve the applicability of conclusions drawn to experimental settings. These refinements are among others (see Robert et al.Citation32 for a more detailed discussion): 1) antibody full VH-VL chains (so far, we can only model CDR-H3-antigen binding), 2) smaller angle grid in the lattice: our framework was limited to integer positions in a 3D grid, 3) addition of constraints at the CDR3 ends in order to reproduce the anchoring of the CDR chains to the framework/conserved domains of the antibody.
Once more experimental data have become available, one may venture into merging simulation and experimental training data. For example, one could perform transfer learning based on antibody sequences with only partially determined experimental labels, thus increasing the biological faithfulness of deep-learning-designed antibody sequences.Citation57 Such a setup may be further augmented in the form of federated learning.Citation58 Furthermore, here we performed deep learning on amino acid sequences and not nucleotide sequences although nucleotide sequences are essential for experimental antibody expression. However, codon usage is often species-specific.Citation59 Therefore, we opted for the more general amino acid encoding. Nevertheless, our deep learning setup would work equally well for nucleotide sequences.
A key property of in silico generative frameworks such as ours is that once trained, it paves the way for large-scale and on-demand generation of antigen-specific and developable immune receptor sequences. The fast production of antibodies has seen continued interest from the field.Citation7,Citation8 Although library-based discoveries have the potential to generate a higher volume of antigen-specific data as compared to crystallography or related approaches, they remain reliant on multiple rounds of selection as well as other experimental heuristics. We approached the discovery process by leveraging deep generative models, which implicitly aim to learn the rules of antibody-antigen binding. Once learned, the generation of vast quantities (virtually limitless) of antibody sequences becomes feasible, abrogating the need for follow-up screening. Rule-based generation also imparts the ability to design (not merely discover) antibody sequences by biasing the deep generative models toward a particular set of developability parameters via reinforcement learning or instance selection.Citation23,Citation60 The combination of near-limitless and fast sequence generation may enable the construction of an on-demand antibody generator where antigen-specific antibody sequences can be obtained at will.
In this work, we did not train on datasets that were selected for both binding and developability therefore not optimizing both antibody design entities at once. This is partly due to the inherent sparsity of the data, although our datasets are the largest currently available. Conditional generation based on several orthogonal sequence and structural propertiesCitation61 in one training dataset is an interesting avenue for future research. Furthermore, we would like to point out that we have not optimized in any way the deep generative architecture used. Therefore, our framework allows for optimizing the generative output of deep learning approaches in future benchmarking studies.Citation11,Citation62,Citation63 In addition, further research is needed to understand the relationship between signal (pattern) complexity, encoding and embedding,Citation20 and the number of sequences needed for achieving satisfactory generation quality.
In closing, naturally occurring proteins represent only a small subset of the theoretically possible protein sequence space. Here, we demonstrate a proof-of-principle that deep learning helps explore a broader sequence and structural space than present in the training data thereby enabling the discovery and the design of antibody sequences with enhanced or novel properties.Citation7,Citation64 Moreover, our ground-truth-based framework may be useful in the establishment of methods for model interpretability.Citation45,Citation64–67
Methods
Reference experimental immunoglobulin and 3D-crystal structure antigen data
Native B-cell receptor (CDR-H3) sequences (, murine origin [we showed in a separate work that murine and human CDR-H3 sequences have similar affinity distributions in the Absolut! antibody-antigen simulation framework]Citation32) were obtained from Greiff and et al.Citation38 Ten antigen 3D-crystal structures were sourced from known antibody-antigen complexes in the Antibody Database (AbDb) ()Citation39 and converted into lattice-based discretized Absolut! format. To annotate each CDR-H3 sequence for antigen specificity, we determined the best bindingCitation32 position of an antibody sequence to an antigen and calculated the corresponding binding affinity via the software suite Absolut! (see below and Robert et al.Citation32). Antigen-specific CDR-H3 sequences were defined as the top 1% affinity-sorted CDR-H3 sequences for each antigen (nseq = 1% times 7 × 106 = 70,000). We chose the top 1% because it selected a sufficiently high number of sequences and ensured high antigen-specific affinity (see Supplementary Fig. S3 for a comparison of the affinity distribution of all 7 million CDR-H3 sequences [“native”] vs the top 1% affinity ones [“native_top”]).
Experimental datasets used for the experimental validation of antibody-design conclusions drawn from ML training on simulated antibody-antigen binding data
The two datasets described in this subsection relate to (as well as the related Supplementary Figures). From the HER2 binder/non-binder dataset published by Mason and colleagues,Citation37 we obtained 11,300 HER2-binders and 27,539 non-binder unique amino acid CDR-H3 sequences of length ten amino acids. From the publication of DeWitt and colleagues,Citation68 we obtained 1,307,472 unique CDR-H3 sequences of length 10 amino acids stemming from naive human B-cells.
Reference CNN model trained on experimental human epidermal growth factor 2 (HER2) CDR-H3 binder and non-binder sequences
CDR-H3 sequences that bind (binders) and do not bind (non-binders) to HER2 were obtained from Mason and colleagues as described previously.Citation37 The sequences were used to train a convolutional neural network (CNN) classifier that assigns an HER2 binding probability to a given input CDR-H3 sequence. The accuracy of this CNN classifier was experimentally validated. We used the CNN classifier to 1) evaluate the HER2 binding probability of CDR-H3 sequences generated by our RNN-LSTM model that was trained on the Mason et al. binder/non-binder dataset, 2) as an oracle to create binder/non-binder datasets from our murine CDR-H3 sequences (the murine CDR-H3s were filtered for length ten amino acids to comply with the input size of the CNN), and 3) to compute the baseline percentages of finding HER2 binders in murine and human CDR-H3s (see and Supplementary Fig. S16).
Generation of lattice-based antibody-antigen binding structures using Absolut!
The Absolut! software was used to compute the binding energy and best binding structure (here termed paratope fold or binding fold) of antibody (CDR-H3) sequences around the antigens in a 3D-lattice space (see Robert et al.Citation32 for a very detailed explanation). Briefly, the antigens of interest, named by their PDB entry (), were transformed into a coarse-grained lattice antigen representation (a step called discretization, performed using the program LatFitCitation69), where each residue occupies one position and consecutive amino acids are neighbors, creating a non-overlapping 3D chain with only 90 degrees angles. In the lattice, a position is encoded as an integer (for instance, [x = 31, y = 28, z = 15] is encoded as a single integer code [x + L*y + L*L*z] where L is the lattice dimension: 64). Further, protein chains are represented as a starting position and a list of moves, for instance, 63263-SUSDLLUR is a peptide starting at position (x = 31, y = 28, z = 15; 31 + 64*28 + 64*64*15 = 63263) with 9 amino acids and following the structure ‘Straight, Up, Straight, Down, Left, Left, Up, Right’ where each ‘turn’ is defined from the previous bond and is coordinate-independent. From each CDR-H3 sequence investigated, all peptides of 11 consecutive amino acids are taken (sliding window with a step size of 1; window size of 11 was chosen to provide the best compromise between computational cost and CDR-H3 length/coverage, see Robert et al.Citation32) and are assessed for binding to the antigen. From exhaustive enumeration of all possible structures of the peptide around the antigen, Absolut! returns the structure minimizing the energy of the complex (Supplementary Fig. S1). Exhaustive enumeration of all possible binding folds (binding structures) of a CDR-H3 sequence enables Absolut! to function as an oracle since it can generate the binding fold as well as evaluate the binding energy of any sequence against the antigen of interest. The energy is computed from neighboring, noncovalent amino acids either between the CDR-H3 and the antigen (binding energy) and within the CDR-H3 (folding energy) using an empirical experimentally estimated potential.Citation70 Among all 11 amino acids peptides for this CDR-H3, the one with the best total (binding + folding) energy is kept and its structure is called the ‘binding structure’ or the ‘paratope fold’ of the CDR-H3 (Supplementary Fig. S1). The paratope in that structure will be the spatial conformation of interacting amino acids on the antibody side and the epitope the spatial conformation of interacting amino acids on the antigen side. In that way, each CDR-H3 sequence is annotated with a 3D binding structure (paratope and epitope) and binding energy (see and Supplementary Fig. S1 for illustration). In summary, using Absolut!, we constructed a dataset of 70 million (7 million CDR-H3 sequences x 10 antigens []) antibody-antigen structures with annotated paratope, epitope, affinity, and antibody developability (see below). The advantages and caveats of the Absolut! simulation suite have been discussed previously by Robert and colleagues.Citation32
Computation of developability parameters
Developability is defined as the “feasibility of molecules to successfully progress from discovery to development via evaluation of their physicochemical properties”.Citation71 Developability parameters (, inspired by the works described in referencesCitation23,Citation37,Citation72) were computed using the module Bio.SeqUtils.ProtParam in BiopythonCitation40 and NetMHCIIpan versions 4.0 and 4.1.Citation73 For NetMHCpan and NetMHCIIpan we used the percent rank (the percentile of the predicted binding affinity compared to the distribution of affinities calculated on a set of random natural peptides) where typically the thresholds for strong binders are defined at 2% and weak binders between 2% and 10%. CDR-H3 sequences were used for the calculation of the aforementioned developability design parameters. We note that for NetMHCpan and NetMHCIIpan, we specified the window size to 11 and calculated the average percent rank for each CDR-H3 sequence.
Deep generative learning using long short-term memory neural networks for generating antibody CDR-H3 sequences
The architecture of the deep generative model used consists of three layers (see and Supplementary Fig. S2): 1) an embedding layer with 256 output-vector dimensions, 2) a recurrent neural network of the type long short-term memory (RNN-LSTM)Citation74 with 1024 units, and finally 3) a fully connected output layer with softmax-activations of 21 (20 amino acids and one whitespace character) output-vector dimensions (see ). Input-target pairs, i.e., sequences and their labels, were obtained by first merging the antibody sequences (CDR-H3s) into a text corpus, sequences were separated by a single whitespace character. A window of size w (w = 42 amino acids) was used to fragment the corpus into chunks of input sequences x of length w. For each input sequence x, a target sequence y was created by sliding a window of size w-1 one step forward. The last character was removed from x, creating an input-target pair (x, y) each with the size w-1. Thus, the LSTM model g(x, θ) is trained to predict the next character of the given sequence using categorical cross-entropy loss L(y, g(x, θ)), where θ is the parameter/weight of the LSTM model. We partitioned the input-target pairs into training (70%), validation (15%), and test (15%) sets. The training was carried out for 20 epochs with Adam optimizer.Citation75 At the end of each epoch, training, and evaluation loss were computed for evaluation. The generation was initiated with a seed string and the hyperparameter temperature was set to 1. Our implementation is based on TensorFlow 2.0.Citation76
Implementation of transfer learning
We leveraged transfer learning to examine whether the generation quality of models trained on lower-sized datasets (data-poor, “low-N,Citation44”) may be improved by transferring learned features from models trained on larger training datasets. For a visualization of the transfer learning setup, see . Prior to any transfer learning experiment, we randomly sampled 1% (nsample = 700) and 10% (7,000) sequences from the set of antigen-specific CDR-H3 sequences, defined as the top 1% affinity-sorted CDR-H3 sequences for each antigen, nsample = 70,000). Sampling was performed 5 times per antigen. Models trained on 70,000 sequences were termed “data-rich” and models trained on 700–7,000 sequences, “data-poor”. In a transfer experiment, a transfer learning architecture was constructed by stacking the pre-trained embedding followed by the pre-trained RNN-LSTM layers from data-rich models and a new fully connected layer (see for network architecture). The training was performed as described in the previous section (“Deep generative learning”). Transfer learning was performed in two ways termed “within-antigen” and “across-antigens” (see ). “Within-antigen” experiments describe transfer-learning between the same antigen (e.g., embedding and RNN-LSTM layers of data-rich models stem from the same antigen that is used to train a data-poor model). “Across-antigens” describes the transfer of layers between different antigens (e.g., the combination of a data-rich model of antigen V and a data-poor model of antigen G). The within-antigen experiments served as positive controls where stronger signals (from a data-rich model) were used to improve the performance of a weaker model.
Sequence similarity, composition, and long-range dependencies
Sequence similarity among generated CDR-H3 sequences was determined by Levenshtein distance and gapped k-mer analysis. Levenshtein distances were computed using the distance function in the package Python-Levenshtein.Citation77 Long-range dependencies were assessed by gapped k-mer analysis using the R package kebabsCitation78 as previously described.Citation79,Citation80
Distance between distributions
The similarity between two CDR-H3 affinity distributions was quantified using the Kolmogorov–Smirnov distance (KSD) using the function ks.diss from the R package Provenance.Citation81 The KSD measures the largest vertical distance between the two examined (cumulative) distributions. A KSD value close to 0 indicates that the distributions are very similar and a larger distance (e.g., 1) indicates larger differences between the distributions.
Mean squared error of positional amino acid frequency matrix
As previously described,Citation82 the difference between two amino acid position-specific frequency matrices () was quantified by the mean squared error (MSE) ,where A is the reference native amino acid frequency matrix, B is the generated amino acid frequency matrix, n is the 20 amino acid alphabet, m is the length of CDR-H3, i is the row index and j is the column index.
Generation of sequences from position-specific weight matrix (PWM)
Lengthwise PWMs were obtained by partitioning the training sequences according to their length and calculating the resulting frequency matrix for each amino-acid position. The frequency matrix was used to sample amino acids for each position to create new PWM sequences (Supplementary Fig. S3).
Graphics
Plots were generated using the R package ggplot2Citation83 and arranged using Adobe Illustrator 2020 (Adobe Creative Cloud 5.2.1.441).
Hardware
Computations were performed on the Norwegian e-infrastructure for Research & Education (NIRD/FRAM; https://www.sigma2.no) and a custom server.
List of abbreviations
| 1D: | = | One dimensional |
| 3D: | = | Three dimensional |
| CDR-H3: | = | Complementarity-determining region 3 of the heavy chain |
| CNN: | = | Convolutional neural network |
| GANs: | = | Generative adversarial networks |
| HER2: | = | Human epidermal growth factor 2 |
| HIV: | = | Human immunodeficiency virus |
| KSD: | = | Kolgomorov-Smirnov distance |
| LD: | = | Levenshtein distance |
| Low-N: | = | Lower-sized training dataset |
| LSTM: | = | Long short-term memory |
| mAb: | = | Monoclonal antibody |
| MHCI: | = | Major histocompatibility complex I |
| MHCII: | = | Major histocompatibility complex II |
| ML: | = | Machine learning |
| OAS: | = | Observed antibody space |
| PDB: | = | Protein data bank |
| RNN: | = | Recurrent neural network |
| SARS-CoV-2: | = | Severe acute respiratory syndrome coronavirus 2 |
| TCRβ: | = | T cell receptor beta |
| VAE: | = | Variational autoencoders |
Data and code availability
Preprocessed datasets, code, and results figures are available at:
https://github.com/csi-greifflab/manuscript_insilico_antibody_generation and https://doi.org/10.5281/zenodo.5211239.
Supplemental Material
Download Zip (18.2 MB)Acknowledgments
We acknowledge generous support by The Leona M. and Harry B. Helmsley Charitable Trust (#2019PG-T1D011, to VG), UiO World-Leading Research Community (to VG), UiO:LifeScience Convergence Environment Immunolingo (to VG, GKS, and IHH), EU Horizon 2020 iReceptorplus (#825821) (to VG), a Research Council of Norway FRIPRO project (#300740, to VG), a Research Council of Norway IKTPLUSS project (#311341, to VG and GKS), a Norwegian Cancer Society Grant (#215817, to VG), and Stiftelsen Kristian Gerhard Jebsen (K.G. Jebsen Coeliac Disease Research Centre) (to LMS and GKS). This work was carried out on Immunohub eInfrastructure funded by University of Oslo and jointly operated by GreiffLab and SandveLab (the authors) in close collaboration with the University Center for Information Technology, University of Oslo, IT-Department (USIT). Finally, we thank Derek Mason and Sai T. Reddy for constructive comments and discussion.
Disclosure statement
E.M. declares holding shares in aiNET GmbH. V.G. declares advisory board positions in aiNET GmbH and Enpicom B.V. V.G. is a consultant for Adaptyv Biosystems, Specifica Inc, and Roche/Genentech.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Lu R-M, Hwang Y-C, Liu I-J, Lee -C-C, Tsai H-Z, Li H-J, Wu H-C. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1. doi:10.1186/s12929-019-0592-z.
- Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus ADME, van Kuppeveld FJM, Haagmans BL, Grosveld F, Bosch B-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):2251. doi:10.1038/s41467-020-16256-y.
- Marasco WA, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25(12):1421–18. doi:10.1038/nbt1363.
- Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Gregg AC, Daniels AD, Jervey S, et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci. 2020;6(3):315–31. doi:10.1021/acscentsci.0c00272.
- Laustsen AH, Bohn M-F, Ljungars A. The challenges with developing therapeutic monoclonal antibodies for pandemic application. Expert Opin Drug Discov. 2022;17(1): 5–8.
- Torjesen I. Drug development: the journey of a medicine from lab to shelf. Pharm J [Internet] 2015; Available from: https://www.pharmaceutical-journal.com/publications/tomorrows-pharmacist/drug-development-the-journey-of-a-medicine-from-lab-to-shelf/20068196.article?firstPass=false
- Narayanan H, Dingfelder F, Butté A, Lorenzen N, Sokolov M, Arosio P. Machine learning for biologics: opportunities for protein engineering, developability, and formulation. Trends Pharmacol Sci [Internet]. 2021;42(3):151–65. doi:10.1016/j.tips.2020.12.004.
- Laustsen AH, Greiff V, Karatt-Vellatt A, Muyldermans S, Jenkins TP. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol [Internet]. 2021;39(12):1263–73. doi:10.1016/j.tibtech.2021.03.003.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit. Nat Rev Drug Discov. 2018;17(3):197–223. doi:10.1038/nrd.2017.227.
- Fischman S, Ofran Y. Computational design of antibodies. Curr Opin Struct Biol. 2018;51:156–62. doi:10.1016/j.sbi.2018.04.007.
- Greiff V, Yaari G, Cowell L. Mining adaptive immune receptor repertoires for biological and clinical information using machine learning. Current Opinion in Systems Biology [Internet] 2020; Available from: http://www.sciencedirect.com/science/article/pii/S2452310020300524
- Brown AJ, Snapkov I, Akbar R, Pavlović M, Miho E, Sandve GK, Greiff V. Augmenting adaptive immunity: progress and challenges in the quantitative engineering and analysis of adaptive immune receptor repertoires. Mol Syst Des Eng. 2019;4:701–36.
- Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform [Internet]. 2019. doi:10.1093/bib/bbz095.
- Graves J, Byerly J, Priego E, Makkapati N, Parish SV, Medellin B, Berrondo M. A review of deep learning methods for antibodies. Antibodies (Basel) [Internet]. 2020;9. Available from. doi:10.3390/antib9020012.
- Csepregi L, Ehling RA, Wagner B, Reddy ST. Immune literacy: reading, writing, and editing adaptive immunity. iScience. 2020;23(9):101519. doi:10.1016/j.isci.2020.101519.
- Pittala S, Bailey-Kellogg C, Elofsson A. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics. 2020;36:3996–4003. doi:10.1093/bioinformatics/btaa263.
- Pertseva M, Gao B, Neumeier D, Yermanos A, Reddy ST. Applications of machine and deep learning in adaptive immunity. 2021 [cited 2021 Apr 26]; Available from: https://www.annualreviews.org/doi/abs/10.1146/annurev-chembioeng-101420-125021
- Wu Z, Johnston KE, Arnold FH, Yang KK. Protein sequence design with deep generative models [Internet]. arXiv [q-bio.QM]2021; Available from: http://arxiv.org/abs/2104.04457
- Horst A, Smakaj E, Natali EN, Tosoni D, Babrak LM, Meier P, Miho E. Machine learning detects anti-DENV signatures in antibody repertoire sequences. Front Artif Intell [Internet]. 2021;4. Available from. https://www.frontiersin.org/articles/10.3389/frai.2021.715462/full.
- Leem J, Mitchell LS, Farmery JHR, Barton J, Galson JD. Deciphering the language of antibodies using self-supervised learning [Internet]. bioRxiv2021 [cited 2021 Nov 18]; 2021.11.10.468064. Available from: https://www.biorxiv.org/content/10.1101/2021.11.10.468064v1
- Ruffolo JA, Gray JJ, Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning [Internet]. arXiv [q-bio.BM]2021; Available from: http://arxiv.org/abs/2112.07782
- Shuai RW, Ruffolo JA, Gray JJ. Generative language modeling for antibody design [Internet]. bioRxiv2021 [cited 2022 Jan 15]; 2021.12.13.472419. Available from: https://www.biorxiv.org/content/10.1101/2021.12.13.472419v1
- Amimeur T, Shaver JM, Ketchem RR, Alex Taylor J, Clark RH, Smith J, Van Citters D, Siska CC, Smidt P, Sprague M, et al. Designing feature-controlled humanoid antibody discovery libraries using generative adversarial networks [Internet]. bioRxiv2020 [cited 2020 May 28]; 2020.04.12.024844. Available from: https://www.biorxiv.org/content/10.1101/2020.04.12.024844v1
- Goodfellow I, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, Bengio Y. Generative Adversarial Nets. In: Ghahramani Z, Welling M, Cortes C, Lawrence ND, Weinberger KQ editors. Advances in neural information processing systems 27. Massachusetts: Curran Associates, Inc.; 2014. p. 2672–80.
- Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J Immunol. 2018;201(8):2502–09. doi:10.4049/jimmunol.1800708.
- Friedensohn S, Neumeier D, Khan TA, Csepregi L, Parola C, de Vries ARG, Erlach L, Mason DM, Reddy ST. Convergent selection in antibody repertoires is revealed by deep learning [Internet]. bioRxiv2020 [cited 2020 May 29]; 2020.02.25.965673. Available from: https://www.biorxiv.org/content/10.1101/2020.02.25.965673v1
- Kingma DP, Welling M. Auto-encoding variational bayes [Internet]. arXiv [stat.ML]2013; Available from: http://arxiv.org/abs/1312.6114v10
- Widrich M, Schäfl B, Pavlović M, Ramsauer H, Gruber L, Holzleitner M, Brandstetter J, Sandve GK, Greiff V, Hochreiter S, et al. Modern hopfield networks and attention for immune repertoire classification. Adv Neural Inf Process Syst [Internet]. 2020;33. Available from. http://proceedings.neurips.cc/paper/2020/hash/da4902cb0bc38210839714ebdcf0efc3-Abstract.html.
- Davidsen K, Olson BJ, DeWitt WS 3rd, Feng J, Harkins E, Bradley P, Matsen FA 4th. Deep generative models for T cell receptor protein sequences. Elife [Internet]. 2019;8. Available from. doi:10.7554/eLife.46935.
- Saka K, Kakuzaki T, Metsugi S, Kashiwagi D, Yoshida K, Wada M, Tsunoda H, Teramoto R. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci Rep. 2021;11(1):5852. doi:10.1038/s41598-021-85274-7.
- Eguchi RR, Anand N, Choe CA, Huang P-S. IG-VAE: generative modeling of immunoglobulin proteins by direct 3D coordinate generation [Internet]. 2020 [cited 2020 Aug 13]; 2020.08.07.242347. Available from: https://www.biorxiv.org/content/10.1101/2020.08.07.242347v1
- Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, Chernigovskaya M, Scheffer L, Slabodkin A, Mehta BB , et al. One billion synthetic 3D-antibody-antigen complexes enable unconstrained machine-learning formalized investigation of antibody specificity prediction. 2021. [Internet]. Available from. doi:10.1101/2021.07.06.451258.
- Robert PA, Meyer-Hermann M. Ymir: A 3D structural affinity model for multi-epitope vaccine simulations. iScience 2021; Available from: doi:https://doi.org/10.1016/j.isci.2021.102979
- Robert PA, Marschall AL, Meyer-Hermann M. Induction of broadly neutralizing antibodies in germinal centre simulations. Curr Opin Biotechnol. 2018;51:137–45. doi:10.1016/j.copbio.2018.01.006.
- Preuer K, Renz P, Unterthiner T, Hochreiter S, Klambauer G. Fréchet chemnet distance: a metric for generative models for molecules in drug discovery. J Chem Inf Model. 2018;58(9):1736–41. doi:10.1021/acs.jcim.8b00234.
- Mathai N, Chen Y, Kirchmair J. Validation strategies for target prediction methods. Brief Bioinform. 2020;21(3):791–802. doi:10.1093/bib/bbz026.
- Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng SM, Ehling RA, Bonati L, Dahinden J, Gainza P, et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat Biomed Eng. 2021;5(6):600–12. doi:10.1038/s41551-021-00699-9.
- Greiff V, Menzel U, Miho E, Weber C, Riedel R, Cook S, Valai A, Lopes T, Radbruch A, Winkler TH, et al. Systems analysis reveals high genetic and antigen-driven predetermination of antibody repertoires throughout B cell development. Cell Rep. 2017;19(7):1467–78. doi:10.1016/j.celrep.2017.04.054.
- Ferdous S, Martin ACR. AbDb: antibody structure database-a database of PDB-derived antibody structures. Database [Internet]. 2018. doi:10.1093/database/bay040.
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–23. doi:10.1093/bioinformatics/btp163.
- Engelhart E, Lopez R, Emerson R, Lin C, Shikany C. Massively multiplexed affinity characterization of therapeutic antibodies against SARS-CoV-2 variants. bioRxiv [Internet] 2021; Available from: https://www.biorxiv.org/content/10.1101/2021.04.27.440939v1.abstract
- Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS. Protein design and variant prediction using autoregressive generative models. Nat Commun. 2021;12(1):2403. doi:10.1038/s41467-021-22732-w.
- Akbar R, Robert PA, Pavlović M, Jeliazkov JR. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Reports [Internet] 2021; Available from: https://www.sciencedirect.com/science/article/pii/S2211124721001704
- Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM. Low-N protein engineering with data-efficient deep learning. Nat Methods. 2021;18(4):389–96. doi:10.1038/s41592-021-01100-y.
- AlQuraishi M, Sorger PK. Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms. Nat Methods. 2021;18(10):1169–80. doi:10.1038/s41592-021-01283-4.
- Ethayarajh K, Jurafsky D. Utility is in the eye of the user: a critique of NLP leaderboards [Internet]. arXiv [cs.CL]2020; Available from: http://arxiv.org/abs/2009.13888
- Isacchini G, Walczak AM, Mora T, Nourmohammad A. Deep generative selection models of T and B cell receptor repertoires with soNNia. Proc Natl Acad Sci U S A [Internet]. 2021:118. Available from. doi:10.1073/pnas.2023141118.
- Semeniuta S, Severyn A, Gelly S. On accurate evaluation of GANs for language generation [Internet]. arXiv [cs.CL]2018; Available from: http://arxiv.org/abs/1806.04936
- Renz P, Van Rompaey D, Wegner JK, Hochreiter S, Klambauer G. On failure modes of molecule generators and optimizers. 2020; Available from: https://chemrxiv.org/articles/On_Failure_Modes_of_Molecule_Generators_and_Optimizers/12213542
- Mensink T, Uijlings J, Kuznetsova A, Gygli M, Ferrari V. Factors of influence for transfer learning across diverse appearance domains and task types [Internet]. arXiv [cs.CV]2021; Available from: http://arxiv.org/abs/2103.13318
- Gelman S, Romero PA, Gitter A. Neural networks to learn protein sequence-function relationships from deep mutational scanning data. bioRxiv [Internet] 2020; Available from: https://www.biorxiv.org/content/10.1101/2020.10.25.353946v1.abstract
- Rao R, Liu J, Verkuil R, Meier J, Canny JF, Abbeel P, Sercu T, Rives A. MSA transformer [internet]. Cold Spring Harbor Laboratory2021 [cited 2021 Feb 18]; 2021.02.12.430858. Available from: https://www.biorxiv.org/content/10.1101/2021.02.12.430858v1
- Kurczab R, Bojarski AJ. The influence of the negative-positive ratio and screening database size on the performance of machine learning-based virtual screening. PLoS One. 2017;12(4):e0175410. doi:10.1371/journal.pone.0175410.
- Kim J, Kim J. The impact of imbalanced training data on machine learning for author name disambiguation. Scientometrics. 2018;117(1):511–26. doi:10.1007/s11192-018-2865-9.
- Pavlović M, Scheffer L, Motwani K, Kanduri C, Kompova R, Vazov N, Waagan K, Bernal FLM, Costa AA, Corrie B, et al. The immuneML ecosystem for machine learning analysis of adaptive immune receptor repertoires. Nat Mach Intell. 2021;3(11):936–44. doi:10.1038/s42256-021-00413-z.
- Gane A, Belanger D, Dohan D, Angermueller C, Vora RDS, Chapelle O, Alipanahi B, Murphy K, Colwell L. A comparison of generative models for sequence design [Internet]. [cited 2021 Oct 24]; Available from: https://research.google/pubs/pub49141.pdf
- Seib V, Lange B, Wirtz S. Mixing real and synthetic data to enhance neural network training – a review of current approaches [Internet]. arXiv [cs.CV]2020; Available from: http://arxiv.org/abs/2007.08781
- Shen J, Nicolaou CA. Molecular property prediction: recent trends in the era of artificial intelligence. Drug Discov Today Technol [Internet] 2020; Available from: http://www.sciencedirect.com/science/article/pii/S1740674920300032
- Athey J, Alexaki A, Osipova E, Rostovtsev A, Santana-Quintero LV, Katneni U, Simonyan V, Kimchi-Sarfaty C. A new and updated resource for codon usage tables. BMC Bioinform. 2017;18(1):391. doi:10.1186/s12859-017-1793-7.
- DeVries T, Drozdzal M, Taylor GW. Instance selection for GANs [Internet]. arXiv [cs.CV]2020; Available from: http://arxiv.org/abs/2007.15255
- Jin W, Wohlwend J, Barzilay R, Jaakkola T. Iterative refinement graph neural network for antibody sequence-structure co-design [Internet]. arXiv [q-bio.BM]2021; Available from: http://arxiv.org/abs/2110.04624
- Chen X, Dougherty T, Hong C, Schibler R, Zhao YC, Sadeghi R, Matasci N, Wu Y-C, Kerman I. Predicting antibody developability from sequence using machine learning [Internet]. 2020 [cited 2020 Oct 9]; 2020.06.18.159798. Available from: https://www.biorxiv.org/content/10.1101/2020.06.18.159798v1.abstract
- Melnyk I, Das P, Chenthamarakshan V, Lozano A. Benchmarking deep generative models for diverse antibody sequence design [Internet]. arXiv [q-bio.BM]2021; Available from: http://arxiv.org/abs/2111.06801
- Gao W, Mahajan SP, Sulam J, Gray JJ. Deep learning in protein structural modeling and design [ Internet]. arXiv [q-bio.BM]2020; Available from: http://arxiv.org/abs/2007.08383
- Jiménez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence [ Internet]. arXiv [cs.AI]2020; Available from: http://arxiv.org/abs/2007.00523
- Preuer K, Klambauer G, Rippmann F, Hochreiter S, Unterthiner T. Interpretable deep learning in drug discovery. In: Samek W, Montavon G, Vedaldi A, Hansen LK, Müller K-R, editors. Explainable AI: interpreting, explaining and visualizing deep learning. Cham: Springer International Publishing; 2019. p. 331–45.
- Ruffolo JA, Sulam J, Gray JJ. Antibody structure prediction using interpretable deep learning [Internet]. bioRxiv2021 [cited 2021 Jul 2]; 2021.05.27.445982. Available from: https://www.biorxiv.org/content/10.1101/2021.05.27.445982v1
- DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, Greenberg PD, Duerkopp N, Emerson RO, Robins HS. A public database of memory and naive B-cell receptor sequences. PLoS One. 2016;11(8):e0160853. doi:10.1371/journal.pone.0160853.
- Mann M, Saunders R, Smith C, Backofen R, Deane CM. Producing high-accuracy lattice models from protein atomic coordinates including side chains. Adv Bioinformatics. 2012;2012:148045. doi:10.1155/2012/148045.
- Miyazawa S, Jernigan RL. Residue–residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996;256(3):623–44. doi:10.1006/jmbi.1996.0114.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020;12(1):1743053. doi:10.1080/19420862.2020.1743053.
- Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proceedings of the National Academy of Sciences 2019; 116:4025–30.
- Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J Proteome Res [Internet]. 2020;19(6):2304–15. doi:10.1021/acs.jproteome.9b00874.
- Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput. 1997;9(8):1735–80. doi:10.1162/neco.1997.9.8.1735.
- Kingma DP, Ba J. Adam: a method for stochastic optimization [Internet]. arXiv [cs.LG]2014; Available from: http://arxiv.org/abs/1412.6980
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. TensorFlow: large-scale machine learning on heterogeneous distributed systems [Internet]. arXiv [cs.DC]2016; Available from: http://arxiv.org/abs/1603.04467
- Ohtamaa DNM Python-levenshtein.Tinkle] https://githubcom/miohtama/python-Levenshtein[Kreiptasi:2016-03-12] [Internet] Available from: https://pypi.org/project/python-Levenshtein/
- Palme J, Hochreiter S, Bodenhofer U. KeBABS: an R package for kernel-based analysis of biological sequences. Bioinformatics. 2015;btv176.
- Weber CR, Akbar R, Yermanos A, Pavlović M, Snapkov I, Sandve GK, Reddy ST, Greiff V. immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking. Bioinf [Internet]. 2020;36(11):3594–96. doi:10.1093/bioinformatics/btaa158.
- Greiff V, Weber CR, Palme J, Bodenhofer U, Miho E, Menzel U, Reddy ST. Learning the high-dimensional immunogenomic features that predict public and private antibody repertoires. J Immunol. 2017;199(8):2985–97. doi:10.4049/jimmunol.1700594.
- Vermeesch P, Resentini A, Garzanti E. An R package for statistical provenance analysis. Sediment Geol. 2016;336:14–25. doi:10.1016/j.sedgeo.2016.01.009.
- Mason DM, Weber CR, Parola C, Meng SM, Greiff V, Kelton WJ, Reddy ST. High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis. Nucleic Acids Res. 2018;46(14):7436–49. doi:10.1093/nar/gky550.
- Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag New York; 2009.