
ABSTRACT
Cluster of differentiation 38 (CD38) is an ecto-enzyme expressed primarily on immune cells that metabolizes nicotinamide adenine dinucleotide (NAD+) to adenosine diphosphate ribose or cyclic ADP-ribose and nicotinamide. Other substrates of CD38 include nicotinamide adenine dinucleotide phosphate and nicotinamide mononucleotide, a critical NAD+ precursor in the salvage pathway. NAD+ is an important coenzyme involved in several metabolic pathways and is a required cofactor for the function of sirtuins (SIRTs) and poly (adenosine diphosphate-ribose) polymerases. Declines in NAD+ levels are associated with metabolic and inflammatory diseases, aging, and neurodegenerative disorders. To inhibit CD38 enzyme activity and boost NAD+ levels, we developed TNB-738, an anti-CD38 biparatopic antibody that pairs two non-competing heavy chain-only antibodies in a bispecific format. By simultaneously binding two distinct epitopes on CD38, TNB-738 potently inhibited its enzymatic activity, which in turn boosted intracellular NAD+ levels and SIRT activities. Due to its silenced IgG4 Fc, TNB-738 did not deplete CD38-expressing cells, in contrast to the clinically available anti-CD38 antibodies, daratumumab, and isatuximab. TNB-738 offers numerous advantages compared to other NAD-boosting therapeutics, including small molecules, and supplements, due to its long half-life, specificity, safety profile, and activity. Overall, TNB-738 represents a novel treatment with broad therapeutic potential for metabolic and inflammatory diseases associated with NAD+ deficiencies.
Abbreviations: 7-AAD: 7-aminoactinomycin D; ADCC: antibody dependent cell-mediated cytotoxicity; ADCP: antibody dependent cell-mediated phagocytosis; ADPR: adenosine diphosphate ribose; APC: allophycocyanin; cADPR: cyclic ADP-ribose; cDNA: complementary DNA; BSA: bovine serum albumin; CD38: cluster of differentiation 38; CDC: complement dependent cytotoxicity; CFA: Freund’s complete adjuvant; CHO: Chinese hamster ovary; CCP4: collaborative computational project, number 4; COOT: crystallographic object-oriented toolkit; DAPI: 4′,6-diamidino-2-phenylindole; DNA: deoxyribonucleic acid; DSC: differential scanning calorimetry; 3D: three dimensional; εNAD+: nicotinamide 1,N6-ethenoadenine dinucleotide; ECD: extracellular domain; EGF: epidermal growth factor; FACS: fluorescence activated cell sorting; FcγR: Fc gamma receptors; FITC: fluorescein isothiocyanate; HEK: human embryonic kidney; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; IgG: immunoglobulin; IFA: incomplete Freund’s adjuvant; IFNγ: Interferon gamma; KB: kinetic buffer; kDa: kilodalton; KEGG: kyoto encyclopedia of genes and genomes; LDH: lactate dehydrogenase; M: molar; mM: millimolar; MFI: mean fluorescent intensity; NA: nicotinic acid; NAD: nicotinamide adenine dinucleotide; NADP: nicotinamide adenine dinucleotide phosphate; NAM: nicotinamide; NGS: next-generation sequencing; NHS/EDC: N-Hydroxysuccinimide/ ethyl (dimethylamino propyl) carbodiimide; Ni-NTA: nickel-nitrilotriacetic acid; nL: nanoliter; NK: natural killer; NMN: nicotinamide mononucleotide; OD: optical density; PARP: poly (adenosine diphosphate-ribose) polymerase; PBS: phosphate-buffered saline; PBMC: peripheral blood mononuclear cell; PDB: protein data bank; PE: phycoerythrin; PISA: protein interfaces, surfaces, and assemblies: PK: pharmacokinetics; mol: picomolar; RNA: ribonucleic acid; RLU: relative luminescence units; rpm: rotations per minute; RU: resonance unit; SEC: size exclusion chromatography; SEM: standard error of the mean; SIRT: sirtuins; SPR: surface plasmon resonance; µg: microgram; µM: micromolar; µL: microliter
KEYWORDS:
Introduction
CD38 is a 45 kDa type II transmembrane protein expressed on immune cells, including macrophages, T cells, B cells, and natural killer (NK) cells, and is prominently upregulated on activated immune cells, thus serving as a biomarker for active infections, inflammatory diseases, autoimmune conditions, and inflammation associated with aging, or “inflammaging”.Citation7,Citation8 CD38 is an ecto-enzyme that hydrolyzes nicotinamide adenine dinucleotide (NAD+) to form adenine diphosphate ribose (ADPR) and nicotinamide (NAM); secondarily, it degrades NAD+ via cyclase activity yielding cyclic adenine dinucleotide (cADPR). Other substrates of CD38 include nicotinamide adenine dinucleotide phosphate (NADP+) and nicotinamide mononucleotide (NMN), a critical precursor of NAD+ in the salvage pathway, preventing its uptake into cells and its conversion to NAD+ intracellularly.Citation4 NAD+ plays a crucial role in several metabolic processes that maintain energy balance, DNA repair, and signaling.Citation9 Decreases in NAD+ are associated with natural aging, and NAD+ boosting is associated with increased life- and health-spans in animals.Citation10 Therefore, TNB-738-mediated inhibition of CD38 represents a promising approach to treat a plethora of acute and chronic conditions associated with NAD+ decline.
CD38 inhibitors are typically either small molecules or antibodies. Small-molecule inhibitors of CD38 include NAD-analogs, flavonoids, and heterocyclic compounds.Citation11 These inhibitors can form either a covalent or non-covalent bond with E226, a crucial residue in the active site of the enzyme. Recent advancements in computational biology and the availability of high-resolution crystal structures of CD38 has led to the design and synthesis of novel inhibitors, such as carba-NAD, Ara-NAD analogs, 4-amino-quinoline derivatives, and other NAD derivatives.Citation12,Citation13 Many naturally occurring compounds, e.g., apigenin, quercetin, leteolindin, kuromanin, luteolin, and rhein, also inhibit CD38 enzymatic activities.Citation14 Although several hundred small molecules are known to inhibit CD38, small molecules are limited in their inhibitory effect due to high dosage requirements, toxic byproducts, poor pharmacokinetics (PK), ability to cross the blood–brain barrier, and off-target binding to other NAD-dependent enzymes.Citation3,Citation11,Citation15,Citation16
Daratumumab (Darzalex, Janssen Pharmaceuticals) and isatuximab (SARCLISA, Sanofi-Aventis) are potent anti-CD38 antibodies developed for the depletion of malignant plasma cells. These antibodies are highly efficacious and safe in the treatment of multiple myeloma. Only isatuximab inhibits CD38 NADase activity.Citation17 Isatuximab is used for the treatment of multiple myeloma by inducing lysis of tumor cells via antibody-dependent cell-mediated cytotoxicity (ADCC), complement-mediated cytotoxicity (CDC), antibody-dependent cell-mediated phagocytosis (ADCP), and direct apoptosis.Citation18 Due to the depletion of CD38-expressing cells, such as plasma cells and activated T cells (notably by Fc-independent mechanisms such as direct apoptosis that cannot be abrogated by Fc silencing), isatuximab is not suitable for the treatment of chronic inflammatory diseases associated with imbalanced NAD+ metabolism.
Existing anti-CD38 antibodies bind to diverse epitopes with variable downstream effects. For instance, the crystal structures of CD38 bound to isatuximab or daratumumab demonstrate that these antibodies bind to two distinct epitopes on CD38, resulting in distinct functional characteristics.Citation17 Isatuximab inhibits CD38 enzyme activity and induces direct apoptosis of CD38 cells in the absence of Fc cross-linking (F(ab’)2 fragments induce apoptosis).Citation19 In contrast, daratumumab does not inhibit CD38 enzyme activity and requires crosslinking via FcγR and FcγRIIb to induce indirect apoptosis.Citation20 Our lab previously identified heavy chain only antibodies (UniAbs) that bind to five different functional epitopes on CD38.Citation21 Each epitope bin comprised a unique set of functional characteristics, including ADCC, CDC, enzyme inhibition, and direct apoptosis. However, no single epitope enabled inhibition of CD38 enzyme activity greater than 50%. We hypothesized that synergistic and complete inhibition of CD38 enzyme activity might be achievable by combining UniAbs that bind to different epitopes.
TNB-738 is a human anti-CD38 antibody generated as a combination of two UniAbs, F11A, and F12A, that bind to two different epitopes on CD38 with high affinity and synergize to achieve maximum CD38 inhibition. UniAbs F11A and F12A were discovered using a sequence-based antibody discovery platform (TeneoSeek) and were paired into bispecific format to form TNB-738. The biparatopic binding of TNB-738 to CD38 enabled near complete inhibition of enzyme activity without depletion of CD38-expressing cells. By preventing the breakdown of NAD+ and NMN by CD38, TNB-738 boosted NAD+ levels in cells; this boost in NAD+ levels led to increased activities of NAD-dependent enzymes, including SIRTs and PARPs, which are involved in protein deacetylation and ADP ribosylation, respectively. The epitope of F11A on CD38 was determined by X-ray crystallography, while the epitope of F12A was predicted using the in-silico epitope mapping tool, MAbTope, and confirmed through binding experiments on mutated CD38 constructs. Overall, TNB-738 has the potential to treat various diseases associated with NAD+ deficiency without the potential side effects of small molecules and cytotoxic antibodies.
Results
Discovery of TNB-738
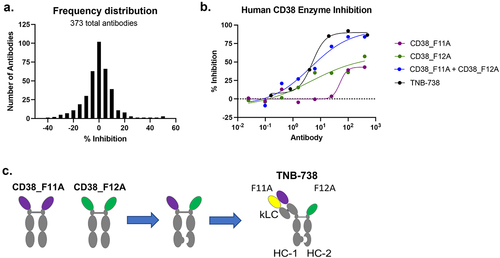
Anti-CD38 UniAbs were identified using a proprietary next-generation sequencing (NGS) approach on immunized UniRats.Citation22 In total, 797 UniAbs were selected for gene assembly, recombinant expression, and functional screening. High-throughput ELISA and cell-binding screens identified 155 CD38-specific binders from 52 unique complementarity-determining region 3 (CDR3) clonotype families. Binders were further evaluated in a CD38 NADase inhibition assay, which identified only partial inhibitors of CD38 ()). No UniAbs inhibited CD38 hydrolase activity greater than 50%. Subsequently, an all-by-all enzyme inhibition experiment composed of the partial inhibitors was performed to identify a synergistic pair that, in combination, strongly inhibits CD38 enzyme activity (data not shown). UniAbs F11A and F12A emerged as a synergistic combination that inhibited CD38 activity greater than 85% ()). TNB-738 was generated by pairing F11A and F12A on an IgG4 Fc using knobs-into-holes technology ()). An inert, germline kappa light chain and CH1 domain were added to the F11A arm of TNB-738 to optimize manufacturability with no loss in its ability to inhibit CD38. The IgG4 Fc of TNB-738 was further engineered to contain L234A and L235A mutations in order to fully silence Fc-mediated effector functions.Citation23 The PK of TNB-738 was evaluated in mice and cynomolgus monkeys (Supplementary Figure S1). Since TNB-738 does not cross-react with CD38 in mice or cynomolgus monkey, the observed linear PK was consistent with nonspecific clearance mechanisms dominating PK, demonstrating stability of TNB-738 in serum.
Figure 1. Discovery of TNB-738. a) 373 anti-CD38 binders were screened for inhibition of CD38 enzyme activity. CHO-HuCD38 cells were treated with antibodies for 15 min at RT. Following incubation, εNAD+ substrate was added to each well and fluorescence was immediately analyzed using a microplate reader for 30 min. A frequency distribution plot of percent inhibition values is shown. b) Dose–response curves of CD38 enzyme inhibition is shown comparing CD38_F11A, CD38_F12A, CD38_F11A + CD38_F12A, and TNB-738. c) TNB-738 was generated by pairing CD38_F11A and CD38_F12A into bispecific format using knobs-into-holes technology on a silenced IgG4 Fc. A germline kappa light chain and CH1 domain were added to CD38_F11A arm of TNB-738 to optimize manufacturability with no loss in its ability to inhibit CD38.
TNB-738 was formulated into 20 mM histidine, 120 mM glycine, 150 mM sucrose, 0.01% PS80, pH 5.8. Differential scanning calorimetry (DSC) was used to assess thermal stability (). Stability studies are ongoing and TNB-738 has been shown to be stable in the current formulation as measured by SEC at 5 ± 3°C for all timepoints tested (). The apparent affinity (avidity) of TNB-738 to human CD38 extracellular domain (ECD) is about 3.8 ± 0.2 to 0.1 ± 0.02 nM, depending on the surface density of the antigen coated on the sensor. By classical affinity measurement, where TNB-738 is loaded on the sensor and human CD38 ECD is flowed, the affinity of TNB-738 to human CD38 is 2.48 nM.
Table 1. a) TNB-738 DSC measurements. b) TNB-738 stability measured by SEC.
TNB-738 binds to CD38 and inhibits CD38 enzymatic activity
Binding of TNB-738 to CD38+ and CD38− cell lines was evaluated by flow cytometry. On-target binding was assessed on Daudi, Ramos, and Chinese hamster ovary (CHO) cells stably transfected to express CD38 (CHO-HuCD38), while off-target binding was assessed on K562, HL-60, HEK 293F, and CHO cells. TNB-738 bound to Daudi, Ramos, and CHO-HuCD38 cells with EC50 values of 39.7, 50.3, and 70.2 nM, respectively ()). No binding was observed on CD38− cells ()), demonstrating target-specificity of TNB-738. Additionally, binding of TNB-738 was evaluated on human peripheral blood mononuclear cells (PBMCs), with similar immune cell subsets bound by TNB-738 compared to daratumumab (Supplementary Figure S2).
Figure 2. TNB-738 binds to CD38 and inhibits CD38 enzyme activity. a) On-target cell binding of TNB-738 was assessed by flow cytometry using Daudi, Ramos, and CHO-HuCD38 cells. b) Off-target cell binding of TNB-738 was assessed by flow cytometry using 293 F, CHO, K562, and HL-60 cells. c) TNB-738-mediated CD38 inhibition was evaluated on Daudi, Ramos, and CHO cells and (d) recombinant CD38 protein. e) Cells were incubated with antibodies at 37°C for 2 hours and the percentage of cells with capping was determined using confocal microscopy. In total, 100 cells were counted.
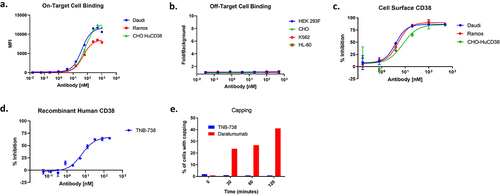
Next, TNB-738-mediated inhibition of CD38 hydrolase activity was evaluated in vitro. CD38-expressing Daudi, Ramos, and CHO-HuCD38 cells were incubated with εNAD+ in the presence of increasing concentrations of TNB-738. Hydrolysis of εNAD+, which leads to an increase in fluorescence at wavelength 310 nm, was measured over time using a microplate reader.Citation24 TNB-738 dose-dependently inhibited cell surface CD38 hydrolase activity with an average maximum percent inhibition of 87 ± 2.3% across the three cell lines tested (). TNB-738 also inhibited recombinant CD38 hydrolase activity with IC50 and maximum percent inhibition of 6.4 nM and 68%, respectively (). Comparison of cell surface and recombinant CD38 inhibition with the small-molecule inhibitor, 78c, is shown in Supplementary Figure S3.
TNB-738-CD38 complexes are not actively internalized by B cells and do not induce cytokine production by CD38+ T cells
Capping and internalization are facilitated by the cytoskeleton, and phosphorylation of several proteins is required.Citation25 Upon internalization, antibody-receptor complexes are transported to lysosomal compartments where these complexes are degraded.Citation25 Studies by Fonaro et al.Citation26 have indicated that capping and internalization of CD38 by antibodies leads to transmembrane signaling and activation of B and T cells. Upon binding of TNB-738 to CD38 on cells, the percentage of cells with capping was assessed by visualizing the TNB-738-CD38 complexes via immunofluorescence. The data demonstrated that TNB-738 did not induce capping on cells in contrast to daratumumab, which induced capping on up to 40% of cells (). Representative immunofluorescence images are shown in Supplementary Figure 4A. Additionally, internalization was assessed by flow cytometry using pH-sensitive fluoroprobes; these pH-sensitive fluoroprobes are non-fluorescent outside of the cell but fluoresce in low pH environments such as the lysosome. Following incubation of Daudi cells with TNB-738, daratumumab, or isotype control coupled to pHrodo iFL (Thermo Fisher), mean fluorescent intensity (MFI) was measured by flow cytometry to measure internalization. TNB-738 induced significantly less internalization compared to daratumumab, as evidenced by the substantial reduction in MFI. (Supplementary Figure 4B). Since TNB-738 neither induces capping nor is actively internalized by CD38+ cells, this suggests that TNB-738 does not activate CD38+ cells, including B and T cells.
To further investigate the effects of TNB-738 on immune cell activation, PBMCs were incubated with TNB-738 and activation markers (CD25 and interferon gamma (IFNγ)) were assessed by flow cytometry. Treatment of CD38+ cells did not result in increased expression of CD25 or IFNγ, demonstrating that TNB-738 does not directly activate T cells (Supplementary Figure S5).
TNB-738-mediated inhibition of CD38 ectoenzyme activity increases NAD+ levels and SIRT1 activity
To assess whether blocking CD38 enzyme activity using TNB-738 led to increased cellular NAD+ levels, CD38-expressing Ramos cells were incubated with TNB-738 for 48 h and intracellular NAD+ concentrations were quantified. As demonstrated in ), TNB-738-treatment resulted in a dose-dependent increase in intracellular NAD+ levels as compared to the isotype control. The downstream effects of increased NAD+ was subsequently assessed by measuring the activity of SIRTs, which are NAD-dependent deacetylases involved in cellular health and metabolic regulation. SIRT1 activity was measured using the SIRT-Glo assay (Promega) following treatment of Ramos cells with TNB-738 or isotype control for 48 h ()). Only in the presence of TNB-738 was an increase in SIRT1 activity observed. In addition to SIRT1, increased activity of other NAD-dependent enzymes, including SIRT3 (Supplementary Figure 6A) and PARP (Supplementary Figure 6B), was observed.
Figure 3. TNB-738-mediated inhibition of CD38 ectoenzyme activity increases NAD+ levels and SIRT1 activity downstream. Ramos cells were treated with a dilution series of TNB-738 or isotype control in the presence or absence of 50 nM NMN for 24 h at 37°C. Following incubation, a) NAD+ levels were measured using the Cell Biolabs NAD+ assay kit, and b) SIRT1 activity was measured using the Promega SIRT-Glo kit.

It is hypothesized that CD38 regulates cellular NAD+ by modulating the availability of NAD+ precursors such as NMN.Citation5Citation6 To test this hypothesis, we evaluated the potentiating effect of NMN supplementation, in the presence or absence of TNB-738-mediated CD38 inhibition, on cellular NAD+ levels and SIRT activity. In all cases, combination of TNB-738 with NMN supplementation further boosted NAD+ levels and SIRT activity compared to TNB-738-treatment alone ()). The increase in NAD+ levels and SIRT activity was dependent on CD38 inhibition; NMN treatment alone without TNB-738 had no effect. In the absence of CD38 inhibition, NMN is likely rapidly degraded by CD38 thus preventing its uptake and conversion to NAD+ intracellularly. Altogether, the data indicate that CD38 inhibition is required to effectively boost NAD+ levels in CD38+ cells.
TNB-738 does not induce lysis of CD38-expressing cells
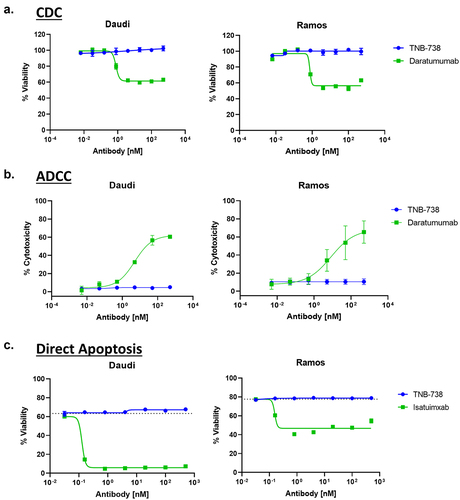
Since the desired mechanism of action of TNB-738 is strictly enzyme inhibition rather than depletion of CD38+ cells, TNB-738 was engineered on a silenced IgG4 Fc. To confirm that TNB-738 does not deplete CD38+ cells, TNB-738 was evaluated in CDC, ADCC, and direct apoptosis assays. CDC is an Fc-mediated effector function in which proteins of the complement cascade are recruited to opsonized cells, leading to formation of the membrane attack complex and lysis of the cell. Incubation of Daudi and Ramos cells with TNB-738 and 5% complement serum did not induce complement-mediated lysis of cells, as measured using Cell-Titer Glo 2.0, compared to the positive control, daratumumab ()). Similarly, incubation of Daudi and Ramos cells with TNB-738 and NK cells did not result in ADCC, an Fc-mediated effector function characterized by NK cell-mediated killing of target cells ()). TNB-738 also did not cause direct apoptosis of cells, as evidenced by the lack of 7-AAD uptake and Annexin V staining on TNB-738-treated Daudi and Ramos cells ()). Cells treated with isatuximab, in contrast, had much lower viability compared to the untreated control. Altogether, the data demonstrated that TNB-738-treatment does not induce lysis of CD38-expressing cells.
Figure 4. TNB-738 does not induce lysis of CD38-expressing cells. a) Daudi and Ramos cells were incubated with increasing concentrations of TNB-738 or daratumumab in the presence of 5% rabbit complement serum for 45 min at 37°C. Cell viability was measured using Cell Titer Glo 2.0. b) NK cells were isolated from PBMCs and incubated with Daudi or Ramos cells and increasing concentrations of TNB-738 or daratumumab for 4 h at 37°C. Tumor killing was evaluated by LDH release. c) Daudi and Ramos cells were treated with TNB-738 or isatuximab for 24 h at 37°C. After incubation, the cells were stained with 7-AAD and Annexin V. Cell viability was analyzed by flow cytometry.
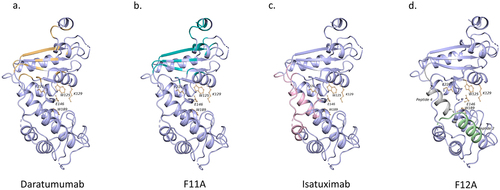
TNB-738 binds to two distinct epitopes on CD38
Epitope binning experiments were performed using surface plasmon resonance (SPR) (BioRad ProteOn). A sandwich format was used to bin F11A, F12A, isatuximab, and daratumumab. The epitope binning results demonstrated that F11A and F12A bind to different epitopes on CD38 (Supplementary Figure 7). F11A and daratumumab compete for binding to CD38 and recognize overlapping epitopes on CD38. Similarly, F12A and isatuximab also recognize overlapping epitopes on CD38 that were distinct from the F11A/daratumumab epitope. Though SPR data suggests that isatuximab and daratumumab compete for binding, crystallography data showed that they bind to distinct epitopes on CD38 (PDB: 7DHA, 4CMH). The apparent competition between isatuximab and daratumumab is caused by the steric collision at the C terminus of CD38, as predicted by in silico superimposition.Citation27
Structure of the CD38-F11A complex
The crystal structure of the CD38-F11A complex was determined by X-ray crystallography. The CD38-F11A complex diffracted to a resolution of 1.9 Å. Protein Interfaces, Surfaces, and Assemblies (PISA) program was used to analyze the interaction between F11A and CD38 (). F11A recognizes 9 residues on CD38 via hydrogen bonds and salt bridges. The data collection and refinement parameters are listed in . Most of the residues that interact with CD38 are present in the CDRs of F11A except for F11A_Y60 and F11A_K65. Interacting residues on CD38 include CD38_Q236, CD38_E292, CD38_E239, CD38_P291, CD38_E299, CD38_Q272, CD38_K276, CD38_N277, CD38_D252 at the C-terminus. Of the nine residues that F11A interacts with on CD38 via hydrogen bonding and salt bridges, daratumumab shares 7. Daratumumab and F11A bind to synonymous residues in the β4, β5, β6 strands and the α9 helix at the C-terminus of extracellular CD38.
Figure 5. Epitope mapping of F11A on CD38. Crystal Structure of the CD38-F11A complex. CD38 ECD is shown in blue with the active site in pink. UniDab_F11A is highlighted yellow.
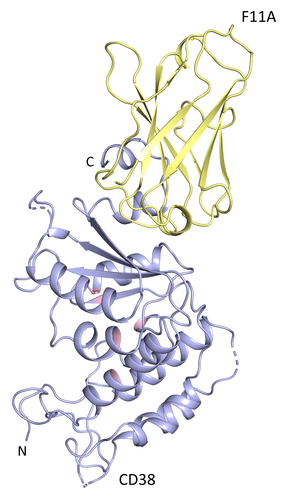
Table 2. Data collection and refinement statistics of the CD38-F11A crystal structure.
F12A epitope prediction
The epitope of F12A was predicted using a docking-based in silico tool called MabTopeCitation28 as attempts to generate and diffract crystal complexes for CD38-F12A were unsuccessful. The residues of CD38 that likely belong to the F12A epitope are highlighted in . They are divided into four categories as a function of their raw probability to belong to the epitope, from violet for the highest probability to cyan for the lowest. These residues constitute the three peptides that were mutated and evaluated for binding to F12A. A fourth peptide was suggested based on the binding site of a small-molecule LX-102 that competes with isatuximab.Citation29 The constructs were named CD38_F12_m1, CD38_F12_m2, CD38_F12_m3, and CD38_F12_m4. The numbering of the mutations corresponds to the sequence of the structural template PDB:2O3S. Mutations were selected among the residues whose lateral chains are exposed to solvent and can thereby interact with F12A. In order to not affect the 3D structure of CD38 proteins, prolines, and other amino acids whose lateral chains were not exposed at the surface were not mutated.
Figure 6. Epitope mapping of F12A on CD38. a) Results of in silico prediction of F12A on CD38 using MAbTope. b) Table summarizing the predicted peptides with suggested mutant residues. c) Binding of F12A and daratumumab antibody to wild-type and mutated CD38. The percent PE+APC+ cells were collected from 4 independent experiments. The amount of PE+APC+ cells were normalized to the total PE+ cells. The results are expressed in mean ± sem of the maximal response. One star (*) indicates significant statistical difference at p ≤ .05, two stars (**) at p ≤ .01.
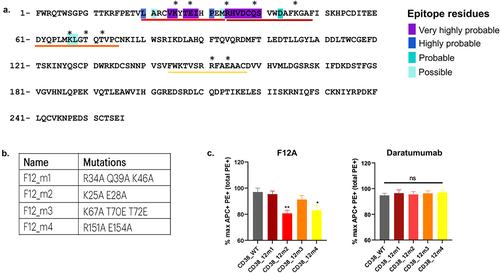
Binding assays on HEK cells overexpressing CD38 mutant constructs were performed to confirm the epitope of F12A that was predicted by MabTope. The constructs were flag tagged (DYKDDDDK) to monitor their expression using a phycoerythrin (PE)-coupled anti-FLAG antibody. Binding of F12A to mutants was monitored using F12A coupled to allophycocyanin (APC). APC-labeled daratumumab was used a positive control. F12A exhibited decreased binding to the mutants, CD38_F12_m2 and CD38_F12_m4, compared to wild-type CD38. Hence, the mutated residues K25, E28, R151, and E154 are part of the epitope. This suggests that F12A has a conformational epitope, as the identified residues are discontinuous. Competition experiments using SPR confirmed that F12A and isatuximab have overlapping epitopes. Ribbon diagrams comparing CD38 epitopes of F11A, F12A, isatuximab, and daratumumab are shown in . Altogether, the results indicate that TNB-738 is an allosteric inhibitor of the enzyme functions of CD38.
Figure 7. Comparison of epitopes on CD38. Epitope on CD38 of daratumumab (a) in yellow, F11A (b) in green, and isatuximab (c) in pink. Peptides predicted using MAbTope for the F12A epitope (d) (green-CD38_F12_m2, gray-CD38_F12_m4). The residues of the active site of CD38 are represented by ball and stick model.
Discussion
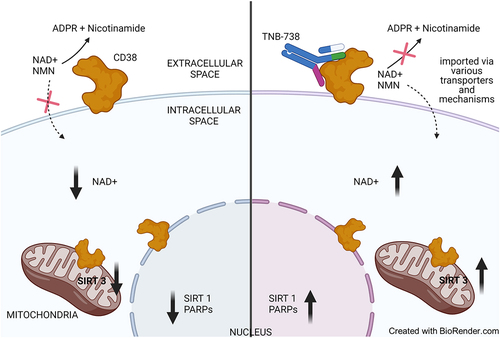
Targeting NAD+ metabolism through CD38 enzyme inhibition has emerged as a promising approach to boost NAD+ levels for the treatment of a wide range of indications associated with NAD+ decline, such as oncologic, metabolic, and inflammatory diseases.Citation4,Citation30 NAD+ is synthesized de novo in cells via the Kynurenine and Preiss-Handler pathways.Citation31 It can also be replenished intracellularly via the salvage pathways that utilize NAM, NMN, and NA to generate NAD+.Citation9 There is no evidence that NAD can be generated extracellularly except during stress/inflammation, where intracellular NAD might move into extracellular space by active exocytosis, diffusion through membrane transporters, or by cell damage and death.Citation32–36 Although extracellular NAD+ is present in low amounts, recent studies show that mammalian cells can import low concentrations of NAD+ across the membrane.Citation29,Citation37–39 This suggests that an unidentified transporter of intact NAD+ might exist.Citation40 The mechanism of direct import of NAD+ into the cells has yet to be ascertained. As shown by others, NMN is an important intermediate that is taken up by cells and converted intracellularly to NAD+.Citation41 Recently, a membrane protein, SLC12A8, that may be responsible for the import of NMN into cells was identified.Citation42 We postulate that the inhibition of extracellular CD38 NADase and NMNase activities by TNB-738 prevents the breakdown of extracellular NAD+ and NMN, which subsequently allows uptake into cells, leading to higher intracellular NAD+ (see for schematic summary).
Figure 8. Schematic representation of intracellular NAD+ boosting by TNB-738-mediated CD38 inhibition.
Currently, several small-molecule CD38 inhibitors have been identified, and two appear most promising, 78c, a 4-amino-quinoline derivative,Citation43 and RBN013209, a heterobicyclic amide. These molecules have been evaluated in animal models of fibrosis and cancer to prevent tissue damage and to restore T cell fitness, respectively. Both small-molecule inhibitors are membrane permeable and inhibit both type II (extracellular) and type III (intracellular) CD38.Citation44 Inhibiting type III CD38 can decrease intracellular cADPR and NAADP levels, which represents a potential safety concern with small-molecule inhibitors due to their ability to cross membranes, including blood–brain barriers. Studies performed in CD38 knockout mice demonstrated that the resultant reduction of cADPR in the brains of knockout mice led to decreased secretion of oxytocin, resulting in marked deficits in maternal nurturing and social behavior.Citation45 Though highly efficacious in pre-clinical models, future studies in humans will be key to understanding the efficacy and safety of these small-molecule inhibitors. Since TNB-738 is an IgG antibody that neither crosses the cell membrane nor the blood–brain barrier, the risks associated with inhibiting CD38 activity in the brain are absent.
NAD+ boosting strategies by supplementation with NMN are actively pursued by several groups and advertised to promote healthy aging.Citation1 However, supplements are readily converted by CD38 and lead only to transient increases of intracellular NAD+.Citation1 Our data suggest that supplementation with NAD+ precursors could be of limited use in cellular environments with high CD38 expression on cells, such as inflamed tissues, due to its rapid consumption by CD38 and other extracellular enzymes.Citation46 In in vitro assays, NMN supplementation alone did not result in increased NAD+ levels in CD38-expressing cells; only in the presence of TNB-738 did NAD+ levels rise, demonstrating that CD38 inhibition is required to effectively boost cellular NAD+.
The desired mechanism of action of TNB-738 is strictly enzyme inhibition without CD38+ cell depletion, thus avoiding side-effects associated with the lysis of subpopulations of immune cells, such as monocytes, effector T cells, and NK cells. Compared to currently available anti-CD38 antibodies daratumumab and isatuximab, TNB-738 binds CD38+ cells with high affinity but does not lyse CD38-expressing cells due to its silenced IgG4 Fc and its distinct epitopes on CD38 (summarized in ).Citation47,Citation48 Both isatuximab and daratumumab are conventional bivalent monoclonal IgG1 antibodies targeting different epitopes on CD38. Only isatuximab inhibits the enzyme activities of CD38, but it also induces CD38+ cell lysis through four independent mechanisms, namely: 1) CDC, 2) ADCC, 3) ADCP, and 4) caspase 3/7-dependent direct apoptosis by crosslinking CD38 via Fc receptors on immune cells. Daratumumab, on the other hand, activates immune effector functions and enhances the enzyme activity of CD38.Citation20 Although TNB-738 competes for binding to CD38 with isatuximab and daratumumab, it potently inhibits cell surface CD38 activity without directly or indirectly lysing cells. Epitope binning experiments demonstrated that daratumumab competes with F11A and isatuximab with F12A. However, mixing of isatuximab and daratumumab did not lead to synergistic inhibition of CD38 activity as observed with mixing F11A and F12A (data not shown). Therefore, biparatopic antibodies based on the variable regions of isatuximab and daratumumab would not lead to similar functionalities as TNB-738. The lack of synergy between daratumumab and isatuximab is likely due to steric collision at the C terminus of CD38 between the variable regions of the isatuximab heavy chain and the variable regions of the daratumumab light chain, as predicted by in silico superimposition.Citation27 TNB-738 binds to two distinct epitopes on CD38, as determined by X-ray crystallography and in silico epitope mapping, which impart its NAD+ boosting qualities without inducing lysis or activation of cells.
Table 3. Comparison of daratumumab, isatuximab, and TNB-738.
Potential diseases that could benefit from TNB-738 treatment are wide ranging. Studies using CD38 knockout animals have shown that CD38 plays a critical role in NAD+ metabolism and that absence or inhibition of CD38 ameliorates disease severity in animal models, including metabolic disorders, fibrosis, and inflammation.Citation49,Citation50 In a mouse model of bleomycin-induced lung fibrosis, CD38 knockout mice had significantly reduced lung fibrosis compared to wild-type mice.Citation40 In addition, we have recently shown that treatment of humanized NOD-scid IL2Rgnull (NSG) mice engrafted with human immune cells with TNB-738 greatly reduced inflammation resulting from graft versus host disease. NAD+ metabolism has also been studied extensively in metabolic and age-related diseases. Many studies have demonstrated that absence or inhibition of CD38 has beneficial effects in animal models of disease, with the effects most pronounced in aged animals. For example, aged mice treated with a small-molecule inhibitor of CD38 ran longer and faster and had better glucose tolerance compared to untreated aged mice.Citation51 Furthermore, CD38-knockout mice were completely protected from high fat diet-induced obesity compared to wild-type mice.Citation52 CD38 inhibition could possibly exacerbate bacterial and viral infections, since it has been hypothesized that NAD+ collapse helps fight bacterial infections and that many viruses rely on NAD-dependent enzymes.Citation53 However, animal studies using CD38 knockout mice and small compounds indicate that inhibition of CD38 has minimal implications for infectious diseases.Citation1Citation2,Citation53
In summary, TNB-738 is a biparatopic anti-CD38 antibody that potently inhibits CD38 enzyme activity, leading to increased NAD+ levels and activities of NAD-dependent enzymes. Importantly, TNB-738 neither depletes nor activates CD38-expressing cells and is minimally internalized. TNB-738 offers benefits over small-molecule inhibitors, and currently available antibodies directed against human CD38. Due to the importance of NAD+ in life- and health-span and the vast number of diseases associated with NAD+ decline, TNB-738 represents a promising approach to shift the equilibrium of the NAD salvage pathway toward NAD+ and away from ADPR and NAM. This could lead to more favorable energy balances, healthier mitochondria, and fewer epigenetic modifications. The broad therapeutic potential of TNB-738 in metabolic and inflammatory diseases warrants further investigation in clinical trials.
Materials and methods
Cloning, expression, and purification of antibodies
Anti-CD38 heavy chain only antibodies (UniAbs) were generated using genetically engineered transgenic rats that express fully human IgG antibodies (UniRat) together with an NGS-based antibody discovery pipeline (TeneoSeek). UniRats were immunized with recombinant human CD38 protein fused to a his-tag (Sino Biological, #10818-H08H) for up to 8 weeks using either Titermax/Ribi or CFA/IFA adjuvant. Draining lymph nodes from all animals were then harvested, and total RNA was collected. cDNA samples containing the full heavy chain variable domain (VH) underwent next-generation sequencing using the MiSeq platform (Illumina) with 2 × 300 paired-end reads. Data from all animals were analyzed, and the most frequent 373 VH sequences were selected for cloning followed by expression in HEK 293 cells.
F11A, F12, isatuximab (KEGG: D11050), and daratumumab (KEGG: D10777) were expressed with ExpiCHO cells using a high titer protocol as described by the manufacturer (Thermo Fisher Scientific, #A29133). Clarified harvests were purified over protein A (MabSelect Sure, Cytiva, #29049104). Neutralized protein A eluate was concentrated and polished by gel filtration (Superdex 200 Increase 10/30 GL) to remove aggregates. All antibodies were formulated in 20 mM citrate, 100 mM NaCl pH 6.2
Stability studies
The sample vials were stored in the controlled temperature reach-in stability chambers at 5 ± 3°C. The samples were injected using a UPLC over the TSKgel UP-SW3000 column (2 µm, 4.6 × 300 mm), (TOSOH, # 23448). 100 mM citrate, 500 mM NaCl, 200 mM L-Arginine, pH 6.2 was used as the mobile phase.
The thermal denaturation experiments for TNB-738 were performed using Capillary-DSC (Malvern). The autosampler was used to fill the reference and sample cells with 400 µL of buffer and 2 mg/mL protein sample, respectively. The sample was analyzed with a scan range of 20–95°C, at scan rate of 1°C /min, and a 16 sec filtering period. DSC scans were analyzed using MicroCal Origin® software.
Affinity measurement
A CM4 sensor chip was installed in a Biacore 4000 and primed with PBS+0.05% Tween-20 at 25°C. All surfaces were activated using standard NHS/EDC for 5 minutes followed by injections of hCD38 diluted to 10 µg/ml in 10 mM sodium acetate pH 5.0 over different surfaces to achieve four different surface densities (800, 600, 200, and 100 RU). After immobilization, the running buffer was switched to PBS + 0.05% p20 with 0.2 mg/ml BSA. Samples were injected at the maximum flow rate of 30 µL/min for 2 mins followed by a dissociation phase of 1800 seconds. Surfaces were regenerated with a 10 sec injection of a 1/200 dilution of phosphoric acid.
For classical affinity measurements, Octet QK-384 (ForteBio) was used. Anti-human IgG Fc Capture (AHC) biosensors (ForteBio, #18-5064) were hydrated in kinetic buffer (1x PBS, 0.1% BSA, 0.02% Tween 20, pH 7.2) and preconditioned in 100 mM Glycine, pH 1.5. A baseline was established for 120 seconds. TNB-738 at 5 µg/µl was immobilized on the AHC sensors for 120 seconds. Another baseline was established for 60 seconds. The antibody-coated sensors were then dipped into a 7-point, two-fold dilution series of the antigen human CD38 ECD (Sino Biological, #10818-H08H) in kinetic buffer starting at 100 nM. Association was observed for 120 seconds, followed by dissociation for 240 seconds. Data analysis was performed using Octet Data Analysis v9.0 (ForteBio). Binding kinetics were analyzed using a standard 1:1 binding model.
Flow cytometry
Cells were combined with antibodies in flow buffer in a 96-well plate (1X phosphate-buffered saline (PBS) + 1% bovine serum albumin (BSA) + 0.1% NaN3) and incubated at 4°C in the dark for 30 minutes. The plate was then washed 2x with flow cytometry buffer and incubated in a 1:100 dilution of goat anti-human IgG PE secondary antibody (Southern Biotech, #2042-09). Following incubation, the cells were washed 2x with flow cytometry buffer, resuspended in flow buffer, and analyzed on the FACSCelesta system (BD Biosciences).
CD38 hydrolase inhibition assay
CD38+ cells (100,000 cells/well) or recombinant CD38 protein (0.05 µg/mL final concentration) were incubated with increasing concentrations of antibodies in hydrolase assay buffer (40 mM Tris, 0.25 M sucrose, 0.8 mg/mL BSA, pH 7.4) in a black, flat-bottom 96-well plate for 15 min at room temperature. Following incubation, nicotinamide 1,N6-ethenoadenine dinucleotide (εNAD+) (Sigma Aldrich, #N2630), an enzymatically active derivative of NAD+ that produces a fluorescence signal hydrolysis, was added to each well at a final concentration of 150 µM. Fluorescence (ex/em: 300/410) was measured over time using a SpectraMax i3x multi-mode plate reader. The enzymatic activity of CD38 (as determined by V0) was measured over the range of antibody concentrations. Percent inhibition was calculated as follows: 100 – [100*(V0 (test article)/V0 (untreated))].
Capping experiment
Daudi cells were incubated with 10 µg/mL TNB-738 or daratumumab for 0, 30, 60, or 120 min at 37°C then fixed and permeabilized (FIX & PERM™ Cell Permeabilization Kit, Thermo Fisher, #GAS003). Cells were then labeled with anti-human IgG FITC (Jackson Laboratories, #109-096-008), anti-CD98 Alexa647 (Abcam, #ab23495, labeled in-house with Alexa647 (Thermo Fisher, #A20186) and Texas Red Phalloidin (Life Technologies, #T7471). Cells were mounted between slide and slip-cover with ProLong™ Gold Antifade Mountant with DAPI (Invitrogen, #P36931). Slides were analyzed with a confocal microscope (A1R, Nikon Imaging Software NIS) with 60x/4 lens. CD98, a transmembrane glycoprotein that forms part of the heterodimeric neutral amino acid transport system, and phalloidin, a bicyclic peptide used to stain actin filaments, were utilized to visualize the intact plasma membrane in order to properly observe capping induced by anti-CD38 antibodies. Cells with and without capping were counted, and percentage capping was graphed for 100 cells per treatment.
Internalization of antibodies
Isotype control IgG1 (Invitrogen, #bgal-mab1), TNB-738, or daratumumab were labeled with Zenon™ pHrodo™ iFL-labeled Fab fragment (Thermo Fisher, #Z25612) for 10 minutes. 0.5 M Daudi cells were incubated with Fc Block (BD Biosciences, #564220) for 30 min at 4°C. Cells were then washed and incubated at 37°C with isotype control IgG1 (R&D), TNB-738, or daratumumab (10 µg/mL) for 60 minutes. Following incubation, cells were washed with cold PBS, stained with DAPI (Thermo Fisher, #D1306), and fluorescence was measured using a FACS Verse cytometer with FACSuite Software version 1.0.6; post-acquisition analysis was performed using FlowJo software.
Assessment of hPBMC activation
Healthy volunteers’ blood was collected at the Etablissement Français du Sang (Nantes, France) from healthy donors. Written informed consent was provided according to institutional guidelines. PBMCs were isolated by Ficoll–Paque density-gradient centrifugation (Eurobio, Courtaboeuf, France). Remaining red blood cells and platelets were eliminated with a hypotonic solution. The cells were then washed and counted. 5 × 105 hPBMC were incubated overnight at 37°C without antibodies or with TNB-738 at 5, 1, or 0.1 µg/mL or with anti CD3 (wells were pre-coated with OKT3 anti-CD3 clone at 10 µg/mL) and anti-CD28 (10 µg/mL, in the supernatant). Following incubation, Brefeldin A (Sigma, #B6542) was added for 4 hours, and the cells were subsequently stained with Fixable Viability Dye eFluor 506 (Thermo Fisher, #65-0866-14), anti-hCD3 PeCy7 (BD Biosciences, #557851), anti-hCD4 PercpCy5.5 (BD Biosciences, #552838), anti-CD16 (clone 3G8, labeled in house with Alexa Fluor 488 IgG labeling kit from Thermo Fisher, #A20181), and anti-CD25 APC Cy7 (BD Biosciences, #557753). Cells were fixed and permeabilized (FIX & PERM Cell Fixation & Cell Permeabilization Kit, Thermo Fisher, #88-8824-00) and stained with anti-hFoxP3 PE (BD Biosciences, #12-4776-42) and anti-hIFNγ V450 (BD Biosciences, #560372). Fluorescence was measured using a FACS Verse cytometer with FACSuite Software version 1.0.6; post-acquisition analysis was performed using FlowJo software.
Complement-dependent cytotoxicity
Antibodies were incubated with tumor cells in a 96-well plate for 10 minutes at room temperature. After incubation, a 1:10 dilution of rabbit complement serum (Sigma Aldrich, #S7764-5ML) was added to each well and incubated for an additional 30 minutes at 37°C and 8% CO2. Cell Titer Glo 2.0 (Promega, #G9241) was then added to each well and luminescence was measured on a SpectraMax i3x plate reader. Percent viability was calculated as follows: 100 * (RLUTest Article/RLUUntreated).
Antibody-dependent cell-mediated cytotoxicity
NK cells were isolated from healthy-donor PBMCs using a commercial NK cell isolation kit (Miltenyi, #130-092-657). The NK cells were then incubated with tumor cells and antibodies in a 96-well plate for 4 hours at 37°C and 8% CO2. Following incubation, the supernatant was harvested and combined with LDH assay substrate (Promega, #J2380) and incubated for 30 minutes at room temperature. After incubation, a stop solution was added to each well. Absorbance (490 nm) was then analyzed on a SpectraMax i3x plate reader. Percent killing was calculated using the following formula: (ODsample well – ODtarget only)/(ODtarget+lysis – OD target only – ODNK only)%.
Direct apoptosis
Antibodies were incubated with tumor cells in a 96-well plate for 24 hours at 37°C and 8% CO2. The cells were then centrifuged and resuspended in a mixture of Annexin V binding buffer, FITC-conjugated Annexin V (BioLegend, #640906), and 7-AAD (BioLegend, #420403) for 10 minutes at room temperature. Following incubation, the cells were analyzed flow cytometry to assess cell viability. A quad gate was used to distinguish between early apoptotic (Annexin V+, 7-AAD-), late apoptotic (Annexin V+, 7-AAD+), and viable cells (Annexin V-, 7-AAD-).
Measurement of NAD+ concentration
Ramos cells were plated in a 96-well plate and incubated with antibody for 24 hours at 37°C and 8% CO2. The cells were then washed once with PBS and resuspended in 1X Extraction Buffer from the Cell Biolab NAD+ assay kit. Cell lysates were made by homogenizing cells followed by centrifugation at 4°C and 14000 rpm for 5 minutes. The supernatant was harvested and NAD+ concentration was measured using Cell Biolabs NAD+ assay kit (#MET-5014), according to manufacturer’s instructions.
Measurement of SIRT activity
Ramos cells were plated in a 96-well plate and incubated with antibody for 24 hours at 37°C and 5% CO2. The cells were then washed once with PBS and resuspended in M-PER Mammalian Protein Extraction Reagent (Thermo Fisher, #78501). Cell lysates were made by homogenizing cells followed by centrifugation at 4°C and 14000 rpm for 5 minutes. The supernatant was harvested and incubated in a 1:30 dilution of anti-SIRT1 (Abcam, #ab32441) or anti-SIRT3 antibody (Abcam, #ab217319) for 4 hours on a rotator at 4°C. Protein A slurry was then added, and samples were incubated for an additional 2 hours at room temperature. Immunoprecipitation buffer was added, followed by centrifugation at 2500 g for 2 minutes. The wash steps with IP buffer were repeated. IgG elution buffer was added for 5 minutes, after which the eluate was centrifuged at 2500 g for 2 minutes. The supernatant was neutralized with 1 M Tris (pH 9) at 1:10. SIRT activity was measured using the SIRT Glo assay kit (Promega, #G6450), according to manufacturer’s instructions.
Epitope binning
Binning experiments were run on BioRad ProteOn SPR biosensor using a GLC sensor chip. F11A, F12A, isatuximab, and daratumumab were amine coupled to the chip surface using a standard 5-minute activation with sulfo-NHS/EDC followed by a 5-minute injection of each mAb at 15 µg/ml in 10 mM sodium acetate pH 5.0 and a 5-minute blocking step with 1 M ethanolamine. Human CD38 was also coupled to a surface as a control. Running buffer contained DPBS pH 7.4 with 0.05% tween-20. All data were collected at 25°C.
Next, a stacking study was run over each of the antibody and huCD38 surfaces. The first injection is the binding of huCD38 at 400 nM for 90 seconds at 100 µl/min, followed by a second injection that contained separately each antibody as well as recombinant huCD38 at 400 nM.
Expression and purification of CD38 and UniDab
CD38 (UniProt: P28907) was cloned in Pichia pastoris-based expression vector to yield the expression of 45–300 amino acid of CD38, which includes mismatches at 5 positions in the CD38 sequence, in fusion with His-tag at its C-terminus. The fusion protein was expressed by transforming in Pichia pastoris strain BICC 9450. The protein was purified from the supernatant by affinity column chromatography. To obtain a homogenous form of the protein, it was further purified by gel filtration chromatography using Superdex-75 column. The protein was eluted in 20 mM HEPES buffer, pH 7.2 with 50 mM NaCl.
UniDab_F11A (antigen-binding fragment of heavy chain only antibodies) expressing construct was generated by cloning the gene fragment in pCDNA3.1 vector. The expression plasmid allows expression of UniDab_F11A in fusion with C-terminal His tag in ExpiCHO cells. The transfection was carried using ExpiFectamine™ CHO Transfection Kit (Thermo Fisher, #A29133) as per manufacturer’s protocol. The resultant clarified supernatant was subjected to affinity column chromatography using Ni-NTA Sepharose (Cytiva, #17526801). The protein obtained after affinity-based purification was further purified using gel filtration chromatography. The protein was eluted in 20 mM HEPES pH 7.2 with 50 mM NaCl.
Crystallization of the CD38-F11A complex, data collection, and structure determination
To obtain the binary complex, CD38 protein was mixed with UniDab_F11A at a molar ratio of 1.2:1. The mixture was incubated at 4°C for 16 hours. The mixture was then loaded onto the Superdex 75 16/60 column and the elute fractions were analyzed. Fractions having both CD38 and UniDab_F11A were pooled and concentrated using a 3 kDa cutoff centrifugal concentrators.
Crystallization of CD38-UniDab_F11A complex was set up using commercially available screens. Crystallization was set up using sitting drop vapor diffusion method with 300 nL drop at 1:2 ratio of protein and reservoir solution. Initial crystals were obtained with 0.1 M Sodium Citrate pH 5.5, and 8% PEG 8000. Crystallization conditions were further optimized by pH and precipitant grid screens using initial crystals as microseeds.
Crystals of CD38-UniDab_F11A complex was cryo-protected in mother liquor containing 25% ethylene glycol and further flash-cooled in liquid nitrogen. The crystals were diffracted, and diffraction data was collected at MX-2 beamline at ANSTO synchrotron facility. The crystals belong to P 32 2 1 space group and have one molecule each of CD38 and UniDab_F11A in the crystallographic asymmetric unit. Diffraction data was processed and integrated using Mosflm.Citation54 Data was scaled and merged; the merged intensities were converted to structure factor using Truncate program from CCP4 Suite.Citation55 The structure was solved using molecular replacement method. PDB 1YH3 chain A for CD38 and PDB 6PZW chain F for UniDab_F11A were used as template for molecular replacement search models.
The model was first refined using rigid-body refinement in Refmac5 of CCP4.Citation56 It was further refined by iterative cycles of manual model building in COOT and positional refinement in Refmac5. The data collection and refinement statistics are summarized in the table. The atomic coordinates were deposited with the Protein Data Bank with accession code 7VKE.
In silico epitope mapping of F12A and confirmation
MAbTope was used to determine the F12A epitope on CD38. Briefly, MAbTope is a docking-based method which generates 5 × 108 poses for the antibody-antigen complex and which, using several scoring functions, filters these poses in order to obtain the 30 best solutions. The interface analysis of these 30 top-ranked solutions allowed identification of the residues that exhibit the highest probability of being implicated in the interaction. For in vitro binding assays, the CD38 sequence disclosed in UniProt ID P28907 was used to construct wild-type and mutant genes. A flag tag was added to the N-terminal end of CD38, and no other modifications were made to the protein. The wild-type and mutated CD38 were designed in silico and reverse translated in DNA using EMBL-EBI emboss – backtranseq tool, using a human codon usage table, and avoiding the Hind III and XhoI restriction sites used for cloning in pcDNA3.1+. A stop codon and the Kozak sequence (GCCACC) were added to the DNA sequence. The genes were synthesized and cloned in pcDNA3.1+ by Twist Bioscience (South San Francisco, CA, USA).
One million HEK293 cells were transiently transfected with 1 µg of either one of the constructs or a mock vector using Metafectene (Biontex Laboratories) according to the manufacturer’s instructions. After 24 hours, the cells were trypsinized, fixed, and permeabilized according to the BD Bioscience CytoFix/CytoPerm kit’s protocol and distributed in 96-well plates. Fifty thousand cells were incubated with 2.5 µg of F12A, daratumumab, or human IgG1 isotype (BioLegend, #BLE403502) in 100 µl of wash buffer (20 mM citrate, 100 mM NaCl, 2 mM EDTA, 1% FBS, pH 6.2) for 1 hour at 4°C. The cells were washed, and the cell pellet was resuspended in 20 µL of 20 mM citrate, 100 mM NaCl, 2 mM EDTA, 1% FCS pH 6.2 with 0.1 µL of an APC-coupled mouse anti-human IgG1 (Miltenyi, #130-119-857) and 0.03 µL of a PE-coupled human anti-Flag antibody (Miltenyi, #130-101-576) and left in dark for 45 min at 4°C. The cells were washed in wash buffer and once again with 20 mM citrate, 100 mM NaCl, 2 mM EDTA, pH 6.2. Fluorescence was then measured using a MACSQ analyzer 10. The initial gatings for cell selection were performed on unstained cells. The experiment was repeated 4 times. Cytometry data was analyzed using FlowJo V10. Graphs and statistical analysis were performed with GraphPad Prism 9.
Supplemental Material
Download Zip (10.1 MB)Acknowledgments
We acknowledge the IBISA MicroPICell facility (Biogenouest), a member of the National Infrastructure France-Bioimaging supported by the French National Research Agency (ANR-10-INBS-04).
Disclosure statement
This research was sponsored by Teneobio, and authors HSU, KD, BB, JA, LMD, RB, SCC, SI, US, WVS, and PD were former employees of Teneobio. HSU, KD, JA, PD are current employees of Amgen. BB, RB, SI, US, and WVS are current employees of Ancora Biotech LLC. SCC and LMD are current employees of Rondo Therapeutics. SNM is a current employee of Aurigene Pharmaceutical Services Limited. RD and ENC served as consultants to Teneobio.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2095949
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in Immunomodulation, cancer, aging, and metabolic diseases. Front Immunol. 2019;10:1187. doi:10.3389/fimmu.2019.01187.
- Lee HC. Structure and enzymatic functions of human CD38. Mol Med. 2006;12(11–12):317–15. doi:10.2119/2006.
- Ichiro IS, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–71. doi:10.1016/j.tcb.2014.04.002.
- Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–47. doi:10.1016/j.cmet.2018.02.011.
- Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23(6):1127–39. doi:10.1016/j.cmet.2016.05.006.
- Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 2019;30(4):630–55. doi:10.1016/j.cmet.2019.09.001.
- Dürig J, Naschar M, Schmücker U, Renzing-Köhler K, Hölter T, Hüttmann A, Dührsen U. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia. 2002;16(1):30–35. doi:10.1038/sj.leu.2402339.
- Chini C, Hogan KA, Warner GM, Tarragó MG, Peclat TR, Tchkonia T, Kirkland JL, Chini E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem Biophys Res Commun. 2019;513(2):486–93. doi:10.1016/j.bbrc.2019.03.199.
- Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22(2):119–41. doi:10.1038/s41580-020-00313-x.
- McReynolds MR, Chellappa K, Baur JA. Age-related NAD+ decline. Exp Gerontol. 2020;134:110888. doi:10.1016/j.exger.2020.110888.
- Chini EN, Chini CCS, Espindola Netto JM, de Oliveira Gc, van Schooten W, de Oliveira GC. The pharmacology of CD38/NADase: an emerging target in cancer and Diseases of aging. Trends Pharmacol Sci. 2018;39(4):424–36. doi:10.1016/j.tips.2018.02.001.
- Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13(9):1331–39. doi:10.1016/j.str.2005.05.012.
- Dong M, Si YQ, Sun SY, Pu XP, Yang ZJ, Zhang LR, Zhang LH, Leung FP, Lam CMC, Kwong AKY, et al. Design, synthesis and biological characterization of novel inhibitors of CD38. Org Biomol Chem. 2011;9(9):3246–57. doi:10.1039/c0ob00768d.
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, Chini EN, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62(4):1084–93. doi:10.2337/db12-1139.
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208–13. doi:10.1126/science.aac4854.
- de Picciotto Ne, Gano LB, Johnson LC, Picciotto NE, Martens CR, Sindler AL, Mills KF, Imai S-I, Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15(3):522–30. doi:10.1111/acel.12461.
- Martin TG, Corzo K, Chiron M, van de Velde H, Abbadessa G, Campana F, Solanki M, Meng R, Lee H, Wiederschain D, et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells. 2019;8(12):E1522. doi:10.3390/cells8121522.
- Zhu C, Song Z, Wang A, Srinivasan S, Yang G, Greco R, Theilhaber J, Shehu E, Wu L, Yang Z-Y, et al. Isatuximab acts through Fc-dependent, independent, and direct pathways to kill multiple myeloma cells. Front Immunol. 2020;11:1771. doi:10.3389/fimmu.2020.01771.
- Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, Dasilva N, Song Z, Yang G, Adrian F, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia. 2016;30(2):399–408. doi:10.1038/leu.2015.240.
- van de Donk NWCJ, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, Lokhorst HM, Parren PWHI. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016;270(1):95–112. doi:10.1111/imr.12389.
- Force Aldred S, Boudreau A, Buelow B, Clarke S, Dang K, Davison L, Harris K, Iyer S, Jorgensen B, Ogana H, et al. Multispecific antibodies targeting CD38 show potent tumor-specific cytotoxicity. JCO. 2018;36(5_suppl):57–57. doi:10.1200/JCO.2018.36.5_suppl.57.
- Clarke SC, Ma B, Trinklein ND, Schellenberger U, Osborn MJ, Ouisse L-H, Boudreau A, Davison LM, Harris KE, Ugamraj HS, et al. Multispecific antibody development platform based on human heavy chain antibodies. Front Immunol. 2018;9:3037. doi:10.3389/fimmu.2018.03037.
- Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi:10.1007/s13238-017-0473-8.
- de Oliveira Gc, Kanamori KS, Auxiliadora-Martins M, Chini CCS, Chini EN, de Oliveira G. Measuring CD38 hydrolase and cyclase activities: 1,N6-ethenonicotinamide adenine dinucleotide (ε-NAD) and nicotinamide guanine dinucleotide (NGD) fluorescence-based methods. Bio Protoc. 2018;8(14):e2938. doi:10.21769/BioProtoc.2938.
- Pierce SK, Morris JF, Grusby MJ, Kaumaya P, Buskirk AV, Srinivasan M, Crump B, Smolenski LA. Antigen-presenting function of B lymphocytes. Immunol Rev. 1988;106(1):149–80. doi:10.1111/j.1600-065x.1988.tb00778.x.
- Funaro A, De Monte LB, Dianzani U, Forni M, Malavasi F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur J Immunol. 1993;23(10):2407–11. doi:10.1002/eji.1830231005.
- Lee HT, Kim Y, Park UB, Jeong TJ, Lee SH, Heo YS. Crystal structure of CD38 in complex with daratumumab, a first-in-class anti-CD38 antibody drug for treating multiple myeloma. Biochem Biophys Res Commun. 2021;536:26–31. doi:10.1016/j.bbrc.2020.12.048.
- Bourquard T, Musnier A, Puard V, Tahir S, Ayoub MA, Jullian Y, Boulo T, Gallay N, Watier H, Bruneau G, et al. MAbTope: a method for improved epitope mapping. J Immunol. 2018;201(10):3096–105. doi:10.4049/jimmunol.1701722.
- Yang L, Li T, Li S, Wu Y, Shi X, Jin H, Liu Z, Zhao Y, Zhang L, Lee HC, et al. Rational design and identification of small-molecule allosteric inhibitors of CD38. Chembiochem. 2019;20(19):2485–93. doi:10.1002/cbic.201900169.
- Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Li C, Shen G, Zou B. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020;5(1):227. doi:10.1038/s41392-020-00311-7.
- Castro-Portuguez R, Sutphin GL. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: targeting tryptophan metabolism to promote longevity and healthspan. Exp Gerontol. 2020;132:110841. doi:10.1016/j.exger.2020.110841.
- Mottahedeh J, Haffner MC, Grogan TR, Hashimoto T, Crowell PD, Beltran H, Sboner A, Bareja R, Esopi D, Isaacs WB, et al. CD38 is methylated in prostate cancer and regulates extracellular NAD. Cancer Metab. 2018;6(1):13. doi:10.1186/s40170-018-0186-3.
- Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001;15(1):10–12. doi:10.1096/fj.00-0566fje.
- Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16(3):177–92. doi:10.1038/nri.2016.4.
- Audrito V, Messana VG, Brandimarte L, Deaglio S. The extracellular NADome modulates immune responses. Front Immunol. 2021;12:704779. doi:10.3389/fimmu.2021.704779.
- Hwang SJ, Durnin L, Dwyer L, Rhee P-L, Ward SM, Koh SD, Sanders KM, Mutafova–Yambolieva VN. β-nicotinamide adenine dinucleotide is an enteric inhibitory neurotransmitter in human and nonhuman primate colons. Gastroenterology. 2011;140(2):608–617.e6. doi:10.1053/j.gastro.2010.09.039.
- Billington RA, Travelli C, Ercolano E, Galli U, Roman CB, Grolla AA, Canonico PL, Condorelli F, Genazzani AA. Characterization of NAD uptake in mammalian cells. J Biol Chem. 2008;283(10):6367–74. doi:10.1074/jbc.M706204200.
- Kulikova V, Shabalin K, Nerinovski K, Dölle C, Niere M, Yakimov A, Redpath P, Khodorkovskiy M, Migaud ME, Ziegler M, et al. Generation, release, and uptake of the NAD precursor nicotinic acid riboside by human cells. J Biol Chem. 2015;290(45):27124–37. doi:10.1074/jbc.M115.664458.
- O’Reilly T, Niven DF. Levels of nicotinamide adenine dinucleotide in extracellular body fluids of pigs may be growth-limiting for actinobacillus pleuropneumoniae and haemophilus parasuis. Can J Vet Res. 2003;67:229–31.
- Buonvicino D, Ranieri G, Pittelli M, Lapucci A, Bragliola S, Chiarugi A. SIRT1-dependent restoration of NAD+ homeostasis after increased extracellular NAD+ exposure. J Biol Chem. 2021;297(1):100855. doi:10.1016/j.jbc.2021.100855.
- Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27(3):513–28. doi:10.1016/j.cmet.2017.11.002.
- Grozio A, Mills KF, Yoshino J, Bruzzone S, Sociali G, Tokizane K, Lei HC, Cunningham R, Sasaki Y, Migaud ME, et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab. 2019;1(1):47–57. doi:10.1038/s42255-018-0009-4.
- Boslett J, Reddy N, Alzarie YA, Zweier JL. Inhibition of CD38 with the Thiazoloquin(az)olin(on)e 78c protects the heart against postischemic injury. J Pharmacol Exp Ther. 2019;369(1):55–64. doi:10.1124/jpet.118.254557.
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, et al. Discovery, synthesis, and biological evaluation of Thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J Med Chem. 2015;58(8):3548–71. doi:10.1021/jm502009h.
- Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature. 2007;446(7131):41–45. doi:10.1038/nature05526.
- Chini CCS, Peclat TR, Warner GM, Kashyap S, Espindola-Netto JM, de Oliveira GC, Gomez LS, Hogan KA, Tarragó MG, Puranik AS, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat Metab. 2020;2(11):1284–304. doi:10.1038/s42255-020-00298-z.
- Park P, Bartle L, Skaletskava A, Golmakher V, Tavares D, Deckert J, Milol V, Blanc V, Sanofi-Aventis. Novel anti-cd38 antibodies for the treatment of cancer. France US 2011/0262454A1. 27 Oct 2011.
- van de Donk NWCJ, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018;131(1):13–29. doi:10.1182/blood-2017-06-740944.
- Shi B, Wang W, Korman B, Kai L, Wang Q, Wei J, Bale S, Marangoni RG, Bhattacharyya S, Miller S, et al. Targeting CD38-dependent NAD+ metabolism to mitigate multiple organ fibrosis. iScience. 2021;24(1):101902. doi:10.1016/j.isci.2020.101902.
- Schneider M, Schumacher V, Lischke T, Lücke K, Meyer-Schwesinger C, Velden J, Koch-Nolte F, Mittrücker H. CD38 is expressed on inflammatory cells of the intestine and promotes intestinal inflammation. PLoS One. 2015;10(5):e0126007. doi:10.1371/journal.pone.0126007.
- Tarragó MG, Chini CCS, Kanamori KS, Warner GM, CarideA, de Oliveira GC, Rud M, Samani A, Hein KZ, and HuangR et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 2018;27(5):1081–1095.e10. doi:10.1016/j.cmet.2018.03.016.
- Chiang SH, Harrington WW, Luo G, Milliken NO, Ulrich JC, Chen J, Rajpal DK, Qian Y, Carpenter T, and Murray R, et al. Genetic ablation of CD38 protects against western diet-induced exercise intolerance and metabolic inflexibility. PLoS One. 2015;10(8):e0134927. doi:10.1371/journal.pone.0134927.
- Singhal A, Cheng CY. Host NAD+ metabolism and infections: therapeutic implications. Int Immunol. 2019;31(2):59–67. doi:10.1093/intimm/dxy068.
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):271–81. doi:10.1107/S0907444910048675.
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP 4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–42. doi:10.1107/S0907444910045749.
- Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3): 240–55. doi:10.1107/S0907444996012255.