
ABSTRACT
To combat the COVID-19 pandemic, potential therapies have been developed and moved into clinical trials at an unprecedented pace. Some of the most promising therapies are neutralizing antibodies against SARS-CoV-2. In order to maximize the therapeutic effectiveness of such neutralizing antibodies, Fc engineering to modulate effector functions and to extend half-life is desirable. However, it is critical that Fc engineering does not negatively impact the developability properties of the antibodies, as these properties play a key role in ensuring rapid development, successful manufacturing, and improved overall chances of clinical success. In this study, we describe the biophysical characterization of a panel of Fc engineered (“TM-YTE”) SARS-CoV-2 neutralizing antibodies, the same Fc modifications as those found in AstraZeneca’s Evusheld (AZD7442; tixagevimab and cilgavimab), in which the TM modification (L234F/L235E/P331S) reduce binding to FcγR and C1q and the YTE modification (M252Y/S254T/T256E) extends serum half-life. We have previously shown that combining both the TM and YTE Fc modifications can reduce the thermal stability of the CH2 domain and possibly lead to developability challenges. Here we show, using a diverse panel of TM-YTE SARS-CoV-2 neutralizing antibodies, that despite lowering the thermal stability of the Fc CH2 domain, the TM-YTE platform does not have any inherent developability liabilities and shows an in vivo pharmacokinetic profile in human FcRn transgenic mice similar to the well-characterized YTE platform. The TM-YTE is therefore a developable, effector function reduced, half-life extended antibody platform.
Introduction
Fc engineering is increasingly used to customize effector functions or half-life of antibody therapeutics. Fc engineering modifications cluster into three main categories: 1) effector function enhancing, 2) effector function silencing, and 3) serum half-life extending. Effector function enhancing modifications are added when improved antibody-mediated cell killing or enhanced immune cell activation via FcγR binding is required (examples of effector function enhancing modifications are afucosylation,Citation1 DLE,Citation2 GAALIE, and others listed Wilkinson et al).Citation3 Effector function silencing modifications are added when the antigen-binding activity is the only desired mechanism of action (MOA). Examples of this include binding and activating an effector cell, as in some immuno-oncology instances,Citation4 or avoiding antibody-dependent enhancement of disease (ADE) in certain viral infections.Citation5 Some commonly used “Fc-silenced” formats are the TM modification (L234F/L235E/P331S),Citation6 LALA-PG (L234A/L235A/P329G),Citation7 removal of the N297 glycan,Citation8 and IgG4P.Citation9 The third category of Fc engineering is used for in vivo serum persistence, which can be achieved by increasing the recycling of an antibody out of the cell and back into the serum through pH-dependent FcRn binding enhancement. To effectively increase the antibody half-life, it is necessary to increase the binding to FcRn at pH 6, while not affecting (or weakening) the binding at pH 7.Citation10 Examples of half-life extending modifications are the YTE modification (M252Y/S254T/T256E),Citation11 N3Y (L432C/H433S/N434Y/H435H/Y436L/T437C),Citation10 DHS (L309D/Q311H/N434S),Citation12 and others listed in Kuo et al.Citation13
While Fc engineering allows the biological activity of therapeutic antibodies to be tailored for specific MOAs and serum half-life, introducing modifications into the Fc region can affect the overall biophysical characteristics of these molecules,Citation14,Citation15 which in turn affects their developability profile. The developability profile of an antibody encompasses multiple properties, including expression titers, propensity for aggregation, solubility and viscosity, thermal stability, nonspecific binding (NSB), reversible self-association (RSA), storage and photostability, post-translational modifications (PTMs), and pharmacokinetic (PK) properties.Citation16,Citation17 All of these properties can substantially impact the length and complexity of the chemistry, manufacturing, and control (CMC) development and therefore affect the overall chances of clinical success. Jain et al. first showed a negative correlation between the number of developability flags and success in the clinic.Citation16 A subsequent analysis of their results also showed that high antibody specificity and the absence of NSB and RSA are more prevalent in approved antibody therapeutics compared with investigational antibodies (i.e., those still in clinical trials).Citation18 Self-interactions can lead to low solubility, aggregation, and high viscosity during antibody production,Citation19,Citation20 while nonspecific binding can lead to off-target binding and fast clearance in vivo.Citation21,Citation22 PTMs, such as oxidation, deamidation, and isomerization, can have multiple effects on an antibody. If found in the complementarity-determining regions (CDRs), these modifications can affect binding affinity.Citation17 In the Fc, oxidation of methionine residues (Met-252 and Met-428) can cause aggregation,Citation23 decrease effector functions,Citation24 and decrease binding to FcRn, and therefore affect molecule half-life.Citation25 These studies, along with accrued experience with the impact of developability issues on CMC activities, have led to an increased emphasis on the developability of molecules moving forward into the clinic.
Despite the resounding success of authorized vaccines against SARS-CoV-2, neutralizing antibodies present a promising complementary approach to protect against infection or prevent disease worsening, especially in immune-compromised individuals who cannot generate a protective immune response. The unprecedented need and urgency caused by this pandemic require neutralizing antibodies, and other therapeutics, to be brought to the clinic faster than ever before. To meet such accelerated timelines, the selected antibody format for SARS-CoV-2 neutralizing antibodies must have desirable developability profiles. AstraZeneca’s Evusheld (AZD7442) is a combination of two SARS-CoV-2 neutralizing antibodies (tixagevimab and cilgavimab) recently granted marketing authorization for prophylaxis of SARS-CoV-2 in immune-compromised individuals in the UK, EU, and Canada and emergency use authorization (EUA) in the US.Citation26–31 The two neutralizing antibodies in Evusheld (AZD7442) each contain two distinct Fc modifications: 1) the YTE modification for increased in vivo half-life, and 2) the TM modification for decreased binding to FcγRs and C1q. The combination of these two sets of Fc modifications is designed to increase the in vivo half-life of the virus-neutralizing antibodies, and to reduce the concern surrounding ADE, which is mediated by antibody:FcγR interactions, respectively. However, some potential concerns were raised regarding developability, as previous analyses of TM-YTE antibodies showed decreased thermostability of the Fc CH2 domain, reducing the Tm1 of the TM-YTE CH2 from 70.1°C to 58.1°C as measured by differential scanning calorimetry (DSC).Citation32,Citation33 One of these studies also showed a slight increase in aggregation propensity, self-association, and a lower apparent solubility compared to its wild type (WT) Fc counterpart. However, that study compared a single variable fragment (Fv) on multiple Fc engineered backbones.
Here, we profiled the biophysical characteristics of a diverse panel of SARS-CoV-2-binding Fvs in the TM-YTE format, providing an in-depth look at the developability profile of the TM-YTE platform. Our study shows that the TM-YTE platform, despite having a lower CH2 Tm1, does not show a propensity for aggregation, does not affect NSB, and is compatible with high concentration protein formulation (HCPF). In addition, the TM-YTE backbone shows a PK profile comparable to the YTE-only backbone, indicating no effect on serum persistence. We also find that the diffusion interaction parameter, kD, acquired by dynamic light scattering (DLS) is more predictive of high concentration viscosity than affinity capture self-interaction nanoparticle spectroscopy (AC-SINS) results. Overall, we show that the TM-YTE platform is a highly developable “Fc-silenced” and half-life extended antibody format.
Results
TM-YTE antibodies exhibit lowered CH2 thermostability
The thermal stability of a panel of SARS-CoV-2 neutralizing antibodies was assessed by DSC. Consistent with our previous study,Citation32 the Tm1 of all TM-YTE antibodies is ~58°C, whereas that of their WT Fc counterparts are 72°C or higher (). The Tm1 represents the unfolding temperature of the CH2 domain. It is unsurprising that CH2 thermostability is affected because all six of the TM-YTE mutation sites lie within the CH2 domain (). The Tm2, often used as a predictor of long-term stability because it represents the antigen-binding fragment (Fab) (and CH3 domain if overlapping), was low in antibodies 2, 6, and 8, but above 74.6°C for the rest of the panel (). Only antibody 7 with a WT Fc had overlapping Tm1 and Tm2 at 75.2°C. shows an overlay of the melting curves of the WT and TM-YTE format of antibody 4 as a representative antibody, with the antibody 4 WT Tm1 at 72°C.
Figure 1. Clustering of the TM-YTE modifications on the IgG1 structure. The TM (L234F/L235E/P331S, shown in dark blue spheres) and YTE (M252Y/S254T/T256E, shown in red spheres) Fc modifications were mapped onto the IgG1 structure (PDB ID 1HZH). The antibody Fab and Fc regions are marked as well as the Fc CH2 and CH3 domains. The antibody Fv is shown in cyan, and the constant domains are shown in different shades of green. Note that while both the TM and YTE modifications are in CH2, the TM modification cluster at the top of the CH2 domain near the hinge region in the binding regions for FcγRs and C1q while the YTE modification cluster near the CH3 domain in the binding region for FcRn. Ribbon diagram of an IgG1 crystal structure with the location of TM and YTE modification sites highlighted as spheres. Both modifications cluster in the Fc CH2 domain.
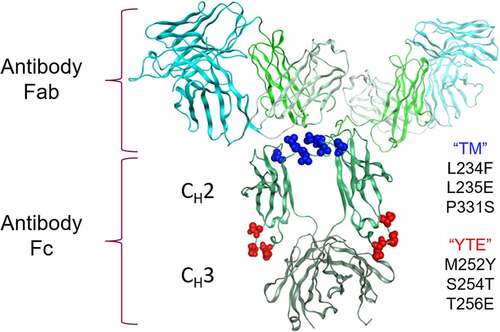
Figure 2. DSC profile overlay of a representative antibody in WT and TM-YTE format. Thermal stability of a representative antibody in WT and TM-YTE format was assessed by DSC. Introduction of TM-YTE modifications results in decreased thermal stability of CH2, as seen by the lower temperature for the first (Tm1) melting peak. A DSC graph plotting the melting temps of Ab 4 in WT format versus TM-YTE format. The Tm1 peak is ~58°C for the TM-YTE format and ~72°C for the WT format.
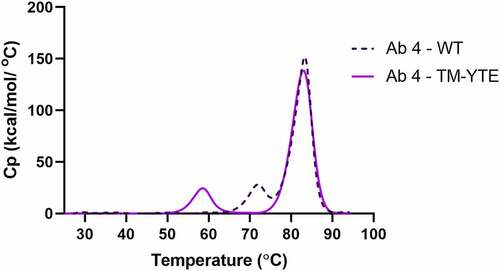
Table 1. Melting temperature of SARS-COV-2 antibody domains by DSC. Thermal stability was assessed by DSC. All antibodies, unless otherwise specified, are in the TM-YTE format and have a lower Tm1 than the corresponding antibody in the WT format. Tm2 varies among the antibody panel.
No aggregation propensity induced by the TM-YTE platform
To test whether the lower thermal stability of CH2 domain in the TM-YTE format affects aggregation or fragmentation propensity, the antibodies were subjected to heat-stress and photo-stress studies. High-performance size-exclusion chromatography (HP-SEC) was used to monitor the level of aggregates and fragments in the antibody samples before and after incubation at 45°C for 14 days, or exposure to 3,000 lux cool white light (CWL) for 7 days. HP-SEC can underestimate the amount of un-separated and large fragments compared to CE-SDS but can still detect the smaller resulting Fab fragments.Citation34,Citation35 In the heat-stress study, no major increase in aggregates was detected for any of the antibodies, and only antibodies 1, 3, and 7 exhibited a low-level increase in fragmentation (, Supplementary Figure 1). The WT version of antibody 7 exhibited similar levels of fragmentation, indicating an Fv-specific cause for this fragmentation. In the photo-stress study, none of the antibodies showed a major increase in aggregation or fragmentation. The control antibody NIP228 was produced in the WT, TM, YTE, and TM-YTE formats and tested alongside in the heat and photo stress assays. No major increase in aggregation or fragmentation was detected with any of the Fc modifications, with levels remaining very low in all formats.
Table 2. Heat-stress and photo-stress studies for TM-YTE SARS-COV-2 antibodies. Heat stress samples were formulated at 1 mg/mL in PBS, pH 7.2, and incubated at 45°C for 14 days. Photo-stress samples were formulated at 2.5 mg/mL in PBS, pH 7.2, and illuminated under 3,000 lux cool white light (CWL) for 7 days. Control samples were formulated at 1 mg/mL in PBS, pH 7.2, and incubated at 4°C. Changes in monomer, aggregate, and fragment content were then analyzed by HP-SEC compared to the control sample.
Peptide mapping analysis () of these photo-stressed samples showed that all antibodies had a similar level of Fc oxidation increase at Met-428 (site important to FcRn binding,Citation36 EU numbering scheme) and that the level was low after photo-stress under CWL for 7 days (<3.5% Met-428 oxidation), indicating that neither the TM-YTE modifications nor the differences in the Fv region significantly affect Fc oxidation in these antibodies. The oxidation level in the different Fv regions varied in the antibody panel, with antibody 6 exhibiting the highest total oxidation increase in the Fv (5.4%), followed by antibody 4 (2.0%). Overall, the TM-YTE platform did not lead to high levels of Fc oxidation, aggregation, or fragmentation after stress.
Table 3. Post-translational modification analysis of photo-stressed and heat-stressed TM-YTE SARS-COV-2 antibodies using peptide mapping by LC-MS. The antibody panel was heat stressed (45°C, 1 mg/ml, 14 days) or photo stressed (CWL, 2.5 mg/ml, 7 days) and then analyzed by reducing tryptic peptide mapping by LC-MS to monitor critical amino acid sites, as well as PTM increase in the Fc and Fv/CDR region.
Heat-stressed antibody samples retain binding to SARS-CoV-2 receptor-binding domain
As part of the accelerated stability study, we wanted to determine if there was any general impact of PTMs on antibody binding caused by the TM-YTE platform. Peptide mapping analysis () showed that compared to photo-stress, heat stress induced higher levels of modifications in the CDR regions. Therefore, the heat-stress samples were deemed the more degraded material to probe for loss of binding.
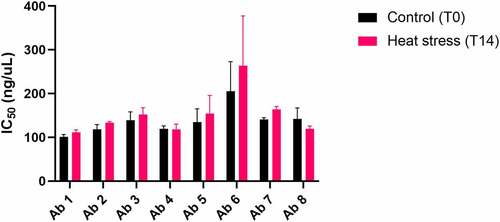
To assess the impact of the accelerated stability heat-stress on the functionality of the antibodies, the heat-stressed samples were tested for loss of binding to SARS-CoV2 receptor-binding domain (RBD) using a sensitive DELFIA. As shown in , no significant difference in IC50 was seen between non-stressed and stressed samples for any of the antibodies under the tested conditions (mAb 6 showed high variability in this assay and differences did not reach significance).
Figure 3. Antibody RBD binding relative potency after 14-day accelerated stability heat stress. Antibody binding to SARS-COV-2 RBD protein before (T0) and after (T14) heat stress at 45°C was compared using a DELFIA. Binding curves were generated for each antibody and used to calculate IC50 values for T0 and T14, which are shown. The average T0 and T14 IC50 values (ng/mL) from both experiments, respectively, are as follows: antibody 1 = (101.1, 111,5); antibody 2 = (118.6, 133.5); antibody 3 = (139.3, 152.3); antibody 4 = (119.4, 118.2); antibody 5 = (134.7, 154.2); antibody 6 = (205.6,263.7); antibody 7 = (140.8, 163.8); antibody 8 = (142.4,119.5). The experiment was run twice. Bars represent standard deviations. T0 and T14 values from both experiments were averaged and compared using a Student’s t-test. None of the antibodies showed a significant difference between T0 and T14. A bar graph plotting the binding potencies of Ab 1–8 before and after heat stress for 14 days. No significant change in potency is observed after heat stress.
TM-YTE antibodies are compatible with HCPF
It was previously reported that an antibody with a TM-YTE Fc showed strong attractive forces as measured by DLS and a lower solubility as measured in a PEG precipitation assay compared with the same antibody with a WT or YTE Fc.Citation32 To maximize its utility, any antibody Fc platform should be compatible with HCPF. To assess RSA, which can lead to solubility and viscosity issues, the TM-YTE SARS-CoV-2 antibody panel was tested by AC-SINS and DLS (). None of the antibodies exhibited a large red shift in the AC-SINS assay, indicating that no RSA was detected. However, measurement of the interaction parameter (kD) by DLS, another predictor for propensity to self-associate, flagged antibodies 1 and 6 with kD values ≤-9 ml/g, a level shown to correlate with strong attractive forces. The antibodies were then concentrated to >125 mg/ml (if possible) and their solubility and viscosity assayed. In this assay antibody 6 could not be concentrated beyond 75 mg/ml and antibody 1 could not be concentrated beyond 110 mg/ml, with antibody 1 showing high viscosity at 100 mg/ml. There was good agreement between the solubility assessment, measured viscosity, and DLS results for flagged antibodies, in contrast to the AC-SINS results, which did not predict RSA for any antibody.
Table 4. Solution property assays of the TM-YTE antibody panel. The TM-YTE SARS-CoV-2 antibody panel was tested for RSA, solubility, and viscosity by AC-SINS, DLS, and a solubility/viscosity assay. NIP228 is a negative control antibody, and IL330396 is a positive control antibody.
TM-YTE antibodies do not exhibit nonspecific binding or reduced FcRn binding
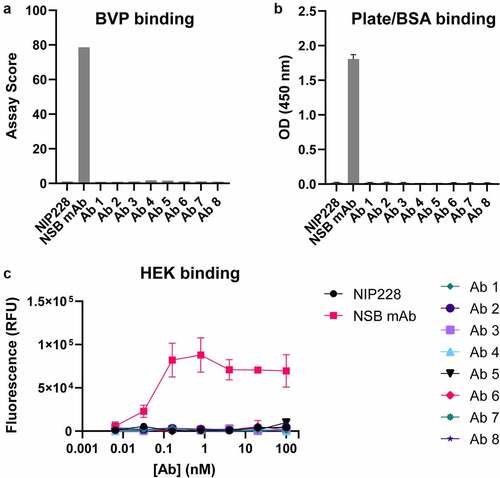
Antibody in-vivo half-life can be affected by NSB and/or by Fc modifications that affect FcRn binding. To evaluate whether the TM-YTE format affects NSB, the antibodies were tested in a baculovirus particle (BVP) ELISA ()) assay and a HEK293 cell binding assay ()). As shown in ), the antibodies do not display any binding to BVPs or binding to bovine serum albumin (BSA) that was immobilized on plates by ELISA. The antibodies also do not show any binding to HEK293 cells ()). Therefore, in both assays, the TM-YTE SARS-CoV-2 antibody panel does not display any NSB properties.
Figure 4. SARS-CoV-2 TM-YTE antibodies do not show Nonspecific Binding (NSB) in BVP ELISA and HEK binding assay. (a+b) The TM-YTE antibody panel was tested for NSB in a BVP ELISA: (a) BVP assay score and (b) BSA or plate binding of the TM-YTE antibody panel. (c) HEK binding assay. NIP228 is a negative control antibody that exhibits low NSB in these assays, and NSB mAb is a positive control antibody that exhibits high NSB in these assays. Assays were repeated three times, representative graphs are shown. Bars represent standard deviations. (a) A bar graph plotting the BVP binding assay score of Ab 1–8 compared to positive control (“NSB mAb”) and negative control (NIP228) antibodies. Assay score for Ab 1–8 is similar to negative control. (b) A bar graph plotting the plate/BSA binding OD (450 nm) of Ab 1–8 compared to positive (“NSB mAb”) and negative control (NIP228) antibodies. The OD of Ab 1–8 is similar to negative control. (c) A line graph plotting the HEK binding assay fluorescent signal versus antibody concentration for Ab 1–8 compared to positive (“NSB mAb”) and negative control (NIP228) antibodies. The fluorescent signal of Ab 1–8 is similar to negative control.
Short in-vivo half-life can also be caused by weak binding or loss of binding to FcRn if residues critical to FcRn binding are directly mutated or undergo increased PTMs. We therefore tested the FcRn binding affinity of the TM-YTE antibody panel using a FcRn column. Antibody is bound to the column at low pH (5.5) and eluted at neutral pH (8.8). A later elution time (long retention time (RT)) indicates stronger FcRn binding. We first assessed the effects of the TM and YTE modifications, separately and in combination, on FcRn binding. ) shows the FcRn affinity column RT profile of antibody 4 with different Fc formats. Comparison of WT and YTE antibody 4 shows that, as expected, adding the YTE modification increases binding to FcRn, as seen by the increased RT. Adding the TM modification onto the YTE Fc did not cause a shift in the FcRn binding profile, while adding the TM modification to the WT Fc caused a very small shift toward a shorter retention time (from 16.3 min to 15.9 min), indicating that the TM modification has little or no effect on FcRn binding. Next, we tested whether the TM-YTE antibodies show any susceptibility to PTMs (deamidation, isomerization, or oxidation) that affect FcRn binding. ) shows the normalized, relative RT of un-stressed, heat-stressed, and photo-stressed antibody samples. First, all tested TM-YTE antibodies retained full binding to the FcRn column after photo and heat stress, showing that inserting the TM-YTE modifications into the Fc does not cause the TM-YTE antibodies to be susceptible to loss of FcRn binding through PTMs. Second, all the tested mAbs showed a relative RT below 1 (0.66–0.81 min), indicating that the entire panel had weaker binding to FcRn than the control antibody NIP228-YTE. However, considering the results from ) indicating that antibody 4 in the YTE and TM-YTE formats show near identical FcRn binding, it would seem that this difference stems from the exceptionally high FcRn binding of our control NIP228-YTE antibody. Strengthening this point, antibodies 5 and 8 showed a longer RT compared to the rest of the panel, indicating stronger FcRn binding through Fv contributions.
Figure 5. FcRn affinity chromatography of the TM-YTE antibody panel. (a) As an example of how TM-YTE modifications affect FcRn binding, the elution profile of antibody 4 in the different Fc formats from an FcRn affinity column is shown. A later elution time indicates stronger FcRn binding. (b) FcRn binding of un-stressed, photo-stressed or heat-stressed antibodies was analyzed using FcRn affinity column. The retention time on the column was normalized between runs as described in the methods section to a well behaved YTE control antibody. A relative retention time of 1 would indicate equal FcRn binding to the control YTE antibody. (a) SEC chromatogram plotting absorbance at 280 nm versus time for Ab4 in WT, TM, YTE, and TM-YTE formats. TM and WT formats elute earlier compared to YTE and TM-YTE formats. (b) A bar graph plotting the relative retention time of Ab 1–8 after no stress, 7 day photo stress, or 14 day heat stress. No significant difference is observed between different conditions.
YTE and TM-YTE antibodies exhibit similar in-vivo PK profiles
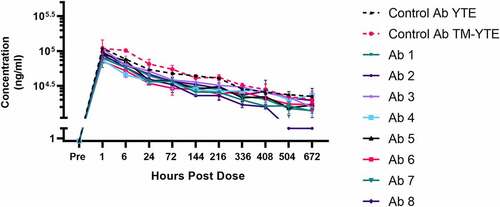
Following intravenous (IV) injection in human FcRn transgenic Tg32 mice, serum antibody concentrations declined in a multi-phasic fashion with a terminal half-life of 2–3 weeks for seven of eight TM-YTE antibodies tested ( and Supplementary Table 1). Seven of eight of the TM-YTE antibodies displayed similar PK profiles, with only antibody 2 showing accelerated clearance. Additionally, the PK of the YTE and TM-YTE versions of the well-characterized control antibody were similar, indicating that adding the TM modification does not significantly alter antibody PK from their respective counterparts that only have the YTE Fc modification.
Figure 6. Serum concentration curves of the TM-YTE antibodies in human FcRn transgenic mice. Mean concentrations of the TM-YTE antibody panel and a well characterized control antibody in human FcRn transgenic Tg32 mouse serum following a single IV injection of 5 mg/kg are shown. Bars represent standard deviations. Pharmacokinetic parameters can be found in supplementary Table 1. A line graph plotting the mean serum concentration vs hours post dose following a single IV injection of 5 mg/kg antibody into human FcRn transgenic Tg32 mice. Test Ab1-8 are compared to control mAbs in YTE and TM-YTE formats. Beginning at 504 hr post dose, Ab 2 begins to show significantly reduced serum concentration compared to the controls and other test Abs.
Discussion
Dozens of potential Fc modifications have been described in the literature,Citation37,Citation38 and over 160 antibodies with FcγR-targeting Fc modifications have entered clinical trials or have been approved.Citation3,Citation39 Many of these Fc modifications have not been extensively characterized for developability, making it difficult to know what downstream impact they may have when incorporated into therapeutics. TM, YTE, and TM-YTE modifications are among the most extensively characterized Fc modificationsCitation6,Citation11,Citation14,Citation32,Citation40,Citation41 and the most clinically advanced. Some representative examples are durvalumabCitation42 and anifrolumabCitation43 (TM only, both approved), nirsevimab (YTE only, recommended by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) for marketing authorization),Citation44 and tixagevimab and cilgavimab (TM-YTE, EUA in the US and authorized in the EU).Citation27,Citation29 Removal of the Fc CH2 glycosylations through either deglycosylation or aglycosylated protein production is also a well-characterized Fc modification and an alternative strategy to disrupt IgG effector functions and complement C1q binding.Citation45 Similarly, to the TM-YTE modifications, CH2 domains lacking the glycosylation at N297 have been shown to have lower thermostability by 6–8°C.Citation46,Citation47 This lower thermostability has been shown to lead to a higher propensity for aggregation under heat stress and greater susceptibility to papain digestion.Citation47 However, an aglycosylated anti-IL6 IgG1 produced in yeast (clazakizumab) exhibited an extended serum half-life of 30 daysCitation48 and was successfully manufactured for use in multiple clinical trials.Citation49,Citation50 A second aglycosylated anti-PD-L1 antibody, atezolizumab, exhibits a normal serum half-life of 3 weeks and is now approved for multiple indications.Citation51–55 It is important to also note that utilizing an Fc modification that does not affect thermostability is not a guarantee that the developability profile will remain unaffected. For example, when the motavizumab Fab was produced on various Fc backbones containing different half-life extending modifications, either the YTE, LS (M428L/N434S),Citation56 or one of several other half-life extending modifications, and tested for thermal stability and propensity for aggregation, the LS-carrying variant, despite having thermostability comparable to WT, showed aggregation rates similar to the other less thermostable Fc variants.Citation57 This is unsurprising because aggregation does not necessarily require protein unfolding and Fc modifications may lead to aggregation and other manufacturing issues in ways unrelated to thermostability. In addition, as seen with our panel of TM-YTE antibodies, the different Fv regions can also have significant effects on the overall molecule characteristics, so every Fv and backbone pairing will need to be critically analyzed. In summary, while it is prudent to aim for similar or improved thermostability as the WT Fc backbone, when designing or selecting new Fc modifications, useful Fc modifications should be fully characterized for developability and should not be overlooked based solely on a slightly lower thermostability.
Determining which biophysical characteristics make an ideal antibody therapeutic has been the focus of intense effort in the antibody engineering field, with several works analyzing which characteristics and assays best differentiate antibodies approved for therapeutic use from those at earlier stages.Citation16,Citation18 Similarly, efforts are being made to determine which early developability assays accurately predict downstream CMC issues, in order to ensure that fully developable clones are selected as lead candidates (and that developable clones are also not accidentally discarded).Citation22,Citation58,Citation59 The AC-SINS assay can be used to detect RSA of antibodies, which at high concentrations may lead to solubility and viscosity issues.Citation59 This assay requires very small amounts of material, making it an attractive method for early-stage screening of large numbers of antibodies. To evaluate how well the AC-SINS assay predicts solubility and viscosity issues, we also tested the self-association of our panel of antibodies by DLS, as well as their actual solubility and viscosity at high concentrations (>125 mg/ml). In AC-SINS, none of the antibodies showed a red-shift that is indicative of RSA. However, during the concentration of these antibodies in clinically relevant formulations (histidine-based buffer at pH 6.0), two antibodies exhibited undesirable solubility attributes. Analyzing the same antibodies by DLS in 20 mM histidine/histidine-HCl (pH 6.0) flagged negative kD values of ≤-9 ml/g, indicating attractive protein–protein interactions that are in good agreement with the observed poor solubility. While the DLS and viscosity assays were run in histidine-based buffers and the AC-SINS assay was run in PBS buffer, the lack of buffering should only make the AC-SINS assay more sensitive to RSA detection. Recently, a framework for screening viscous and opalescent solution behavior has been described utilizing kD in histidine-based buffers as a singularly predictive determinant with a high rate of success, in agreement with these results.Citation60 That study, however, did not include the AC-SINS assay for correlation. Although the high-throughput and low material demand aspects of AC-SINS make it an attractive early developability screening option compared to DLS, clear gaps have been identified in its ability to predict RSA at high concentrations. Combining AC-SINS, DLS, and viscosity assays is therefore the recommended approach when screening the top antibody candidates for high concentration solution properties.
IgG molecules are recycled back into the serum through their interaction with FcRn. PTMs in residues that affect FcRn binding generated either during storage or in the serum, can affect antibody half-life.Citation25 The most common PTM that affects FcRn binding in WT IgG molecules is oxidation at residues Met-252 and Met-428 (EU numbering), with Met-252 oxidation exhibiting a greater impact on FcRn binding than Met-428 oxidation.Citation36 The YTE modification (M252Y/S254T/T256E), introduced to extend serum half-life of IgGs, mutates Met-252 to a tyrosine, therefore leaving Met-428 as the only susceptible oxidation site in the Fc region that could affect FcRn binding. As shown by peptide mapping analysis (), the Met-428 oxidation rate across the entire TM-YTE antibody panel was similarly low after 7 days of CWL photo-stress. This demonstrates that Fc oxidation in the TM-YTE platform is generally low and not affected by the Fv region, and under normal manufacturing and storage conditions, is not likely to reach a level sufficient to affect PK. The extended serum half-life of the TM-YTE antibody platform recently also received clinical validation from an analysis of initial time points from the Phase 1 trial of Evusheld (AZD7442).Citation61 The analysis showed that the TM-YTE mAbs comprising Evusheld (AZD7442; tixagevimab and cilgavimab) had a serum half-life of approximately 90 days, similar to that seen for the anti-RSV motavizumab that contains only the YTE modification.Citation40
Peptide mapping is an essential tool to assess the degradation pathways of an antibody at the primary sequence level. In addition to de-risking the impact of the TM-YTE mutation on the critical Fc Met-428 oxidation levels, peptide mapping can be used to assess general PTM levels. To evaluate if any sequence liability is present in the CDR, antibodies are stressed in a pH 7.2 PBS buffer at 45°C for up to 14 days. A 7-day timepoint was also taken and analyzed to allow us to plot the modification rate (time 0, 7, and 14 days). With these data, the potential degradation pathways during long-term storage and whether they pose a challenge to liquid formulation stability can be predicted. However, the results for this analysis need to be evaluated carefully and are best combined with an evaluation of the impact on antibody potency. Even when a modification in the CDR is identified, it does not necessarily lead to the candidate being eliminated or requiring sequence optimization if the modification has no effect on antibody potency. Even if the modification is found to affect antibody potency, it may be amenable to a control strategy. For example, most Asn sites show an accelerated deamidation rate at alkaline pH (i.e., in PBS buffer at pH 7.2), but this is suppressed at acidic pH. Therefore, even if an Asn site was found to have a moderate deamidation rate in PBS heat-stressed samples, it is still likely to be controlled by a pH 6.0 formulation buffer. Similarly, Asp isomerization rate is known to be accelerated by high temperature. If an Asp site was found to isomerize in PBS heat-stressed samples, it will also likely increase in pH 6.0 formulation buffer.Citation62,Citation63 However, as monoclonal antibody therapeutics are stored either frozen or at 2–8°C refrigerated temperature, with limited time out of refrigeration, isomerization can generally be controlled at levels that will not affect potency during storage. In summary, understanding primary sequence modifications and their impact by combining peptide mapping with potency assays provides valuable insights during the early developability assessment, and allows an early assessment of the criticality of a modification. This early read-out of potential critical quality attributes is especially valuable to initiate an early method development and control strategy planning.
In addition to FcRn binding, the PK of an antibody can be affected by multiple parameters, some of which are difficult to fully predict or de-risk. Target-mediated clearance, Fab isoelectric point (as well as the balance of charge within the CDRs), glycosylation state, RSA, and NSB have all been shown to potentially affect the clearance of antibodies.Citation22,Citation64–66 In vitro assays, such as BVP ELISA, HEK cell binding or DNA and insulin-binding assays, can de-risk antibodies for NSB, and AC-SINS and DLS examine RSA. It has been suggested that in vitro assays screening for NSB, RSA, and FcRn binding should be used as a triaging step to select favorable clones, which are then further validated in Tg32 FcRn humanized mice.Citation64 This approach would seem to be validated here, as our antibody panel did not flag in any of the NSB assays or in AC-SINS. Even so, following IV injection in human FcRn transgenic Tg32 mice, one of the eight antibodies exhibited fast clearance (antibody 2). This indicates that the initial in vitro screening step is effective, but may still miss some antibodies that exhibit fast clearance in vivo. Interestingly, both antibodies that displayed strong attractive forces in DLS (antibodies 1 and 6) did not show accelerated clearance in vivo.
The SARS-CoV-2 TM-YTE neutralizing antibody panel is our first in-depth look at the developability profile of a diverse panel of TM-YTE antibodies. Introducing both the half-life extending YTE and the effector function reducing TM modification to the same IgG1 Fc has previously been reported to lower the thermal stability of the Fc to 58°C.Citation32 It was unclear, however, if the lower thermal stability of the TM-YTE Fc would translate into a less stable or developable molecule, in particular, due to changes in conformational stability and aggregation propensity. In the panel of eight antibodies tested here, none of the antibodies showed a significant increase in aggregation propensity, with only three antibodies showing a small increase in fragmentation under heat stress. Importantly, for one of these and for the control antibody NIP228, the levels of aggregation/fragmentation observed between WT Fc and TM-YTE Fc were almost identical. Our second greatest concern was that instabilities in the Fc might lead to NSB, but none of the antibodies were flagged in the BVP ELISA or the HEK cell binding assay, two assays that detect NSB. The solubility assay also did not show any problems with concentrating six of eight of these antibodies to high concentrations (>125 mg/ml) nor did the rest of the assays point to any problem related to the Fc. Overall, our scrutiny of the diverse panel of TM-YTE antibodies did not reveal any developability issues precluding clinical use of the TM-YTE platform. This conclusion is further strengthened by the recent marketing authorizations and emergency use authorization granted to Evusheld (AZD7442), which is a combination of two TM-YTE antibodies.Citation26–29
Materials and methods
Antibody and protein production
Antibody VH and VL DNA fragments were cloned into a proprietary CMV-driven mammalian expression plasmid containing human IgG1 Fc with the YTE serum half-life extending and TM effector function-ablating modifications. The plasmids were sequence verified by Sanger sequencing. Antibodies were expressed transiently in HEK293F cells using 293 Fectin (Gibco #12347019) according to the manufacturer’s instructions and grown in Freestyle medium (Gibco #12338018) for 6 days. At 6 days, the media was clarified by centrifugation, filtered, and purified using MabSelect SuRe resin (Cytiva #11-0034-94). Antibody purity was determined using HP-SEC as described below, and the correct mass of antibodies was verified by mass spectrometry. SARS-CoV-2 RBD (residues 334–526) was cloned with an N-terminal CD33 leader sequence and C-terminal GSSG linker, AviTag, GSSG linker, and 8xHisTag, expressed in FreeStyle 293 cells (Thermo Fisher) and isolated by affinity chromatography using a HisTrap column (GE Healthcare), followed by size-exclusion chromatography with a Superdex 200 column (GE Healthcare). Purified proteins were analyzed using SDS-PAGE to ensure purity and appropriate molecular weights. Endotoxin levels were measured using Charles River Endosafe® cartridge (Charles River) and found to be below 1EU/mg.
NIP228 and “NSB mAb” control antibodies
NIP228 is a monoclonal antibody against 4-hydroxy-3-iodo-5-nitrophenylacetic acidCitation67 that historically has been shown not to exhibit aggregation/fragmentation propensities under similar heat/photo stress conditions and is used as a negative control in select developability assays. The “NSB mAb” is a monoclonal antibody against an undisclosed target not related to SARS-CoV-2 that shows high BVP scores and plate binding in the Baculovirus ELISA and high HEK293 cell binding in the HEK binding assay and is used as a positive control antibody in these assays. Unless otherwise stated, both control antibodies are on a WT human IgG1 Fc. Both control antibodies were discovered and produced in-house.
HP-SEC
Antibody samples were analyzed using HP-SEC to determine levels of aggregate, monomer, and fragment. Samples (100 μg in PBS buffer) were injected on an Agilent 1200 series high-performance liquid chromatography (HPLC) instrument and separated using a TSKgel G3000SWxl size-exclusion column (Tosoh Bioscience #08541). The mobile phase was 100 mM sodium phosphate (pH 6.8), and sample flow rate was 1 mL/min. Ultraviolet (UV) detection was performed at 280 nm.
Baculovirus ELISA
BVP ELISA was performed essentially as described elsewhere,Citation22 with some modifications. Briefly, a 1% BV suspension in 50 mM sodium carbonate buffer (pH 9.6) was used to coat half of 96-well ELISA plates (Nunc Maxisorp) overnight at 4°C, while the second half of the ELISA plates was left uncoated to test the antibodies for plate binding. All following steps were performed at room temperature. The next day the wells were washed with Dulbecco’s PBS (DPBS) and then incubated with blocking buffer (Dulbecco’s PBS with 0.5% BSA) for 1 h, followed by three washes with DPBS. Next, test antibodies at 100 nM and 10 nM in blocking buffer were added to both the BVP coated and uncoated wells and incubated for 1 h, followed by three washes with DPBS. Next, goat anti-human IgG-horseradish peroxidase (HRP) secondary antibodies (1:5000 dilution, Sigma-Aldrich #A0170) in blocking buffer were added to the wells and incubated for 1 h, followed by three washes with DPBS. Finally, 3,3’,5,5’-tetramethylbenzidine substrate (SeraCare #5120-0075) was added to each well and incubated for 2 minutes. The reactions were stopped by adding an equal volume of 0.2 M sulfuric acid to each well. The absorbance was read at 450 nm. BVP score and plate binding were determined by normalizing absorbance to control wells with no test antibody.
HEK binding assay
Nonspecific HEK cell binding was measured using a Mirrorball Fluorescence Cytometer (SPT Labtech). First, 10 µL of Alexa Fluor 647 goat anti-human IgG (H + L) antibody (Invitrogen #A-21445) diluted to 16 nM in Mirrorball buffer (Hanks’ Balanced Salt solution with 0.5% BSA) was added to wells of a 384-well, clear bottom plate. Next, 10 µL of test antibody serially diluted in Mirrorball buffer was added to the wells. Finally, 20 µL of HEK293f cells diluted to 250,000 cells/mL in Mirrorball buffer was added to the wells. The plate was incubated at room temperature for 2 h, and the fluorescence of each well was measured using the Mirrorball Fluorescence Cytometer.
AC-SINS
AC-SINS was performed essentially as described elsewhere,Citation68 with some modifications. Briefly, both whole goat IgG (Jackson ImmunoResearch #005-000-003) (non-capture) and polyclonal goat anti-human IgG Fc (Jackson ImmunoResearch #109-005-098) (capture) antibodies were dialyzed into 20 mM potassium acetate (pH 4.3) buffer, and then conjugated to 20 nm gold nanoparticles (Innova Biosciences #3201-0100) at a 3:2 ratio of capture:non-capture antibodies. Antibodies were incubated with gold nanoparticles at a 9:1 ratio for 1 h at room temperature, and then blocked by the addition of 0.1 μM poly-(ethylene glycol) methyl ether thiol (2000 MW, Sigma-Aldrich #729140) for 1 h. The coated and blocked nanoparticles were concentrated 12.5-fold by centrifugation and stored at 4°C. To assess self-association, 5 μL of nanoparticles were mixed with 45 μL of purified antibody at 50 ug/mL in PBS, pH 7.2 or HSA buffer [20 mM histidine, 120 mM sucrose, 80 mM arginine, pH 6] in a 384-well plate. Nanoparticles were mixed with buffer only (no antibody) as a control. Absorbance was measured on a SPECTROstar Nano UV/vis plate reader from 490 to 700 nm. The wavelength of peak absorbance was calculated in the MARS data analysis software and used to determine the wavelength shift compared to the nanoparticle-only control.
Accelerated stability heat stress study
For accelerated stability testing, samples were diluted to 1 mg/mL in PBS (pH 7.2) and incubated for 2 weeks at either 4°C or 45°C. Samples were then analyzed by HP-SEC, peptide mapping, and DELFIA binding assay as described in those sections. The monomer, aggregate, and fragment percentages for each sample were calculated based on curve integration using the HPLC ChemStation software (Agilent). The change in monomer, aggregate, and fragment content was calculated from the difference between each sample incubated at 45°C versus 4°C.
Photostability assay
For photostability testing, antibodies were formulated at 2.5 mg/mL in PBS (pH 7.2), filled into 1cc Schott glass vials, stoppered/sealed, and placed into an ICH-compliant photostability chamber (Caron Model 6545-2). Samples were exposed to cool white light at 3000 lux over the course of 1 week, for a total exposure of approximately 500,000 lux hours. Samples were analyzed by HP-SEC and peptide mapping as described in those sections.
DSC
Thermal melting transitions were determined by using Microcal VP differential scanning calorimetry system (Malvern, PA). Monoclonal antibody solutions were diluted to 1 mg/ml in the final buffer and change in the heat capacity (Cp) was measured as the samples were heated from 20° to 95°C using a temperature ramp of 90°C/h. Normalized heat capacity data were obtained after subtracting to the buffer blank and normalizing to the concentration of the monoclonal antibody. Data were analyzed using Microcal LLC origin software to calculate the thermal melting transitions associated with the unfolding of the distinct domains.
Solubility assay
Each antibody was dialyzed into the intended histidine-based platform formulations at pH 6.0. Samples were then concentrated in Amicon Ultra centrifugal filters (Millipore Sigma UCF9030) at 4,000 g. Each sample was mixed every 1–2 minutes during centrifugation and assessed visually for any signs of sample precipitation. Protein concentration was also routinely measured by absorbance at 280 nm using UV–vis spectrophotometer (Trinean DropSense 96, Unchained Labs Pleasanton, CA) with respective antibody’s extinction coefficients and corrected for density when necessary. Any early precipitation events were noted and samples centrifuged further with diligent monitoring of protein concentration; precipitating samples showed minimal further increase in protein concentration upon continued centrifugation.
Viscosity
Concentrated antibody solutions were diluted in the intended histidine-based platform formulations to 100 mg/mL and filtered using 0.22 um filters. Each sample was subjected to rotational shear stresses of 1000 s−1 at 23°C on an MCR301 rheometer using the CP20-1 cone and plate system (Anton Parr, Part 3274). Five measurements were taken each minute over the course of data collection and averaged to provide viscosity values
DLS
The antibody panel was reformulated into histidine/histidine-HCl buffer, pH 6.0, to match buffer composition/pH common to potential platform formulations. Z-average apparent diffusion coefficient measurements were carried out using Dynapro plate reader (Wyatt technology, Santa Barbara, CA) equipped with a laser source at 833 nm wavelength. The scattered light was collected in a back scattering mode at an angle 153°. For each antibody, 40 ul at respective concentrations 2, 4, 6, 8, and 10 mg/ml was distributed in triplicate into a low volume 384-well plate (Corning, Tewksbury, MA), covered with a paraffin film, and spun at 3000 RPM for 1 minute to remove any air bubbles. The sample chamber was equilibrated for 1 h prior to measurements, and data were collected for 10 acquisitions of 10 seconds and averaged for each well. The Z-average translational diffusion coefficient was determined from cumulant analysis of the autocorrelation function and modeled as
D= D0(1+ kDC) to obtain “kD” diffusion virial coefficient (interaction parameter).
Peptide mapping
Reduced tryptic peptide mapping was performed by first denaturing and reducing 100 µg of antibody sample in a guanidine hydrochloride buffer with dithiothreitol (20291, Thermo Scientific) at 37°C for 30 minutes. The sample was then alkylated with iodoacetamide (786–078, G-Biosciences) for 30 minutes in the dark at room temperature. Subsequently, the sample mixture was dialyzed into 6 M Urea, and then diluted with Tris pH 7.5 buffer to allow for trypsin digestion. Trypsin (V5280, Promega) was added at 1:20 protease:protein ratio and incubated at 37°C for 4 h. The reaction was quenched by adding trifluoroacetic acid (TFA, T6508, Sigma-Aldrich). Digests of the sample were analyzed using a Fusion Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) connected with an AQUITY ultra-performance liquid chromatography (UPLC; Waters). An AQUITY UPLC BEH300 C18 column (1.7 μm, 2.1 × 150 mm; Waters) was used for separation. The column temperature was held at 55°C. Mobile phase A was 0.02% TFA in water, and mobile phase B was 0.02% TFA in acetonitrile. Digested peptides were eluted from the column with a 0–35% linear gradient and the chromatographic profile was monitored by using UV absorbance at 220 nm and MS. MS data were processed by BiopharmaFinder 3.0 (Thermo Fisher Scientific).
DELFIA binding assay
A DELFIA® (PerkinElmer) method was used to measure antibody binding to the RBD of the SARS-CoV-2 spike protein before and after heat stress. Nunc MaxiSorp™ 96-well plates were coated overnight with purified RBD protein (Sino Biological). Assay plates were then washed with 1X TBS-Tween buffer (PerkinElmer) and blocked with BSA (Sigma). Antibody samples were serially diluted and added to the plate for 1 h. Bound antibody was detected with DELFIA® Europium-N1 anti-human IgG (PerkinElmer), and the signal quantified via time-resolved fluorescence on an Envision plate reader (PerkinElmer). IC50 was calculated with a four-parameter logistic (4PL) curve using GraphPad Prism v 8.0. IC50 of the stressed sample was compared to a reference sample to generate a percent relative potency.
FcRn affinity chromatography
Approximately 40 µg/40 µL of antibody was loaded onto a 1 ml huFcRN-coupled Sepharose affinity column using an Agilent HPLC, followed by a 3CV linear gradient from buffer A (20 mM MES, 150 mM NaCl, pH5.5) to 40% buffer B (20 mM tris + 150 mM NaCl, pH8.8) and a 18CV linear gradient from 40% to 100% buffer B. The experiment was performed at a flow rate of 0.5 ml/min at room temperature using the Agilent-DAD to measure the A280 of the elution profile. Retention time was normalized to the relative retention time with the following formula and using the well-behaved control antibody NIP228 in the WT and YTE format: Relative RT = ([sample RT – NIP228 RT]/[NIP228-YTE RT – NIP228 RT]).
PK studies in human FcRn transgenic mice
PK studies were conducted in human FcRn transgenic Tg32 mice. Mice were fed a standard rodent chow diet and maintained on a 12/12 hour light cycle in microisolator cages. Vendor and facility health monitoring programs identified the mice free of all commonly tested rodent pathogens. All animal studies were approved by AstraZeneca’s Institutional Animal Care and Use Committee and were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility. Mice (6/group) were IV injected with 5 mg/kg antibody in PBS. Blood samples were collected at 1 h, 6 h, and 24 h, and on days 3, 6, 9, 14, 17, 21, and 28 post-injection.
PK ELISA
The concentration of antibodies in mouse serum samples was determined using a qualified ELISA assay. The method procedure was a stepwise format in which wash steps follow each incubation step. Microtiter plates were coated with a sheep anti-human IgG antibody (AU003.M, The Binding Site). A standard curve of each antibody was prepared in 10% pooled normal mouse serum. Three levels of quality control (QC) samples were prepared in 100% pooled mouse serum and were stored frozen. Following a blocking step with Casein buffer, standard curve calibrators, QCs, and samples diluted to the method minimum required dilution (MRD) of 1:10 were added to the microtiter plates and incubated for 1 hr with shaking. After a wash, HRP-labeled goat anti-human IgG antibody (A80-219P, Bethyl Laboratories) was added to the plate and incubated for 1 h with shaking. Chromogenic color development is directly related to levels of analyte in the sample. Reaction was stopped with acid, and the plate was read on a spectrophotometer at 450 nm, and data analyzed with SoftMax® Pro (SMP), version 5.4. The standard curve was established using a 4-parameter logistic curve fit model without weighting provided by the software program SMP v5.4. The quantitative range of this assay was 31.25 ng/mL (lower limit of quantitation [LLOQ]) to 1000 ng/mL (upper limit of quantitation [ULOQ]).
PK analysis
Noncompartmental PK analysis was performed with Phoenix WinNonlin 6.3 (Pharsight, Sunnyvale, CA, USA) on the composite serum antibody concentration data following a single IV administration of 5 mg/kg dose. Nominal doses and sampling times were used. Maximum concentration after dosing (Cmax) was observed. The area under the concentration–time curve from time 0 to infinity (AUC0-∞) was calculated with the log-linear trapezoidal method. Systemic clearance (CL) after dosing was estimated using dose/AUC0-∞. The terminal elimination half-life (t½) was calculated using ln(2)λz, where λz is the first-order terminal rate constant estimated by log-linear regression of the terminal log-linear decay phase. The steady-state volume of distribution (Vss) was estimated using Vss = MRTinf . CL, where MRTinf is the mean residence time determined by area under the first moment curve and AUC0-∞.
List of Abbreviations
AC-SINS Affinity capture self-interaction nanoparticle spectroscopy
ADE Antibody-dependent enhancement
BVP Baculo-virus particles
CMC Chemistry, manufacturing, and control
CHMP Committee for Medicinal Products for Human Use
COVID-19 Corona virus disease 2019
CWL Cool white light
DLS Dynamic light scattering
DSC Differential scanning calorimetry
ELISA Enzyme-linked immunosorbent assay
EMA European Medicines Agency
EUA Emergency use authorization
EU European Union
Fab Fragment antigen binding
FcRn Neonatal Fc receptor
Fc Fragment crystallizable region
FcγR Fragment crystallizable gamma receptor
FDA Federal drug administration
Fv Fragment variable region
HCPF High concentration protein formulation
HEK Human embryonic kidney
HP-SEC High-performance size-exclusion chromatography
IV Intravenous
mAb Monoclonal antibody
MOA Mechanism of action
NSB Non-Specific Binding
PBS Phosphate buffered saline
PK Pharmacokinetics
PTM Post-translational modifications
RT Retention time
RSA Reversible Self Association
SARS-CoV-2 Severe acute respiratory syndrome coronavirus 2
SD Standard deviation
UK United Kingdom
WT Wild type
Supplemental Material
Download Zip (320.3 KB)Acknowledgments
The authors would like to thank all researchers involved in this study. We would like to acknowledge Linda Xu for helping with vector design and antibody generation, Sathish Hasige for insightful guidance and review, Rebecca Holcomb for developing physicochemical assays, Meagan Prophet for evaluating the antibodies for co-formulation feasibility and Shihua Lin for developing the RBD binding potency assay.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2152526
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A. 2011;108(31):12669–15. doi:10.1073/pnas.1108455108.
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103(11):4005–10. doi:10.1073/pnas.0508123103.
- Wilkinson I, Hale G. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international nonproprietary names. MAbs. 2022;14(1):2123299. doi:10.1080/19420862.2022.2123299.
- Chen X, Song X, Li K, Zhang T. FcgammaR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front Immunol. 2019;10:292. doi:10.3389/fimmu.2019.00292.
- Eroshenko N, Gill T, Keaveney MK, Church GM, Trevejo JM, Rajaniemi H. Implications of antibody-dependent enhancement of infection for SARS-CoV-2 countermeasures. Nat Biotechnol. 2020;38(7):789–91. doi:10.1038/s41587-020-0577-1.
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64(6):700–04. doi:10.1107/S0907444908007877.
- Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJ, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. 2017;292(9):3900–08. doi:10.1074/jbc.M116.767749.
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–601.
- Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, Pedley B, Adair JR. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol. 1993;30(1):105–08. doi:10.1016/0161-5890(93)90432-B.
- Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall’Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015;290(7):4282–90. doi:10.1074/jbc.M114.603712.
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281(33):23514–24. doi:10.1074/jbc.M604292200.
- Lee CH, Kang TH, Godon O, Watanabe M, Delidakis G, Gillis CM, Sterlin D, Hardy D, Cogne M, Macdonald LE, et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat Commun. 2019;10(1):5031. doi:10.1038/s41467-019-13108-2.
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs. 2011;3(5):422–30. doi:10.4161/mabs.3.5.16983.
- Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. MAbs. 2015;7(1):84–95. doi:10.4161/19420862.2014.985494.
- Thakkar SV, Sahni N, Joshi SB, Kerwin BA, He F, Volkin DB, Middaugh CR. Understanding the relevance of local conformational stability and dynamics to the aggregation propensity of an IgG1 and IgG2 monoclonal antibodies. Protein Sci. 2013;22(10):1295–305. doi:10.1002/pro.2316.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. doi:10.1080/19420862.2018.1553476.
- Starr CG, Tessier PM. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotechnol. 2019;60:119–27. doi:10.1016/j.copbio.2019.01.008.
- Yadav S, Liu J, Shire SJ, Kalonia DS. Specific interactions in high concentration antibody solutions resulting in high viscosity. J Pharm Sci. 2010;99(3):1152–68. doi:10.1002/jps.21898.
- Connolly BD, Petry C, Yadav S, Demeule B, Ciaccio N, Moore JM, Shire SJ, Gokarn YR. Weak interactions govern the viscosity of concentrated antibody solutions: high-throughput analysis using the diffusion interaction parameter. Biophys J. 2012;103(1):69–78. doi:10.1016/j.bpj.2012.04.047.
- Datta-Mannan A, Lu J, Witcher DR, Leung D, Tang Y, Wroblewski VJ. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. MAbs. 2015;7(6):1084–93. doi:10.1080/19420862.2015.1075109.
- Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4(6):753–60. doi:10.4161/mabs.22189.
- Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL Jr. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47(18):5088–100. doi:10.1021/bi702238b.
- Mo J, Yan Q, So CK, Soden T, Lewis MJ, Hu P. Understanding the impact of methionine oxidation on the biological functions of IgG1 antibodies using hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2016;88(19):9495–502. doi:10.1021/acs.analchem.6b01958.
- Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48(6–7):860–66. doi:10.1016/j.molimm.2010.12.009.
- U.S. Food and Drug Administration. Coronavirus (COVID-19) Update: FDA authorizes new long-acting monoclonal antibodies for pre-exposure prevention of COVID-19 in certain Individuals. December 8, 2021 press release [accessed September 18, 2022]. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-long-acting-monoclonal-antibodies-pre-exposure.
- Medicines and Healthcare products Regulatory Agency. Evusheld approved to prevent COVID-19 in people whose immune response is poor. 17 March, 2022 press release [accessed September 18, 2022]. https://www.gov.uk/government/news/evusheld-approved-to-prevent-covid-19-in-people-whose-immune-response-is-poor.
- Health Canada. Health Canada authorizes Evusheld for the prevention of COVID-19 in immune compromised adults and children. April. 2022; 14. press release accessed September 18, 2022. https://www.canada.ca/en/health-canada/news/2022/04/health-canada-authorizes-evusheld-for-the-prevention-of-covid-19-in-immune-compromised-adults-and-children.html
- European Medicines Agency. EMA recommends authorisation of COVID-19 medicine Evusheld. March 24, 2022 press release [accessed September 18, 2022]. https://www.ema.europa.eu/en/news/ema-recommends-authorisation-covid-19-medicine-evusheld
- Montgomery H, Hobbs FDR, Padilla F, Arbetter D, Templeton A, Seegobin S, Kim K, Campos JAS, Arends RH, Brodek BH, et al. Efficacy and safety of intramuscular administration of tixagevimab-cilgavimab for early outpatient treatment of COVID-19 (TACKLE): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2022;10(10):985–96. doi:10.1016/S2213-2600(22)00180-1.
- Levin MJ, Ustianowski A, De Wit S, Launay O, Avila M, Templeton A, Yuan Y, Seegobin S, Ellery A, Levinson DJ, et al. Intramuscular AZD7442 (Tixagevimab-Cilgavimab) for Prevention of Covid-19. N Engl J Med. 2022;386(23):2188–200. doi:10.1056/NEJMoa2116620.
- Borrok MJ, Mody N, Lu X, Kuhn ML, Wu H, Dall’Acqua WF, Tsui P. An “Fc-silenced” IgG1 format with extended half-life designed for improved stability. J Pharm Sci. 2017;106(4):1008–17. doi:10.1016/j.xphs.2016.12.023.
- Edgeworth MJ, Phillips JJ, Lowe DC, Kippen AD, Higazi DR, Scrivens JH. Global and local conformation of human IgG antibody variants rationalizes loss of thermodynamic stability. Angew Chem Int Ed Engl. 2015;54(50):15156–59. doi:10.1002/anie.201507223.
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs. 2011;3(3):253–63. doi:10.4161/mabs.3.3.15608.
- Cordoba AJ, Shyong BJ, Breen D, Harris RJ. Non-enzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818(2):115–21. doi:10.1016/j.jchromb.2004.12.033.
- Gao X, Ji JA, Veeravalli K, Wang YJ, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al. Effect of individual Fc methionine oxidation on FcRn binding: met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J Pharm Sci. 2015;104(2):368–77. doi:10.1002/jps.24136.
- Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296. doi:10.3389/fimmu.2019.01296.
- Gunn BM, Lu R, Slein MD, Ilinykh PA, Huang K, Atyeo C, Schendel SL, Kim J, Cain C, Roy V, et al. A Fc engineering approach to define functional humoral correlates of immunity against Ebola virus. Immunity. 2021;54(4):815–28 e5. doi:10.1016/j.immuni.2021.03.009.
- The antibody society. Antibody therapeutics approved or in regulatory review in the EU or US. Webpage updated September 2 2022 [accessed September 18, 2022]. https://www.antibodysociety.org/resources/approved-antibodies/.
- Robbie GJ, Criste R, Dall’acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53. doi:10.1128/AAC.01285-13.
- Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol. 2009;46(8–9):1750–55. doi:10.1016/j.molimm.2009.01.026.
- US Food and Drug Administration. FDA approves durvalumab after chemoradiation for unresectable stage III NSCLC. February. 2018; 16. press release accessed September 18, 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-durvalumab-after-chemoradiation-unresectable-stage-iii-nsclc
- US Food and Drug Administration. Novel Drug Approvals for 2021. May 13 2022 press release [accessed September 18, 2022]. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2021.
- European Medicines Agency. Meeting highlights from the committee for medicinal products for human use (CHMP) 12-15 September 2022. September 16 2022 press release [accessed September 18 2022]. https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-12-15-september-2022.
- Jefferis R, Lund J. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol Lett. 2002;82(1–2):57–65. doi:10.1016/S0165-2478(02)00019-6.
- Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, Lund J, Jefferis R. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol Immunol. 2000;37(12–13):697–706. doi:10.1016/S0161-5890(00)00105-X.
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3(6):568–76. doi:10.4161/mabs.3.6.17922.
- Eskandary F, Durr M, Budde K, Doberer K, Reindl-Schwaighofer R, Waiser J, Wahrmann M, Regele H, Spittler A, Lachmann N, et al. Clazakizumab in late antibody-mediated rejection: study protocol of a randomized controlled pilot trial. Trials. 2019;20(1):37. doi:10.1186/s13063-018-3158-6.
- Mease P, Strand V, Shalamberidze L, Dimic A, Raskina T, Xu LA, Liu Y, Smith J. A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis. 2012;71(7):1183–89. doi:10.1136/annrheumdis-2011-200704.
- Mease PJ, Gottlieb AB, Berman A, Drescher E, Xing J, Wong R, Banerjee S. The efficacy and safety of clazakizumab, an anti-interleukin-6 monoclonal antibody, in a phase IIb study of adults with active psoriatic arthritis. Arthritis Rheumatol. 2016;68(9):2163–73. doi:10.1002/art.39700.
- European Medicines Agency. Tecentriq EPAR. September 29, 2017 press release [accessed September 18, 2022]. https://www.ema.europa.eu/en/medicines/human/EPAR/tecentriq.
- U.S. Food and Drug Administration. FDA approves new, targeted treatment for bladder cancer. May 18, 2016 press release [accessed September 18, 2022]. https://www.fda.gov/news-events/press-announcements/fda-approves-new-targeted-treatment-bladder-cancer.
- US Food and Drug Administration. FDA approves atezolizumab for BRAF V600 unresectable or metastatic melanoma. July 30, 2020 press release [accessed September 18, 2022]. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-braf-v600-unresectable-or-metastatic-melanoma.
- US Food and Drug Administration. FDA approves atezolizumab plus bevacizumab for unresectable hepatocellular carcinoma. May 29, 2020 press release [accessed September 18, 2022]. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-plus-bevacizumab-unresectable-hepatocellular-carcinoma.
- US Food and Drug Administration. FDA approves atezolizumab for first-line treatment of metastatic NSCLC with high PD-L1 expression. May 18, 2020 press release [accessed September 18, 2022]. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-first-line-treatment-metastatic-nsclc-high-pd-l1-expression.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28(2):157–59. doi:10.1038/nbt.1601.
- Booth BJ, Ramakrishnan B, Narayan K, Wollacott AM, Babcock GJ, Shriver Z, Viswanathan K. Extending human IgG half-life using structure-guided design. MAbs. 2018;10(7):1098–110. doi:10.1080/19420862.2018.1490119.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020;12(1):1743053. doi:10.1080/19420862.2020.1743053.
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 2014;6(2):483–92. doi:10.4161/mabs.27431.
- Kingsbury JS, Saini A, Auclair SM, Fu L, Lantz MM, Halloran KT, Calero-Rubio C, Schwenger W, Airiau CY, Zhang J, et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6(32):eabb0372. doi:10.1126/sciadv.abb0372.
- Loo YM, McTamney PM, Arends RH, Abram ME, Aksyuk AA, Diallo S, Flores DJ, Kelly EJ, Ren K, Roque R, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in non-human primates and has an extended half-life in humans. Sci Transl Med. 2022:eabl8124.
- Yan Q, Huang M, Lewis MJ, Hu P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 2018;10(6):901–12. doi:10.1080/19420862.2018.1478646.
- Lu X, Nobrega RP, Lynaugh H, Jain T, Barlow K, Boland T, Sivasubramanian A, Vasquez M, Xu Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs. 2019;11(1):45–57. doi:10.1080/19420862.2018.1548233.
- Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. doi:10.1080/19420862.2017.1417718.
- Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. MAbs. 2015;7(3):483–93. doi:10.1080/19420862.2015.1016696.
- Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2018;9(1):15–32. doi:10.1007/s13238-017-0408-4.
- Webster CI, Hatcher J, Burrell M, Thom G, Thornton P, Gurrell I, Chessell I. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain. 2017;158(4):660–68. doi:10.1097/j.pain.0000000000000810.
- Geng SB, Wu J, Alam ME, Schultz JS, Dickinson CD, Seminer CR, Tessier PM. Facile preparation of stable antibody-gold conjugates and application to affinity-capture self-interaction nanoparticle spectroscopy. Bioconjug Chem. 2016;27(10):2287–300. doi:10.1021/acs.bioconjchem.6b00207.