
ABSTRACT
Single-chain fragment variable (scFv) domains play an important role in antibody-based therapeutic modalities, such as bispecifics, multispecifics and chimeric antigen receptor T cells or natural killer cells. However, scFv domains exhibit lower stability and increased risk of aggregation due to transient dissociation (“breathing”) and inter-molecular reassociation of the two domains (VL and VH). We designed a novel strategy, referred to as stapling, that introduces two disulfide bonds between the scFv linker and the two variable domains to minimize scFv breathing. We named the resulting molecules stapled scFv (spFv). Stapling increased thermal stability (Tm) by an average of 10°C. In multiple scFv/spFv multispecifics, the spFv molecules display significantly improved stability, minimal aggregation and superior product quality. These spFv multispecifics retain binding affinity and functionality. Our stapling design was compatible with all antibody variable regions we evaluated and may be widely applicable to stabilize scFv molecules for designing biotherapeutics with superior biophysical properties.
Introduction
The US Food and Drug Administration first approved a monoclonal antibody (mAb) in 1986, and since then the development of antibody-based therapies has grown exponentially, resulting in the availability of over 100 mAbs that treat a wide range of diseases. Citation1–3 There has also been rapid development in numerous complex molecular formats and classes, including bi- and multi-specific antibodies and chimeric antigen receptor T cells, that explore new mechanisms to specifically meet the needs of patients with the highest probability of success.Citation4–8 A mAb recognizes its target antigen through its variable fragment (Fv), which is composed of interacting variable domains, the light chain (LC) variable region (VL) and heavy chain (HC) variable region (VH). In the late 1980s, Bird et al.Citation9 designed a single-chain Fv (scFv) as a genetic fusion of VL and VH with a flexible linker in either VL-linker-VH (LH, “light-heavy”) or VH-linker-VL (HL, “heavy-light”) orientations. The scFv is the minimal binding unit that recapitulates the antigen-binding specificity of its parental mAb. These scFv molecules have played a pivotal role as building blocks in current biotherapeutics, as well as detection/diagnostic reagents.
The development of scFv molecules is challenging because of their low stability and a tendency to aggregate (reviewed in Ref.Citation10,Citation11). Conversion of an antigen-binding fragment (Fab) of a mAb into an scFv involves removal of the two constant domains (CH1 and CL of the heavy and light chains, respectively). The Fab interface between the heavy and light chains comprises VL/VH and CH1/CL interactions. These two independent sets of interactions provide synergistic stabilizing effects. In addition, the V/C (variable region/constant region) junctions contribute to some stabilization.Citation12 In comparison, an scFv is maintained by VL/VH interface interactions only. The flexible scFv linker weakly tethers the two variable regions together in space but does not contribute significantly to the thermal stability of the scFv.Citation13 Short linkers will lead to scFv dimers and higher-order oligomers, whereas longer linkers lead to higher monomer content. However, the thermal stability of scFvs with varying linker lengths remains nearly identical.Citation13 Generally, loss of the stabilizing interactions from the constant regions often translates to a loss of thermal stability by 10 ºC or more in Tm.Citation14 Lower thermal stability may lead to domain unfolding, which is a well-known mechanism of protein aggregation. Even in the absence of domain unfolding, the apparently weaker VL/VH interactions alone can allow transient separation (“breathing”) and swapping of the domains in different molecules, particularly at high concentrations, leading to scFv-mediated aggregation without protein unfolding.Citation11 As a result of these two mechanisms, scFv biologics are often associated with substantial development hurdles , which affect product qualityCitation15,Citation16 and sometimes efficacy and safety.Citation17 Thus, strategies to improve scFv stability and aggregation are of great interest.
A number of strategies to improve scFv biophysical properties have been attempted.Citation11,Citation18–26 These strategies include introducing disulfide bonds between VL/VH domains,Citation25 improving VL/VH domain stability and/or interface interactions using different experimental methods,Citation19–22 using additional dimerization motifsCitation18 and complementarity-determining region transfer to frameworks known to have higher stability,Citation27 or transfer of key stabilizing framework residues to antibodies of low stability.Citation28 Of these various approaches, the inter-VL/VH chain disulfide bonds, first introduced to stabilize an Fv (dsFv),Citation25,Citation26 have been the most widely applied. However, stabilization of scFv using this strategy has not been consistent.Citation29
We have recently generated multispecific antibodies having a single-Fab arm and single or multi- scFv domains using a set of previously identified ‘knob and hole’ Fc-mutations in the CH3 domain.Citation30,Citation31 Analysis of long-term stability for several bi- and tri-specific antibody samples at high concentrations revealed poor stability and a high ability to aggregate after several weeks when incubated at higher temperatures (data summarized below). These findings indicate the challenges of scFv multispecific development.
Here, we report a novel and widely applicable scFv stapling strategy that significantly improves stability compared with unstapled versions. The “stapled” scFv molecules (spFv) generally have an increase of about 10 ºC in Tm for scFv molecules with both kappa and lambda light chains. In a set of anti-CD3 scFv/spFv and anti-BCMA Fab bispecific molecules, the results show that stapling the anti-CD3 scFv significantly improved the yields and quality of the bispecific monomer, whereas some scFv bispecifics were a mixture of monomer and oligomers. The spFv retained binding affinity to CD3 compared with the corresponding scFv bispecifics, and the scFv and spFv bispecific proteins activated CD4+ and CD8+ T cells equally with similar killing of BCMA+ tumor cells. The spFv bispecifics also displayed minimal aggregation upon heat stress at high concentrations, whereas the corresponding scFv molecules displayed significant aggregation. This was also true for a number of other spFv bi- and tri-specifics proteins. Thus, spFv can lead to equally potent biotherapeutics with significantly improved developability. Further, stapling can also increase the success of scFv conversion, allowing more spFv molecules to be available as molecular building blocks for therapeutic constructs.
Results
Stapling scFv: concept and design
The scFv stapling strategy to minimize aggregation due to scFv instability and “breathing” is shown in . In this strategy, we attempted to engineer in disulfide bonds (SSs) between the two Cys residues placed in the flexible linker and the Cys residues introduced into the VL and VH domains (anchor positions, one each). The scheme of using linker-anchor point disulfide bonds mimics the effects of a staple and is thus termed “stapling” (). We hypothesized that these SS bonds will restrict their transient and reversible “breathing” and inter-molecular swapping but not negatively impact the small movements between the two variable domains, which may be important for antigen binding.Citation32 In addition, stapling will reduce the overall conformational entropy of the stapled scFv (or spFv) as well as that of the flexible linker. This will lead to an improvement in spFv stability.
Figure 1. Stapling of scFv. (a) scFv stapling to improve low stability and minimize breathing-mediated aggregation. (b) Cartoon schematic of the stapling scheme using HL configuration as example. A similar scheme is valid for the LH construct. Pink dashed lines indicate flexible linkers connecting the C-terminus of leading variable region and the N-terminus of the trailing variable region to the stapling ‘CPPC’ motif. The segment labeled CPPC in the middle of the linker indicates one possible design of a staple which occurs naturally in the IgG1 hinge. Anchor points (labeled C(APL) and C(APH)) that are mutated to Cys residues in VH and VL are shown in stick. Short lines (yellow) between the staple and anchor point Cys residues indicate disulfide bond formation. Distances dstaple and dAP (yellow, dashed line) are labeled (c) Graphical illustration of anchor point selection geometry consideration (HL configuration) mapped onto the structure of an Fv from a human germline antibody (PDB ID 5I19, GLk1). Cter: C terminus of leading domain; Nter: N terminus of trailing domain; APH: anchor position on leading domain; APL: anchor position on trailing domain; dAP: distance between APH and APH; d1-d4: various distances as defined in the figure. Similar illustrations can be drawn for the LH orientation (not shown). Anchor points for HL orientation are Chothia position 43 for VH (H43C) and position 100 for VL (L100C); for LH: Chothia position 42 in VL (L42C) and 105 in VH (H105C). (d, e) Cβ(Cys1)-Cβ(Cys2) distance between Cys residues in hinge CPPC motifs in structures of human IgG4 (PDB 5DK3; (d) and mouse IgG2a (PDB 1IGT; (e). These distances range from about 7 Å to 9 Å.
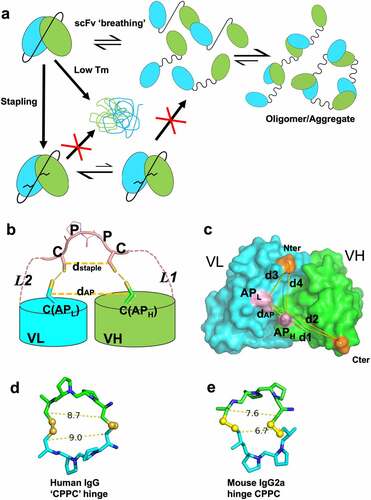
For stapling to correctly form and to be widely applicable, a number of structural conditions need to be satisfied. First, the Fv anchor positions are exposed, structurally conserved and mutation to Cys will not affect folding or binding. The distance (dAP) () between them should have a narrow distribution and a low probability of forming an inter-VL/VH disulfide bond directly. For the chosen anchor positions (Figure S1a, ChothiaCitation33 positions 42 of VL and 105 of VH for LH and positions 43 of VH and 100 of VL for HL), the distances, Cα (dAP, Cα) and Cβ (dAP, Cβ) and geometry () have a rather narrow and similar distribution in antibody structures (Figure S2b-e). The LH and HL Cα distances have an average of 8.2 Å (range of 7–9 Å) and 6.9 Å (range of 6–8 Å), respectively (Figure S2b). The Cβ distances are 8.7 Å (range of 7–10.0 Å) and 8.2 Å (range of 7–10.0 Å), respectively (Figure S2c). These distances are much longer than the typical Cα and Cβ distances (4–6.8 Å, Figure S2b and 3.5–4.8 Å, Figure S2c, respectively) of disulfide bonds. Thus, for most Fvs, the two anchor positions likely will not efficiently form disulfide bonds directly.
Second, the distance between the two linker Cys residues (dstaple) is compatible with (dAP), and these Cys residues have a low probability of forming a disulfide loop. We selected a short sequence of “CPPC” (Cys-Pro-Pro-Cys) as one possible stapling motif because this sequence natively occurs in human IgG1 hinge and some rodent IgGs. The structures of humanCitation34 and mouse IgGCitation35 mAb molecules show that the Cβ(Cys1)-Cβ(Cys2) distances in the human and mouse IgG hinges range from 7 to 9 Å (). This range is comparable to the distances between the two anchor points in both LH and HL orientations (, S2c). In addition, the CPPC motif was found to have the slowest rate of SS loop closureCitation36 due to the relative conformational rigidity of the Pro-Pro residues. Thus, the CPPC staple will likely be of the right geometry for stapling, i.e., correctly forming disulfide bonds to the anchor point Cys residues.
Third, the linker Cys residues need to be positioned close to the respective anchor Cys simultaneously to allow proper stapling disulfide formation and to prevent scrambling, i.e., formation of SS between unintended Cys residues. The stapling Cys positioning is a result of the linker segments L1 (from C terminus of the leading domain to first linker Cys) and L2 (from second linker Cys to trailing domain N terminus). Antibody structure analysis and molecular modeling suggest that L1 lengths of 7 to 9 residues long and L2 lengths of 4 to 5 residues would be suitable for the geometry and distances (dAP and d1-d4) between the anchor points and the termini of the VL and VH domains (, Figure S3a-c). As the exact L1 and L2 lengths are difficult to predict accurately due to Fv structural variability, we experimentally sampled the range to define the optimal lengths (see below) for a linker of equal composition that works for both LH and HL constructs.
Model and therapeutic spFv molecules are significantly more stable
In order to assess the stapling designs, we selected several antibodies to generate in scFv and corresponding spFv formats: two antibodies with kappa light chains (GLk1 and GLk2) from synthetic phage antibody libraries;Citation37 a lambda-containing antibody (CAT2200) that binds IL-17A;Citation38 and two scFv variants, Cris7a and Cris7b, derived from anti-CD3 mAb Cris7,Citation39 which have potential use in CD3-based T-cell redirecting. We constructed and expressed the scFv and spFv molecules in both LH and HL orientations. A standard (GGGGS)4 linker was used for the scFv constructs. For the spFv constructs, different linker lengths within the L1 and L2 ranges above were sparsely sampled. The SDS-PAGE showed that the reduced scFv and spFv proteins migrate identically, but the non-reduced spFv molecules migrate faster than the corresponding scFv (), indicating that additional disulfide bonds in spFvs are formed as expected.
Figure 2. Stapling design improves the thermal stability of Cris7a/b domains. (a) SDS-PAGE of scFv and spFv proteins of Cris7a/Cris7b in LH orientation. (b) Thermal stability of Cris7a scFv/spFv and Cris7b scFv/spFv domains by DSC. Parameters related to protein design and enthalpy features from analysis are listed in Table S1.
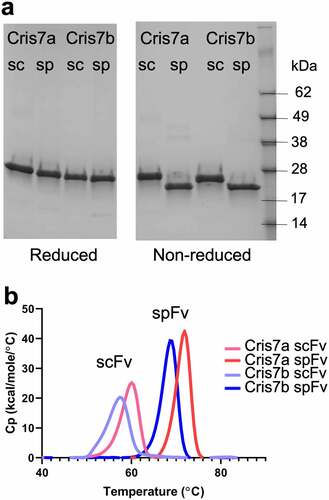
The thermal stability of the scFv and spFv molecules was investigated by differential thermal calorimetry (DSC). The data are shown in Table S1 with DSC profiles for Cris7a/b shown in . Comparison of the corresponding scFv and spFv proteins indicates a roughly 10°C increase in Tm upon stapling, regardless of the Tm of the starting scFv (Table S1). For example, the Tm of the Cris7a/b LH scFvs is 59.7°C and 57.1°C, respectively (). The corresponding spFv proteins have Tm of 71.6°C and 68.6°C, respectively. This indicates a stabilization (ΔTm) of more than 11°C (, Table S1). Glk1 spFv has a Tm of ~80°C, an increase of 9°C relative to its scFv (Table S1). This is comparable to the Tm of its Fab counterpart.Citation40 In addition to Tm changes, thermal melting of spFv proteins involves 20% or more increase in enthalpy change (ΔH) relative to the corresponding scFv proteins (Table S1). The Tm and ΔH values of both LH and HL configurations of the same VL/VH pair are very similar (Table S1), indicating that scFv configuration does not significantly affect thermal stability. The results also show that L1 and L2 lengths did not impact ΔTm. Overall, these results indicate that L1 and L2 lengths of 7–9 and 4–5 residues, respectively, are compatible with significant stabilization of an spFv.
To select a stapling linker that is most widely applicable, we further explored the impact of linker segment length on the thermal stability of spFv constructs. Using two VH/VL pairs, we varied the L1 (7, 8 and 9 residues) and L2 (4, 5 residues) segment lengths in all combinations in LH only. The thermal stability of these scFv/spFv molecules was assessed with fluorescence using UNCLE (Unchain Labs, CA). The Tm data are shown in Table S1b. The results indicate that the Tm and ΔTm are essentially identical for constructs having L1 lengths of 8 and 9 residues. For several spFv proteins with a 7 residue L1, the Tm appears to be slightly lower, suggesting that shorter L1 might be less favorable for thermal stabilization. The trailing segment L2 lengths of 4 and 5 residues did not lead to any significant differences in Tm. Thus, we selected the longer L1 = 9 and L2 = 5 for better compatibility with a wide range of scFv molecules.
Structures of spFvs reveal proper stapling and VL/VH pairing
To confirm the proper formation of stapling disulfide bonds and to reveal any structural consequences, we determined the crystal structures of several scFv and spFv molecules, some in complex with the target protein (Table S2). The overall structures of the unbound spFv and some scFv/spFv:antigen complexes are shown in . These structures are consistent with typical Fv structures with both VL and VH domains packing against each other. In all spFv structures, some residues of the linker are ordered and resolved in the electron density maps. The disulfide bonds between the linker and the anchor points are generally well ordered in both LH and HL orientations (). A representative electron density map of the stapling linker region for Glk2 is shown in . The two stapling SS bonds and the linker CPPC motif are well ordered, indicating that stapling indeed occurred as designed. In contrast, no linker residues are ordered in the only scFv containing the complex of CAT2200 and IL-17 ().
Figure 3. Structures and comparison of various scFv/spFv domains. In all structures, VL is colored blue and VH is colored green. The linker segments are colored pink. (a) GLk1 spFv LH. (b) GLk1 spFv HL. (c) GLk2 spFv HL. (d) 2mfo-dFc electron density contoured at 1.5 σ about the CPPC staple motif and anchor points for Glk2 spFv HL. Circles in orange indicate the stapling disulfide density. (e) CAT2200b spFv HL. (f) unbound CAT2200b spFv HL compared with CAT2200a scFv LH bound to IL-17 (silver). (g) front and back views of unbound CAT2200b spFv HL compared with CAT2200a spFv LH bound to IL-17 (silver).
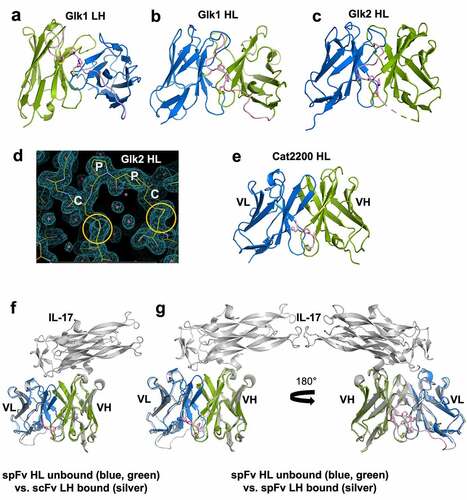
For Glk1, we obtained structures of LH and HL spFv (). There are four independent copies of spFv in the LH crystal and two independent copies in the HL crystal. Within LH, the pairwise Cα root-mean-square deviations (rmsds) including the stapling linkers between spFv copies range from 0.22 Å to 0.40 Å. The pairwise Cα rmsd between the two HL spFv molecules is 0.20 Å. When the LH and HL spFv structures are compared, the pairwise Cα rmsds are slightly higher (from 0.53 to 0.77 Å, average 0.61 Å) excluding the linker for 195 to 219 Cα atoms (average 213). We also compared the LH and HL spFv with the Fv fragment in its corresponding Fab (pdb id 5i19).Citation40 The rmsds between LH spFv and Fab Fv are from 0.41 to 0.64 Å (average 0.57 Å) for 203 to 215 Cα atoms. The rmsds between HL spFv and Fab Fv are 0.56 to 0.71 Å (average 0.64 Å) for ~210 Cα atoms. These values indicate that the structures of the LH and HL spFvs and the Fab Fv are very similar. These small differences are likely the result of crystal packing. Similar observations were true for GLk2.
To identify any structural impact on antigen binding, we crystallized CAT2200 scFv and spFv molecules in a complex with IL-17. For the CAT2200 scFv and spFv variants crystallized, the structures are nearly identical with and without a bound target (). They are also identical regardless of orientation and presence or absence of stapling. The rmsd for all matching Cα atoms between pairs of structures is very small: 0.41 Å between unbound spFv (HL) and antigen-bound scFv (LH) (), 0.46 Å between unbound spFv (HL) and bound spFv (LH) (), and 0.37 Å between bound scFv and bound spFv (LH/HL). These structural data show that stapling does not affect the domain structures of VL and VH or relative VL/VH packing.
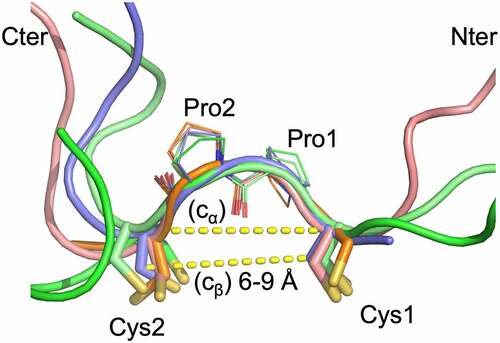
As noted above, the CPPC motif and the disulfides between the staple and anchor points are generally well ordered in the structures (). The CPPC structures are very similar with main chain atom rmsd of 0.37 Å (range 0.15 to 0.5 Å). The Cβ (Cys1)–Cβ (Cys2) distances range from 6 to 9 Å, and Cα (Cys1)-Cα (Cys2) distances are from ~ 6.5 Å to 9 Å. These distances are comparable to those in the IgG hinge structures. In the structures, the two Pro residues adopt the trans conformation, and the highly similar CPPC motif structures in the different crystals indicate that Pro-Pro motif is relatively rigid. This rigidity likely serves to strengthen the VL and VH domain interaction. At the same time, the observed range of Cβ (Cys1)-Cβ (Cys2) distances also indicates that some flexibility is still allowed for proper VL/VH orientation. The linker residues beyond the CPPC motif typically are either disordered or have different conformations (). The L1 and L2 segments typically do not have specific interactions with the main VL and VH domains except for occasional H bonds. For example, in the GLK2 spFv structure, the side chain of trailing VL domain residue E1 forms H bond interactions with the trailing linker segment main chain (GLK2 HL spFv, Figure S4). However, this appears to have little effect on the spFv structures.
Figure 4. The staple and linker conformations in 5 spFv structures (bright green, GLk1 spFv LH; light pink, GLk1 spFv HL; purple, GLk2 spFv HL; light green, CAT2200 spFv LH bound to IL-17; orange, CAT2200 spFv HL unbound). The CPPC motif has been re-labeled as Cys1, Pro1, Pro2, Cys2 for clarity. The structures are superimposed on the mainchain of the CPPC motif. The dashed lines indicate Cα-Cα and Cβ-Cβ distances between the Cys1 and Cys2 residues. The range of Cβ-Cβ distances observed in all copies of the linker staple Cys residues are indicated. N-termini are indicated with ‘Nter’, C-termini are indicated with ‘Cter’.
spFv bispecifics (BCMA Fab x CD3 scFv/spFv) show improved protein quality
We generated several sc/spFv × Fab bispecific constructs (illustrated in ) to further study the activity, biophysical properties and translatability of the stapled scFv molecules in a more relevant therapeutic molecular background. Expanding from the prior work with Cris7 domains, we paired two unique anti-CD3 scFv/spFv binding arms, Cris7b as described above and CD3sp34v1, which was derived from SP34,Citation41 and combined them with two related anti-BCMA Fab moieties using the knob-in-hole heterodimerization platform.Citation42 For the scFv constructs, we also included the Bird linkerCitation9 to assess the impact of linker composition. The anti-BCMA Fab arm, mAb1 and mAb1h are from a murine mAb and its humanized variant (Figure S5). Pairing the two BCMA Fab arms with two different anti-CD3s each in three formats (scFv with Bird linker, scFv with (G4S)4 linker and spFv) generated 12 distinct BCMA-targeting bispecific molecules (Figure S6). The constructs are listed in Table S3. All bispecific samples were expressed at a small scale (40 mL culture) from Expi-CHO mammalian cell culture and purified through a two-step process as described in Methods. Analytical size-exclusion chromatography (aSEC) was performed to assess sample purity post-purification and revealed significant differences between the scFv and spFv bispecific molecules (Figure S6). In scFv-containing samples, the overall yield was lower for the desired monomer. Additionally, in proteins with Cris7b scFv arm, there was a notable higher molecular weight species in the purified samples (Figure S6, upper plots). In contrast, the spFv containing constructs usually have higher yields and >98% bispecific monomer (Figure S6). These results demonstrate that stapling in either CD3 scFv arm significantly improved bispecific product yields and quality. The identity of the flexible linker, either (G4S)4 or Bird, did not show any appreciable differences.
Figure 5. Bispecifics with spFv show improved yields, product quality and expected disulfide formation in the stapled linker. (a) Schematic of BCMA (Fab) x CD3 (scFv/spFv) bispecific molecular architecture. HK in Fc regions indicate the knob-in-hole (K, knob; H, hole) mutations for Fc heterodimerization. RF (H435R and Y436F) mutations in the Fab containing heavy chain are introduced for purification to prevent binding to Protein A of RF containing chain monomers or homodimers. (b) SEC profiles post-CH1 of scFv/spFv Cris7b containing molecules with mAb1 indicate presence of oligomer species (labeled O) in scFv proteins that is absent in spFv proteins (monomer, M). (c) MS2-HCD spectrum of the peptide derived from non-reduced proalanase digestion representing the expected stapled disulfide linkage between Cys119-Cys237. The b- and – y type backbone fragments from each peptide half are annotated in the sequence map that is composed of Pep1 and Pep2 connected via the disulfide bridge.
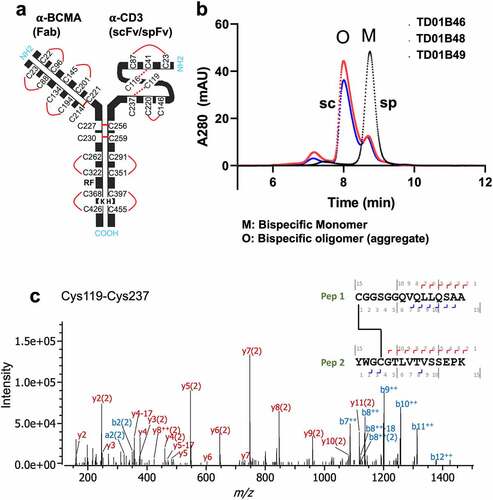
We chose one set of constructs to move into larger scale expression and purification to generate large batches of highly purified recombinant antibodies that could be used in a panel of analyses to further test the stapling technology. Based on the data described above, we chose three Cris7b-containing molecules, 1) TD01B46 (mAb1 × Cris7b spFv); 2) TD01B48 (mAb1 × Cris7b scFv G4S); and 3) TD01B49 (mAb1 × Cris7b scFv Bird), for large-scale expression. Purification of these bispecific samples revealed more pronounced trends in product quality and yield (), as was seen in the small-scale purified samples. The scFv bispecific samples (TD01B48, TD01B49) show a large, higher molecular weight oligomeric peak (“O”) as dominating the post-CH1 purified sample, which is even more pronounced than the corresponding small-scale expression (Figure S6). In comparison, the spFv bispecific (TD01B46) remains a dominant peak that corresponds to the desired monomeric species. Disulfide mapping by liquid chromatography-mass spectrometry experiments was conducted and confirmed that all the expected disulfide bonds, including those of the spFv moiety, are correctly formed with negligible scrambling (, Fig S7). These data together emphasize the improvement shown in protein production and quality when the stapling is applied.
spFv bispecifics show significantly improved thermal stability
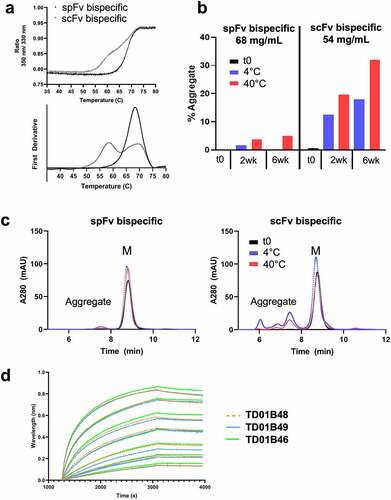
We evaluated the impact of stapling on the thermal stability of the bispecifics using advanced differential scanning fluorimetry (nanoDSF) measurements (, Table S4). For Cris7b bispecifics, the scFv-containing construct has a first melting transition with Tm of ~59.0°C and Tonset from 50°C to 56°C. The scFv Tm is thus likely similar to that of the isolated scFv (Table S1). For the corresponding spFv bispecifics (, Table S4), this low Tm transition is not observed; rather the first transition has a Tm of ~68.3°C, which is apparently convoluted with the transitions of the other domains of the bispecifics. Moreover, the Tonset of this spFv bispecific protein increased to ~61°C (, Table S4). Together, these findings are interpreted to indicate an approximate 10°C or higher stability improvement for the spFv moiety. Similar improvements in stability are also observed for the CD3sp34v1 spFv constructs (Table S4). These data indicate that the thermal stabilization seen in isolated scFv/spFv constructs (Table S1) is also present when they are incorporated into therapeutic constructs.
Figure 6. Cris7b spFv bispecific proteins with mAb1 are stable and retain binding affinity to CD3. (a) NanoDSF traces of Cris7b scFv/spFv bispecifics with mAb1 (TD01B49, TD01B46) show~10°C transition to higher Tm on incorporation of stapling mutations. (b, c) Cris7b spFv bispecific proteins are resistant to heat-induced aggregation. SEC traces (b) and quantification of aggregate levels (c) show that Cris7b spFv bispecifics have a dramatic reduction in heat-induced aggregation over 6 week time frame at either 4°C or 40°C. (d) BLI binding traces show comparable binding features (association and dissociation) for binding to recombinant CD3. Green: Cris7b spFv, TD01B46; blue: Cris7b scFv Bird, TD01B49; Orange dashed lines: Cris7b G4S, TD01B48.
spFv bispecifics are resistant to heat stress-induced aggregation
We further evaluated the impact of spFv on aggregation induced by heat stress as a predictive indicator of protein shelf stability at 4°C.Citation43 The highly purified Cris7b scFv/spFv bispecifics (TD01B49 and TD01B46, respectively) (purity >98.5%) were concentrated in Dulbecco’s phosphate-buffered saline (DPBS) to ~60 mg/ml. The concentrated samples were then incubated at 4°C and 40°C. At two-week intervals, a small aliquot of each sample was diluted to 1 mg/ml and then run on aSEC. The results are shown in . Over a six-week incubation period at 4°C, the spFv bispecific TD01B46 ( left panels) remained a monomer, while at 40°C it showed a modest increase to about 5% dimer species at 6 weeks. By contrast, over the same period at either 4°C or 40°C, the scFv bispecific TD01B49 (, right panels) showed a large increase to about 18% and 32% aggregate species (dimers and higher-order oligomers), respectively. These data demonstrate that spFv bispecifics are much more resistant to heat-induced aggregation at high protein concentration.
spFv bispecifics retain binding affinities
Binding was performed using bio-layer interferometry (BLI) and enzyme-linked immunosorbent assay (ELISA) with highly purified bispecific samples (TD01B46, TD01B48 and TD01B49) to determine whether stapling impacts binding to target antigens for either the sc/pFv or Fab arms. Recombinant CD3ε/δ heterodimer protein was used to assess the binding of the scFv/spFv anti-CD3 arms. Binding measurements show that the scFv and spFv bispecifics bind CD3 similarly, indicating that incorporation of stapling in the scFv did not alter CD3 binding (; Figure S8a). The sensorgrams from BLI () show similar binding responses, association and dissociation profiles, indicating that scFv and spFv bispecifics have similar binding kinetics for CD3. These results are consistent with the ELISA binding data (Figure S8a). The scFv linker (Bird and G4S) also has no significant impact on affinity for the target protein. The anti-BCMA Fab arm, identical in either the scFv or spFv bispecific molecules, also shows a similar binding affinity to the recombinant BCMA (Figure S8b,c). Together, these data indicate that the spFv exhibits a binding affinity similar to that of the corresponding scFv protein and that incorporation of the stapling mutations does not affect binding of the partner domain in the bispecific molecules of interest.
spFv bispecifics show potent killing in CD3 target assays
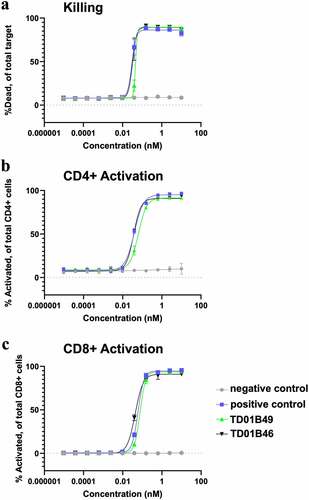
To determine the effect of BCMA-targeting bispecific molecules on T cell activation and tumor cell-killing potential, H929-Fluc-GFP cells were used as target cells for two human donor pan-T cells. Detection of T cell activation and killing and were assessed at 72 h by flow cytometry. All bispecific proteins potently killed BCMA+ H929-GFP+ cells in a cytotoxicity assay with a very similar EC50 (), whereas a negative control bispecific with a Cris7b scFv/non-targeting Fab (negative control) showed no killing activity. All bispecific constructs with either an scFv or spFv domain also activated CD4+ and CD8+ T cells with similar EC50s, whereas the negative control molecule did not activate T cells (). This indicates that the H929 cell killing was the result of T cell activation by CD3 redirection. Taken together, these data suggest that spFv fully retains scFv function in therapeutic constructs.
Figure 7. spFv bispecific molecules function similarly to their non-stapled counterparts. (a) spFv bispecific (TD01B46) has potent killing activity of BCMA+ cancer cells. (b, c) scFv (TD01B49)/spFv bispecifics (TD01B46) activate CD4+/CD25+ (b) and CD8+/CD25+ (c) T cells with similar potency. The negative control has no killing or T cell activating activity.
Stapling significantly improves biophysical properties of potential therapeutic multispecific molecules
We applied stapling to a number of proprietary scFv bi- and tri-specific therapeutic antibodies that previously showed poor biophysical properties. While the scFv-containing molecules displayed obvious aggregation upon heat stress at high concentrations, the spFv counterparts showed minimal aggregation under similar conditions (). These molecules still maintain their respective target binding. In some cases, antigen binding is slightly improved (). The improvement in biophysical properties is very beneficial for downstream therapeutic development, which may also lead to improved product quality and therapeutic outcomes.
Table 1. Comparison of biophysical properties of scFv/spFv bi- and tri-specifics.
Discussion
In this work, we report on the use of our “stapling” method to significantly improve scFv stability and reduce its tendency to aggregate. Stapling is achieved by forming specific disulfide bonds between the otherwise flexible linker and two conserved anchor positions on the VL and VH domains. Disulfide bonds in proteins contribute to their stability. In fact, each VL and VH domain contains a conserved intra-domain disulfide bond that confers considerable domain stability. Disulfides increase protein stability by covalently linking distant structural elements together to increase the compactness of protein structure.Citation44,Citation45
Stapling, as herein described, improves scFv stability and reduces aggregation by several mechanisms. First, in contrast to scFv molecules, the linker between the VL and VH domains in spFv molecules is generally found to be ordered, particularly the stapling elements and intervening sequence, indicating a reduction in the conformational entropy of the linker. Second, the short distance between the two stapling disulfide bonds (about 6–9 Å in determined spFv structures) limits the extent to which the VL and VH domains can separate from each other, further reducing the conformational entropy of the spFv compared with scFv. Interestingly, the structures indicated that there is very little direct interaction between the ordered linkers and VL and VH domains except for the two stapling disulfide bonds. Stapling has also been shown to not impact the relative orientation of the VL/VH domains. Thus, stapling did not provide new interactions between VH and VL, but rather effectively increased existing native interactions. Both entropy reduction and increased tethering contribute to the significant increase in Tm of the spFv format, which in turn reduces domain unfolding, a common factor in scFv protein aggregation. In contrast, the long linker (typically 15 aa or longer) between the VL and VH domains in an scFv does not provide such restraints. Thus, stapling is an effective scheme to improve the scFv stability and reduce aggregation, leading to superior developability of spFv-containing biotherapeutics.
The anchor positions selected for stapling are structurally well conserved in all Fv domains of either kappa or lambda light chains. The geometry of the two sets of anchor positions for LH and HL spFv has a relatively narrow range of variation (Figure S2b,c). Thus, stapling by a simple motif such as CPPC is likely applicable to nearly all Fv fragments. This scheme is superior to a previous disulfide-mediated scFv stabilization approach, i.e., forming direct disulfide bonds between VL and VH domains (L43-H105, DS1, and L100-H44, DS2). The stabilizing effects of DS1 and DS2 have been inconsistent,Citation29 and as such they have not been widely applied in current therapeutics incorporating scFv moieties. Formation of a disulfide bond between two positions in proteins requires a relatively narrow range of geometry.Citation46 The Cα-Cα distance range is 4.8–6.8 Å (peak at 5.6 Å), and Cβ-Cβ distances are in the range of ~3.4–4.8 Å (peak at ~3.8 Å) from analyses of protein structures (Figure S2d,e).Citation46 These parameters, along with environments of the two positions, determine the allowed SS bond conformations, which are critical for successful formation. Analysis of 2501 high-resolution Fab/scFv structures shows that most Fv structures do not have the geometry compatible with SS bond formation. For example, the Cβ-Cβ distances are distributed around 5.2 Å and 5.8 Å for DS1 and DS2, respectively. These distances are much greater than those in typical SS bonds in proteins. While protein structural flexibility may allow a subset of these Fvs to form SS bonds, it is also likely that a disulfide bond between these positions with unfavorable geometry may not form or result in structural strain when formed in a large proportion of Fv domains. On the other hand, the two legs of the stapling motif are external to the Fv domains and can easily position properly to satisfy the linker-anchor point disulfide geometry. Our analysis showed that the distances between the two stapling legs are comparable to the distances of the two sets of anchor points for LH and HL (Figure S2b,c). Thus, as shown in the multiple examples reported in this work, stapling is much more widely applicable.
In summary, we presented a novel and widely applicable strategy of stapling scFv to enhance stability and reduce scFv-mediated aggregation. With improved stability over and structural and functional identities to scFv, spFv can be used anywhere a corresponding scFv is used, including multispecifics, CAR-Ts and other molecular architectures. Stapling will likely improve the successful conversion rate of Fab/mAb to scFv so that more spFv moieties are available for incorporation into fit-for-purpose molecular entities. The improved biophysical properties resulting from the use of spFv instead of scFv will allow construction of biotherapeutics of superior developability, thus enabling faster development and improving drug quality, efficacy and safety.
Materials and methods
Expression and purification of scFv/spFv proteins and bispecific antibodies
All protein expression constructs were cloned into a CMV promoter-driven mammalian expression vector and produced from either Expi293 or ExpiCHO cells using manufacturer protocols and purified using affinity chromatography. The BCMA protein was generated similarly and biotinylated using a commercial biotinylation kit. The details are described in Supplemental Information (SI) methods.
SDS-PAGE analysis of protein samples
Cris7a and Cris7b scFv and spFv domain proteins were prepared at 0.3 mg/mL in 1× dPBS with the addition of 1× NuPAGE LDS Sample Buffer (Invitrogen). Samples were split in half and to one set 1 mM dithiothreitol was added to allow for reduction. All samples were heated at 90°C 5 m prior to loading. All samples were loaded at 20 mL volumes into the wells of a 4–12% Bis-Tris NuPAGE Gel (Invitrogen), along with a SeeBlue Plus2 Pre-stained Ladder (Invitrogen) and run at 180 V for 45 min. The final gel was stained with SimplyBlue SafeStain (Invitrogen) for 1 h at room temperature (RT) and then destained overnight in ddH2O.
Differential scanning calorimetry
The scFv and spFv proteins were dialyzed overnight against 1× DPBS (Gibco) for GLk1 and CAT2200a/CAT2200b or MES (25 mM MES, pH 6.0, 100 mM NaCl) for GLk2. The dialysis buffer was then 0.22 micron filtered and used as the reference solution and for buffer–buffer blanks in the DSC experiment. Proteins were diluted to ~0.5 mg/mL in the filtered buffer, and 400 μL of each protein or buffer sample was loaded into a 96-deep well plate (MicroLiter Analytical Supplies, 07–2100) and kept at 4°C in the autosampler drawer over the course of the experiment. A MicroCal Capillary DSC with Autosampler (Malvern) was used to perform the DSC experiments. DSC scans were performed from 25°C to 95°C at a 60°C/h scan rate with no sample rescans. No feedback was selected, and the filtering period was set at 15 s. After each sample, the cells were cleaned with a 10% Contrad-70 solution and a buffer–buffer blank was run. Data analysis was performed using Origin 7.0 with the MicroCal VP-Capillary DSC Automated Analysis add-on (Malvern). The baseline range and type were manually chosen and then subtracted. The previous buffer blank was subtracted from the sample curve followed by concentration-dependent normalization. The thermal melting profiles were analyzed using the non-two-state transition model. Iterative curve fitting was performed to derive thermodynamic parameters associated with the melting, e.g., thermal stability and enthalpy, which are reported in Table S1A.
Thermal stability by uncle
Protein stability of several scFv/spFv paired samples was evaluated incorporating intrinsic fluorescence and static light scattering (SLS) analyses on the Uncle instrument (Unchained Labs, Pleasanton, CA, USA). Thermal melting mid-point (Tm) was determined by intrinsic fluorescence measured through with blue wavelength acquisition (Blue − 473 nm, filter 4) and thermal aggregation (Tagg) by SLS through UV acquisition (UV − 226 nm, filter 3) settings. Sample concentrations ranged from 0.1 to 0.5 mg/mL in DPBS, and analysis was performed in duplicate using 8.8 mL sample volume in the Uncle Uni cuvettes. Thermal melt profiles were collected using a linear heating ramp of 0.3°C/minute between 20 ~ 85°C with a 60 s incubation period and 30 s plate hold period. Data were collected and analyzed concurrently with Uncle software and Tm and Tagg (Tonset) values were directly exported.
Protein structure determination
Protein crystals were produced using the hanging drop vapor diffusion methods. All X-ray diffraction data were processed with XDSCitation47 and CCP4.Citation48 Phases were determined by molecular replacement (MR) using PhaserCitation49 with homology models generated in MOE (Montreal, Canada) except for scFv CAT2200a scFv LH/IL-17 complex, for which the structure of pdb id 2vxsCitation38 was used as search models. The models were refined in PhenixCitation50 and manually adjusted in Coot.Citation51 Figures were generated in PyMOL (www.schrodinger.com). The X-ray diffraction data and refinement statistics are given in Table S2. For detailed methodology, see SI Methods.
Disulfide mapping analysis: sample preparation, instrument parameters and data analysis
Disulfide mapping by LC-MS experiments was conducted to confirm the disulfide linkages. The spFv bispecific antibody (TD01B46) was digested with a two-enzyme combination as described in SI methods and disulfide linkages were determined based on accurate m/z, intact mass of the disulfide complex (MS1) and masses of the peptide backbone fragments generated via tandem mass spectrometry (MS2). The details are the methods are described in SI Methods.
Differential scanning fluorimetry of bispecific variants
Conformational stability of bispecific proteins with Cris7b/CD3sp34v1 scFv/spFv paired with two anti-BCMA Fab arms was measured using advanced differential scanning fluorimetry (nanoDSF) technology by monitoring the intrinsic fluorescence of tryptophan upon thermal unfolding. The unfolding was measured by loading each sample into 24 well capillary (NanoTemper, Cat# PR-AC002) from a 384 well sample plate (ThermoNunc, Cat# 264573) with a heating ramp of 1°C/minute between 20 ~ 95°C using the Prometheus NT.48 instrument (NanoTemper Technologies GmbH). Each sample was measured at 0.5 mg/ml in phosphate buffer saline (PBS) in duplicate. The intrinsic fluorescence of each sample at 330 and 350 nm was used to monitor unfolding during the temperature ramp and recorded as changes in fluorescence intensity over time. Data were collected and saved as projects and processed using the PR.StabilityAnalysis v1.0.2 software. The processed data contained integrated thermal melting profiles, first derivatives for fluorescence at 330 nm, 350 nm, 330/350 ratio, and light scattering data for all the samples. Thermal melting mid-point (Tm) values as well as the onset of aggregation (Tagg) were identified and reported.
Bio-layer interferometry of Cris7 bispecific molecules
Binding of the Cris7b scFv and stapled spFv bispecific molecules to recombinant CD3 antigen (human CD3 epsilon and CD3 delta heterodimer protein, Acro Biosystems) and recombinant BCMA antigen were measured by BLI using an Octet HTX instrument (Sartorius, formerly ForteBio). To evaluate BCMA binding, Streptavidin (SA) capture sensors (Sartorius) were loaded with biotinylated-BCMA protein to ~1 nm signal in PBS. Bispecific samples were loaded to antigen coated SA sensors at seven antibody concentrations starting at 100 nM in 2-fold dilution (100 nM ~1.5 nM), diluted in 1× DPBS with 0.05% tween-20 to prevent nonspecific interactions. The association and dissociation times were 900 s, respectively. To evaluate CD3 binding, anti-human IgG Fc (AHC) capture biosensors (Sartorius) were loaded with 3 ug/mL bispecific sample of interest in PBS. After loading, sensor tips were washed in 1× DPBS with 0.02% Tween 20 and 1 mg/mL bovine serum albumin (BSA) for blocking. Recombinant CD3 antigens were loaded to antibody coated sensors at seven concentrations starting at 100 nM in 2-fold dilutions (100 nM ~ 1.5 nM), diluted in 1× DPBS with 0.02% tween 20 and 1 mg/mL BSA to prevent nonspecific interactions. The association was monitored for 1800 s and dissociation for 900 s, respectively. All the measurements were performed at 30°C with agitation at 1,200 rpm. Sensorgrams were referenced for buffer effects and then analyzed using the ForteBio Data Analysis HT software (V. 12.0.1.55). Kinetic responses were baseline subtracted, aligned and globally fit using a 1:1 fitting model or 2:1 heterogeneous ligand-binding model to obtain values for association (Kon), dissociation (Koff) rate constants and the equilibrium dissociation constants (KD).
ELISA of bispecific molecules
Bispecific antibodies were analyzed for binding to either a recombinant biotinylated BCMA or a recombinant biotinylated CD3 antigen (human CD3 epsilon and CD3 delta heterodimer protein, Acro Biosystems Catalog # CDD-H82W6). ELISAs were carried out according to standard protocols and plates were washed three times with TBS containing 0.05% Tween 20 (TBS-T) between each incubation step. Ninety-six-well Maxisorp plates (Nunc) were coated with 1 ug/mL of Streptavidin, diluted in 1× dPBS for 18 h at 4°C. All plates were coated with 20 nM of antigen of interest for 1 h at room temperature and then blocked with 3% BSA in PBS-T (1× dPBS with 0.05% tween 20) for 1 h at room temperature. After blocking, the plates were incubated with serial dilution of purified bispecific samples in PBS-T for 1 h at RT, and were then incubated with goat anti-human IgG F(ab’2)2 HRP (Jackson ImmunoResearch, Catalog # 109-035-006) for 1 h at RT. After washing, BM Chemiluminescence ELISA Substrate (POD) was added (Millipore) and plates were immediately read on an Envision plate reader (Perkin Elmer) using ultrasensitive illuminesence. Raw data was exported to GraphPad Prism where curves were generated and analyzed with a nonlinear regression curve fit.
High concentration and heat stress study
Concentratability studies were performed using Amicon Ultra 4 centrifugal filtration devices with 30 kDa MWCO membrane (Catalog# UFC803096. MilliporeSigma, Burlington, MA). First, the spin columns were filled with water and spun at 4200 g for 6 min to equilibrate the membrane. TD01B46 (2 mg/ml) and TD01B49 proteins (0.5 mg/ml) were each loaded in pre-washed spin columns and centrifuged at 4200 g at 15-min time intervals. At the end of each 15-min centrifugation step, the concentrators were removed from the centrifuge and a visual estimate of the remaining sample volume was recorded. The concentration step was repeated until a sufficiently large volume of the concentrated sample was available for profiling by SEC for purity analysis. At the end of the centrifugation process, the concentrated samples were recovered, and the protein concentration was determined using NanoDrop ND1000 (ThermoFisher Scientific, Waltham MA). The final concentrations were 68 mg/ml and 54 mg/ml for TD01B46 (stapled) and TD01B49 (unstapled) bispecific proteins, respectively. The concentrated samples were split into two equal parts, one stored at 4°C and the other stored at 40°C. Aliquots from the incubated samples were taken at different time points (t0, t-1 wk, t-2 wk and t-6 wk) and diluted to 1 mg/mL with 1× dPBS for purity analysis by SEC. For SEC, 20 ug of the 1 mg/mL sample was loaded onto an analytical size-exclusion HPLC (TSKgel BioAssist G3SWxl, 7.8 mm ID × 30 cm H, 5um, TOSOH; guard column: TSKgel BioAssist SWxl guard column, 6 mm ID × 4 cm H, 7um, TOSOH; Agilent HPLC system) and monitored for separation of the sample at UV 280 nm, at 1 mL/min for 20 min using 200 mM sodium phosphate (pH 6.8) as the running buffer.
Cytotoxicity assay
Intellicyt iQue3 and ForeCyt software (Sartorius) were used to determine the effect of BCMA-targeting test molecules upon T cell activation and tumor cell-killing potential. H929-Fluc-GFP cells served as target cells for two human donor Pan T cells (Hemacare). The assay was set up in a 96-well plate at a T cell-to-target ratio of 3:1. Test molecules were added at a starting concentration of 10 nM and serially diluted at 1:4 in complete media. All molecules were tested in duplicate at minimum. Detection of killing and T cell activation status was assessed 72 h later by flow cytometry. Endogenous GFP expressed in H929 was used to separate T cells from target cells. Cytotoxicity was measured using Near-IR Live/Dead stain (ThermoFisher), while activation in CD4 and CD8 T cells was assessed with anti-human CD25-BV650 (BD Biosciences Catalog # 563719), anti-human CD4-BV510 (Biolegend, Catalog # 317443) and anti-human CD8-PE/Cy7 (Biolegend, Catalog # 344711). Using Prism software (GraphPad), cytotoxicity and CD25 MFI data were exported, log-transformed, and four parameter logistically fit to generate regression curves for reporting of EC50.
Cell binding
Briefly, H929 wildtype and knockout cells were counted and stained with CFSE (BD Pharmingen, Catalog# C34554) and/or Cell Trace violet proliferation dyes (BD Pharmingen, Catalog# C34557), as well as near IR live/dead stain (ThermoFisher, Catalog# L10119). Each cell population was quenched with fetal bovine serum (Gibco, Catalog# 16000–036), Fc-blocked with Human TruStain FcX blocking reagent (Biolegend, Catalog# 422301) and then plated together in 96-well plates with 50K total cells per well. The cells were then incubated with serial dilutions of test molecules for 1 h at 37°C (1/2 log serial dilutions starting at 2 µM). The cells were washed 2× in fluorescence-activated cell sorting (FACS) buffer (Becton Dickinson, cat# 554657) and then incubated with 1 µg/mL AF647-labeled goat anti-human Fc (Jackson IR, cat# 109-606-098) detection reagent for 30 min at 4°C. The cells were again washed 2× in FACS buffer and then analyzed on the Intellicyt iQue 3 (Sartorius) high throughput flow cytometer. The raw data were exported and analyzed in GraphPad Prism.
Abbreviations
| aSEC | = | Analytical size-exclusion chromatography |
| BLI | = | Bio-layer interferometry |
| CAR-NK | = | Chimeric antigen receptor NK-cells |
| CAR-T | = | Chimeric antigen receptor T-cells |
| CR | = | Constant region |
| DSC | = | Differential thermal calorimetry |
| dsFv | = | Disulfide Fv |
| ELISA | = | Enzyme-linked immunosorbent assay |
| Fab | = | Fragment of antigen-binding |
| FDA | = | U.S. Food and Drug Administration |
| Fv | = | Variable fragment |
| HC | = | Heavy chain |
| HL | = | Heavy-light orientation |
| LC | = | Light chain |
| LC-MS | = | Liquid chromatography-mass spectrometry |
| LH | = | Light-heavy orientation |
| mAb | = | Monoclonal antibody |
| mL | = | milliliter |
| scFv | = | Single-chain fragment variable |
| spFv | = | Stapled scFv |
| Tm | = | Thermal stability |
| VH | = | Variable heavy |
| VL | = | Variable light |
| VR | = | Variable region |
Contributions
JL devised and supervised the project. LEB generated scFv/spFv constructs and crystallized and determined all spFv structures. EGP designed and characterized the BCMA/CD3 BsAbs. MF and MD generated scFv/spFv proteins for analysis by AG. FY, RN, S-JW, TL, ERL and SJ carried out biophysical studies. NK, BDR, BW, PA and RD generated all bispecific proteins. SH, JT and NM performed cell binding and killing studies. EG, TL, AM, HN and HG performed mass spectrometry analysis. AAA, AT, CH contributed to ideation. AAA crystallized and determined the structures of scFv/spFv antigen complexes. AZ, PC, WCC and JL supervised many activities. JL, LEB and EGP wrote the manuscript. PC provided extensive guidance and critical editing. All coauthors contributed to the manuscript preparation.
Supplemental Material
Download MS Word (3.9 MB)Acknowledgments
The authors thank the following for their technical and other assistance. Daniel Grau, Michael Kane, Laetitia Eugene, Hau Truong, Sitilia Rencheli and Sidharth Mohan in protein expression and purification; Xiefan Lin-Schmidt for providing the BCMA sequence; and Advance Photon Source (APS) for X-ray data collection.
Disclosure statement
JL, LEB, MF, MD, AAA, AT & CH are co-inventors in a provisional patent application (US20210047435A1). This application covers the scFv stapling technology and its potential applications in multispecific antibodies and other therapeutic or diagnostic protein modalities and detection reagents wherever scFv can normally be used.
Data availability statement
Atomic coordinates and structure factors of spFv and scFv/spFc:antigen complexes have been deposited in the Protein Data Bank, www.rcsb.org, under the ID codes 8DY0, 8DY1, 8DY2, 8DY3, 8DY4 and 8DY5. All other data are included in the main text and SI.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2195517
Additional information
Funding
References
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi:10.1186/s12929-019-0592-z. PMID: 31894001.
- Senior M. Fresh from the biotech pipeline: fewer approvals, but biologics gain share. Nat Biotechnol. 2023;41:174–15. doi:10.1038/s41587-022-01630-6. PMID: 36624153.
- Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. mAbs. 2023;15:2153410. doi:10.1080/19420862.2022.2153410. PMID: 36472472.
- Carter PJ, Rajpal A. Designing antibodies as therapeutics. Cell. 2022;185:2789–805. doi:10.1016/j.cell.2022.05.029. PMID: 35868279.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182–212. doi:10.1080/19420862.2016.1268307. PMID: 28071970.
- Brinkmann U, Kontermann RE. Bispecific antibodies. Science. 2021;372:916–17. doi:10.1126/science.abg1209.
- Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585–608. doi:10.1038/s41573-019-0028-1.
- June CH, O’connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–65. doi:10.1126/science.aar6711.
- Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–26. PMID: 3140379 http://www.ncbi.nlm.nih.gov/pubmed/3140379.
- Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347:773–89. doi:10.1016/j.jmb.2005.01.053. PMID: 15769469.
- Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi:10.1006/jmbi.2000.4265. PMID: 11162109.
- Fenn S, Schiller CB, Griese JJ, Duerr H, Imhof-Jung S, Gassner C, Moelleken J, Regula JT, Schaefer W, Thomas M, et al. Crystal structure of an anti-Ang2 CrossFab demonstrates complete structural and functional integrity of the variable domain. PLoS One. 2013;8:e61953. PMID: 23613981. doi:10.1371/journal.pone.0061953.
- Long NE, Sullivan BJ, Ding H, Doll S, Ryan MA, Hitchcock CL, Martin EW Jr., Kumar K, Tweedle MF, Magliery TJ. Linker engineering in anti-TAG-72 antibody fragments optimizes biophysical properties, serum half-life, and high-specificity tumor imaging. J Biol Chem. 2018;293:9030–40. doi:10.1074/jbc.RA118.002538. PMID: 29669811.
- Toughiri R, Wu X, Ruiz D, Huang F, Crissman JW, Dickey M, Froning K, Conner EM, Cujec TP, Demarest SJ. Comparing domain interactions within antibody Fabs with kappa and lambda light chains. mAbs. 2016;8:1276–85. doi:10.1080/19420862.2016.1214785. PMID: 27454112.
- Roberts CJ. Protein aggregation and its impact on product quality. Curr Opin Biotechnol. 2014;30:211–17. doi:10.1016/j.copbio.2014.08.001. PMID: 25173826.
- Jiskoot W, Randolph TW, Volkin DB, Middaugh CR, Schoneich C, Winter G, Friess W, Crommelin DJ, Carpenter JF. Protein instability and immunogenicity: roadblocks to clinical application of injectable protein delivery systems for sustained release. J Pharm Sci. 2012;101:946–54. doi:10.1002/jps.23018. PMID: 22170395.
- Pham NB, Meng WS. Protein aggregation and immunogenicity of biotherapeutics. Int J Pharm. 2020;585:119523. doi:10.1016/j.ijpharm.2020.119523. PMID: 32531452.
- Arndt KM, Muller KM, Pluckthun A. Helix-stabilized Fv (hsFv) antibody fragments: substituting the constant domains of a Fab fragment for a heterodimeric coiled-coil domain. J Mol Biol. 2001;312:221–28. doi:10.1006/jmbi.2001.4915. PMID: 11545598.
- Asial I, Cheng YX, Engman H, Dollhopf M, Wu B, Nordlund P, Cornvik T. Engineering protein thermostability using a generic activity-independent biophysical screen inside the cell. Nat Commun. 2013;4:2901. doi:10.1038/ncomms3901. PMID: 24352381.
- Gil D, Schrum AG. Strategies to stabilize compact folding and minimize aggregation of antibody-based fragments. Advances in bioscience and biotechnology. 2013;4:73–84. 10.4236/abb.2013.44A011. PMID: 25635232.
- Monsellier E, Bedouelle H. Improving the stability of an antibody variable fragment by a combination of knowledge-based approaches: validation and mechanisms. J Mol Biol. 2006;362:580–93. doi:10.1016/j.jmb.2006.07.044. PMID: 16926023.
- Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annual review of chemical and biomolecular engineering. Annu Rev Chem Biomol Eng. 2012;3:263–86. PMID: 22468604. doi:10.1146/annurev-chembioeng-062011-081052.
- Tiller KE, Tessier PM. Advances in Antibody Design. Annu Rev Biomed Eng. 2015;17:191–216. doi:10.1146/annurev-bioeng-071114-040733. PMID: 26274600.
- Zhao JX, Yang L, Gu ZN, Chen HQ, Tian FW, Chen YQ, Zhang H, Chen W. Stabilization of the single-chain fragment variable by an interdomain disulfide bond and its effect on antibody affinity. Int J Mol Sci. 2010;12:1–11. doi:10.3390/ijms12010001. PMID: 21339972.
- Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci U S A. 1993;90:7538–42. doi:10.1073/pnas.90.16.7538. PMID: 8356052.
- Jung SH, Pastan I, Lee B. Design of interchain disulfide bonds in the framework region of the Fv fragment of the monoclonal antibody B3. Proteins. 1994;19:35–47. doi:10.1002/prot.340190106. PMID: 8066084.
- Kugler M, Stein C, Schwenkert M, Saul D, Vockentanz L, Huber T, Wetzel SK, Scholz O, Pluckthun A, Honegger A, et al. Stabilization and humanization of a single-chain Fv antibody fragment specific for human lymphocyte antigen CD19 by designed point mutations and CDR-grafting onto a human framework. Protein Eng Des Sel. 2009;22:135–47. PMID: 19188138. doi:10.1093/protein/gzn079.
- Schaefer JV, Pluckthun A. Transfer of engineered biophysical properties between different antibody formats and expression systems. Protein Eng Des Sel. 2012;25:485–506. doi:10.1093/protein/gzs039. PMID: 22763265.
- Weatherill EE, Cain KL, Heywood SP, Compson JE, Heads JT, Adams R, Humphreys DP. Towards a universal disulphide stabilised single chain Fv format: importance of interchain disulphide bond location and vL-vH orientation. Protein Eng Des Sel. 2012;25:321–29. doi:10.1093/protein/gzs021. PMID: 22586154.
- Nesspor TC, Kinealy K, Mazzanti N, Diem MD, Boye K, Hoffman H, Springer C, Sprenkle J, Powers G, Jiang H, et al. High-Throughput generation of bipod (Fab × scFv) bispecific antibodies exploits differential chain expression and affinity capture. Sci Rep. 2020;10:7557. PMID: 32372058. doi:10.1038/s41598-020-64536-w.
- Spreter Von Kreudenstein T, Lario PI, Dixit SB. Protein engineering and the use of molecular modeling and simulation: the case of heterodimeric Fc engineering. Methods. 2014;65:77–94. doi:10.1016/j.ymeth.2013.10.016. PMID: 24211748.
- Fransson J, Teplyakov A, Raghunathan G, Chi E, Cordier W, Dinh T, Feng Y, Giles-Komar J, Gilliland G, Lollo B, et al. Human framework adaptation of a mouse anti-human IL-13 antibody. J Mol Biol. 2010;398:214–31. PMID: 20226193. doi:10.1016/j.jmb.2010.03.004.
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–17. doi:10.1016/0022-2836(87)90412-8. PMID: 3681981.
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015;22:953–58. doi:10.1038/nsmb.3129. PMID: 26595420.
- Harris LJ, Larson SB, Hasel KW, McPherson A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry. 1997;36:1581–97. doi:10.1021/bi962514. PMID: 9048542.
- Zhang RM, Snyder GH. Dependence of formation of small disulfide loops in two-cysteine peptides on the number and types of intervening amino acids. J Biol Chem. 1989;264:18472–79. PMID: 2808384 https://www.ncbi.nlm.nih.gov/pubmed/2808384.
- Shi L, Wheeler JC, Sweet RW, Lu J, Luo J, Tornetta M, Whitaker B, Reddy R, Brittingham R, Borozdina L, et al. De Novo selection of high-affinity antibodies from synthetic fab libraries displayed on phage as pIX fusion proteins. J Mol Biol. 2010;397:385–96. PMID: 20114051. doi:10.1016/j.jmb.2010.01.034.
- Gerhardt S, Abbott WM, Hargreaves D, Pauptit RA, Davies RA, Needham MR, Langham C, Barker W, Aziz A, Snow MJ, et al. Structure of IL-17A in complex with a potent, fully human neutralizing antibody. J Mol Biol. 2009;394:905–21. doi:10.1016/j.jmb.2009.10.008. pii PMID: 19835883.
- Alberola-Ila J, Places L, de la Calle O, Romero M, Yague J, Gallart T, Vives J, Lozano F. Stimulation through the TCR/CD3 complex up-regulates the CD2 surface expression on human T lymphocytes. J Immunol. 1991;146:1085–92. PMID: 1671400 https://www.ncbi.nlm.nih.gov/pubmed/1671400.
- Teplyakov A, Obmolova G, Malia TJ, Luo J, Muzammil S, Sweet R, Almagro JC, Gilliland GL. Structural diversity in a human antibody germline library. mAbs. 2016;8:1045–63. doi:10.1080/19420862.2016.1190060. PMID: 27210805.
- Pessano S, Oettgen H, Bhan AK, Terhorst C. The T3/T cell receptor complex: antigenic distinction between the two 20-kd T3 (T3-delta and T3-epsilon) subunits. Embo J. 1985;4:337–44. PMID: 2410254 https://www.ncbi.nlm.nih.gov/pubmed/2410254.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21. doi:10.1093/protein/9.7.617. PMID: 8844834.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. mAbs. 2020;12:1743053. PMID: 32249670. doi:10.1080/19420862.2020.1743053.
- Gekko K, Kimoto A, Kamiyama T. Effects of disulfide bonds on compactness of protein molecules revealed by volume, compressibility, and expansibility changes during reduction. Biochemistry. 2003;42:13746–53. doi:10.1021/bi030115q. PMID: 14622021.
- Betz SF. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2:1551–58. doi:10.1002/pro.5560021002. PMID: 8251931.
- Dani VS, Ramakrishnan C, Varadarajan R. MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 2003;16:187–93. doi:10.1093/proeng/gzg024. PMID: 12702798.
- Kabsch W. Xds. Acta crystallographica section D, biological crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. PMID: 20124692. doi:10.1107/S0907444909047337.
- Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;53:240–55. doi:10.1107/S0907444994003112.
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–82. PMID: 11567148 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11567148.
- Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Radiat. 2004;11:53–55. http://scripts.iucr.org/cgi-bin/paper?S090904950302413010.1107/S0909049503024130
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi:10.1107/S0907444910007493. PMID: 20383002.