
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Immune checkpoint inhibitors that overcome T cell suppressive mechanisms in tumors have revolutionized the treatment of cancer but are only efficacious in a small subset of patients. Targeting suppressive mechanisms acting on innate immune cells could significantly improve the incidence of clinical response by facilitating a multi-lineage response against the tumor involving both adaptive and innate immune systems. Here, we show that intra-tumoral interleukin (IL)-38 expression is a feature of a large frequency of head and neck, lung and cervical squamous cancers and correlates with reduced immune cell numbers. We generated IMM20324, an antibody that binds human and mouse IL-38 proteins and inhibits the binding of IL-38 to its putative receptors, interleukin 1 receptor accessory protein-like 1 (IL1RAPL) and IL-36R. In vivo, IMM20324 demonstrated a good safety profile, delayed tumor growth in a subset of mice in an EMT6 syngeneic model of breast cancer, and significantly inhibited tumor expansion in a B16.F10 melanoma model. Notably, IMM20324 treatment resulted in the prevention of tumor growth following re-implantation of tumor cells, indicating the induction of immunological memory. Furthermore, exposure of IMM20324 correlated with decreased tumor volume and increased levels of intra-tumoral chemokines. Together, our data suggest that IL-38 is expressed in a high frequency of cancer patients and allows tumor cells to suppress anti-tumor immunity. Blockade of IL-38 activity using IMM20324 can re-activate immunostimulatory mechanisms in the tumor microenvironment leading to immune infiltration, the generation of tumor-specific memory and abrogation of tumor growth.
Introduction
An overwhelming amount of data demonstrate that cancer cells hijack normal physiological mechanisms to inhibit antitumor immune responses in the tumor microenvironment (reviewed in ref.Citation1). T cell-targeted immune checkpoint inhibitors, such as anti-CTLA-4, anti-PD-1 and anti-PD-L1, can reverse such inhibitory mechanisms and show great promise in a wide spectrum of cancer types.Citation2 Despite remarkable clinical responses to these therapies within individual patients, most do not experience meaningful or sustained efficacy, either due to limited infiltration of immune cells, low tumor mutational burden (reviewed in ref.Citation3) or the lack of a sustained immune response.Citation4 Therefore, identification of additional pathways that restrain anti-tumor immune responses and can be modulated to deliver differentiated efficacy, either alone or in combination with current immune checkpoint inhibitors, is urgently needed. One of the most promising areas of research is focused on the reversal of inhibitory mechanisms that regulate innate immune cell populations, particularly macrophages and dendritic cell populations, which orchestrate adaptive immune responses and lymphocyte recruitment within the tumor microenvironment.Citation5 In light of the limited success observed with systemic dosing of broad innate immune activators TLR7/9Citation6 or STING agonists,Citation7,Citation8 focused therapies that release tumor-dependent suppressive mechanisms would be preferred due to the decreased risk of life-threatening systemic pro-inflammatory responses.
The significance of B cell responses in cancer is an active area of investigation, and multiple studies associate their presence in the tumor with a positive prognosis. A number of studies have demonstrated the presence of tumor-specific antibodies within the B cell repertoires of oncology patients.Citation9,Citation10 Thus, we interrogated patient memory B cell repertoires to identify novel cancer targets using Immunome’s internal discovery platform, which combines high throughput hybridoma generation and multiplexed cell-based screening to capture the memory B cell repertoire of treatment-naïve human cancer patients.Citation11,Citation12 Identification of antigens recognized by the patient’s humoral immune system facilitates the development of therapeutic antibodies against cancer-associated antigens on malignant cells and within the tumor microenvironment. Analysis of the B cell repertoire from a treatment-naïve, stage IIb head and neck cancer patient meeting our sample selection criteria yielded an antibody specific for interleukin (IL)-38 that was identified by phenotypic screening and protein microarray.
Interleukin IL-38 is a member of the IL-1 family of cytokines, which are comprised of 11 members (reviewed in ref.Citation13). IL-38 functions as an antagonist for IL-36R and IL-1 receptor accessory protein-like 1 (IL1RAPL1), limiting the activation of innate immune cells, and inhibiting the secretion of chemotactic and stimulatory factors that are critical for the induction of adaptive immunity.Citation14–16 IL-38 is predominantly expressed in skinCitation17 and has limited expression in other organs during normal tissue homeostasis (reviewed in ref.Citation15), but can be released by epithelial cells immediately following tissue injury to suppress inflammation. Consistent with a role in negatively regulating inflammation in the skin, IL-38 knockout mice demonstrate delayed resolution of imiquimod (IMQ)-induced psoriasis in an IL-17-dependent manner, which can be rescued with the administration of recombinant IL-38, but do not otherwise demonstrate an abnormal phenotype suggesting that IL-38 antagonism would be well-tolerated.Citation18 Similarly, IL-38 knockout mice are more sensitive to dextran sulfate sodium (DSS)-induced colitis compared to wild-type mice due to activation of the NLRP3 inflammasome, consistent with a role for IL-38 in immunosuppression following tissue insult.Citation19 Clinically, psoriatic patients exhibit increased levels of circulating IL-38 consistent with a role for IL-38 in controlling skin inflammation.Citation17 In the context of cancer, overexpression of IL-38 results in the acceleration of tumor growth in the Lewis Lung model of lung cancer in vivo and reduced intra-tumoral T cell infiltrates.Citation20 In lung adenocarcinoma patients, high IL-38 expression is associated with poor prognosis, correlates with PD-L1, and negatively correlates with CD8+ T cell density by histology.Citation20,Citation21 While the cellular source of IL-38 is unclear, the release of IL-38 is observed in vitro following apoptosis of tumor cells suggesting that IL-38 may function as a negative feedback loop to suppress intra-tumoral immunity.Citation16
Here, we describe the identification of IL-38 as a promising target in cancer and the development of a potent antibody therapeutic capable of blocking function. The identification of cancer patient-derived B cells targeting this protein prompted expression analyses demonstrating that IL-38 is frequently expressed in multiple cancer indications and is associated with immunologically cold tumors. Using a monoclonal antibody that binds to human and mouse IL-38, we show that IL-38 antagonism restrains tumor growth, enhances intra-tumoral immune activation, and leads to development of antitumor immunological memory in syngeneic mouse models of cancer. To our knowledge, this is the first report outlining IL-38 antagonism as a therapeutic intervention strategy to inhibit tumor growth and confer long-term protection against cancer.
Results
IL-38 is expressed in multiple cancers and correlates with poor immune infiltration
Using Immunome’s internal discovery platform,Citation11 an antibody clone, IMM20130, was identified from a patient-derived hybridoma library. This library was generated by the fusion of human memory B cells from the draining lymph node of a treatment-naïve head and neck cancer patient and screened for binding to tumor cell lines comprising multiple oncology indications. Target identification studies demonstrated that IMM20130 bound specifically to IL-38 among >21,000 native proteins spotted on human protein microarray (). Subsequent flow cytometry analyses showed that IMM20130 bound human cancer cell lines derived from multiple cancer indications (Suplementary Figure S1) including A549, which have been previously shown to secrete IL-38 under apoptotic conditions.Citation16 These studies suggest that IL-38 expression was not only a feature of the original patient but is also relevant to other patients across multiple cancer types.
Figure 1. IL-38 is targeted by B cells in cancer patients and is highly expressed in multiple tumor types. (a) Identification of IL-38 as the specific target of IMM20130 assessed by protein microarray. Binding signal and S-Scores shown for the top 200 proteins based on binding signal. (b) Assessment of IL-38 transcript expression in tumor and normal tissue using TCGA RNAseq data. Samples were normalized for IL-38 expression and subdivided based on high (brown) and low (green) expression using thresholds set according to a value of 10–15% of normal tissues. Numbers in each plot represent frequency of IL-38 positive samples in all tumor samples using this threshold. (c) Immunohistochemical analysis of IL-38 expression in three cancer types. Head and neck squamous carcinoma (HNSC), lung tumors comprising multiple subtypes including adenocarcinoma and squamous cell carcinoma, and cervical squamous carcinoma (CESC) were stained with IMM20324 or isotype control antibodies to detect IL-38 expression. Pathological scoring was performed according to staining density. Representative examples of specimens with a range of expression intensity within each indication are shown. (d) Average intensity of IL-38 expression across cancer indications tested. Intensity scoring analysis was performed using a 7-point scale, with 0 corresponding to no expression and 6 being strong staining. Scoring was performed in both cytoplasmic and nuclear compartments and averaged to calculate final scores.
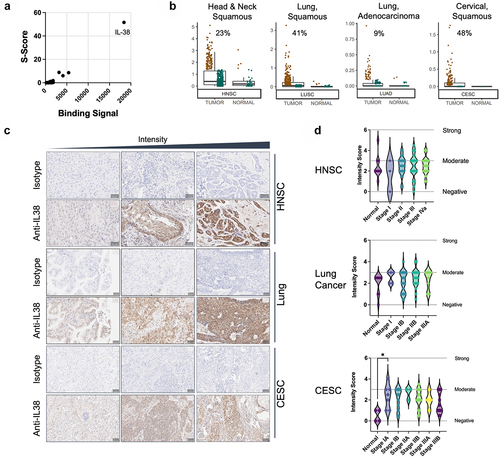
To confirm IL-38 expression in primary patient tumors, we examined IL-38 mRNA expression data from The Cancer Genome Atlas (TCGA). IL-38 expression was significantly higher in head and neck squamous carcinoma (HNSC) compared to normal tissues (), confirming the relevance of IL-38 in the cancer type in which it was initially identified. Interestingly, IL-38 transcripts were also detectable in multiple squamous tumor types among the 30 types tested. In lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and cervical squamous cell carcinoma (CESC), significant IL-38 expression was observed in >40% of samples tested (). Expression was also observed in esophageal carcinoma and skin cutaneous melanoma (Supplementary Figure S2). Interestingly, carcinomas with squamous cell morphology exhibited the highest IL-38 expression and highest frequency of IL-38 positive patient samples.
To confirm the TCGA expression and further investigate the prevalence of IL-38 in cancer, we assessed IL-38 protein expression in tumor microarrays comprising primary HNSC, lung (both LUAD and LUSC) and CESC primary tumor sections. Our internal antibody, IMM20324, was used for detection of IL-38 in primary tumor sections, which were scored for expression intensity using a scale of 0 (negative) to 6 (strong) across clinical stages (). In all three types of cancer tested, IL-38 was expressed in the majority of tumors. For head and neck samples, only 2 of 70 sections were negative for IL-38 and the remainder exhibited weak to moderate expression. For both lung (n = 68) and cervical squamous cancers (n = 70), all tumors showed some level of expression at a weak to moderate intensity. Interestingly, expression was observed both in the cytoplasmic and nuclear compartments, similar to other IL-1 family members.Citation22,Citation23 IL-38 expression was also detected in some normal tissues tested, including normal lung and head and neck tissues, suggestive of a role in normal tissue homeostasis and in alignment with previous reports.Citation15 In CESC, IL-38 expression in normal cervical tissue was lower compared to cervical tumors, with expression in stage I CESC tumor expression being significantly higher than normal tissue (p = 0.0396). Overall, these data demonstrate that IL-38 expression is a feature of the tumor microenvironment in a large percentage of patients across multiple indications.
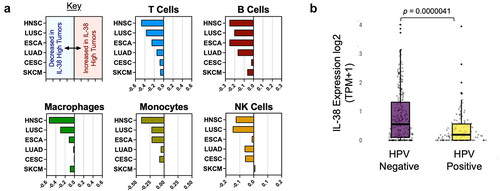
Recent studies found that IL-38 expression is associated with the suppression of inflammationCitation24 and can modulate innate and adaptive immunity.Citation16,Citation20 We, therefore, used datasets from the TCGA to identify the effects of IL-38 expression on immune cell lineage-specific gene expression. We saw that high IL-38 expression was associated with low ImSig lineage-specific enrichment scores, previously defined by Nirmal et al.Citation25 This was true for multiple lineages, including T cells, B cells, natural killer (NK) cells, monocytes, and dendritic cells (DCs), and consistent with the proposed role of IL-38 in suppressing immune infiltration (). Moreover, these negative associations were evident in multiple tumor types, and were more marked in head and neck, lung squamous, and esophageal cancers, where a high incidence of IL-38 expression was noted.
Figure 2. IL-38 expression is associated with reduced immune infiltration and HPV status in head and neck squamous carcinoma. (a) Signature analysis of immune lineages in IL-38 high versus IL-38 low head and neck tumors. Negative values represent reduced expression of lineage-associated genes and lower infiltration. Positive values represent increased infiltration of indicated lineages. HNSC: head and neck squamous cell carcinoma; LUSC: lung squamous cell carcinoma; ESCA: esophageal carcinoma: LUAD: Lung adenocarcinoma; CESC: cervical squamous cell carcinoma and endocervical adenocarcinoma: SKCM: skin cutaneous melanoma. (b) IL-38 expression in HPV-negative and HPV-positive populations of head and neck cancers.
To validate the findings from the TCGA dataset, additional analyses were performed using a proprietary commercial dataset.Citation26 Among 67 cancer subtypes profiled, gastroesophageal squamous carcinoma, HNSC, and LUSC were confirmed as top cancer types with high frequency of IL-38 positive samples (). Proprietary immune cell population-specific gene setsCitation27 were utilized to estimate the infiltration of different immune cell populations in the tumor samples. Consistent with the TCGA analysis and previous reports,Citation20 IL-38 mRNA expression was negatively correlated with CD8+ T cells, macrophages, and NK cells in HNSC, LUSC and gastroesophageal squamous carcinoma ().
Table 1. IL-38 expression in selected cancer types using Tempus data.
Table 2. IL-38 expression is associated with immune cell infiltration in selected cancers.
Human papillomavirus (HPV) status of head and neck cancer is a major determinant of immune activity, with negative HPV status correlating with lower immune activity and less immune infiltration.Citation28 Therefore, we investigated IL-38 expression in HPV− and HPV+ HNSC tumors using the proprietary commercial dataset.Citation26 We found that IL-38 was significantly higher (p = 0.0000041) in HPV− tumors, consistent with IL-38 expression being associated with lower immune activity ().
Taken together, the systematic assessment of patient-derived memory B cell specificity led to the discovery of IL-38 as a protein of interest in a patient with head and neck cancer. Subsequent analyses demonstrated highly penetrant IL-38 expression across multiple cancer subtypes and a negative association with pan-immune cell infiltration. These findings are consistent with a role for IL-38 as an immunosuppressive cytokine regulating both innate and adaptive arms of the immune system.
Generation and development of anti-IL-38 antibodies
To identify neutralizing anti-IL-38 antibodies with therapeutic potential, mouse hybridomas were generated from B cells isolated from the spleens of CD-1 mice immunized with recombinant human IL-38. Among multiple IL-38 binders, one IgG2a antibody was selected due to its high affinity to human IL-38, in vitro neutralization potency, and binding to IL-38 from multiple species, including murine IL-38. Similar to our patient-derived antibody, this clone exhibited strong specificity for IL-38 in a native human protein microarray with no binding to other IL-1 family members or other non-related proteins and was designated IMM20331 (). IMM20331 immunoglobulin (Ig) heavy- and light-chain sequences were recombinantly expressed using a wild-type mouse Fc IgG2a construct to generate IMM20324, a recombinant version of the hybridoma-derived antibody. Binding analysis of IMM20331 antibody purified from hybridoma supernatant demonstrated equivalent human IL-38 binding compared to its recombinant counterpart IMM20324 in a direct IL-38 ELISA (Supplementary Figure S3) and is subsequently referred to as IMM20324H for clarity. Both versions of IMM20324 were used for investigating function.
Figure 3. IMM20324 is specific for IL-38, binds human and mouse protein with different affinities and neutralizes binding to putative human IL-38 receptors. (a) Identification of IL-38 as the specific target of IMM20324H assessed by protein microarray. Binding signal and S-scores shown for the top 200 proteins based on binding signal. (b) (Top) Binding of recombinantly produced IMM20324 mIgG2a antibody to recombinant human and mouse IL-38 using BLI. Two-fold titrations (1.52–0.095 µM) were performed to generate KD values for binding to species-specific proteins. (Bottom) Values shown are average of two experiments. (c) ELISA-based binding analysis of IMM20324 to recombinant full-length human and mouse IL-38 using direct ELISA. (d) Flow cytometry-based binding to human and mouse IL-38 overexpressing CHO cells. Cells were fixed and permeabilized to assess binding of intracellular IL-38. Mock-transfected controls, in addition to isotype staining controls for human (gray) and mouse (black) cells also shown on graph. (e) Inhibition of IL-38 receptor binding to its receptors. Inhibition of binding to IL-36R is shown in the circles, IL1RAPL1 is shown in squares. Isotype control-treated samples are in blue, while samples treated with IMM20324 are shown in red.
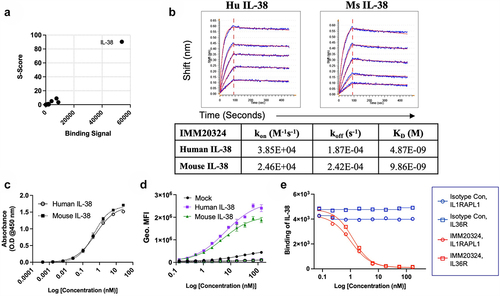
Binding of IMM20324 antibody to human and mouse IL-38 was characterized using several methods. Binding to soluble recombinant human and murine IL-38 proteins was tested using biolayer interferometry (BLI) on a Gator Plus instrument. IMM20324 was loaded onto anti-mouse Fc biosensors and dipped into wells containing either human- or mouse-soluble IL-38 protein. IMM20324 bound to recombinant human and mouse IL-38 proteins with comparable affinity (KD = 4.87 and 9.86 nM, respectively). A slightly faster Kon and slower Koff rates of the IMM20324 interaction with human IL-38 resulted in KDs within two-fold difference between human and mouse IL-38 (). Comparable binding of IMM20324 to human and mouse proteins was also confirmed in a plate-bound ELISA () and intracellular flow cytometry-based binding experiments using HEK293 cells overexpressing human and mouse IL-38 (). In these experiments, relative affinity to mouse and human IL-38 was similar. Thus, data from multiple assays confirmed IMM20324 binds to mouse IL-38 protein.
Strikingly, IMM20324 readily neutralized binding of recombinant human IL-38 to its putative receptors IL-1RAPL1 and IL-36R.Citation29 Addition of IMM20324 inhibited IL-38 interaction with plate-bound human IL1RAPL1-Fc and human IL-36R-Fc in a dose-dependent manner, with IC50 values of 1.267 nM and 1.4667 nM, respectively (). Together, these data demonstrate that IMM20324 specifically and selectively binds to human and mouse IL-38, and prevents binding of IL-38 to its putative receptors.
IMM20324H demonstrates a favorable PK profile and is well tolerated in non-tumor-bearing mice
Since IMM20324 demonstrated binding to mouse IL-38, fully immunocompetent syngeneic mouse models were used for in vivo testing. In order to accelerate in vivo assessment, IMM20324H antibody was used for testing. First, to assess pharmacokinetics (PK), non-tumor-bearing C57BL/6 mice were administered a single dose of 10 mg/kg IMM20324H either intravenous (IV) or intraperitoneal (IP) and serum concentration was determined by direct IL-38 ELISA at the indicated timepoint. A single IP 10 mg/kg dose of IMM20324H resulted in a Cmax of 80.131 µg/mL at 24 h and had a half-life of 146.566 h, while an IV dose resulted in a Cmax of 192.288 µg/mL at 5 min and had a half-life of 91 h ( and Supplementary Table S1). Repeated dosing of non-tumor bearing C57BL/6 mice was then performed over a period of 21 d using a weekly (QW) or twice-weekly (Q2W) dosing schedule. Compared to the single-dose PK profile, plasma levels of IMM20324H were further increased when multiple doses were administered using both dosing schedules (). Importantly, plasma concentrations of IMM20324H correlated with the dosing schedule, as the highest plasma levels were detected in groups that received higher and more frequent doses of IMM20324H.
Figure 4. IMM20324H exhibits favorable PK profile and is well tolerated in non-tumor-bearing mice. (a) In vivo pharmacokinetic analysis of IMM20324H following administration of antibody by intravenous (IV) and intraperitoneal (IP) routes performed in C57BL/6 mice. Dose shown is 10 mg/kg. (b) Analysis of serum IMM20324H concentrations on D21 following administration of antibody by IP injection. Groups were dosed once weekly (QW) or twice weekly (Q2W) with the labeled dose in mg/kg. (c) Average body weight of animals from each treatment group was monitored throughout the duration of the study (d) Average spleen weights of animals comprising each treatment group were assessed upon study termination on D21. (e) Assessment of circulating cytokine levels in serum following administration of IMM20324H. Blood was sampled following study termination. Data presented as log2 transformation of the cytokine concentration+1. Dotted line indicates the lower limit of quantification (LLOQ) at 10 pg/mL.
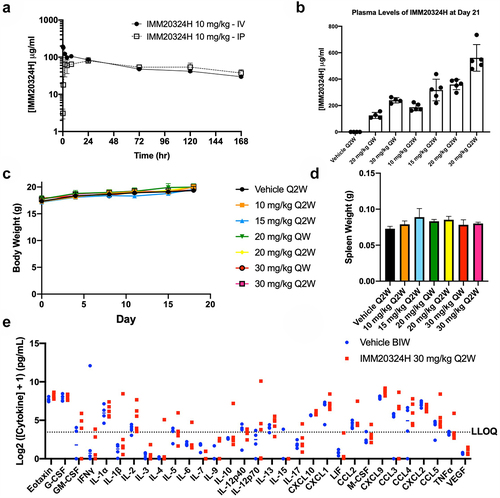
At this stage, it was also important to evaluate whether IL-38 blockade leads to systemic inflammation in naïve mice, given that IL-38 knockout mice demonstrated an inability to resolve induced inflammation in disease models. Following dosing in naïve animals, IMM20324H was well tolerated, as all mice maintained normal body weight throughout the study (). On D21, mice were euthanized and gross analysis of the organs revealed no obvious abnormalities (data not shown), including enlargement of the spleen or gross skin pathology (). Finally, to determine whether any systemic upregulation of inflammatory cytokines occurred in IMM20324H-treated mice following neutralization of IL-38, a Luminex-based multiplex analysis of plasma was performed in the highest dose group. Importantly, no significant dose-dependent induction of inflammatory cytokines compared to vehicle controls was observed () supporting observations in naïve IL-38 knockout miceCitation30 that loss of function does not induce systemic inflammation. Together, these data demonstrate that IMM20324H has a PK profile of a typical IgG antibody following both IV and IP administration and does not lead to immunostimulatory effects that could be associated with inhibition of an immunosuppressive pathway.
Anti-IL-38 treatment induces complete tumor regression in a subset of EMT6 tumor-bearing mice and protects against tumor rechallenge
Ex vivo syngeneic mouse models were screened for IL-38 expression by immunohistochemistry (IHC) and the EMT6 breast cancer model was selected for in vivo efficacy studies based on detectable IL-38 expression (Supplementary Figure S4). Mice were dosed with IMM20324H twice weekly for 3 weeks. IMM20324H treatment did not lead to a significant reduction in mean tumor volume up to D17, the final day which all mice were on study (). However, a subset of mice from each treatment group clearly responded to therapy, with 30% of IMM20324H-treated tumors exhibiting a 70% reduction in tumor volume compared to the mean of the vehicle group. This strong response in a subset of mice was evident when looking at tumor volume throughout the duration of the study (). Linear mixed effects (LME) modeling revealed that this strong response in 30% of mice was sufficient to achieve a significant difference at all three doses tested (). Upon study completion, tumors had completely regressed in 4 of 30 (13.3%) treated with IMM20324H (). To determine whether anti-IL-38 treatment induced a protective anti-tumor memory response, these mice were rechallenged with EMT6 tumor cells in the opposite flank without additional antibody dosing. Strikingly, complete regression of secondary tumors was observed in anti-IL-38-treated responding mice () that resulted in a marked increase in survival compared to naïve age-matched controls (). Importantly, tumors from this rechallenge were initially detectable, indicating successful inoculation and seeding of tumor cells in recipient mice.
Figure 5. Targeting of IL-38 inhibits tumor growth and elicits immunological memory against EMT6 breast tumors in vivo. (a–e) Mice were randomized at D1 based on tumor volume and treated with three different doses of IMM20324H or a PBS vehicle control twice per week for 6 total doses. (a) Mean tumor volume up to D17 (b) Tumor volume on D17, the final day all mice were on study. (c) Individual growth curves for each mouse in the indicated group up to D45. (d–e) EMT6 cells were implanted into the opposite flank of IMM20324H-treated mice following full regression of the primary tumor. No additional antibody was administrated during secondary challenge. (d) Secondary EMT6 tumor growth curves compared to untreated age-matched mice. (e) Survival curves of rechallenged mice showing % survival per group. *p <0.05.
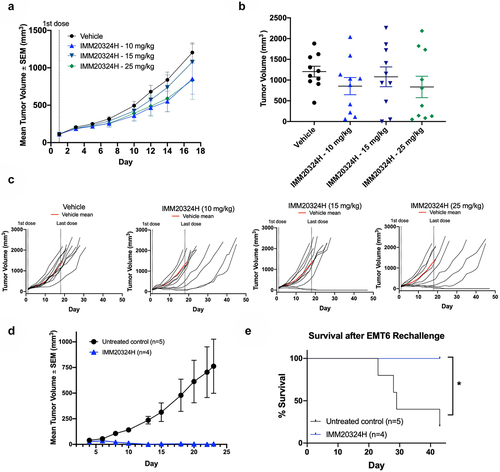
Table 3. Linear mixed effects modeling of IMM20324H treatment in the EMT6 syngeneic model.
Taken together, these data indicate that anti-IL-38 treatment can suppress tumor growth in a syngeneic model of breast cancer and that successful responses are associated with the generation of immunological memory that inhibits subsequent tumor regrowth in the absence of additional therapy.
IMM20324 inhibits tumor growth in the B16.F10 syngeneic mouse model and induces immunostimulatory chemokine expression in the tumor microenvironment
For subsequent in vivo studies, the B16.F10 model was selected based both on positive IL-38 expression (Supplementary Figure S4), and because anti-PD-1/PD-L1 treatments demonstrate limited efficacy in this immunologically cold tumor model.Citation31 Mice were treated with IMM20324 IP every 3days starting when tumor volume reached 60–65 mm3. Treatment with IMM20324 at both 10 and 50 mg/kg resulted in a reduction of mean tumor volume throughout the study (). At D 10, the final day that all mice were on study, IMM20324 treatment resulted in a statistically significant reduction in mean tumor volume in both 10 mg/kg (64.4% reduction) and 50 mg/kg (55.9% reduction) groups (). Furthermore, this delayed tumor growth persisted beyond D10, as indicated in the spider-grams of individual tumors (). To assess the antitumor effect of IMM20324 throughout the duration of the study, tumor growth curves were generated for each animal using an exponential model of growth. These curves were used to assess the growth rate of tumors, which was significantly inhibited by IMM20324 treatment (). This reduction in tumor growth rate also led to delayed progression of tumors to 1500 mm3, which was significantly reduced in the 10 mg/kg IMM20324 treatment (). Lastly, analysis using an LME model to compare isotype control group and IMM20324 groups revealed that treatment at 10 mg/kg significantly impeded tumor growth compared to the isotype control group (), corroborating the results observed in the EMT6 syngeneic model.
Figure 6. IMM20324 inhibits growth of B16.F10 tumors in vivo. (a–e) Mice were randomized at D0 based on tumor volume and treated with 10 or 50 mg/kg dose of IMM20324 or isotype control intraperitoneally every 3days for six total doses. Tumor volumes were measured every 3 days. Tumor growth curves were fitted with exponential model. (a) Mean tumor volume up to D 10. (b) Interim tumor volumes of individual animal on D10. (c) Growth curves of individual animal from the indicated group over the duration of the study. (d) Calculated average tumor growth rate over the duration of study. (e) Calculated days when tumor volume reaches 1500 mm3 based on fitting of tumor growth curve. * p < 0.05, ** p < 0.01, ns = not significant.
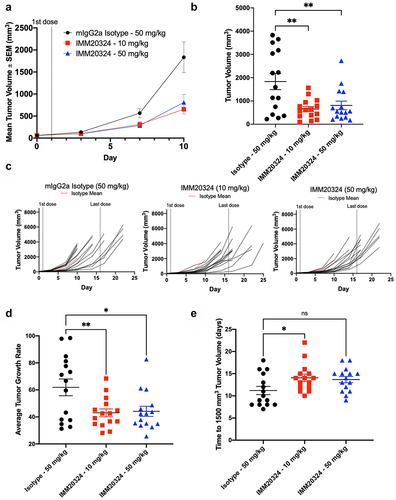
Table 4. Linear mixed effects modeling of IMM20324 treatment in the B16.F10 syngeneic model.
To evaluate the pharmacodynamic and PK profile of IMM20324 in tumor-bearing mice and investigate the mechanism of action, IMM20324 dosing was performed at 3 mg/kg, 10 mg/kg, and 30 mg/kg in an additional study. Single-dose IP injection of IMM20324 in B16.F10 tumor-bearing mice demonstrated a dose-dependent increase in exposure, with a Cmax of 33, 66, and 333 ug/mL after a 3, 10, and 30 mg/kg dose, respectively (Supplementary Table S2 and ). After dosing every 3 days for four repeated doses, IMM20324 showed a dose-dependent increase of exposure in terminal serum 24hours post last dose (). Notably, the final tumor volumes had a significant negative correlation with terminal exposures of IMM20324.
Figure 7. IMM20324 treatment decreases tumor volume and increases intra-tumoral chemokines in a dose-dependent manner. (a) B16.F10 tumor-bearing mice given a single dose of IMM20324 at 3, 10, or 30 mg/kg and mice were bled at indicated timepoints. IMM20324 exposure was measured by ELISA and plotted over time (n = 3 mice per timepoint). (b) Mice were randomized at D0 based on tumor volume and treated indicated dose of IMM20324 or isotype control intraperitonially every 3 days for four total doses. Mice were taken down on D11, 24 hours after the final dose. Terminal exposure of IMM20324 was plotted against tumor volume on D11. (c) Intra-tumoral chemokine concentrations detected by Luminex analysis. Mice in all IMM20324 treatment groups are shown. r and p values were calculated using Pearson correlation co-efficients.
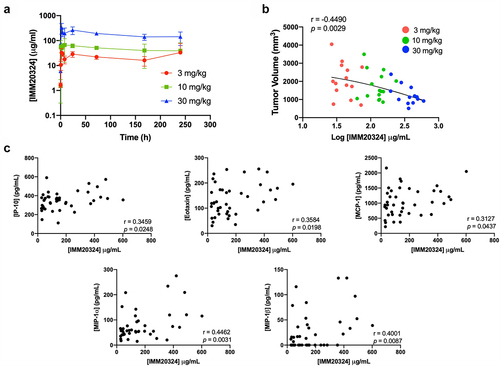
Luminex analysis was then performed on tumor lysates to investigate the relationship between reduced tumor size and intra-tumoral immune activity. These data demonstrated that concentrations of multiple chemokines showed significant positive correlation with exposure of IMM20324, including MIP-1α/CCL3 and MIP-1β/CCL4 (p < 0.01) and IP-10/CXCL10, Eotaxin/CCL11, and MCP-1/CCL2 (p < 0.05) (). Thus, reduction in tumor growth is associated with increased intra-tumoral chemokines upon IMM20324 treatment.
Overall, these data demonstrate that anti-IL-38 treatment leads to reduced tumor growth in the immunologically cold B16.F10 melanoma syngeneic tumor model, and that increased intra-tumoral IMM20324 concentrations correlate with a dose-dependent induction of immune activity.
Discussion
In this report, we describe the generation and testing of IMM20324, an antibody targeting a novel immunosuppressive cytokine that controls activation of the myeloid cell compartment and is involved in orchestrating the early phases of immune responses. IMM20324 binds to human and mouse IL-38 and robustly inhibits binding to its putative receptors IL-36R and IL1RAPL1, leading to in vivo inhibition of tumor growth and production of immune-stimulatory chemokines. Although IL-38 has mostly been studied in the context of innate immunity, neutralization of IL-38 modulates long-term immunity against tumor-specific antigens after reimplantation. The identification of IL-38 through the systematic interrogation of cancer patient antibody repertoires demonstrates its utility as a tool for the discovery of novel targets involved in the maintenance and evolution of tumors.
We identified IL-38 as an oncology target through the isolation of an anti-IL-38-specific memory B cell from the lymph node of a head and neck cancer patient. Since IL-38 has been detected systemically in patients with autoimmune disease,Citation32 it is unclear why normal tolerance mechanisms allowed this clone to escape positive and negative selection by the patient’s immune system. Nonetheless, our RNA expression profiling of primary tumors not only demonstrated high incidence of IL-38 expression in head and neck cancer but also substantial expression in several other indications, including lung, esophageal and cervical cancers. There is precedence for expression of IL-38 in cancer,Citation20,Citation21 but we believe that ours is the first study to demonstrate high prevalence of IL-38 expression in HNSC, lung, esophageal and CESC. Our RNA expression analysis identified high IL-38 expression in squamous cancers. While the reason for this is not clear, high IL-38 levels may reflect the constitutive expression in the epithelial cells from which the tumor cells are derived. Previously reported expression of IL-38 in tissues associated with barrier function, such as skin,Citation17 colonic epithelium,Citation33 and salivary glandCitation34 indicate that IL-38 may be constitutively expressed in these organs for immediate release if required. This notion aligns with data demonstrating that IL-38 can reduce inflammatory cytokine production by keratinocytesCitation17 and respiratory epithelial cells,Citation35 and juxtaposes with in vivo data in mouse models of psoriasisCitation18 and asthmaCitation36 where IL-38 treatment ameliorates disease. Additional evidence for an immunoregulatory protective effect for IL-38 comes from studies demonstrating IL-38 upregulation following trauma. For example, IL-38 is upregulated following ischemic strokeCitation37 where higher IL-38 serum levels correlate with improved prognosis, providing anecdotal evidence that IL-38 mediates protection of tissues immediately following trauma.Citation38 Our data investigating the expression of IL-38 in tumors extend current literature and suggest that, through the adoption of processes used during normal tissue homeostasis, cancer cells exploit IL-38-dependent mechanisms to dampen immune activity within the tumor microenvironment and evade tumor-specific immune responses. It is also possible that transformation of cells that express IL-38 may confer a selective advantage through their resistance to immune attack. Additional work will be required to understand the relationship between IL-38 and sensitivity to immunosurveillance.
High expression of IL-38 led to a marked reduction in immune cell signatures in head and neck, lung squamous and esophageal tumors. This was also observed to a lesser extent in lung adenocarcinoma, where previous data demonstrated an inverse correlation between IL-38 expression and infiltrating T cells,Citation20 as well as cervical and skin cancer. The reason for this discrepancy is not clear, although IL-38 has been shown to lead to immune activation in some contexts.Citation16 Importantly, we did not observe increased infiltration of immune cells in IL-38 high tumors in any of the indications tested, in agreement with the immunosuppressive role of this cytokine. Moreover, the elevated levels of IL-38 in HPV− head and neck cancers, where patient prognosis is less favorable than their HPV+ counterparts,Citation39 allude to a negative correlation that could be exploited to improve outcomes within this patient population.
We saw a clear correlation between high IL-38 expression and reduced infiltration of multiple lineages including T, B, NK and myeloid cells. These observations align with previous data demonstrating that enforced IL-38 expression inhibits the migration of CD4 and CD8 T cells into tumors in vivo and that cases of lung adenocarcinoma with high IL-38 expression show reduced lymphocyte infiltration.Citation20 Weinstein et al.Citation40 also demonstrated that the transfer of IL-36γ-secreting cells into tumors induced the formation of tertiary lymphoid structures (TLS) supporting the notion that relief of IL-36 antagonistic mechanisms could aid the initiation of a coordinated immune response. Given the importance of TLS as indicators of good prognosis in solid tumorsCitation41,Citation42 and as predictors of response to immune-oncology (IO) therapy,Citation43,Citation44 it is feasible that strategies which induce formation of TLS in pre-clinical models will translate into robust clinical responses and improve patient outcomes. Due to their accelerated growth rates compared to tumors in human patients, syngeneic models are, however, not suitable for exploring mechanisms driving formation of TLS.Citation45 Extended analyses of expression in primary tumor samples, in tandem with identification of the specific cellular source of IL-38 in each may help elucidate the underlying basis of this observation.
IMM20324 was selected due to its exquisite specificity to IL-38, functional capacity to block both IL-36R- and IL1RAPL1-specific binding, and binding to mouse IL-38. IL-38-mediated inhibition of IL-36 binding to IL-36R prevents multiple events associated with the initiation of immunity including maturation of dendritic cells, M1 polarization of macrophages, and skewing of CD4+ T cells toward a TH1 phenotype.Citation46 IL-38 also inhibits IL1RAPL1 signaling of macrophages, reducing inflammatory cytokine production that leads to the secretion of IL-17 by T cells.Citation16,Citation18 We found that RNA expression of IL-36R and IL1RAPL1 receptors was elevated in squamous cancers, including CESC and lung tumors and that IL-36R expression correlated with IL-38 expression in HNSC, CESC and LUSC (data not shown), supporting the idea that modulation of signaling through these receptors could be relevant to tumor biology. We hypothesize that the combined effect of restoring stimulatory activity through both receptors will facilitate the initiation of a coordinated, robust immune response against the tumor. The evaluation of IL-38 neutralization in an immunocompetent mouse model was essential for studying these immune-stimulatory effects. However, given the approximately two-fold lower relative affinity for mouse versus human IL-38, we believe that our humanized version of IMM20324 will show higher potency in human subjects in the clinic. Importantly, IMM20324 demonstrated a favorable PK profile and was well-tolerated after administration of efficacious doses. Given there were no overt infection or immune related findings after prolonged IL-38 blockade, we hypothesize that there are other redundant mechanisms, such as interferons, tumor necrosis factor (TNF) or IL-17, maintaining homeostasis of skin and gut microbiome, in accordance with the lack of phenotype under unchallenged conditions in IL-38 knockout mice.Citation30
Since IL-38 functions as an inhibitory cytokine to suppress inflammation, we proposed that treatment with an anti-IL-38 antibody may be beneficial in an immunologically cold tumor model such as B16.F10, which is poorly immunogenic and is characterized by the absence of lymphocyte infiltration, as well as minimal macrophage and MDSC infiltration.Citation47 Indeed, treatment with IMM20324 led to significantly reduced B16.F10 tumor volume due to delayed tumor growth and increased levels of intra-tumoral cytokines. These findings were also observed in the EMT6 model, which has marginally more immune cell infiltration compared to the B16.F10 model.Citation47 In this model, complete inhibition of tumor growth was observed in multiple mice following anti-IL-38 treatment, while other mice within the group did not exhibit delayed tumor growth compared to the isotype control group. Whether the intra-group variation in responses seen in the EMT6 model is related to immune status prior to treatment, as has been shown for T cell-based checkpoint therapies,Citation48,Citation49 or differences in tumor cell susceptibility to IL-38-dependent mechanisms based on microenvironmental factors, will require further study. Similarly, whether the differences in response rates when comparing the two syngeneic models tested are reflective of distinct immune compositions, tumor model characteristics, or kinetic considerations related to differential growth rates in the two models cannot be determined in this study. However, these observations suggest that, similar to other currently available immune-oncology treatments, patient selection methods may be required for identification of patients that would benefit most from IL-38 blockade. Nonetheless, the complete regression observed in some mice afforded us the opportunity to assess the long-term effect of IL-38 blockade. The remarkable lack of tumor growth observed in any of the previously cured mice, even in the absence of subsequent treatment, suggests that the inhibition of IL-38 facilitated the generation of immune memory, rather than inducing short-term inflammation. As such, we postulate that IL-38 blockade rebalances the patient immune system in favor of normal anti-tumor activity.
Targeting of IL-38 is differentiated from current IO therapies in several ways. Therapies where the mechanism of action relies on the patients’ immune response to be sufficiently developed to be able to reactivate existing intra-tumoral T cells,Citation44,Citation50,Citation51 such as those targeting the PD-1/PD-L1 axis, demonstrate excellent clinical results in the patients who meet these criteria, but are ineffective in those where their intrinsic immune response is nascent or absent. Anti-IL-38 therapeutics may share similar limitations. However, since we postulate that IL-38 suppression targets myeloid cells, which act upstream of T cells during the execution of a physiological immune response, IL-38 modulation could provide treatment options for a greater cross-section of patients, including those without pre-existing adaptive antitumor responses. Further, beyond IL-38 monotherapy, blockade of this pathway could be used in combination with other checkpoint therapies to kickstart adaptive immunity and increase the probability of favorable responses in these patients. Interestingly, PD-L1+ lung adenocarcinomas have higher IL-38 expression than PD-L1− tumors,Citation21 suggesting that targeting of multiple pathways may be necessary to overcome the combined inhibitory mechanisms that suppress immunity. Chemotherapy may induce IL-38 production from apoptotic cancer cells (ref.16 and data not shown). Therefore, combination of anti-IL-38 with chemotherapy could counteract IL-38-dependent resistance mechanisms linked to decreased immune cell infiltration. This may be further enhanced by combinations with other immune modulatory therapies. In contrast to other therapies targeting innate immune cells with agonists of innate receptors (reviewed in ref.Citation52) that may have undesired side effects due to strong universal activation of immune cells, IL-38 blockade more closely parallels classical checkpoint inhibitors which target suppressive mechanisms and may therefore have a reduced risk of serious adverse events associated with clinical efficacy.
In summary, screening of memory B cells from the immune repertoire of a cancer patient identified an antibody against IL-38. Subsequent studies demonstrated that IL-38 is expressed in multiple cancer indications at high prevalence and is inversely correlated with immune cell infiltration, consistent with its reported role as a suppressor of immune activity. IMM20324 was highly selective for binding to IL-38 and blocked binding of IL-38 to its putative receptors. The ability to bind to mouse IL-38 facilitated testing in mouse syngeneic models where treatment led to inhibition of tumor growth. Dosing of tumor-naïve animals did not lead to measurable immune activation. Markedly, successful in vivo responses resulted in long-term protection against tumor regrowth and were evident in models that respond poorly to current checkpoint therapies. Our data support the investigation of IMM20324 as a promising therapy to overcome a suppressive mechanism mediated by tumor cells to impede anti-cancer immune responses.
Materials and methods
Cell lines and animals
Cell lines were sourced from commercial vendors and maintained in culture according to supplier recommendations. Cells were maintained for at least 3 passages prior to analysis by flow cytometry. C57BL/6 and BALB/c mice were purchased from the Jackson Laboratory at 6–8 weeks old. Mice were given food and water ad libitum and all protocols were approved by the respective Institutional Animal Care and Use Committee.
Analysis of IL-38 expression and immune cell infiltration in TCGA cohort
RNAseq data from multiple tumor types in the TCGA cohort were downloaded from the recount2 project.Citation53–55 IL-38 expression in individual samples is depicted in jitter points box plots. The IL-38 expression thresholds in different tumor types are chosen based on expression level of IL-38 in 90th percentile normal samples. Samples with an IL-38 expression level above the threshold are considered as IL-38 positive samples. Conversely, samples with an IL-38 expression level below the threshold are considered as IL-38 negative samples. Gene Set Enrichment Analysis (GSEA), with ImSig R package, was carried out to understand immune cell-type infiltration in these tumors stratified by IL-38 expression thresholds. ImSig uses a set of gene signatures and their co-expression to indicate the relative abundance of that particular immune cell type in transcriptomic data. Enrichment of ImSig enrichment scoresCitation25 for each immune cell population are calculated for IL-38 positive and IL-38 negative samples.
Analysis of IL-38 expression and immune cell infiltration in Tempus database
Tumor samples were clinically profiled at a College of American Pathologists-accredited, Clinical Laboratory Improvement Amendments-certified laboratory (Tempus Labs, Chicago, IL). De-identified clinical and molecular data were analyzed from U.S. pan-cancer patient records structured and stored in the Tempus database. RNA sequencing data, along with matched clinical data, were used to assess IL-38 gene expression and estimate immune infiltration by cancer subtype. Descriptive statistics regarding the distribution of RNA expression across clinical and demographic conditions pan-cancer were generated in Tempus multimodal xT data. Parametric hypothesis tests of log2(TPM+1) normalized RNA expression differences were conducted between populations of interest, and results were reported as significant at a threshold of p = 0.05. Immune cell infiltration was estimated using a neural network-based approach which integrates both gene expression and visual texture features (NEXT). The NEXT framework demonstrated increased accuracy in predicting presence of specific immune cell types, including B cells, CD4 T cells and macrophages in solid tumors.Citation27 All statistical analyses were performed in R Studio v2022.2.1 (R 3.3.0).
Anti-IL-38 antibodies
IMM20331 mouse IgG2a/k antibody was purified from a mouse hybridoma supernatant. Its recombinant version, IMM20324, was expressed in EXPI293 cells using two separate vectors encoding heavy and light chains of IMM20331 antibody. IMM20331 and IMM20324 were equivalent for binding to human IL-38 (Supplemental ). For simplicity, IMM20331 is referred to as IMM20324H to denote hybridoma supernatant-derived material and to differentiate it from its recombinant counterpart.
Human proteome array
IMM20324 was incubated at 1 µg/mL overnight at 4°C against the native CDI HuProt array V4 (CDI Laboratories, Inc.). Slides were washed and antibody binding was detected with an AlexaFluor-647-conjugated anti-H+L secondary antibody (Jackson ImmunoResearch #709-606-149). Nonspecific hits bound by the secondary antibody were eliminated from the analysis. Selective binding to target proteins was analyzed by Z-score to determine specificity and reproducibility of binding to replicates on each slide.
Affinity of anti-IL-38 antibodies to IL-38 measured by biolayer interferometry
Affinity of IMM20324 was evaluated using BLI on a GatorBio Plus instrument according to the manufacturer’s recommendations. Assay buffer (K Buffer) was supplied by GatorBio and contained phosphate-buffered saline (PBS), supplemented with 138 mM NaCl, 2.7 mM KCl, 0.02% BSA, 0.002% Tween-20, 0.005% ProClin 300, pH 7.4 at 25°C. Purified antibody sample loading was normalized to 5 µg/mL. Recombinant human and mouse IL-38 proteins were serially diluted twofold with assay buffer in 1.52–0.095 µM range. GatorBio anti-Mouse IgG Fc (MFC) probes were pre-soaked in assay buffer for a minimum of 10 min prior to start of each experiment. Reagents were prepared at RT and transferred to a black polystyrene 96-well plate. Experimental parameters were defined as following: Baseline (120 s), Loading (60 s), Baseline2 (120 s), Association (90 s), Dissociation (360 s) at 1000 RPM. New probes were used for each assay background normalization was performed using a blank per the manufacturer’s recommendations. The experiment was repeated twice. Data were processed using GatorBio’s Analysis software version 2.10.4.0713. Signal from reference wells was subtracted from the raw response of each sample. After background correction, data were aligned to association (i.e., leveled at Y-axis). Savitzky–Golay filtering was used to process the data. The processed data corresponding to the association and dissociation steps were globally fitted using a 1:1 model to generate linked binding curves. Kon, Koff and KD values were calculated for each separate experiment, and average values were reported.
ELISA-based antibody binding to IL-38
Recombinant full-length human (generated in-house) or mouse IL-38 (Adipogen #AG-40A-0191-C050) was diluted to a final concentration of 0.5 µg/mL in PBS, dispensed into high binding plates (Corning, #9018) and incubated overnight at 4°C. The plate was then washed three times with wash buffer (PBS+0.05% Tween) and blocked with blocking buffer (PBS+2% BSA) at room temperature (RT) for 1 h. Anti-IL-38 antibodies were incubated for 1 h at RT followed by three washes with wash buffer. The horseradish peroxidase (HRP)-conjugated rabbit anti-mouse secondary antibody (Jackson ImmunoResearch, #309-035-082) was incubated at RT for 1 h followed by three washes with wash buffer. O-phenylenediamine dihydrochloride (OPD) substrate (Sigma, #P8287) was incubated for 10–15 min in dark at RT prior to the addition of stop solution (2N sulfuric acid, R&D systems, #DY994). The amount of hu-IL-38 binding to sample/standard was determined by measuring absorbance at 450 nm using an EnSpire plate reader (model number: 2300–0000, PerkinElmer).
Flow cytometry
Flow cytometry was performed to assess binding to tumor cell lines. Briefly, adherent cells were harvested using non-enzymatic detachment buffer (CellStripper; Corning, # 25–056-CI) and incubated at 37°C for 10–30 min. Following release, cells were washed in complete media and stained with fixable live/dead dye (Life Technologies, #L34965). Primary antibody binding was performed at 4°C followed using IMM20324. Detection was performed by staining with a polyclonal AF647 goat anti-mouse IgG2a secondary antibody (Jackson ImmunoResearch, #115-607-186). Samples were analyzed on an Attune NxT flow cytometer (Life Technologies) and analyzed using FlowJo X (BD BioSciences). Binding curves were generated in Prism V9 using the sigmoidal, 4PL, log function.
Neutralization of human IL-38 binding to IL1RAPL1 or IL-36R
Human IL1RAPL1-Fc (Sino Biological, #10177-H02H) and human IL-36R-Fc (R&D Systems, #872-RP) were diluted in coating Buffer (50 mM Carbonate-Bicarbonate Buffer, pH 9.4) at final concentration of 0.5 µg/mL and coated on MSD high bind plates (MSD) for approximately 2 h at RT. Recombinant full length human IL-38 (Adipogen, #AG-40A-0191-C050) was diluted in assay Diluent (1% BSA, 0.1% Tween 20, in 1× PBS). For antibody testing, anti-IL-38 antibodies were pre-incubated with IL-38 for approximately 1 h at RT. IL-38 or antibody-IL-38 mixture was transferred to pre-coated MSD plates and incubated for approximately 2 h at RT. After washing, the MSD plate was incubated with biotinylated mouse anti-human IL-38 (clone H127C, Thermo Fisher Scientific, #14-7385-82) at final concentration of 100 ng/mL and Sulfo-Tag-Streptavidin (MesoScale Discovery, #L15×B) at final concentration of 100 ng/mL for 1 h at RT. Binding of IL-38 to plate-bound IL1RAPL1-Fc or human IL-36 R-Fc was detected using the Meso Sector S 600 plate reader. Data are plotted using GraphPad Prism version 9.0.1. Dose response curves were fitted with “[Inhibitor] vs. response – Variable slope (four parameters)” method.
Immunohistochemical analysis of IL-38 expression
Tissues were formalin fixed and embedded in paraffin and sectioned using standard protocols. Antigen retrieval was performed using sodium citrate and staining was performed using either IMM20324 for human tissues or a rabbit polyclonal (LS Bio, #LS-C201139) for staining of mouse syngeneic ex vivo tumors. Detection was performed using an anti-rabbit polyclonal HRP-conjugated secondary antibody. Imaging was performed on a NanoZoomer-HT 2.0 image system.
Pharmacokinetic and tolerability analysis of IMM20324
For single-dose PK analysis, 6–7-week-old C57BL/6 female mice were given a 10 mg/kg dose of IMM20324H either IP or IV (n = 9 per group). At each timepoint, blood from three mice was collected retro-orbitally or by cardiac puncture in K2-EDTA tubes (Becton Dickinson, #366643) and plasma was isolated. For multidose tolerability study, 5- to 6-week-old C57BL/6 female mice were randomly assigned to groups of 4–5 mice, dosed as indicated with IMM20324H or PBS vehicle control, and body weight was measured twice per week. On D21, blood was collected in K2-EDTA tubes and plasma was isolated. Spleens from all mice were removed and weighed.
Plasma concentration of IMM20324 was determined by a direct IL-38 ELISA. And 0.1 µg/mL human full-length IL-38 (Adipogen, #AG-40A-0191-C050) or NovoPro, # 519753) was coated to each well of a high binding 96-well plate (Corning, #3369) overnight at 4°C. Wells were blocked with PBS + 2% BSA at room temperature for 1 h then washed thrice with PBS + 0.05% Tween. Plasma samples were diluted in PBS, added to each well for 2 h at RT, and then wells were washed 3 times with PBS + 0.05% Tween. Anti-mouse IgG-HRP (Southern Biotech # 6170–05) was added to each well for 2 h at RT, and then wells were washed thrice with PBS + 0.05% Tween. OPD substrate (Sigma, #P8287) diluted in phospho-citrate/sodium perborate buffer was added per well, developed for 5–30 min, and absorbance was measured at 450 nm.
EMT6 syngeneic tumor model
For this model, 5 × 10Citation5 EMT6 cells were implanted into the flank for 6- to 8-week-old BALB/c female mice. When average tumor volume reached 110–115 mm3 approximately 8 d later, mice were randomly assigned to indicated treatment groups (n = 10 per group) and dosing was initiated. For the duration of the study, tumors were measured with a digital caliper twice per week and tumor volume was calculated by (longest diameter) × (shortest diameter)Citation2 × 0.52. Mice were sacrificed when tumor volume reached 2000 mm3. For rechallenge studies, cured mice from this study (n = 4 per group) were paired with aged-matched BALB/c mice (n = 5) and inoculated with 5 × 10Citation5 EMT6 cells in the opposite flank of the initial inoculation. Tumor volume and body weight were measured twice per week and mice were sacrificed when tumor volume reached 2000 mm3 or at the conclusion of the study on D43. Average tumor volumes were compared using one-way ANOVA with Dunnett’s post hoc test. Significant increases in survival were assessed using log-rank test.
B16.F10 syngeneic tumor model
For this model, 2.5 × 10Citation5 B16.F10 cells were implanted into the flank for 7- to 8-week-old C57BL/6 female mice. When average tumor volume reached 60–65 mm3 approximately 6–7 d later, mice were randomly assigned to indicated treatment groups (n = 15 per group) and dosing was initiated. For the duration of the study, tumors were measured with a digital caliper twice per week and tumor volume was calculated by (longest diameter) × (shortest diameter)2/2. Mice were sacrificed either when tumor volume reached 3000 mm3 or on D11, 24 h after the final dose of IMM20324 depending on the study. Average tumor volume, growth rate, and time-tumor-volume were compared using one-way ANOVA with Dunnett’s post hoc test.
Analysis of tumor growth rate, time-tumor-volume, and linear mixed effects modeling
Tumor growth curves for each animal were fitted using the exponential model of growth (tumor volume = a*exp(b*days)), which provided the highest average R2 and lowest average Akaike Information Criterion (AIC) value as compared to other models tested. Tumor growth rate in percentages were calculated using a difference in time with lag and difference in tumor volume with lag using the following equations:
Difference in Days = Timepoint – lag(Timepoint)
Difference in Growth = Volume – lag(Volume)
Tumor growth rate % = (Difference in Growth/Difference in Days)/lag(Volume) * 100
Finally, the tumor volume comparison between treatment arms was analyzed using the LME model using tumor volume as the outcome and treatment groups as the fixed variable absorbing the variance of time.Citation53-55 As this was a nested experimental design, individual animals were controlled for as a random effect. Results allow the comparison of each treatment arm compared to control with and without absorbing the variance of time. This LME analysis was completed with all the time points.Citation54,Citation55
Generation of tumor homogenates and intra-tumoral cytokine analysis
Twenty-four hours after the final dose of IMM20324, B16.F10 tumors were harvested, flash frozen, and stored at −80°C. To generate tumor homogenates, flash-frozen tumors were placed in 2 mL Bead Ruptor Pre-filled Bead Tubes (Omni International Catalog # 19–628) in lysis buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, and 0.5% NP-40. A bead mill homogenizer (Bead Ruptor Elite, Omni International Catalog # 19-042E) was used to perform two 30 s bead beating cycles. Tubes were spun at >10,000 × g for 10 min and the lysate was transferred to a new tube. Protein concentration was measured using the Pierce BCA Protein Assay kit (Thermo Scientific Catalog # 23225) and all tumor homogenates were normalized to 5 mg/mL. Multiplexing analysis was performed using the Luminex™ 200 system (Luminex, Austin, TX, USA) by Eve Technologies Corp. (Calgary, Alberta). Thirty-two markers were simultaneously measured in the samples using Eve Technologies’ Mouse Cytokine 32-Plex Discovery Assay® (MilliporeSigma, Burlington, Massachusetts, USA) according to the manufacturer’s protocol. The 32-plex consisted of Eotaxin, G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1β, MIP-2, RANTES, TNFα, and VEGF. Assay sensitivities of these markers range from 0.3 to 30.6 pg/mL for the 32-plex. Individual analyte sensitivity values are available in the MilliporeSigma MILLIPLEX® MAP protocol.
Abbreviations
IL-38, interleukin 38; IL1RAPL1, interleukin 1 receptor accessory protein-like 1; IL-36R, interleukin 36 receptor; HNSC, head and neck squamous carcinoma; CESC, cervical squamous carcinoma; LUSC, lung squamous cell carcinoma; TCGA, The Cancer Genome Atlas; IV, intravenous; IP, intraperitoneal; QW, weekly dosing; Q2W, twice-weekly dosing.
Supplemental Material
Download MS Word (1.4 MB)Acknowledgments
The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Disclosure statement
Authors are employees and shareholders of Immunome, Inc. The described antibodies and data have been included in patent applications.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2212673
Additional information
Funding
References
- Esfahani K, Elkrief A, Calabrese C, Lapointe R, Hudson M, Routy B, Miller WH, Calabrese L. Moving towards personalized treatments of immune-related adverse events. Nat Rev Clin Oncol. 2020;17(8):504–17. doi:10.1038/s41571-020-0352-8.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. doi:10.1038/s41577-020-0306-5.
- Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–50. doi:10.1038/s41568-019-0116-x.
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–55. doi:10.1126/science.aar4060.
- Ginefra P, Lorusso G, Vannini N. Innate immune cells and their contribution to T-Cell-Based Immunotherapy. Int J Mol Sci. 2020;21(12):4441. doi:10.3390/ijms21124441.
- Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, Varner JA, Pu M, Messer KS, Guiducci C, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2(18):e93397. doi:10.1172/jci.insight.93397.
- Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. doi:10.1111/imr.12765.
- Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, Tse A, Khilnani A, Marabelle A, Golan T. LBA15 Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29(viii 712):viii712. doi:10.1093/annonc/mdy424.015.
- Pero SC, Rosenfeld AM, Shukla GS, Mei L, Sun Y, Meng W, Fournier DJ, Harlow SP, Robinson MK, Krag DN, et al. Diversification and shared features of tumor‐binding antibody repertoires in tumor, sentinel lymph node and blood of three patients with breast cancer. Clin Transl Immunol. 2022;11(8):e1409. doi:10.1002/cti2.1409.
- Khan TA, Friedensohn S, de VA, Straszewski J, Ruscheweyh H-J, Reddy ST. Accurate and predictive antibody repertoire profiling by molecular amplification fingerprinting. Sci Adv. 2016;2(3):e1501371. doi:10.1126/sciadv.1501371.
- DiMuzio JM, Heimbach BC, Howanski RJ, Dowling JP, Patel NB, Henriquez N, Nicolescu C, Nath M, Polley A, Bingaman JL, et al. Unbiased interrogation of memory B cells from convalescent COVID-19 patients reveals a broad antiviral humoral response targeting SARS-CoV-2 antigens beyond the spike protein. Vaccine: X. 2021;8:100098. doi:10.1016/j.jvacx.2021.100098.
- Nikitin PA, DiMuzio JM, Dowling JP, Patel NB, Bingaman-Steele JL, Heimbach BC, Henriquez N, Nicolescu C, Polley A, Sikorski EL, et al. IMM-BCP-01, a patient-derived anti–SARS-CoV-2 antibody cocktail, is active across variants of concern including Omicron BA.1 and BA.2. Sci Immunol. 2022;7(75):eabl9943. doi:10.1126/sciimmunol.abl9943.
- Baker KJ, Houston A, Brint E. IL-1 family members in cancer; two sides to every story. Front Immunol. 2019;10:1197. doi:10.3389/fimmu.2019.01197.
- van Devan de VF, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, Joosten LAB, van der MJ, Hao R, Kalabokis V, et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc National Acad Sci. 2012;109(8):3001–05. doi:10.1073/pnas.1121534109.
- Mora J, Schlemmer A, Wittig I, Richter F, Putyrski M, Frank A-C, Han Y, Jung M, Ernst A, Weigert A, et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol. 2016;8(5):426–38. doi:10.1093/jmcb/mjw006.
- Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, Gubinelli E, Mazzanti C, Cavani A, Ruvo M, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death & Disease. 2018;9(11):1104. doi:10.1038/s41419-018-1143-3.
- Han Y, Mora J, Huard A, da SP, Wiechmann S, Putyrski M, Schuster C, Elwakeel E, Lang G, Scholz A, et al. IL-38 ameliorates skin inflammation and limits IL-17 production from γδ T cells. Cell Rep. 2019;27(3):835–46.e5. doi:10.1016/j.celrep.2019.03.082.
- de GD, Wang RX, Amo-Aparicio J, Lee JS, Dowdell AS, Tengesdal IW, Marchetti C, Colgan SP, Joosten LAB, Dinarello CA. IL-38 gene deletion worsens murine colitis. Front Immunol. 2022;13:840719. doi:10.3389/fimmu.2022.840719.
- Kinoshita F, Tagawa T, Akamine T, Takada K, Yamada Y, Oku Y, Kosai K, Ono Y, Tanaka K, Wakasu S, et al. Interleukin-38 promotes tumor growth through regulation of CD8+ tumor-infiltrating lymphocytes in lung cancer tumor microenvironment. Cancer Immunol Immunother. 2021;70(1):123–35. doi:10.1007/s00262-020-02659-9.
- Takada K, Okamoto T, Tominaga M, Teraishi K, Akamine T, Takamori S, Katsura M, Toyokawa G, Shoji F, Okamoto M, et al. Clinical implications of the novel cytokine IL-38 expressed in lung adenocarcinoma: possible association with PD-L1 expression. PLos One. 2017;12(7):e0181598. doi:10.1371/journal.pone.0181598.
- Luheshi NM, Rothwell NJ, Brough D. The dynamics and mechanisms of interleukin-1α and β nuclear import. Traffic Cph Den. 2008;10(1):16–25. doi:10.1111/j.1600-0854.2008.00840.x.
- Diaz-Barreiro A, Huard A, Palmer G. Multifaceted roles of IL-38 in inflammation and cancer. Cytokine. 2022;151:155808.
- Xie L, Huang Z, Li H, Liu X, Zheng S, Su W. IL-38: a new player in inflammatory autoimmune disorders. Biomol. 2019;9(8):345. doi:10.3390/biom9080345.
- Nirmal AJ, Regan T, Shih BB, Hume DA, Sims AH, Freeman TC. Immune cell gene signatures for profiling the microenvironment of solid tumors. Cancer Immunol Res. 2018;6(11):canimm.0342.2018. doi:10.1158/2326-6066.CIR-18-0342.
- Beaubier N, Bontrager M, Huether R, Igartua C, Lau D, Tell R, Bobe AM, Bush S, Chang AL, Hoskinson DC, et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol. 2019;37(11):1351–60. doi:10.1038/s41587-019-0259-z.
- Reiman D, Sha L, Ho I, Tan T, Lau D, Khan AA. Integrating RNA expression and visual features for immune infiltrate prediction. Pac Symposium Biocomput Pac Symposium Biocomput. 2019;24:284–95.
- Gameiro SF, Ghasemi F, Barrett JW, Koropatnick J, Nichols AC, Mymryk JS, Vareki SM. Treatment-naïve HPV+ head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology. 2018;7(10):e1498439. doi:10.1080/2162402X.2018.1498439.
- Veerdonk FL, Graaf DM, Joosten LA, Dinarello CA. Biology of IL‐38 and its role in disease. Immunol Rev. 2017;281(1):191–96. doi:10.1111/imr.12612.
- Palomo J, Troccaz S, Talabot-Ayer D, Rodriguez E, Palmer G, Budunova I. The severity of imiquimod-induced mouse skin inflammation is independent of endogenous IL-38 expression. PLos One. 2018;13(3):e0194667. doi:10.1371/journal.pone.0194667.
- Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J Clin Invest. 2018;128(2):805–15. doi:10.1172/JCI96113.
- Rudloff I, Godsell J, Nold‐petry CA, Harris J, Hoi A, Morand EF, Nold MF. Brief report: interleukin‐38 exerts antiinflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(12):3219–25. doi:10.1002/art.39328.
- Xie C, Yan W, Quan R, Chen C, Tu L, Hou X, Fu Y. Interleukin-38 is elevated in inflammatory bowel diseases and suppresses intestinal inflammation. Cytokine. 2020;127:154963. doi:10.1016/j.cyto.2019.154963.
- Ciccia F, Accardo‐palumbo A, Alessandro R, Alessandri C, Priori R, Guggino G, Raimondo S, Carubbi F, Valesini G, Giacomelli R, et al. Interleukin-36α axis is modulated in patients with primary Sjögren’s syndrome. Clin Exp Immunol. 2015;181(2):230–38. doi:10.1111/cei.12644.
- Gao X, Chan PKS, Lui GCY, Hui DSC, Chu I-T, Sun X, Tsang M-M, Chan BCL, Lam C-K, Wong C-K. Interleukin-38 ameliorates poly(I: c) induced lung inflammation: therapeutic implications in respiratory viral infections. Cell Death & Disease. 2021;12(1):53. doi:10.1038/s41419-020-03283-2.
- Sun X, Hou T, Cheung E, Iu T-T, Tam V-H, Chu I-T, Tsang M-M, Chan P-S, Lam C-K, Wong C-K. Anti-inflammatory mechanisms of the novel cytokine interleukin-38 in allergic asthma. Cell Mol Immunol. 2019;17(6):1–16. doi:10.1038/s41423-019-0300-7.
- Rafie MZ, Esmaeilzadeh A, Ghoreishi A, Tahmasebi S, Faghihzadeh E, Elahi R. IL-38 as an early predictor of the ischemic stroke prognosis. Cytokine. 2021;146:155626. doi:10.1016/j.cyto.2021.155626.
- Lambertsen KL, Finsen B, Clausen BH. Post-stroke inflammation—target or tool for therapy? Acta Neuropathol. 2019;137(5):693–714. doi:10.1007/s00401-018-1930-z.
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus–related and –unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26(4):612–19. doi:10.1200/JCO.2007.14.1713.
- Weinstein AM, Chen L, Brzana EA, Patil PR, Taylor JL, Fabian KL, Wallace CT, Jones SD, Watkins SC, Lu B, et al. Tbet and IL-36γ cooperate in therapeutic DC-mediated promotion of ectopic lymphoid organogenesis in the tumor microenvironment. Oncoimmunology. 2017;6(6):e1322238. doi:10.1080/2162402X.2017.1322238.
- Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-Cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res. 2016;22(12):3005–15. doi:10.1158/1078-0432.CCR-15-2762.
- Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K, Wanibuchi H, Ohira M. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J Surg Res. 2017;215:74–82. doi:10.1016/j.jss.2017.03.033.
- Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, Becht E, Katsahian S, Bizouard G, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Resp Crit Care. 2014;189(7):832–44. doi:10.1164/rccm.201309-1611OC.
- Petitprez F, de RA, Keung EZ, Chen T-W, Sun C-M, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougoüin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):1–5. doi:10.1038/s41586-019-1906-8.
- Domblides C, Rochefort J, Riffard C, Panouillot M, Lescaille G, Teillaud J-L, Mateo V, Dieu-Nosjean M-C. Tumor-associated tertiary lymphoid structures: from basic and clinical knowledge to therapeutic manipulation. Front Immunol. 2021;12:698604. doi:10.3389/fimmu.2021.698604.
- Chelvanambi M, Weinstein AM, Storkus WJ. Tumor Microenvironment, the Role of Interleukins – Part a. Adv Exp Med Biol. 2020;1240:95–110.
- Yu JW, Bhattacharya S, Yanamandra N, Kilian D, Shi H, Yadavilli S, Katlinskaya Y, Kaczynski H, Conner M, Benson W, et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLos One. 2018;13(11):e0206223. doi:10.1371/journal.pone.0206223.
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. 2018;175(4):1014–30.e19. doi:10.1016/j.cell.2018.09.030.
- Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, Becher B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–53. doi:10.1038/nm.4466.
- Cabrita R, Lauss M, Sanna A, Donia M, Larsen MS, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–65. doi:10.1038/s41586-019-1914-8.
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–55. doi:10.1038/s41586-019-1922-8.
- Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting innate immunity in cancer therapy. Vaccines (Basel). 2021;9(2):138. doi:10.3390/vaccines9020138.
- Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B, Leek JT. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017;35(4):319–21. doi:10.1038/nbt.3838.
- Benzekry S, Lamont C, Beheshti A, Tracz A, Ebos JML, Hlatky L, Hahnfeldt P, Mac Gabhann F. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol. 2014;10(8):e1003800. doi:10.1371/journal.pcbi.1003800.
- Zavrakidis I, Jóźwiak K, Hauptmann M. Statistical analysis of longitudinal data on tumour growth in mice experiments. Sci Rep-Uk. 2020;10(1):9143. doi:10.1038/s41598-020-65767-7.
- Vaidya VG, Alexandro FJ. Evaluation of some mathematical models for tumor growth. Int J Biomed Comput. 1982;13(1):19–35. doi:10.1016/0020-7101(82)90048-4.