
ABSTRACT
Host cell proteins (HCPs) are process-related impurities derived from the manufacturing of recombinant biotherapeutics. Residual HCP in drug products, ranging from 1 to 100 ppm (ng HCP/mg product) or even below sub-ppm level, may affect product quality, stability, efficacy, or safety. Therefore, removal of HCPs to appropriate levels is critical for the bioprocess development of biotherapeutics. Liquid chromatography-mass spectrometry (LC-MS) analysis has become an important tool to identify, quantify, and monitor the clearance of individual HCPs. This review covers the technical advancement of sample preparation strategies, new LC-MS-based techniques, and data analysis approaches to robustly and sensitively measure HCPs while overcoming the high dynamic range analytical challenges. We also discuss our strategy for LC-MS-based HCP workflows to enable fast support of process development throughout the product life cycle, and provide insights into developing specific analytical strategies leveraging LC-MS tools to control HCPs in process and mitigate their potential risks to drug quality, stability, and patient safety.
Introduction
During the biotherapeutics production process, cells from organisms, such as Escherichia coli (E. coli) and Chinese hamsters (Cricetulus griseus, CRIGR) are engineered to produce biologic drug products. These cells also express host cell proteins (HCPs), the endogenous proteins unrelated to the intended therapeutic recombinant product that have a wide range of physicochemical properties (such as isoelectric point, molecular weight, and hydrophobicity). HCPs are process-related impurities and considered critical quality attributes (CQAs), since residual HCPs in the drug product can affect product quality, stability, and safety.Citation1 While there is no universal standard for specific HCP limit levels in biotherapeutics, HCPs should be actively monitored by sensitive and robust analytical methods during the bioprocess and in the final drug product to confirm the removal of impurities.Citation2–5 The current US Food and Drug Administration (FDA) guidelines apply for protein therapeutics, vaccines and gene therapies regarding the HCP characterization with no difference in expected characterization and validation across modalities. HCP monitoring occurs at various stages of the biotherapeutics production workflow, starting from early upstream processing and including in-process pools and drug products from preclinical, clinical stages and the final commercialized product.Citation6,Citation7 If a high-risk HCP is detected during the bioprocess in drug substance, a toxicity risk control strategy should be applied to monitor and remove HCP to ensure patient safety.Citation1 Most antibodies and many therapeutic proteins are generated from Chinese hamster ovary (CHO) or E. coli cells so the majority of analytical methods are developed for HCPs of biotherapeutics from these two types of cell lines. New therapeutic modalities might involve the use of other cell lines, such as insect,Citation8 or yeast.Citation9 Different HCP patterns might be expected with different cell lines. However, for HCP studies regardless of cell lines used, we recommend starting with the established analytical methods for HCPs from CHO or E. coli, and optimizing the methods as needed for HCPs from other sources.
In general, most HCPs are likely safe in a total level under 100 ppm (ng HCP/mg product) measured by enzyme-linked immunosorbent assays (ELISAs).Citation10 This is supported by a recent study that demonstrated the manufacturing process capability for 29 commercially marketed monoclonal antibody (mAb) molecules containing individual HCPs with an average of 20 ppm and no more than 100 ppm by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.Citation11 However, low levels of specific HCP can be responsible for or contribute to drug instability, toxicity, immunogenicity, or anti-drug antibody responses.Citation1 Some high-risk HCPs, such as hydrolases, may cause polysorbate degradation and affect drug stability at low ppm levels.Citation12 This has driven the need for increasingly sensitive methods for HCP detection and quantitation, due to the wide dynamic range of the HCP population, with more than six orders of magnitude difference in different process pools.Citation7,Citation13
A variety of analytical techniques have been applied to develop methods for HCP measurement and control. One- and two-dimensional polyacrylamide gel electrophoresis were once commonly used for HCP profiling in bioprocess development, but such gel-based methods are laborious, have low sensitivity toward certain proteins, may miss individual low abundant proteins, and cannot reveal identity.Citation14,Citation15 ELISAs and MS are now the major analytical methods used to measure HCPs during process development. For total HCP detection, ELISA is widely used and considered the gold standard assay due to its combination of sensitivity, selectivity, dynamic range, sample throughput capacity and cost efficiency.Citation6 Since ELISA is a technology based on immunoreactivity to detect a specific or the total HCP level, it cannot detect non-immunoreactive proteins or identify individual proteins. In contrast, LC-MS-based methods are orthogonal, complementary analytical methods that can obtain individual protein identity and quantity, and are independent of immunoreactivity, enabling a comprehensive analysis of the HCP population.Citation1 Here, we provide a literature review of the application of LC-MS/MS-based methods for HCP identification and quantitation and recent LC-MS method development, challenges, and strategies. We also present our strategies and considerations for HCP characterization and process monitoring by LC-MS/MS through case studies.
Application of LC-MS/MS-based methods for HCP identification and quantitation
Over 100 therapeutic antibody drugs are currently approved by the FDA.Citation16 Recent studies used LC-MS/MS-based methods as an approach orthogonal to ELISA to characterize HCPs in mAb drugs in support of bioprocess development,Citation17 drug safety assessment and quality control.Citation18,Citation19 In addition, the application of LC-MS/MS methods for HCP studies supports ELISA critical reagents characterization, as well as gene therapy and vaccine production.Citation20,Citation21
Supporting mAb drug manufacturing process control
LC-MS/MS HCP identification and quantitation methods can be applied to upstream and downstream process samples, allowing manufacturers to closely monitor the levels of hundreds to thousands of HCPs in support of process development, as summarized in . Early applications of LC-MS/MS methods with limited sensitivity (~20 ppm) were useful for tracking abundant HCPs during process-development, including co-purifying HCPs.Citation26,Citation27 Through HCP enrichment during sample preparation, we have been able to improve the overall analysis sensitivity and identify critical HCPs, such as those involved with polysorbate degradation.Citation12 In parallel, both lower and high sensitivity LC-MS/MS methods can be applied to the characterization of upstream bioprocesses and knockout cell lines.Citation22–25 In addition to the increased sensitivity, the industry has also improved overall method robustness, by demonstrating recovery of specific high-risk proteinsCitation7,Citation28,Citation29 or set of multiple HCP standards.Citation30 With robustness shown for a single method, the applications of LC-MS/MS methods are expanding into HCP comparability assessmentCitation10,Citation17 and good manufacturing practice (GMP) release testing.Citation30 Fundamental challenges still remain regarding how to adequately control for such a heterogenous population and very low-abundant proteins. We can further leverage the data of HCP populations and profiles by linking to pivotal clinical data, allowing us to better support process design, process operations, and risk assessments.
Table 1. Cross-industry studies demonstrating the application LC-MS/MS methods in support of process development.
Understanding HCP population and interaction mechanism to improve purification process
Trace-level HCPs can lead to undesirable product instability, immune responses, or formulation changes, and they need to be removed to a sufficient level. Process development to mitigate these risks requires sensitive methods to understand the HCP population and the interaction between HCP and antibody.Citation30,Citation31 LC-MS/MS-based proteomics approaches to profile HCPs from harvested cell culture fluid (HCCF) to the final drug substance allow us to understand how abundant or problematic HCPs are removed across different process steps. Generally, HCP patterns in HCCF are relatively similar across CHO cell lines, while HCP patterns during purification and product concentration become more divergent; therefore, MS can be valuable to detect individual HCPs in downstream processes and guide purification development efforts.Citation17,Citation32 For example, protein identification and quantitation by size exclusion chromatography (SEC) fractionation coupled to LC-MS/MS showed HCPs (like high-risk hydrolytic enzymes) co-eluting with high molecular weight (HMW) species – this indicated the co-purification of HCPs with mAb and that reducing HMW species in drug substance might significantly decrease the levels of some HCPs.Citation33
MS methods can also identify different post-translational modifications and protein–protein interaction sites. Native LC-MS involves the characterization of predominantly protein complexes and enables the detection of protein–protein interactions. Recently, non-covalent mAb-lipase complexes were studied by native MS to demonstrate the direct role of HCP lipase glycosylation in its binding to mAb drugs.Citation34 The study indicated purification strategies could be implemented to remove lipases, such as using lectins to remove high-mannose HCP glycoforms or specific washes to disrupt electrostatic or hydrophobic interactions. In addition, fast photochemical oxidation of proteins LC-MS provided higher order structure details to confirm the interaction sites between mAb and lipases, suggesting rationally designed mutations in the interaction domain could also decrease the co-purification. All these methods provide guiding information for controlling HCP expression and purification in upstream and downstream bioprocess.
Characterizing critical reagents for total anti-HCP ELISA
MS-based methods have been widely used to assess the suitability of ELISA critical reagents for measuring HCP content. These critical reagents include anti-HCP polyclonal antibodies and HCP reference standards produced by the host cells as antigens to generate anti-HCP. Two‐dimensional (2D) difference gel electrophoresis and MS-based approaches are the recommended methods to qualify HCP reference standards and coverage of anti-HCP antibodies.Citation31,Citation32 However, the 2D Western blot (2D-WB) has challenges with imaging low abundance or low molecular weight proteins in the gel, as they are typically less immunogenic than higher molecular weight proteins.Citation30 Additionally, denaturing conditions for western blotting can alter HCP epitopes and reduce their binding affinity to anti-HCP antibodies, resulting in decreased anti-HCP antibody coverage. Post-translational modifications affected measured HCP patterns more when analyzed by 2D-gel than by MS analysis. To this point, orthogonal MS-based methods, such as antibody affinity-based MS were applied to better characterize anti-HCP antibodies and to identify uncovered 2D-WB spots.Citation35,Citation36 Compared to 2D-WB methods, MS approaches can cover a high dynamic range of protein abundances, isoelectric points, and molecular weights, allowing higher coverage and the detection of antibodies against low molecular weight HCPs.Citation35 Control antibodies can be used to reduce false positives of nonspecific-binding proteins that interact with anti-HCP antibodies.Citation30,Citation37 Alternatively, Seisenberger et al. established a method to overcome the limitations of nonspecific binding of HCPs to the matrix by conjugating chemically cleavable linkers with antibodies to facilitate the separation of proteins nonspecifically interacting with matrix from the specific binding of HCPs to antibodies.Citation38
Analyzing HCPs in gene therapy and vaccine studies
Characterization of HCPs during the development of emerging drug modalities, such as during viral vector production for gene therapy, is still in its early stage.Citation20 2D electrophoresis-coupled LC-MS analysis, which uses a 2D gel to visualize proteins and MS analysis of the excised-gel to identify specific proteins, demonstrated HCP differences across host expression platforms for viral vector production.Citation39 LC-MS/MS analysis also confirmed the co-purification of specific N-glycosylated HCPs and adeno-associated virus serotype 8 (AAV8), suggesting AAV8 likely interacted with terminal N-glycans within HCPs.Citation40 The complexity of gene therapy platforms using different vectors and expression systems, as well as different locations of HCPs inside viral vectors, make HCP analysis for gene therapies more challenging than for mAbs. Improved extraction of membrane proteins, and other analytical methods like SEC,Citation41 can potentially be used in combination with MS for viral vector HCP analysis.
HCPs are also potential impurities in cell culture-based vaccines. Recombinant protein vaccines can be made by expressing virus antigens in yeast or bacterial cells, and adenoviral vector-based vaccines may be based on chimpanzee or human adenovirus.Citation21 In these cases, proteins from the hosts may find their way in the final product if not efficiently removed. Adverse effects from HCPs in vaccines may be less of a concern than in biotherapeutics due to infrequent dosing schedules (typically months or years apart), as this translates to lower exposure over time. The European Medicines Agency (EMA) also sets the limit for HCP content per vaccine dose at 400 ng.Citation21 However, it is still important to monitor HCPs to keep their levels low, show that those present do not pose a safety risk to patients, and confirm manufacturing consistency.Citation21,Citation42 To translate the limit for HCP content per vaccine dose to ng HCP/mg product, the information on the mg of product is needed. For gene therapy and vaccine studies, the mg of product will not be limited to protein only, as the product contains other components in addition to protein, which is different from ppm level calculations for antibody drugs. The tryptic digest-based LC-MS methods used for HCP analysis in recombinant mAb antibodies are also applicable to vaccines. For example, in a recent study from the University of Ulm, MS analysis was performed on three lots of the ChAdOx1 vaccine for COVID-19 from AstraZeneca to compare the process impurities.Citation21 This vaccine was based on Chimpanzee Adenovirus Y25 and produced in human T-REx-293 cells. The study indicated that in the tested lots of this vaccine, as much as 70% of the total detected protein content was of human origins, exceeding the EMA’s HCP specification limits.
HCP LC-MS method development, challenges, and strategies
Proteomic bottom-up LC-MS/MS approaches have been commonly established in the last decade for host cell protein identification as orthogonal methods to the more traditional ELISA methods.Citation13,Citation43–45 However, the differences between bioprocess samples and proteomic samples soon resulted in method development specifically designed for HCP samples. Proteome samples are a complex mixture of thousands of proteins, whereas bioprocess samples contain a huge excess of drug product and very low levels (sub ppm to ppm) of HCPs, requiring an analysis method spanning a wide dynamic range of >6 orders of magnitude. Furthermore, the dynamic range increases throughout the purification process. Despite advances in instrumentation and the availability of fast-scanning high-resolution mass spectrometers, a regular one-dimensional LC-MS/MS method reaches its limits when attempting to resolve sample components or peptides across abundance differences of >5 orders of magnitude.Citation45 The method optimization concerns all steps in a typical LC-MS/MS method: sample preparation, chromatographic separation, MS acquisition, and data analysis. Here, we will review recent developments and strategies toward tackling the high dynamic range challenges of HCP analysis by LC-MS/MS. The overall strategies can be categorized in the following three areas: 1) HCP enrichment 2) advances in LC separation, and 3) advanced MS technology and analysis.
HCP enrichment strategies
A typical bottom-up sample preparation workflow consists of sample denaturation, reduction of disulfide bonds, alkylation, and subsequently a complete tryptic digestion overnight.Citation46 To increase method sensitivity, one approach is the removal of therapeutic protein matrix prior to digestion by offline affinity depletion with a Protein A chromatography.Citation47–49 Alternatively, HCPs can be enriched by anion exchange for HCP capture,Citation50 or Protein A in combination with uniquely developed and produced anti-CHO-HCP chromatography.Citation12 Even though all of these efforts have been proven to work effectively, they are also very labor-intensive with high cost and therefore, less suitable for routine testing. Huang et al. developed the native digestion for enhanced HCP identificationCitation51 by mAb removal strategy. In this method, trypsin was added directly to the sample without a denaturing step. The native antibody is hardly digested and can be subjected to heat precipitation after digestion, drastically decreasing the amount of antibody peptides present in the sample injected into the MS. This simple and effective protocol became very popular and was broadly used across the industry.Citation7,Citation52–54 This new concept created room for further development and optimization based on non-denaturing incubation conditions. Complete digestion of all proteins within one sample was no longer desirable, instead finding the optimal balance between a preferably complete digestion of HCPs present and as little digestion as possible of the antibody. One approach was to optimize the incubation time. Kufer et al. and Yang et al. described the benefits of a shorter incubation time of 2 h.Citation7,Citation52 The enzymatic reaction is very fast and occurs mostly in the first few minutes of the incubation time,Citation55 thus a short incubation time results in less digested mAb while still sufficient for HCP digestion. Another parameter of the native digestion, which influences the optimal HCP digestion, is the trypsin-to-substrate mass ratio. Multiple studies demonstrated that a lower ratio is beneficial for the sensitivity of HCP detection due to lower mAb digestion.Citation7,Citation54,Citation56 The native digestion also provides a new platform that can be combined with advances in LC separation and MS instrumentation to showcase super-sensitive HCP identification methods. Li et al. presented a mild digestion method, sodium deoxycholate (SDC)-aided, as an alternative without antibody depletion to the native digestion.Citation57 More recently, new efforts have emerged applying alternative enrichment or separation techniques before the tryptic digestion: hydrophilic interaction liquid chromatography (HILIC) enrichment,Citation58 SEC fractionationCitation59 and ProteoMiner enrichment of low abundant species.Citation54,Citation60,Citation61
shows a list of recent publications, including those on these HCP enrichment strategies, and compares method characteristics such as sample handling time, LC flow, gradient length, estimated sample throughput per day (based on MS run time), published sensitivity and number of NIST HCPs identified in RM 8671. The sample handling time is shown in the following categories: “low” for sampling handling time below 8 h, “medium” for sample preparation time between 15 and 20 h, and “high” for sample preparation methods including numerous manual steps in addition to overnight incubation. To further simplify the table, LC flow is indicated by “analytical” for 100–300 µl/min, “micro” for 0.5–50 µl/min and “nano” for 0.2–0.3 µl/min. Gradient length is indicated as “short” for 20–30 min, “medium” for 40–65 min and “long” for 100–260 min. Additionally, we estimated the sample throughput per day for MS measurement based on the gradient length. As several studies did not include wash and equilibration times used, we added 60 min for wash steps, equilibration and blank runs to each gradient length to estimate a realistic sample throughput per day, except for the study presented by Ma et al., which specifically mentions the sample throughput.Citation64
Table 2. Recent studies and trend of LC-MS based analysis in HCP characterization.
Advances in chromatographic separation of HCP peptides
The standard LC setup for HCP LC-MS/MS analysis is a one-dimensional C18 reversed phase (RP) separation method. Multiple studies examined the use of an additional and/or orthogonal LC separation method to resolve co-eluting mAb and HCP peptides.Citation45,Citation52,Citation62 These approaches included the use of high-pH RP coupled with low-pH RP separation in the second dimension,Citation45,Citation50,Citation65,Citation66 high/low-pH 2D with an additional concatenation pooling step,Citation62 and mixed mode (SDB-RPS) or strong cation exchange (SCX) chromatography with low-pH RP separation.Citation52 Even though ready-to-use stage tips enabled relatively fast and simple 2D methods, offline fractionation adds time-consuming steps due to the use of MS incompatible matrices. While less suitable for routine testing, for applications requiring in-depth analysis (e.g., for DIA spectral library generation), 2D-LC-MS/MS approaches offered an effective way to build comprehensive libraries.Citation67 Another approach to increase sensitivity by LC conditions is the use of much lower flow rates in the micro to nano flow range. Microflow had not yet attracted much attention in the HCP field, until Evosep introduced an alternative LC system.Citation64,Citation68 In brief, the Evosep system combines a low-pressure elution from specific-stage tips with a high pressure analytical gradient. The main advantages are minimized carry over and increased throughput due to the parallel operation of the steps between injections without affecting performance or robustness.Citation64,Citation68 So far, with antibody production on the scale of several thousand liters, there has been no urgent demand to reduce the analyzed sample volumes. Nevertheless, with more industry efforts toward more sustainable and ecological workflows, micro flow can offer an attractive alternative to analytical flow due to increased MS signal, reduced source contamination, and less solvent consumption.Citation69 Additionally, micro flow has been validated in a regulated bioanalytical laboratory.Citation70 Nanoflow methods have been shown in several recent publications to offer superior sensitivity for HCP identification while using very low sample amounts.Citation7,Citation53,Citation56 Thus, nano flow methods are very valuable for applications requiring deep HCP profiling, such as root cause analysis of PS20 degradation through CHO enzymes,Citation7 and for impurity testing of new modalities such as gene therapy products, where the sample amount will be very limited compared to antibody production.Citation71,Citation72 While nanoLC-MS provides great value for HCP analysis that requires sub-ppm to single-digit ppm level sensitivity, it often suffers with compromised operational robustness and reduced sample throughput,Citation73–75 As a result, several HCP studies using nano flow were only performed with a limited set of samples and replicates, which was not sufficient to demonstrate method robustness required for method validation.Citation76 Thus, nano flow methods are currently not broadly used for routine process support compared to established 1D-LC-MS/MS methods operated at analytical flow. summarizes all advantages and limitations of the described methods and compares general LC separation strategies.
Table 3. Comparison of different LC and MS methods with their advantages and limitations for HCP analysis.
Advanced MS techniques for HCP identification and quantitation
Three classes of MS detection methods are generally available: data-dependent acquisition (DDA), data independent acquisition (DIA), and targeted MS. compares advantages and limitations for each method. For both protein discovery and HCP identification studies, DDA is the method of choice and provides an unbiased platform requiring no prior knowledge about proteins present in the analyzed sample.Citation13,Citation46 DIA offers an untargeted approach for HCP identification and quantification by acquisition of full MS2 fragment spectra over a specified mass range. Selected or multiple reaction monitoring (SRM/MRM) and parallel reaction monitoring (PRM) are targeted MS methods used for absolute quantification of selected HCPs based on the analysis of previously specified transitions pairs.
In more detail, DDA is a tandem MS method selecting precursors for fragmentation based on their abundance in MS1 survey scans.Citation46 Only the top N most intense precursors are fragmented, resulting in relatively clean MS2 peptide spectra, which are then surveyed against a sequence database for protein identification. In contrast, DIA relies on the fragmentation of all precursors in a specified m/z window regardless of their intensity.Citation77,Citation78 Zhang et al. provided a helpful overview of all currently available DIA acquisition schemes, including DIA, MSE, SWATH (sequential window acquisition of all theoretical fragment ion spectra), BoxCar DIA, and ScanningSWATH.Citation79 While providing complete data sets, MS2 spectra derived from multiple precursors have the downside of challenging computational deconvolution. Nevertheless, DIA has great potential to serve as a generic platform method for quantification of single HCPs by the “top 3” method.Citation13,Citation80 Numerous studies over the last decade demonstrate the successful application of the top 3 method to HCP quantification in bioprocess samples.Citation13,Citation81–84 Additionally, Hamaker et al. presented an alternative application of DIA for HCP characterization: quantification of differential expression changes in HCCF samples to provide more insights on the influence of cell culture conditions on the HCP profile.Citation23 Advanced capabilities available on the MS instruments, such as gas phase separationCitation45,Citation63 and BoxCar acquisitionCitation56 can also be applied to address high dynamic range issues and improve the HCP analysis sensitivity. Compared to DIA, MRM and PRM are more straightforward and traditional approaches for protein quantification.Citation85,Citation86 MRM assays are usually performed on triple quadrupole MS systems by analyzing transitions of precursor and 3 to 5 fragment ions.Citation85,Citation87 MRM methods are refined in an iterative process to compile a precursor-transition list, which represents the target proteins best, while focusing on sensitivity and selectivity. In contrast to MRM, PRM methods are performed on a high-resolution hybrid mass spectrometer.Citation88 Instead of selecting a limited number of transitions, PRM mode acquires all possible fragment ions to confirm the target peptides.Citation86 Therefore, PRM is highly specific and reduces the expenses of assay development compared to MRM.Citation89,Citation90 Both MRM and PRM have been shown to provide comparable sensitivity, linearity, dynamic range, and reproducibility.Citation90,Citation91 The ability to retrieve absolute quantitative information from targeted methods depends on the use of internal standards, synthetic stable-isotope labeled peptides or proteins, due to their identical physicochemical properties compared to the corresponding endogenous peptides. Thus, the initial set up of targeted assays is quite time and cost intensive. However, once set up, a targeted MS assay affords highly sensitive and robust quantification of HCPs of interest as reported by several studies.Citation13,Citation24,Citation92
HCP data analysis
To identify proteins present in a sample, the acquired tandem mass spectra (DDA) are commonly searched against a protein sequence database in FASTA format. This sequence database should contain all theoretically possible host cell protein, product, and contaminant sequences. Two common sources for reference proteomes are Uniprot.org and RefSeq. All databases vary in regard to completeness, degree of redundancy and quality of sequence annotation.Citation93 Proteins of the prevalent host expression systems for therapeutic protein production, CHO cells and E. coli cells, are usually referred to by the Uniprot accession number.Citation1,Citation94 Interestingly, even though CHO cells have been broadly used, the CHO proteome information is annotated poorly on Uniprot. Uniprot, composed of Swiss-Prot (manually reviewed high quality entries) and its supplement TrEMBL (unreviewed entries), provides three different CRIGR reference proteomes or alternatively a full protein list based on species affiliation. As of September 2022, Uniprot lists 78,360 CHO entries, containing 243 Swiss-Prot entries and 78,117 TrEBML entries, so only 0.3% of all sequences are reviewed and non-redundant protein annotations. The total number of entries grew from 35,000 in 2015 to over 59,000 in 2020 and 78,000 in 2022, in contrast to the number of Swiss-Prot entries (manually reviewed high-quality entries), which remained consistent over this period. Comparing the number of entries to other recent references describing 26,530 CHO genes and 47,829 unique protein sequences,Citation1 it becomes apparent that a significant proportion of Uniprot entries contains redundant sequences, while the use of only one of the CHO reference proteomes is insufficient due to incompleteness. Thus, HCP analysis can suffer from a regular update of a locally saved Uniprot sequence database, which leads to a larger sequence database without an increase in high-quality entries. As an alternative, several studies used RefSeq/NCBI protein entries.Citation17,Citation23,Citation95 However, the majority of publications mentioned HCPs based on their Uniprot accession number.Citation1,Citation7,Citation13,Citation84,Citation96,Citation97 Poor correlation (only Swiss-Prot entries) of Uniprot and RefSeq complicates comparisons of published results. The protein Peroxiredoxin-1 (PRDX1) serves as an example for redundant entries and problematic descriptions in different databases is. PRDX1 has two related Uniprot entries: Q9JKY1 Peroxiredoxin-1 (sp) and G3GYP9 Peroxiredoxin-1 (tr). The same sequences are listed in RefSeq by NP_001233694.1 peroxiredoxin-1 and XP_007608925.1 peroxiredoxin-1 isoform X1, respectively. Both sequences only differ in a single amino acid (Q140R). Due to a sequence identity of 99.5%, almost all peptides appear as “shared peptides” when both entries are present in the search database (e.g., by CRIGR Uniprot download). In addition, if no peptide with the point mutation is detected, search engines cannot distinguish between both entries and will favor one of them randomly. In this case, both entries will be displayed as a protein group, which is derived from redundancies rather than true isoforms. In the literature, PRDX1 is referenced with the accession Q9JKY1 by several studies,Citation1,Citation52,Citation98,Citation99 mentioned with the accession G3GYP9,Citation12,Citation100 or even described by an outdated NCBI number 81,917,543.Citation17,Citation94
Several publicly available software tools and the functionality of different search engines are well-described by Nesvizhskii et al.Citation93 The predominantly used software tools in HCP-related publications are Proteome Discoverer (Thermo Fisher Scientific) with Sequest and Mascot, and Protein Pilot (Sciex). The gold standard criterion for a positive protein identification across the HCP community is the detection of at least two unique peptides with a confidence of >95%. To exclude incorrect identifications, most studies rely on the community-approved empirical threshold of 1–5% false discovery rate (FDR), estimated by the target-decoy search strategy.Citation101,Citation102 The 1–5% FDR is much higher than most proteomics analyses, which have more than thousands of protein identifications, for a few reasons. First, it was recognized that the target-decoy strategy is efficient and accurate for medium-to-large proteomic data sets, but the accuracy of FDR estimation is greatly diminished and exceedingly optimistic for very small datasets (e.g., < 20 identifications), which are of particular interest for final product HCP samples.Citation101,Citation102 Kreimer et al. also mentioned the limitations of the target-decoy strategy for purified HCP samples and described the use of a higher FDR rate of 5% instead of 1% FDR.Citation81 Because it is very important for patients’ safety and product stability to thoroughly identify HCPs in an ultrafiltration and diafiltration (UF/DF) sample, we recommend using a less stringent FDR cutoff such as 5 or 10% and reviewing identifications manually for false positive identifications case-by-case for drug product samples.
Compared to DDA, DIA data analysis is more complex due to multiplexed MS2 spectra, which are more challenging to deconvolute computationally. DIA analysis requires MS2 spectral libraries containing information about variables such as precursor and fragment masses, fragmentation pattern, and retention times.Citation78 These spectral libraries can be generated either experimentally or in silico by prediction algorithms (library-free approaches). Comprehensive libraries are needed for sensitive peak identification since only spectra present in the library can be matched with acquired peaks for HCP identification and quantitation.Citation95,Citation103,Citation104 However, one needs to consider the balance between comprehensiveness and sensitivity based on the two major challenges that extensive libraries present. They require a very robust chromatographic setup and statistical correction for the large number of spectra in the library.Citation78,Citation105 From the HCP perspective, comprehensive libraries are very valuable for DIA analysis of medium- to large-sized sample data sets, such as null strain fermentations, HCCF or early process pools. In the case of more downstream process samples and purified HCP samples, we recommend using a smaller sample-specific library. A very practical option is the use of DDA runs of prior process steps or null strain fermentations instead of extensive fragmentation techniques. The main advantage is that usually already-acquired data can be used without additional measurements, while containing higher-quality spectra than the purified sample itself (as they would be used for library-free approaches). Zhang et al. provide a very detailed overview and comparison of workflows with recently available DIA software tools for peak identification and scoring.Citation79
The most widely used software tools for quantitative analysis of HCPs are Skyline, Analyst, and Spectronaut.Citation78,Citation106 From the users’ perspective, Skyline is easily accessible as open source software for DIA and targeted data, provides ongoing improvements, and offers a high degree of freedom as numerous parameters can be edited.Citation107 However, the manual review requires experienced users, and therefore Skyline might not offer the best platform for routine high throughput data analysis. Analyst is software limited to Sciex instrument data, and allows the user to do rapid targeted data processing and ensure compliance. Spectronaut offers from our perspective a user-friendly frontend, various options for DIA data filtering and plotting, and predefined data analysis schemes that facilitate routine use.Citation108 In general, the users of the bioinformatics tools for HCP analysis should be aware of potential challenges, which are primarily linked to the comprehensiveness of CHO protein annotations, the quality of accessible sequence databases, as well as statistical strategies for an appropriate error rate control. As described above, the most common approach for targeted quantification is based on the use of heavily labeled surrogate peptides. For quantification of target proteins, measured fragment ion peak areas of endogenous peptides are summed and compared to the summed transition peak areas of the heavyly labeled reference peptides.Citation87,Citation89 Thus, the unknown peptide amount is simply calculated by proportion. For absolute quantification of HCPs, we recommend using at least two peptides per protein to ensure reliable and accurate results. Numerous software tools used for targeted assays are available either open source or commercially for assay development, peak integration, quality assessment, and statistical analysis.Citation109 Among these, Skyline is the software tool, mentioned the most in HCP studies, along with MS vendor-specific software tools.Citation13,Citation24,Citation83,Citation92
Evaluation of assay performance and system suitability testing
To date, data from HCP analysis by LC-MS/MS is often used as additional characterization information to support process development rather than directly submitted to health authorities. To evaluate the method performance, one can follow the ICH guidance documents containing recommendations for submission of analytical data and methods validation.Citation76 Briscoe et al. described an approach specifically eligible for bioanalytical LC-MS/MS.Citation110 First, in an instrument qualification, the suitability of the used equipment is demonstrated to fit the intended use. The next step is conducting a method validation for a particular procedure to examine different characteristics such as specificity, accuracy, repeatability, intermediate precision, reproducibility, detection limit, quantitation limit, and robustness.Citation76,Citation111 Importantly, accuracy and reproducibility investigations should include a minimum of three replicates of the full analytical procedure (not injection replicates) plus testing by different analysts on different days and/or in different laboratories.
Concerning robustness, parameters, such as digestion temperature, digestion time, stability of stored samples, influence of different trypsin lots, LC column lots, and mobile phases should be considered for HCP LC-MS/MS methods. Due to observed variations in LC-MS analysis, e.g., signal drifts, and regularly low limit detection of HCPs, priming injections of a solution with known masses and calibration prior to each measurement is recommended.Citation110 One of the major quality checks are blank runs to determine carry over, contamination, and interferences. To prevent false positive identifications through carryover, we recommend in addition to running blank runs, to also run process samples in order from most purified to least purified. Second and particularly important for MS-based methods, is the implementation of system suitability test (SST) samples into every batch of test samples to ensure continued high quality and reliability of results. SST samples should mimic test samples through similarity (e.g., low level spiked drug product samples), be stable, homogenous, and available in sufficient amounts.Citation112 We recommend measuring SST samples before and after the test samples, or systematically throughout the batch if the sample set is large. Monitoring SST samples over a longer period can help to track instrument and assay performance, as several references mention in their studies.Citation13,Citation51,Citation52,Citation113 For example, Walker et al. described the generation of a method control sample for an HCP identification assay by DDA using a purified recombinant protein spiked into antibody matrix at 50 ppm to ensure a reliable protein identification above this level.Citation13 Bereman et al. reported an automated pipeline to monitor instrument performance by analyzing SST runs with the open source software Skyline and the add-on Panorama AutoQC.Citation114 Tools such as AutoQC offer an easy and fast way to monitor system suitability once set up. However, the fact that Panorama operates server-based could deter biopharma consumers from using it.
Strategies for HCP characterization and process monitoring by LC-MS/MS
While ELISA is a robust workhorse for monitoring HCPs from in-process pools to the drug products, MS is a valuable orthogonal tool to investigate HCPs whenever inconsistent ELISA results or major process changes occur. In addition, MS is routinely used for HCP profiling as a control strategy for product quality and safety. We currently use various LC-MS/MS methods for the identification and quantitation of individual HCPs. Our HCP identification toolbox consists of methods with various sample preparation strategies and LC-MS conditions, with different limits of detection (LODs) for HCP identification, as shown in . The major challenge in HCP analysis is the required broad dynamic range, and that HCPs are not equally critical due to their varied impacts on drug quality and patient safety at different levels. Therefore, the listed methods can be good starting points to develop appropriate methods to screen whether individual high abundance HCPs are present in HCCF and/or in-process pools. shows an example of a decision tree for a workflow supporting process development with HCP characterization by LC-MS/MS. LC-MS samples are prepared by either denaturing or native digestion during the development stage, as described in .
Figure 1. General workflow for HCP characterization by LC-MS/MS analysis.
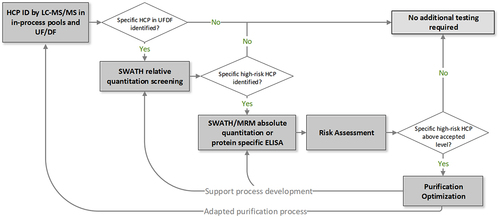
Table 4. Our commonly used HCP identification methods by LC-MS/MS analysis.
To develop and evaluate these methods, a model sample consisting of purified reference mAb spiked with known levels of 48-protein Universal Proteomics Standard (UPS-1; Sigma Aldrich) was established to simulate trace-level HCPs in a product. With this control sample, the LOD of HCP identification can be determined by the ability to detect most if not all the spiked proteins at and above the threshold defining the LOD. The detection limit for a specific method should be robust and reproducible based on multiple replicates and across runs, as the method may be relied on as a general platform to demonstrate the clearance of individual HCPs.Citation13,Citation52 If a specific HCP is identified in UF/DF pools, we would confirm it with a quantitative MS-based method and may start a risk assessment based on prior knowledge of the identified protein.Citation1,Citation19 For a known high-risk protein, a customized protein-specific quantitative method would be developed to cover the target minimum level recommended in the risk assessment.
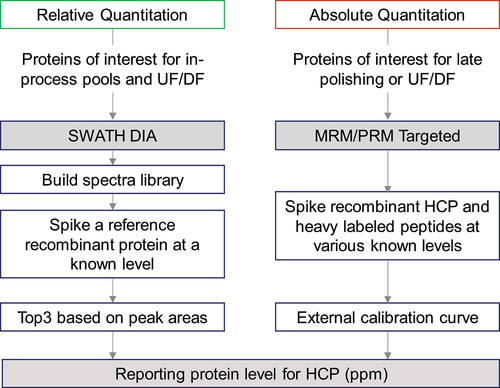
Quantitative methods including protein-specific ELISA, SWATH-MS, or targeted MRM-MS may be used to monitor and confirm the specific HCP during process development. As described in , we use DIA/SWATH MS for quantitation of identified HCPs to guide their optimal removal conditions during purification.Citation13 For optimizing upstream processing and early purification step conditions, relative quantitation by SWATH is generally convenient and sufficient due to high reproducibility and robustness. For testing samples from late purification steps or UF/DF pools, absolute quantification by targeted MRM analysis confirms the target HCP levels. However, if peptides of interest consistently co-elute with other peptides across LC runs, SWATH-based quantitation may not be selective or accurate enough; sample preparation or LC-MS conditions would need to be optimized accordingly. Other necessary steps for protein quantitation by SWATH include confirming digestion conditions for consistency and screening appropriate reference proteins for quantitation accuracy. Targeted MS analysis is used for absolute quantitation of HCPs in late purification steps and drug product to confirm their levels and ensure their acceptable removal. This is because targeted MS analysis (PRM or MRM) has high selectivity, specificity, and accuracy across different types of samples. When using PRM-MS, we use the corresponding recombinant protein or internal peptides from the drug molecule as the reference protein/peptides for absolute quantitation. When using MRM, the selection of representative peptides for quantitation is essential, as the corresponding heavy-labeled internal standard peptides are based on the selected peptides.Citation92 Generally, we only consider unique peptides by in-silico search, whereas miscleaved peptides and peptides with labile-modified amino acids are excluded. For absolute quantification of HCPs, we recommend using at least two peptides per protein to ensure reliable and accurate results. Each internal standard peptide should be optimized with the LC-MS/MS instrument to ensure good detection and no interference from other peptides in a blank or purified sample. Based on the efforts involved, we do not recommend quantitation by MRM/PRM for early-stage development work.
Figure 2. Workflow for HCP quantitation by SWATH and targeted (MRM/PRM) MS analysis.
In later development stages, an improved process with significantly reduced HCP levels is developed for the clinical product. During the ongoing clinical trials, drug quality, and patient health is closely monitored. For monitoring potential unknown HCPs after process changes, in-process pools of the adapted process are again subjected to HCP characterization to ensure the removal of all residual HCPs across different runs in the final UF/DF pools. When product-related issues (polysorbate degradation, adverse clinical effects) or assay-related issues (nonlinear or high total, inconsistent HCP readings from ELISA assay) occur, new and highly sensitive LC-MS/MS methods for identification and quantitation can be developed. For example, specific workflow parameters, such as sample load, tryptic digestion conditions, and LC-MS/MS instrumentation, could be optimized to measure HCPs at <0.1 ppm.Citation7
Case studies of LC-MS/MS-based HCP analysis
Supporting the development of an HCP-specific ELISA for PLBL2
LC-MS/MS analysis can not only identify and quantify individual HCPs, but also support anti-CHOP (Chinese hamster ovary proteins) ELISA troubleshooting and development. In an early HCP study, abnormal non-linear dilution response observed in our anti-CHOP ELISA method was investigated by complementary in-gel digestion coupled with MS. MS analysis enabled confident identification of phospholipase B-like 2 (PLBL2), an HCP known to elicit immune response in patients, as the major protein impurity to show the ELISA dilution nonlinear response, and it confirmed the consequence of excessive PLBL2 in anti-CHOP ELISA assay.Citation19,Citation26 The MS analysis confidently identified excess PLBL2 as the antigen responsible for the abnormal ELISA readings. This was further confirmed by low CHOP values, no more PLBL2 identified by MS, and linear dilution responses when samples were pre-treated with anti-PLBL2 antibodies. Therefore, we developed a PLBL2-specific ELISA to assess PLBL2 levels in our mAb portfolio biotherapeutics, including both commercial products and those in clinical development. PLBL2 was detected below 1 ppm in all of the products after implementing purification improvements to remove PLBL2.Citation26 Ultimately, the identification of PLBL2 by LC-MS/MS analysis not only revealed the root cause for abnormal anti-CHOP ELISA readings, but also led to the development of a new PLBL2-specific ELISA.
Supporting downstream process development using a sensitive LC-MS/MS method
For highly active HCPs that can affect product stability at very low levels (such as 1 ppm level), it is necessary to use highly sensitive LC-MS/MS analysis to evaluate clearance efficiency and inform process development. To enable a 1 ppm detection limit, we developed a method starting with high-load materials (e.g., ~20 mg mAb with analytical flow LC-MS) at native digestion conditions with low enzyme-to-protein ratio. An example of enzyme-to-protein ratio optimization for improving HCP identification can be found in our previous study.Citation7 The method sensitivity was specifically optimized and evaluated for the molecule by measuring the UPS-1 spiked into the mAb1. At the process development stage, HCPs were reproducibly identified in the UF/DF pools with multiple replicates per sample across different process runs. For positive controls to confirm assay performance, we used a system suitability sample with certain spiked proteins at a known level as well as a representative UF/DF pool sample with high HCP levels. This method was used to confirm, in multiple replicates across different process runs, that an added bioprocessing step successfully cleared HCPs below the 1 ppm detection limit. The analysis demonstrated the capacity and reproducibility of this direct LC-MS method and the benefits of adding an extra purification step into the existing process for HCP clearance.
Identification of low-level polysorbate degradation enzymes
Polysorbates (PS) are the most common class of surfactants in biopharmaceutical formulations and have set the benchmark to stabilize protein drugs. Degradation of PS by HCP hydrolytic enzymes in drug products may affect the protein-stabilizing properties of PS and lead to long-term drug stability issues. Historically, we used MRM-based targeted MS methods to identify and quantify proteins of interest, such as high-risk HCPs and specific hydrolytic enzymes.Citation13 To support process development and mitigate the PS degradation, we need to explore and discover more potential enzymes involved in the PS degradation process. For one product that has PS degradation challenges, we used anti-CHOP affinity-chromatography to enrich HCPs and LC-MS/MS analysis to identify low abundant proteins and enhance the CHOP-to-product ratio from <1 ppm in final drug substance to >34,000 ppm in anti-CHOP column eluate by ELISA measurement, and confirmed the presence of additional PS-degrading enzymes like lysosomal acid lipase and palmitoyl-protein thioesterase 1. The enzymes of interest were then recombinantly expressed and their PS degradation potential was characterized by enzyme activity assays and LC-MS-based PS content and free fatty acid methods.Citation12 We further developed an ultra-sensitive LC-MS/MS platform for robust identification of HCPs at 0.1 ppm, with the potential for higher detection and quantitation sensitivity at 0.01 ppm for specific enzymes of interest using targeted PRM-MS, to demonstrate the clearance of these HCPs by a modified purification process.Citation7 With this platform, the optimized digestion condition improved the identification of HCPs; addition of SDC during sample preparation disrupted the association of hydrophobic hydrolases with antibody drugs;Citation57 and nano-LC-MS/MS analysis further enhanced the sensitivity and detection dynamic range. Our work using different strategies filled a significant gap in HCP analysis for detecting particular extra low-level hydrolases in drug process development. More recently, Li et al. developed an activity-based protein profiling approach to identify active enzymes responsible for polysorbate degradation. It is a function-related assay that reacts target types of enzymes with chemical probes and subsequently identifies them by LC – MS/MS.Citation115 The identification of hydrolases alone provides a global proteome view of possible causes for PS degradation, but there is no direct link between PS-caused instability and identified proteins. Therefore, to study potentially high-risk enzymes, we need to take a holistic look and use orthogonal approaches, such as enzyme expression, activity assays, and PS content and free fatty acids assays, to study function, mechanism and potential risks of the hydrolases and ensure drug quality and safety.Citation12,Citation34,Citation115
Using quantitative LC-MS/MS methods (SWATH and MRM) to support process development removal of a high-risk HCP
PRDX1 is considered a high-risk HCP due to its potential to cause immunogenic responses. PRDX1 is a hitch-hiker protein that sticks to many mAb drugs,Citation1 and thus is often identified in drug substances. Generally, PRDX1 is well-controlled during purification, with less than 10 ppm in most launched mAb drugs and up to 100 ppm in a few.Citation11 We identified PRDX1 by LC-MS/MS in UF/DF pool samples for a specific mAb molecule (mAb2) at the development stage. High levels of PRDX1 at ~30–150 ppm across different process runs were determined by using the SWATH-based relative quantitation method.Citation13 Therefore, a toxicology assessment was needed to establish an acceptance level during the process and ensure drug safety for a potential high-dose drug. Based on the in-silico method and prior knowledge, toxicology experts could be engaged to further perform the risk assessment that links the HCP and any potential adverse clinical effects for later development. During the purification development optimization, we used SWATH-based relative quantitation to inform the optimal conditions with higher throughput (accuracy generally within 2 folds); and in the final purification pools, we confirmed the reduction of PRDX1 below a conservative level (e.g., < 5 ppm) by both SWATH-based relative quantitation and MRM-based absolute quantitation (accuracy generally 80–120%). Additionally, we tested some commercial ELISA kits to measure PRDX1 and observed dilution nonlinearity and a low spike-recovery rate only in mAb2, indicating a strong association of PRDX1 with this specific drug. It may require a substantial amount of time to develop a molecule-specific PRDX1 ELISA assay, so MS serves as a complementary tool to facilitate method development. Quantitative LC-MS/MS methods are not typically required for routine HCP process testing for batch release; however, they can provide useful information and may be necessary for specific high-risk HCPs during process development, especially when an ELISA assay is not available yet.
Conclusions and outlook
HCPs are considered a critical quality attribute for therapeutic proteins, especially mAbs, and should be monitored to mitigate drug quality and safety risks. They can be thoroughly profiled by LC-MS/MS methods for an overall broad dynamic range of HCP identification and quantitation. We recommend using LC-MS/MS-based methods for HCP identification from the early process development stage because some HCPs may not show immunoreactivity in conventional ELISA assays for specific molecules, even with low anti-HCP values. In general, we report total HCP levels by ELISA to demonstrate HCP profile consistency for downstream process development. However, when HCP levels are high or abnormal, LC-MS/MS-based strategies should be necessary instead of complementary. There is no consensus for how to detect and measure any specific high-risk HCPs. Sample preparation, LC conditions, and MS analytical methods need to be developed case-by-case to best detect HCPs during process development.
For individual HCPs that may have adverse clinical effects, tiered toxicology assessment of in vitro/in vivo evaluation and clinical observation is critical to link the HCP to clinical drug safety and determine the acceptable level of residual HCP. Therefore, developing fast and accurate MS-based quantitative methods for individual HCPs will support targeted HCP characterization to mitigate the risk posed by the presence of high-risk HCPs in drug products. Beyond direct characterization, LC-MS analysis has shown a broader application in the field of HCP study, especially using affinity purification and LC-MS-based methods to evaluate the anti-HCP antibody’s coverage. The method can further be used to select the optimal antibody for high-risk HCP clearance based on the coverage and profiling of identified HCPs.
Compared to ELISA-based HCP analysis, LC-MS/MS analysis has developed into a highly sensitive platform for HCP detection with the power to directly identify individual HCPs. However, MS technology has its own implementation challenges, and there is still a need to make MS-based methods more robust and higher throughput. Extra efforts may be needed to validate such assays by tailored method development and design. Additionally, reproducibility of MS analysis should be confirmed by performing multiple replicates from different runs for multiple representative protein standards, using discreet data analysis with proper false discovery rates and manually checking MS data to achieve regulatory requirements. Method robustness will be required when using LC-MS/MS for HCP monitoring and quantitation in quality control, with the increasing popularity of using LC-MS-based multi-attribute method.Citation116 The strategic combination of both identification and quantitation of individual HCPs allows for better drug quality control during process development and reduces potential HCP-related risks.
Acknowledgments
The authors would like to thank Christopher Yu for kindly reviewing our manuscript draft and giving feedback. The authors acknowledge the following colleagues for their support and collaboration: Vikas Sharma, Guoying Jiang, Margaret Lin, Lori O’Connell, Shelley Elvington, Inn Yuk, Amy Shen, and John Stults.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Jones M, Palackal N, Wang F, Gaza-Bulseco G, Hurkmans K, Zhao Y, Chitikila C, Clavier S, Liu S, Menesale E, et al. “High-risk” host cell proteins (HCPs): a multi-company collaborative view. Biotechnol Bioeng. 2021;118(8):2870–15. doi:10.1002/bit.27808.
- FDA. Points to consider in the manufacture and testing of monoclonal antibody products for human use. FDA-1994-D-0318 1994.
- EMA. DNA and host cell protein impurities, routine testing versus validation studies. 1997; CPMP/BWP/382/97.
- Pharmacopeia US. 2016. Residual host cell protein measurement in biopharmaceuticals. USP. Published General Chapter 1132. 39:1416–36.
- Pharmacopeia EU. Host-cell protein assays. European pharmacopoeia monograph 2.6.34. 2017;4:20634.
- Zhu-Shimoni J, Yu C, Nishihara J, Wong RM, Gunawan F, Lin M, Krawitz D, Liu P, Sandoval W, Vanderlaan M. Host cell protein testing by ELISAs and the use of orthogonal methods. Biotechnol Bioeng. 2014;111(12):2367–79. doi:10.1002/bit.25327.
- Yang F, Li D, Kufer R, Cadang L, Zhang J, Dai L, Guo J, Wohlrab S, Greenwood-Goodwin M, Shen A, et al. Versatile LC–MS-Based workflow with robust 0.1 ppm sensitivity for identifying residual HCPs in biotherapeutic products. Anal Chem. 2022;94(2):723–31. doi:10.1021/acs.analchem.1c03095.
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13(16):1935–43. doi:10.1089/10430340260355347.
- Kulagina N, Besseau S, Godon C, Goldman GH, Papon N, Courdavault V. Yeasts as biopharmaceutical production platforms. Front Fungal Biol. 2021;2. doi:10.3389/ffunb.2021.733492.
- Reisinger V, Toll H, Mayer RE, Visser J, Wolschin F. A mass spectrometry-based approach to host cell protein identification and its application in a comparability exercise. Anal Biochem. 2014;463:1–6. doi:10.1016/j.ab.2014.06.005.
- Molden R, Hu M, Yen ES, Saggese D, Reilly J, Mattila J, Qiu H, Chen G, Bak H, Li N. Host cell protein profiling of commercial therapeutic protein drugs as a benchmark for monoclonal antibody-based therapeutic protein development. MAbs. 2021;13(1):1955811. doi:10.1080/19420862.2021.1955811.
- Graf T, Tomlinson A, Yuk IH, Kufer R, Spensberger B, Falkenstein R, Shen A, Li H, Duan D, Liu W, et al. Identification and characterization of polysorbate-degrading enzymes in a monoclonal antibody formulation. J Pharm Sci. 2021;110(11):3558–67. doi:10.1016/j.xphs.2021.06.033.
- Walker DE, Yang F, Carver J, Joe K, Michels DA, Yu XC. A modular and adaptive mass spectrometry-based platform for support of bioprocess development toward optimal host cell protein clearance. MAbs. 2017;9(4):654–63. doi:10.1080/19420862.2017.1303023.
- Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): implications for downstream process development. Biotechnol Bioeng. 2010;105(2):306–16. doi:10.1002/bit.22532.
- Hogwood CEM, Bracewell DG, Smales CM. Measurement and control of host cell proteins (HCPs) in CHO cell bioprocesses. Curr Opin Biotechnol. 2014;30:153–60. doi:10.1016/j.copbio.2014.06.017.
- The antibody society, Inc. Antibody therapeutics approved or in regulatory review in the EU or US. [assessed Mar 8, 2023]. www.antibodysociety.org/antibody-therapeutics-product-data.
- Falkenberg H, Waldera-Lupa DM, Vanderlaan M, Schwab T, Krapfenbauer K, Studts JM, Flad T, Waerner T. Mass spectrometric evaluation of upstream and downstream process influences on host cell protein patterns in biopharmaceutical products. Biotechnol Prog. 2019;35(3):e2788. doi:10.1002/btpr.2788.
- Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol Bioeng. 2015;112(9):1727–37. doi:10.1002/bit.25628.
- Vanderlaan M, Zhu-Shimoni J, Lin S, Gunawan F, Waerner T, Van Cott KE. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog. 2018;34(4):828–37. doi:10.1002/btpr.2640.
- Bracewell DG, Smith V, Delahaye M, Smales CM. Analytics of host cell proteins (HCPs): lessons from biopharmaceutical mAb analysis for gene therapy products. Curr Opin Biotechnol. 2021;71:98–104. doi:10.1016/j.copbio.2021.06.026.
- Krutzke L, Rosler R, Allmendinger E, Engler T, Wiese S, Kochanek S. Process- and product-related impurities in the ChAdOx1 nCov-19 vaccine. Elife. 2022;11:11. doi:10.7554/eLife.78513.
- Yuk IH, Nishihara J, Walker D Jr., Huang E, Gunawan F, Subramanian J, Pynn AFJ, Yu XC, Zhu-Shimoni J, Vanderlaan M, et al. More similar than different: host cell protein production using three null CHO cell lines. Biotechnol Bioeng. 2015;112(10):2068–83. doi:10.1002/bit.25615.
- Hamaker NK, Min L, Lee KH. Comprehensive assessment of host cell protein expression after extended culture and bioreactor production of CHO cell lines. Biotechnol Bioeng. 2022;119(8):2221–38. doi:10.1002/bit.28128.
- Chiu J, Valente KN, Levy NE, Min L, Lenhoff AM, Lee KH. Knockout of a difficult-to-remove CHO host cell protein, lipoprotein lipase, for improved polysorbate stability in monoclonal antibody formulations. Biotechnol Bioeng. 2017;114(5):1006–15. doi:10.1002/bit.26237.
- Carver J, Kern M, Ko P, Greenwood-Goodwin M, Yu XC, Duan D, Tang D, Misaghi S, Auslaender S, Haley B, et al. A ribonucleoprotein-based decaplex CRISPR/Cas9 knockout strategy for CHO host engineering. Biotechnol Prog. 2022;38(1):e3212. doi:10.1002/btpr.3212.
- Vanderlaan M, Sandoval W, Liu P, Nishihara J, Tsui G, Lin M, Gunawan F, Parker S, Wong RM, Low J , et al. Hamster phospholipase B-like 2 (PLBL2): A host-cell protein impurity in therapeutic monoclonal antibodies derived from Chinese hamster ovary cells. BioProcess International. 2015;13(4):18–29.
- Zhang Q, Goetze AM, Cui H, Wylie J, Trimble S, Hewig A, Flynn GC. Comprehensive tracking of host cell proteins during monoclonal antibody purifications using mass spectrometry. MAbs. 2014;6(3):659–70. doi:10.4161/mabs.28120.
- Gilgunn S, El-Sabbahy H, Albrecht S, Gaikwad M, Corrigan K, Deakin L, Jellum G, Bones J. Identification and tracking of problematic host cell proteins removed by a synthetic, highly functionalized nonwoven media in downstream bioprocessing of monoclonal antibodies. J Chromatogr A. 2019;1595:28–38. doi:10.1016/j.chroma.2019.02.056.
- Chen Y, Xu CF, Stanley B, Evangelist G, Brinkmann A, Liu S, McCarthy S, Xiong L, Jones E, Sosic Z, et al. A highly sensitive LC-MS/MS method for targeted quantitation of lipase host cell proteins in biotherapeutics. J Pharm Sci. 2021;110(12):3811–18. doi:10.1016/j.xphs.2021.08.024.
- Pilely K, Johansen MR, Lund RR, Kofoed T, Jorgensen TK, Skriver L, Mørtz E. Monitoring process-related impurities in biologics–host cell protein analysis. Anal Bioanal Chem. 2022;414(2):747–58. doi:10.1007/s00216-021-03648-2.
- Wohlrab S, Wiedmann M, Aschner M, Reusch D, Bulau P, Haindl M. Tracking host cell proteins while biopharmaceutical manufacturing: advanced methodologies to improve product quality. Am Pharm Rev. 2018;21:36–39.
- Gunawan F, Nishihara J, Liu P, Sandoval W, Vanderlaan M, Zhang H, Krawitz D. Comparison of platform host cell protein ELISA to process-specific host cell protein ELISA. Biotechnol Bioeng. 2018;115(2):382–89. doi:10.1002/bit.26466.
- Hu M, Molden R, Hu Y, Huang Y, Qiu H, Li N. Host cell protein identification in monoclonal antibody high molecular weight species. J Chromatogr B Analyt Technol Biomed Life Sci. 2022;1210:123448. doi:10.1016/j.jchromb.2022.123448.
- Hecht ES, Mehta S, Wecksler AT, Aguilar B, Swanson N, Phung W, Dubey Kelsoe A, Benner WH, Tesar D, Kelley RF, et al. Insights into ultra-low affinity lipase-antibody noncovalent complex binding mechanisms. MAbs. 2022;14(1):2135183. doi:10.1080/19420862.2022.2135183.
- Seisenberger C, Graf T, Haindl M, Wegele H, Wiedmann M, Wohlrab S. Questioning coverage values determined by 2D western blots: a critical study on the characterization of anti-HCP ELISA reagents. Biotechnol Bioeng. 2021;118(3):1116–26. doi:10.1002/bit.27635.
- Waldera-Lupa DM, Jasper Y, Kohne P, Schwichtenhovel R, Falkenberg H, Flad T, Happersberger P, Reisinger B, Dehghani A, Moussa R, et al. Host cell protein detection gap risk mitigation: quantitative IAC-MS for ELISA antibody reagent coverage determination. MAbs. 2021;13(1):1955432. doi:10.1080/19420862.2021.1955432.
- Henry SM, Sutlief E, Salas-Solano O, Valliere-Douglass J. ELISA reagent coverage evaluation by affinity purification tandem mass spectrometry. MAbs. 2017;9(7):1065–75. doi:10.1080/19420862.2017.1349586.
- Seisenberger C, Graf T, Haindl M, Wegele H, Wiedmann M, Wohlrab S. Toward optimal clearance: a universal affinity-based mass spectrometry approach for comprehensive ELISA reagent coverage evaluation and HCP hitchhiker analysis. Biotechnol Prog. 2022;38(3):e3244. doi:10.1002/btpr.3244.
- Rumachik NG, Malaker SA, Poweleit N, Maynard LH, Adams CM, Leib RD, Cirolia G, Thomas D, Stamnes S, Holt K, et al. Methods matter: standard production platforms for recombinant AAV produce chemically and functionally distinct vectors. Mol Ther Methods Clin Dev. 2020;18:98–118. doi:10.1016/j.omtm.2020.05.018.
- Aloor A, Zhang J, Gashash EA, Parameswaran A, Chrzanowski M, Ma C, Diao Y, Wang P, Xiao W. Site-specific N-glycosylation on the AAV8 capsid protein. Viruses. 2018;10(11):644. doi:10.3390/v10110644.
- Johnson S, Wheeler JX, Thorpe R, Collins M, Takeuchi Y, Zhao Y. Mass spectrometry analysis reveals differences in the host cell protein species found in pseudotyped lentiviral vector. Biologicals. 2018;52:7. doi:10.1016/j.biologicals.2017.12.005.
- Trauchessec M, Hesse AM, Kraut A, Berard Y, Herment L, Fortin T, Bruley C, Ferro M, Manin C. An innovative standard for LC-MS-based HCP profiling and accurate quantity assessment: application to batch consistency in viral vaccine samples. Proteomics. 2021;21(5):e2000152. doi:10.1002/pmic.202000152.
- Doneanu CE, Chen W. Analysis of host-cell proteins in biotherapeutic proteins by LC/MS approaches. Methods Mol Biol. 2014;1129:341–50. doi:10.1007/978-1-62703-977-2_25.
- Levy NE, Valente KN, Choe LH, Lee KH, Lenhoff AM. Identification and characterization of host cell protein product-associated impurities in monoclonal antibody bioprocessing. Biotechnol Bioeng. 2014;111(5):904–12. doi:10.1002/bit.25158.
- Doneanu CE, Anderson M, Williams BJ, Lauber MA, Chakraborty A, Chen W. Enhanced detection of low-abundance host cell protein impurities in high-purity monoclonal antibodies down to 1 ppm using ion mobility mass spectrometry coupled with multidimensional liquid chromatography. Anal Chem. 2015;87(20):10283–91. doi:10.1021/acs.analchem.5b02103.
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. doi:10.1038/nature19949.
- Bomans K, Lang A, Roedl V, Adolf L, Kyriosoglou K, Diepold K, Eberl G, Mølhøj M, Strauss U, Schmalz C, et al. Identification and monitoring of host cell proteins by mass spectrometry combined with high performance immunochemistry testing. PLos One. 2013;8(11):e81639. doi:10.1371/journal.pone.0081639.
- Thompson JH, Chung WK, Zhu M, Tie L, Lu Y, Aboulaich N, Strouse R, Mo WD. Improved detection of host cell proteins (HCPs) in a mammalian cell-derived antibody drug using liquid chromatography/mass spectrometry in conjunction with an HCP-enrichment strategy. Rapid Commun Mass Spectrom. 2014;28(8):855–60. doi:10.1002/rcm.6854.
- Madsen JA, Farutin V, Carbeau T, Wudyka S, Yin Y, Smith S, Anderson J, Capila I. Toward the complete characterization of host cell proteins in biotherapeutics via affinity depletions, LC-MS/MS, and multivariate analysis. MAbs. 2015;7(6):1128–37. doi:10.1080/19420862.2015.1082017.
- Soderquist RG, Trumbo M, Hart RA, Zhang Q, Flynn GC. Development of advanced host cell protein enrichment and detection strategies to enable process relevant spike challenge studies. Biotechnol Prog. 2015;31(4):983–89. doi:10.1002/btpr.2114.
- Huang L, Wang N, Mitchell CE, Brownlee T, Maple SR, De Felippis MR. A novel sample preparation for shotgun proteomics characterization of HCPs in antibodies. Anal Chem. 2017;89(10):5436–44. doi:10.1021/acs.analchem.7b00304.
- Kufer R, Haindl M, Wegele H, Wohlrab S. Evaluation of peptide fractionation and native digestion as two novel sample preparation workflows to improve HCP characterization by LC–MS/MS. Anal Chem. 2019;91(15):9716–23. doi:10.1021/acs.analchem.9b01259.
- Chen IH, Xiao H, Daly T, Li N. Improved host cell protein analysis in monoclonal antibody products through molecular weight cutoff enrichment. Anal Chem. 2020;92(5):3751–57. doi:10.1021/acs.analchem.9b05081.
- Zhang S, Xiao H, Li N. Ultrasensitive method for profiling host cell proteins by coupling limited digestion to ProteoMiner technology. Anal Biochem. 2022;657:114901. doi:10.1016/j.ab.2022.114901.
- Deng Y, Gruppen H, Wierenga PA. Comparison of Protein Hydrolysis Catalyzed By Bovine, Porcine, And Human Trypsins. J Agric Food Chem. 2018;66(16):4219–32. doi:10.1021/acs.jafc.8b00679.
- Nie S, Greer T, O’Brien Johnson R, Zheng X, Torri A, Li N. Simple and sensitive method for deep profiling of host cell proteins in therapeutic antibodies by combining ultra-low trypsin concentration digestion, long chromatographic gradients, and boxcar mass spectrometry acquisition. Anal Chem. 2021;93(10):4383–90. doi:10.1021/acs.analchem.0c03931.
- Li D, Farchone A, Zhu Q, Macchi F, Walker DE, Michels DA, Yang F. Fast, robust, and sensitive identification of residual host cell proteins in recombinant monoclonal antibodies using sodium deoxycholate assisted digestion. Anal Chem. 2020;92(17):11888–94. doi:10.1021/acs.analchem.0c02258.
- Wang Q, Slaney TR, Wu W, Ludwig R, Tao L, Leone A. Enhancing host-cell protein detection in protein therapeutics using HILIC enrichment and proteomic analysis. Anal Chem. 2020;92(15):10327–35. doi:10.1021/acs.analchem.0c00360.
- Zhao B, Abdubek P, Zhang S, Xiao H, Li N. Analysis of host cell proteins in monoclonal antibody therapeutics through size exclusion chromatography. Pharm Res. 2022;39(11):3029–37. doi:10.1007/s11095-022-03381-0.
- Mörtstedt H, Makower Å, Edlund P-O, Sjöberg K, Tjernberg A. Improved identification of host cell proteins in a protein biopharmaceutical by LC–MS/MS using the ProteoMiner™ Enrichment Kit. J Pharm Biomed Anal. 2020;185:113256. doi:10.1016/j.jpba.2020.113256.
- Chen IH, Xiao H, Li N. Improved host cell protein analysis in monoclonal antibody products through ProteoMiner. Anal Biochem. 2020;2020:113972. doi:10.1016/j.ab.2020.113972.
- Yang F, Walker DE, Schoenfelder J, Carver J, Zhang A, Li D, Harris R, Stults JT, Yu XC, Michels DA. A 2D LC-MS/MS strategy for reliable detection of 10-ppm level residual host cell proteins in therapeutic antibodies. Anal Chem. 2018;90(22):13365–72. doi:10.1021/acs.analchem.8b03044.
- Johnson ROB, Greer T, Cejkov M, Zheng X, Li N. Combination of FAIMS, protein a depletion, and native digest conditions enables deep proteomic profiling of host cell proteins in monoclonal antibodies. Anal Chem. 2020;92(15):10478–84. doi:10.1021/acs.analchem.0c01175.
- Ma J, Sensitive KG. Rapid, rOBUST, AND REPRODUCIBLE WORKFLOW FOR HOST CELL PROTEIN PROFILING IN BIOPHARMACEUTICAL PROCESS DEVELOpment. J Proteome Res. 2020;19(8):3396–404. doi:10.1021/acs.jproteome.0c00252.
- Farrell A, Mittermayr S, Morrissey B, Mc Loughlin N, Navas Iglesias N, Marison IW, Bones J. Quantitative host cell protein analysis using two dimensional data independent LC–MSE. Anal Chem. 2015;87(18):9186–93. doi:10.1021/acs.analchem.5b01377.
- Jawa V, Joubert MK, Zhang Q, Deshpande M, Hapuarachchi S, Hall MP, Flynn GC. Evaluating immunogenicity risk due to host cell protein impurities in antibody-based biotherapeutics. Aaps J. 2016;18(6):1439–52. doi:10.1208/s12248-016-9948-4.
- Hansen FM, Tanzer MC, Brüning F, Bludau I, Stafford C, Schulman BA, Robles MS, Karayel O, Mann M. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. Nat Commun. 2021;12(1):254. doi:10.1038/s41467-020-20509-1.
- Bache N, Geyer PE, Bekker-Jensen DB, Hoerning O, Falkenby L, Treit PV, Doll S, Paron I, Müller JB, Meier F, et al. A novel LC system embeds analytes in pre-formed gradients for rapid, ultra-robust proteomics. Molecular & Cellular Proteomics: MCP. 2018;17(11):2284–96. doi:10.1074/mcp.TIR118.000853.
- Bian Y, Zheng R, Bayer FP, Wong C, Chang Y-C, Meng C, Zolg DP, Reinecke M, Zecha J, Wiechmann S, et al. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC–MS/MS. Nat Commun. 2020;11(1):157. doi:10.1038/s41467-019-13973-x.
- Christianson CC, Johnson CJ, Needham SR. The advantages of microflow LC–MS/MS compared with conventional HPLC–MS/MS for the analysis of methotrexate from human plasma. Bioanalysis. 2013;5(11):1387–96. doi:10.4155/bio.13.73.
- Satkunanathan S, Wheeler J, Thorpe R, Zhao Y. Establishment of a novel cell line for the enhanced production of recombinant adeno-associated virus vectors for gene therapy. Hum Gene Ther. 2014;25(11):929–41. doi:10.1089/hum.2014.041.
- Strobel B, Miller FD, Rist W, Lamla T. Comparative analysis of cesium chloride- and iodixanol-based purification of recombinant adeno-associated viral vectors for preclinical applications. Hum Gene Ther Methods. 2015;26(4):147–57. doi:10.1089/hgtb.2015.051.
- Vialaret J, Picas A, Delaby C, Bros P, Lehmann S, Hirtz C. Nano-flow vs standard-flow: which is the more suitable LC/MS method for quantifying hepcidin-25 in human serum in routine clinical settings? J Chromatogr B. 2018;1086:110–17. doi:10.1016/j.jchromb.2018.04.003.
- Zhang M, An B, Qu Y, Shen S, Fu W, Chen YJ, Wang X, Young R, Canty JM, Balthasar JP, et al. Sensitive, high-throughput, and robust trapping-micro-LC-MS strategy for the quantification of biomarkers and antibody biotherapeutics. Anal Chem. 2018;90(3):1870–80. doi:10.1021/acs.analchem.7b03949.
- Qu M, An B, Shen S, Zhang M, Shen X, Duan X, Balthasar JP, Qu J. Qualitative and quantitative characterization of protein biotherapeutics with liquid chromatography mass spectrometry. Mass Spectrom Rev. 2017;36(6):734–54. doi:10.1002/mas.21500.
- FDA. ICH Q2B validation of analytical procedures: methodology. FDA-1996-D-0169 1997.
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & Cellular Proteomics: MCP. 2012;11(6):11. doi:10.1074/mcp.O111.016717.
- Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol. 2018;14(8):e8126. doi:10.15252/msb.20178126.
- Zhang F, Ge W, Ruan G, Cai X, Guo T. Data-independent acquisition mass spectrometry-based proteomics and software tools: a glimpse in 2020. Proteomics. 2020;20(17–18):1900276. doi:10.1002/pmic.201900276.
- Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Molecular & Cellular Proteomics: MCP. 2006;5(1):144. doi:10.1074/mcp.M500230-MCP200.
- Kreimer S, Gao Y, Ray S, Jin M, Tan Z, Mussa NA, Tao L, Li Z, Ivanov AR, Karger BL. Host cell protein profiling by targeted and untargeted analysis of data independent acquisition mass spectrometry data with parallel reaction monitoring verification. Anal Chem. 2017;89(10):5294–302. doi:10.1021/acs.analchem.6b04892.
- Heissel S, Bunkenborg J, Kristiansen MP, Holmbjerg AF, Grimstrup M, Mørtz E, Kofoed T, Højrup P. Evaluation of spectral libraries and sample preparation for DIA-LC-MS analysis of host cell proteins: a case study of a bacterially expressed recombinant biopharmaceutical protein. Protein Expr Purif. 2018;147:69–77. doi:10.1016/j.pep.2018.03.002.
- Husson G, Delangle A, O’Hara J, Cianferani S, Gervais A, Van Dorsselaer A, Bracewell D, Carapito C. Dual data-independent acquisition approach combining global HCP Profiling and absolute quantification of key impurities during bioprocess development. Anal Chem. 2018;90(2):1241–47. doi:10.1021/acs.analchem.7b03965.
- Pythoud N, Bons J, Mijola G, Beck A, Cianférani S, Carapito C. Optimized sample preparation and data processing of data-independent acquisition methods for the robust quantification of trace-level host cell protein impurities in antibody drug products. J Proteomics Res. 2021;20(1):923–31. doi:10.1021/acs.jproteome.0c00664.
- Picotti P, Aebersold R. Selected reaction monitoring–based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–66. doi:10.1038/nmeth.2015.
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteom. 2012;11(11):1475–88. doi:https://doi.org/10.1074/mcp.O112.020131.
- Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23. doi:10.1016/j.aca.2017.01.059.
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Molecular & Cellular Proteomics: MCP. 2012;11(12):1709–23. doi:10.1074/mcp.O112.019802.
- Rauniyar N. Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int J Mol Sci. 2015;16(12):28566–81. doi:10.3390/ijms161226120.
- Ronsein GE, Pamir N, von Haller PD, Kim DS, Oda MN, Jarvik GP, Vaisar T, Heinecke JW. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J Proteomics. 2015;113:388–99. doi:10.1016/j.jprot.2014.10.017.
- Schiffmann C, Hansen R, Baumann S, Kublik A, Nielsen PH, Adrian L, von Bergen M, Jehmlich N, Seifert J. Comparison of targeted peptide quantification assays for reductive dehalogenases by selective reaction monitoring (SRM) and precursor reaction monitoring (PRM). Anal Bioanal Chem. 2014;406(1):283–91. doi:10.1007/s00216-013-7451-7.
- Gao X, Rawal B, Wang Y, Li X, Wylie D, Liu Y-H, Breunig L, Driscoll D, Wang F, Richardson DD. Targeted host cell protein quantification by LC–MRM enables biologics processing and product characterization. Anal Chem. 2020;92(1):1007–15. doi:10.1021/acs.analchem.9b03952.
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4(10):787–97. doi:10.1038/nmeth1088.
- Valente KN, Lenhoff AM, Lee KH. Expression of difficult-to-remove host cell protein impurities during extended Chinese hamster ovary cell culture and their impact on continuous bioprocessing. Biotechnol Bioeng. 2015;112(6):1232–42. doi:https://doi.org/10.1002/bit.25515.
- Sim KH, Liu L-Y, Tan HT, Tan K, Ng D, Zhang W, Yang Y, Tate S, Bi X. A comprehensive CHO SWATH-MS spectral library for robust quantitative profiling of 10,000 proteins. Sci Data. 2020;7(1):263. doi:10.1038/s41597-020-00594-z.
- Joucla G, Le Senechal C, Begorre M, Garbay B, Santarelli X, Cabanne C. Cation exchange versus multimodal cation exchange resins for antibody capture from CHO supernatants: identification of contaminating host cell proteins by mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;942-943:126–33. doi:10.1016/j.jchromb.2013.10.033.
- Liu X, Chen Y, Zhao Y, Liu-Compton V, Chen W, Payne G, Lazar AC. Identification and characterization of co-purifying CHO host cell proteins in monoclonal antibody purification process. J Pharm Biomed Anal. 2019;174:500–08. doi:10.1016/j.jpba.2019.06.021.
- Müller B, Heinrich C, Jabs W, Kaspar-Schönefeld S, Schmidt A, Rodrigues de Carvalho N, Albaum SP, Baessmann C, Noll T, Hoffrogge R. Label-free protein quantification of sodium butyrate treated CHO cells by ESI-UHR-TOF-MS. J Biotechnol. 2017;257:87–98. doi:10.1016/j.jbiotec.2017.03.032.
- Zhang S, Xiao H, Molden R, Qiu H, Li N. Rapid polysorbate 80 degradation by liver carboxylesterase in a monoclonal antibody formulated drug substance at early stage development. J Pharm Sci. 2020;109(11):3300–07. doi:10.1016/j.xphs.2020.07.018.
- Chiverton LM, Evans C, Pandhal J, Landels AR, Rees BJ, Levison PR, Wright PC, Smales CM. Quantitative definition and monitoring of the host cell protein proteome using iTRAQ – a study of an industrial mAb producing CHO-S cell line. Biotechnol J. 2016;11(8):1014–24. doi:10.1002/biot.201500550.
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi:10.1038/nmeth1019.
- He K, Fu Y, Zeng W-F, Luo L, Chi H, Liu C, Qing L-Y, Sun R-X, He S-M. A theoretical foundation of the target-decoy search strategy for false discovery rate control in proteomics. arXiv. 2015. doi:10.48550/arXiv.1501.00537.
- Midha MK, Kusebauch U, Shteynberg D, Kapil C, Bader SL, Reddy PJ, Campbell DS, Baliga NS, Moritz RL. A comprehensive spectral assay library to quantify the Escherichia coli proteome by DIA/SWATH-MS. Sci Data. 2020;7(1):389. doi:10.1038/s41597-020-00724-7.
- Blattmann P, Stutz V, Lizzo G, Richard J, Gut P, Aebersold R. Generation of a zebrafish SWATH-MS spectral library to quantify 10,000 proteins. Sci Data. 2019;6(1):190011. doi:10.1038/sdata.2019.11.
- Rosenberger G, Bludau I, Schmitt U, Heusel M, Hunter CL, Liu Y, MacCoss MJ, MacLean BX, Nesvizhskii AI, Pedrioli PGA, et al. Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat Methods. 2017;14(9):921–27. doi:10.1038/nmeth.4398.
- Navarro P, Kuharev J, Gillet LC, Bernhardt OM, MacLean B, Röst HL, Tate SA, Tsou C-C, Reiter L, Distler U, et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat Biotechnol. 2016;34:1130–36. doi:10.1038/nbt.3685.
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, McCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–68. doi:10.1093/bioinformatics/btq054.
- Bruderer R, Bernhardt OM, Gandhi T, Miladinović SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Molecular & Cellular Proteomics: MCP. 2015;14:1400–10. doi:10.1074/mcp.M114.044305.
- Colangelo C, Chung L, Bruce C, Cheung K. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods (San Diego, Calif). 2013:61. doi:10.1016/j.ymeth.2013.05.004.
- Briscoe CJ, Stiles MR, Hage DS. System suitability in bioanalytical LC/MS/MS. J Pharm Biomed Anal. 2007;44:484–91. doi:10.1016/j.jpba.2007.03.003.
- FDA. Guidance for industry: analytical procedures and methods validation for drugs and biologics. 2015: FDA-2015-N-0007.
- Thompson M, Wood R. Harmonized guidelines for internal quality control in analytical chemistry laboratories (Technical Report). Pure Appl Chem. 1995;67:649–66. doi:10.1351/pac199567040649.
- Trauchessec M, Hesse AM, Kraut A, Berard Y, Herment L, Fortin T, Bruley C, Ferro M, Manin C. An innovative standard for LC-MS-based HCP profiling and accurate quantity assessment: application to batch consistency in viral vaccine samples. PROTEOMICS. 2021;21:2000152. doi:10.1002/pmic.202000152.
- Bereman MS, Beri J, Sharma V, Nathe C, Eckels J, MacLean B, McCoss MJ. An automated pipeline to monitor system performance in liquid chromatography–tandem mass spectrometry proteomic experiments. J Proteome Res. 2016;15:4763–69. doi:10.1021/acs.jproteome.6b00744.
- Li X, Chandra D, Letarte S, Adam GC, Welch J, Yang RS, Rivera S, Bodea S, Dow A, Chi A, et al. Profiling active enzymes for polysorbate degradation in biotherapeutics by activity-based protein profiling. Anal Chem. 2021;93:8161–69. doi:10.1021/acs.analchem.1c00042.
- Ren D. Advancing mass spectrometry technology in cGMP environments. Trends Biotechnol. 2020;38:1051–53. doi:10.1016/j.tibtech.2020.06.007.