
ABSTRACT
Aging is usually characterized with inflammation and disordered bile acids (BAs) homeostasis, as well as gut dysbiosis. The pathophysiological changes during aging are also sexual specific. However, it remains unclear about the modulating process among gut microbiota, BA metabolism, and inflammation during aging. In this study, we established a direct link between gut microbiota and BA profile changes in the liver, serum, and four intestinal segments of both sexes during aging and gut microbiota remodeling by co-housing old mice with young ones. We found aging reduced Actinobacteria in male mice but increased Firmicutes in female mice. Among the top 10 altered genera with aging, 4 genera changed oppositely between male and female mice, and most of the changes were reversed by co-housing in both sexes. Gut microbiota remodeling by co-housing partly rescued the systemically dysregulated BA homeostasis induced by aging in a sex- and tissue-specific manner. Aging had greater impacts on hepatic BA profile in females, but intestinal BA profile in males. In addition, aging increased hepatic and colonic deoxycholic acid in male mice, but reduced them in females. Moreover, muricholic acids shifted markedly in the intestine, especially in old male mice, and partially reversed by co-housing. Notably, the ratios of primary to secondary BAs in the liver, serum, and all four intestinal segments were increased in old mice and reduced by co-housing in both sexes. Together, the presented data revealed that sex divergent changes of gut microbiota and BA profile in multiple body compartments during aging and gut microbiota remodeling, highlighting the sex-specific prevention and treatment of aging-related disorders by targeting gut microbiota-regulated BA metabolism should particularly be given more attention.
Introduction
Aging-related chronic inflammation, also called inflammaging, is a hallmark and a risk factor for the development of age-associated pathologies such as cardiovascular diseases, stroke, insulin resistance, diabetes, Alzheimer’s disease, and Parkinson’s diseases.1–Citation4 Although the cause of chronic inflammation remains poorly understood, emerging evidence implicates aging-associated alterations in the composition of bile acids (BAs) and gut microbiota play critical roles in modulating host inflammation and metabolism.Citation5–Citation11
BAs are synthesized from cholesterol in the hepatocyte and pass into the small intestine where they promote intestinal absorption of dietary lipids prior to the reabsorption into the liver through enterohepatic circulation or excretion in the feces.Citation12,Citation13 In addition to the well-established role for dietary lipid absorption and maintaining cholesterol homeostasis, BAs also function as signaling molecules regulating glucose, lipid, energy metabolism, and immunity through BA receptors such as nuclear farnesoid X receptor (FXR) and membrane Takeda G protein receptor 5 (TGR5).Citation13–Citation18 In the liver, hepatic enzymes generate free and conjugated primary BAs. In the intestine, BAs restrict bacterial proliferation, whereas bacterial enzymes bile salt hydrolase (BSH) and 7α-dehydroxylase deconjugate BAs and convert primary BAs into secondary BAs, respectively.Citation19–Citation21 Given the joint co-metabolism of both host and gut microbiota on BAs homeostasis, and the diversified binding affinities on BA receptors among free and conjugated BAs, eubiosis is essential for maintaining BA homeostasis and health.
Gut microbiota plays a pivotal role in maintaining metabolic homeostasis of host.Citation5–Citation8,Citation22 However, the composition, diversity, and function of the microbiota are not always constant during the lifetime of the host.Citation5,Citation6,Citation23 Therefore, the shifted microbial composition and its related dysregulation of host metabolism contribute to the development of various diseases such as metabolic diseases and aging-associated disorders.Citation5,Citation24,Citation25 Moreover, various microbiota-targeted interventions have shown favorable effects on host health.Citation26–Citation29 Interestingly, fecal microbiota transplantation is effective in extending mouse lifespan by restoring secondary BA synthesis.Citation30 Therefore, exploration of host BA pool homeostasis during aging is of great significance for better understanding the physiological process of aging and its related disorders. Meanwhile, there are significant sexual differences in terms of the susceptibility to aging and the occurrence of aging-related disorders such as insulin resistance, energy balance, organ dysfunctions, and even death rate.Citation31–Citation34 However, the pathophysiological mechanisms underlying the sexual differences in longevity and aging-related diseases are quite complex and poorly understood. Our previous study reveals the sex dissimilarity in metabolism is closely associated with the differences in both BAs and microbiota profiles.Citation35 Therefore, the systemic investigation on the compositional changes of BA pool in old male and female mice bears much significance for elucidating the sexual differences during aging and susceptibility to aging-related disorders.
In our current study, we performed a systemic investigation on BA pool profiles in liver tissue, serum, and four different segments of intestinal contents (jejunum, ileum, cecum, and colon) in both male and female of young and old mice with targeted BA metabolomics, as well as the composition of gut microbiota. Moreover, we further evaluated the impacts of microbiota remodeling on aging-related disorders and BA pool homeostasis by co-housing young and old mice of both sexes, respectively. Our results showed that aging caused gut dysbiosis and comprehensively dysregulated BA homeostasis with increased systemic inflammation in a sex-specific manner. Gut microbiota remodeling by co-housing partially rescued the above changes in old mice. Therefore, our current study highlights that gut microbiota is a potential target for improving the health of the old population by restoring BA homeostasis, and sex-specific strategy should particularly be given more attention.
Results
Aging causes hepatic inflammation and splenomegaly
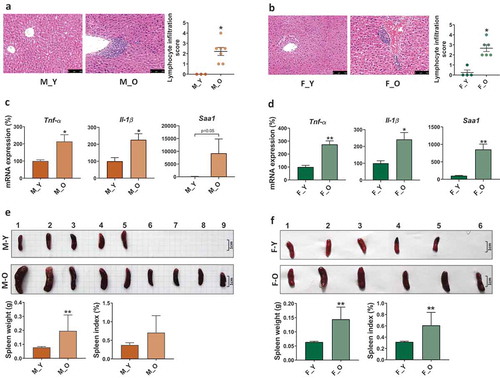
Since inflammaging is the common character for most aging-related disorders,Citation1,Citation36 we first compared the liver histology and the gene expression of pro-inflammatory cytokines between young (3-month-old) and old (26-month-old) mice of both sexes. The results showed that old mice had massive hepatic lymphocyte infiltration in both sexes, but not in young mice (). The mRNA levels of Tnf-α (tumor necrosis factor-alpha) and Il-1β (interleukin 1 beta), as well as Saa1 (serum amyloid A1), which is highly expressed in response to inflammation and tissue injury, were significantly higher in old mice than young mice suggesting the aging-related hepatic inflammation in old mice of both sexes (). In addition, we also found that old mice had enlarged spleen size and increased spleen to body weight ratio (), which is a probable physiological response to bacterial infection.Citation37 Together, the data indicated that old mice were characterized by higher systemic inflammation than young mice.
Figure 1. Histological and phenotypic changes in young and old mice of both sexes.
Aging alters hepatic BA profile sex-specifically
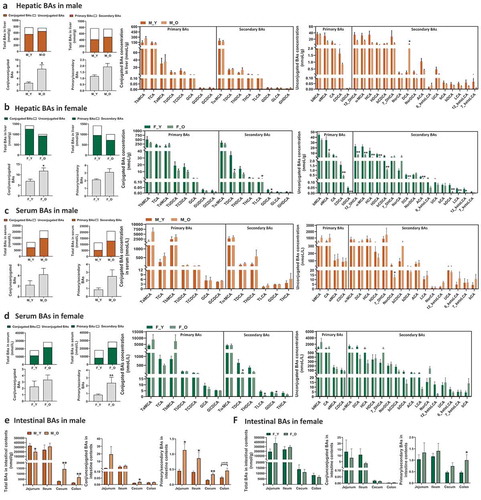
As BAs are synthesized in the liver and play important roles in regulating host metabolism and inflammation, we first analyzed BA composition in liver tissue in the young and old mice of both sexes. The quantity of total BAs in liver remained relatively constant with aging in male mice but reduced in female mice (). The old male mice showed increased ratios of conjugated to unconjugated and primary to secondary BAs, while the total hepatic BA, and ratios of conjugated to unconjugated and primary to secondary BAs were much higher in females than males of either young or old mice (). Regarding the individual BAs, many unconjugated hepatic BAs showed decreasing trend with aging in male mice; however, only DCA level was significantly higher in old male mice at 15-fold than young ones. In contrast, there were 16 BAs that were markedly lower in old female mice than young mice, and most of them were unconjugated primary and secondary BAs. Interestingly, the DCA levels in old male and female were oppositely altered in the context of similar levels of CA between old and young mice regardless of their sexes (), suggesting the differential expression of bacterial 7α-dehydroxylase in these mice. Additionally, the proportions of BAs such as TβMCA and TαMCA were also differently changed with aging in male or female mice, respectively (Figure S1 A). These results suggested that aging divergently altered hepatic BA composition in a sex-specific manner with a more dramatic variation in female mice than male mice.
Figure 2. BA profile in the liver, serum, and intestine of young and old mice.
Aging alters BA profile in serum sex-specifically
To further assess the aging-related dysregulation on BA homeostasis, we quantified the BA composition in serum of these mice. We found that both old male and female mice had 1.8-fold and 1.6-fold increase in total BAs in serum compared with young mice of same sex, respectively (). The increase in total BAs in serum of old mice was mainly due to an increase in the conjugated primary BAs, such as TαMCA in male mice and Tα+βMCA in female mice. Moreover, increased ratios of conjugated to unconjugated, and primary to secondary BAs were observed in serum of old mice of both sexes. Aging reduced NorDCA in male mice, and conjugated secondary BAs, such as TDCA, TLCA, and GDCA in female mice (). The proportions of TαMCA, TβMCA, and TωMCA were differentially changed in the two sexes during aging (Figure S1 B). The results suggested that aging resulted in sex-specific alterations in serum BA profiles.
Aging alters BA profiles in different intestinal segments sex-specifically
Since intestinal tract is the main site for BA functions for either facilitating dietary lipids absorption or regulation on host metabolism through intestinal BA receptors,Citation38–Citation40 we then quantified the BA profiles in the contents of four intestinal segments, including jejunum, ileum, cecum, and colon. BAs are mainly reabsorbed into the portal vein at ileum, and approximately 5% excreted from feces, so the total BAs concentration was reduced dramatically in cecum and colon contents in all mice ()). In male mice, the total BA concentrations were lower in jejunum, but higher in cecum and colon contents than young mice. Moreover, the ratios of primary to secondary BAs were significantly higher in old mice than those of young mice ()). In contrast, the old and young female mice showed comparable levels of either total BAs or the ratios of conjugated to unconjugated, and primary to secondary BAs in the four intestinal segments, except for the significant higher ratio of primary to secondary BAs in colon contents ()), suggesting the abnormally sex-specific BA synthesis, transport, or metabolism during aging.
The concentrations of individual BAs were altered with aging in a segment- or sex-dependent manner. For example, 9 out of 19 detected unconjugated BAs were reduced in the jejunum and 7 unconjugated BAs were increased in the cecum of old male mice, whereas most unconjugated BAs were relatively constant in other two intestinal segments of male mice (Figure S2). In female mice, unconjugated BAs were relatively constant in the intestinal segments with the exception of ileum (Figure S3). In old male mice, βMCA tended to be consistently increased in the ileum, cecum, and colon, while secondary BAs DCA and LCA were consistently increased in cecum and/or colon (Figure S2). In contrast, βMCA was only elevated in the ileum in old female mice (Figure S3). Moreover, Tα+βMCA, which are FXR antagonists, as well as TωMCA and TDCA had increased trend in the cecum and colon in old male mice but not in old female mice (Figure S2 and S3). In general, more BAs were changed in the intestine of male mice than female mice, especially in the jejunum, cecum, and colon. Importantly, MCAs were significantly shifted in the intestine of male mice but not female mice (Figure S2 and S3). These findings indicated divergent changes in intestinal BA composition sex-specifically during aging, which might lead to different metabolic effects.
Aging alters gut microbiota composition sex-specifically
Given the intricate role of gut microbiota in aging-related disorders and maintaining BAs pool homeostasis, we further profiled the compositional changes of gut microbiota during aging in these mice. First of all, the Unweighted UniFrac PCoA showed that old mice of both sexes had dramatically different patterns of gut microbiota with their young counterparts ()), which is consistent with previous reports.Citation41 Surprisingly, aging had opposite effects on shifting Shannon index in the two sexes ()), suggesting the sex-specific impacts of aging on bacterial α diversity. At the phylum level, the most significant change caused by aging in male mice was the reduction of Actinobacteria ()). However, in female mice, the abundance of Actinobacteria as well as Firmicutes was increased, while Proteobacteria was reduced during aging ()). At the genus level, the top 10 significantly altered genera with aging in both sexes were summarized ()). Specifically, there were five increased genera including Lachnospiraceae_NK4A136_group, norank_f_Lachnospiraceae, unclassified_f_Ruminococcaceae, Oscillibacter, and Blautia, and five decreased genera including norank_f_Erysipelotrichaceae, Faecalibaculum, Enterohabdus, Bifidobacterium, and [Eubacterium]_fissicatena_group in old male mice ()). Interestingly, the majority of bacteria among the top 10 significantly changed genera were decreased in old female mice including Desulfovibrio, norank_f_Lachnospiraceae, norank_f_Bacteroidales_S24-7_group, unclassified_f_Lachnospiraceae, Lachnospiraceae_NK4A136_group, Blautia, Roseburia, Lachnospiraceae_UCG-006, and norank_f_Ruminococcaceae, except for norank_f_Erysipelotrichaceae ()). Four genera were oppositely changed with aging between male and female mice, such as norank_f_Erysipelotrichaceae, Lachnospiraceae_NK4A136_group, norank_f_Lachnospiraceae, and Blautia.
Figure 3. Aging induces gut microbiota disbalance in both sexes.
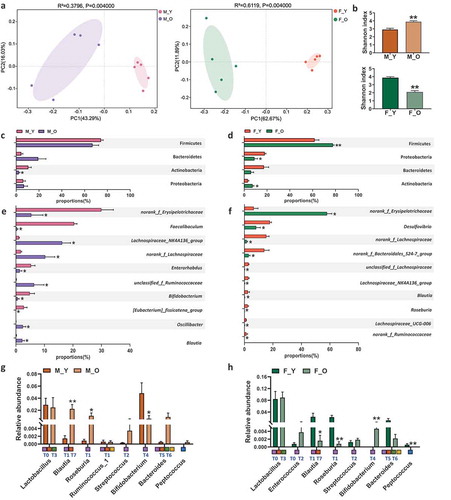
Since many bacterial species are involved in BAs metabolism, such as BA deconjugation,Citation20,Citation40,Citation42 epimerization,Citation43 7α-dehydroxylation,Citation44–Citation46 and desulfation,Citation47 bacterial genera that contained the main species with BA metabolizing functions were further studied. Based on previous phylogenetic analysis with human gut microbiota, the eight phylotypes of BSH (T0-T7) were also labeled under each genus in ) to define their deconjugation activity.Citation21 Old male mice had increased Blautia and Roseburia and reduced Bifidobacterium when compared with young male mice ()). Some species under Blautia and Roseburia contain BSH-T1, which had the highest abundance of BSHs in the gut microbiota of human.Citation21 It is worth noting that old female mice had reduced Blautia and Roseburia and increased Bifidobacterium, which were totally opposite to the changes in males ()). These data indicated that the alternation of gut microbiota composition during aging was sex-specific which might account for, at least partially, the disparity of aging-related disorders including the imbalanced BAs metabolism between males and females.
Co-housing with young mice reduces hepatic inflammation and splenomegaly in old mice
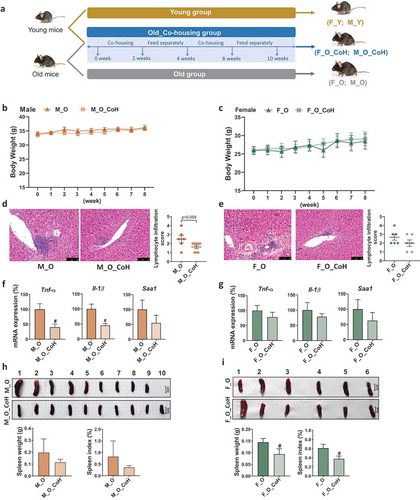
To test whether the compositional alteration of gut microbiota is causative for aging-related disorders, we remodeled the gut microbiota by co-housing old mice with their young partners of the same sex periodically for 10 weeks to minimize the impact of lifestyle between old and young mice (See experiment design in )). First, co-housing experiment did not impact the body weight of old mice in both sexes (). However, co-housing reduced the hepatic lymphocyte infiltration score and mRNA expression level of Tnf-α, Il-1β, and Saa1, especially in old male mice (). Moreover, co-housing also reduced the spleen weight and index, especially in old female mice (). These results indicated co-housing old mice with young mice exerted beneficial effects to different extent in male and female old mice implying the sex-specific impacts of gut microbiota on aging.
Figure 4. Histological and phenotypic changes in old mice after co-housing.
Co-housing with young mice remodels gut microbiota composition of old mice sex-specifically
To determine the role of gut microbiota in improving aging-related disorders by co-housing, we profiled the composition of gut microbiota based on 16 S rRNA gene sequencing. First, Unweighted UniFrac PCoA showed distinct clustering of intestinal microbe communities of each group of both sexes (). In specific, we observed that young mice were separated away from old mice alongside PC1 at 30.1% and 46.1% interpretation power for male and female mice, respectively. The co-housed old male mice were clustered between young and old mice, but separated with old mice by PC2, while the co-housed old female mice were closer to young female mice, indicating that the microbiota profile in old mice were shifted significantly after co-housing ()). Meanwhile, we observed that co-housing caused significant restoration in bacterial α diversity in old female mice, but not in male ones compared to their old partners ()). Then, we further analyzed the relative abundance of bacteria at different levels. At the phylum level, co-housing reversed the decreased abundance of Actinobacteria in old male mice, but showed no significant impacts in old female mice (). At genus level, more significant impacts of co-housing on gut microbiota were observed with sex-specific alterations. For example, co-housing significantly reversed the relative abundance of Faecalibaculum, Bifidobacterium, unclassified_f_Ruminococcaceae, Blautia, and Ruminiclostridium in old male mice ()), as well as norank_f_Erysipelotrichaceae, norank_f_Lachospiraceae, unclassified_f_Lachospiraceae, Lachospiraceae_NK4A136_group, Lachnospiraceae_UCG-006, Roseburia, and Lachnoclostridium in old female mice ()). Notably, co-housing led to a dramatic reduction in the abundance of norank_f_Erysipelotrichaceae from 58% to 12%, which was comparable to the level of young female mice ()).
Figure 5. Co-housing improves gut microbiota disbalance induced by aging in both sexes.
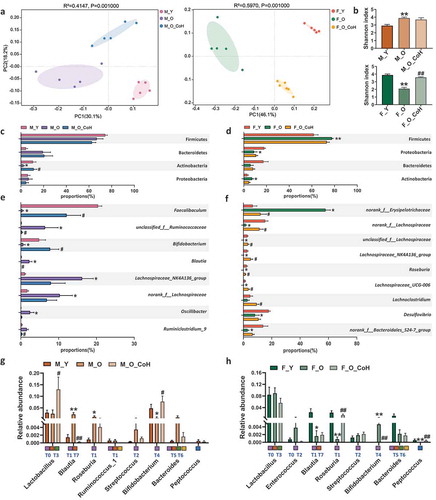
In addition, we further compared the impacts of co-housing on BA metabolism-related genera. Co-housing significantly reversed the aging-associated changes of Blautia (BSH-T1 and T7) and Bifidobacterium (BSH-T4) in male mice. Moreover, Lactobacillus (BSH-T0 and T3) was increased in co-housed old male mice ()). In females, co-housing normalized the abundance of Roseburia (BSH-T1), Bifidobacterium (BSH-T4), and Peptococcus ()). In addition to the gut microbiota remodeling, the sex-specific difference in gut microbiota composition was also observed in young mice including norank_f_Erysipelotrichaceae, Faecalibaculum, Bifidobacterium, Lachospiraceae_NK4A136_group, norank_f_Lachospiraceae, Desulfovibrio, nonrank_f_Bacteroidales_S24-7_group, and Roseburia. However, these sex-related differences were abolished or even reversed with aging, while co-housing reverted some of them (Figure S4). Taken together, these data indicated that co-housing old mice with young ones remodeled their gut microbiota composition sex-specifically.
Gut microbiota remodeling alters BA profile in the liver, serum, and intestine
Given the important crosstalk between gut microbiota and BA metabolism, we further tested whether gut microbiota remodeling with co-housing alters BA pool profiles in sex-specific manner. The BA profiles in liver, serum, and different segments of intestine were quantified in both old male and female mice with or without co-housing. In general, co-housing increased total hepatic BAs levels but did not significantly alter the ratios of conjugated to unconjugated, or primary to secondary BAs in both sexes ()). Co-housing resulted in different extent of alterations in hepatic BA signatures between male and female mice. Specifically, co-housing significantly reversed the hepatic concentrations of βDCA and 12-ketoLCA in old male mice ()), and TDCA, TLCA, and βDCA in old female mice ()). Notably, co-housing mainly changed the concentrations of secondary BAs, instead of primary BAs in both sexes, which is consistent with the role of gut microbiota in BAs metabolism. Additionally, the proportions of most individual BAs remained relatively constant in response to gut microbiota remodeling (Figure S5).
Figure 6. BA profile in the liver, serum, and intestine of male and female mice after co-housing.
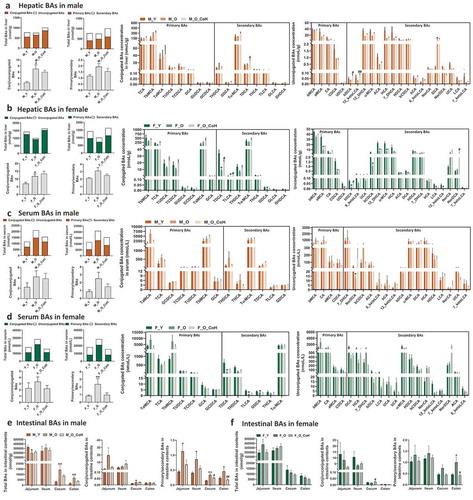
In serum, co-housing reversed the increased total BA levels in old mice of both sexes (), and in particular the ratios of conjugated to unconjugated, primary to secondary BAs in old female mice ()). Although not statistically significant, co-housing resulted in reversed changes of some unconjugated BAs such as 7-DHCA, NorDCA, βCDCA, 6-ketoLCA, and βCA in males and HCA, 7-DHCA, βDCA, LCA, and βCA in females in concentrations (). In contrast to the concentration, co-housing also led to compositional changes of either conjugated or unconjugated BAs sex-specifically. For example, co-housed male mice showed decreased TαMCA, and increased TβMCA and TωMCA in composition, but increased TωMCA, and decreased TαMCA and TβMCA were observed in co-housed females (Figure S5B). In addition, co-housing caused decreased composition in 7-DHCA, and increased DCA, ωMCA in males, whereas decreased βMCA, and increased ωMCA, NorDCA in females (Figure S5B).
Intestinal tract is the main site for BAs functions where the gut microbiota modulates BA homeostasis through a series of bacterial enzymes and the majority of BAs are then reabsorbed at ileum,Citation48 so the total BA concentrations were depleted in the cecal and colon contents in all mice ()). In male mice, co-housing reduced the ratio of primary to secondary BAs in all four intestinal segments, which rescued the increased levels caused by aging ()). In female mice, similar changes were observed, but only the ratios of conjugated to unconjugated in cecal, and primary to secondary BAs in colon were significantly reversed by co-housing ()). Moreover, there were a number of individual BAs that were altered to different extent in concentration upon co-housing sex-specifically (Figure S6). However, there were only two unconjugated secondary BAs that were significantly reversed by co-housing, i.e. 6-ketoLCA and 12-ketoLCA in jejunum of male mice (Figure S6), whereas more significantly altered BAs were observed such as βMCA, βCA, and 7-DHCA in ileum and 12-ketoLCA, 7-DHCA, and NorCA in colon of co-housed old female mice (Figure S7). Since the eight BSH phylotypes have different deconjugation activity on various substrates,Citation21 we further studied the effects of age and gut microbiota remodeling on the ratio of free to conjugated BAs. The results indicated that aging significantly decreased the ratios of DCA/GDCA and DCA/TDCA, but increased CDCA/GCDCA and CDCA/TCDCA, and in which only CDCA/GCDCA and CDCA/TCDCA were significantly reversed by co-housing in male mice (Figure S8). No significant differences were observed in the ratios of these free to conjugated BAs in female mice (Figure S8).
Altogether, these results indicated that systemic BA profiles were greatly influenced by varying degrees by the gut microbiota remodeling through co-housing in a sex-specific manner, which suggests that the inoculation of bacteria from young partners holds benefits for attenuating aging-related disorders through re-balancing BA homeostasis.
Gut microbiota remodeling alters BA-related gene expression
To test whether gut microbiota remodeling could exert comprehensive impact on hepatic gene expression, liver transcriptome was performed in the old male mice with or without co-housing. In total, eighty-one genes were upregulated and 793 genes were downregulated in the co-housed old male mice compared to those of old mice with Q value < 0.05 and fold change ≥ 2 or ≤0.5 used as the cutoff criteria (Figure S9A). Using the Database for Annotation, Visualization and Integrated Discovery (DAVID), we further found more than 10 out of 30 altered pathways were related to infection-induced diseases, including Staphylococcus aureus infection, leishmaniasis, malaria, and tuberculosis (Figure S9B). Additionally, seven downregulated pathways were associated with immune response and inflammation, such as NOD-like receptor signaling pathway, cytokine-cytokine receptor interaction, TNF signaling pathway, and NF-kappa B signaling pathway.
To further explore the co-housing-affected genes, Gene Set Enrichment Analysis (GSEA)Citation49 was employed to identify significantly enriched biological pathways on the basis of normalized enrichment score (NES) ranking. Compared with separated housed old male mice, co-housed old mice significantly activated the expression of “Bile acid and bile salt metabolism” and “TCA cycle and respiratory electron transport” gene sets and inhibited the expression of “Interferon gamma signaling” and “Innate immune system” gene sets (Figure S9 C-D). These data suggest that gut microbiota remodeling by co-housing significantly affect the expression of BA metabolism and inflammation-related genes by the gut-liver axis, which is consistent with the observations on the inflammatory phenotypes and BA profiles of these mice.
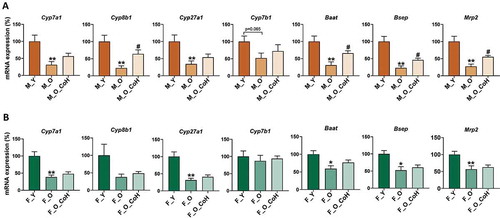
Next, BAs hemostasis-related genes were analyzed by qPCR in the liver and ileum during aging and after co-housing in both sexes. In male mice, the mRNA levels of hepatic Cyp7a1, Cyp8b1, Cyp27a1, Cyp7b1, Baat, Bsep, and Mrp2 were reduced in old mice, while co-housing reversed Cyp8b1, Baat, Bsep, and Mrp2 levels ()). In female mice, aging reduced most of the above genes. However, co-housing had limited effect on reversing those changes ()). It should be noted that the mRNA level of Cyp7b1, which is involved in alternative BA synthesis, was not changed during aging or co-housing in female mice. Additionally, the expression of ileal BA transporters had reduced trend in old male mice but co-housing did not reverse thus changes. On the contrary, in female mice, aging had no effect on altering these gene expression, but co-housing had the trend to reduce them (Figure S10A-B).
Figure 7. Hepatic BAs metabolism-related gene expression in male (a) and female (b) mice. *p< .05 and **p < .01 compared with young group; # p < .05 compared with old group. n = 5–8 mice per group.
Sex-related BA profiles at different age stages
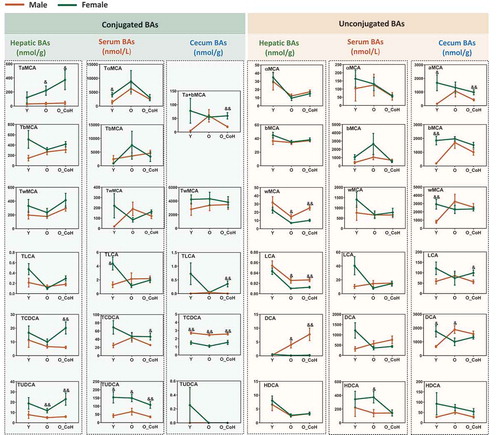
In addition to the shifted BA profiles during aging and after co-housing in two sexes, the concentrations of individual BA were also significantly different between male and female mice (). In general, compared with the male mice, female mice had higher level of hepatic conjugated BAs including all conjugated MCA, TLCA, TCDCA, and TUDCA. On the contrary, male mice had higher levels of hepatic free BAs, such as ωMCA, LCA, and DCA. Additionally, aging increased the sex difference of TαMCA, TUDCA, and DCA but narrowed the sex difference of TβMCA and TLCA in liver. Moreover, aging narrowed the sex difference of TLCA, LCA, and DCA in the serum, free, and conjugated αMCA and βMCA in the cecum. Compared with the male mice, female mice had higher concentrations of TCDCA and TUDCA in the liver and serum at young, old, and/or co-housed conditions. Hepatic DCA levels were increased with age and co-housing in male mice, while it remained very low and relatively constant with age in females (). These data suggested BA levels were sex different and there were divergent changes of BAs during aging and co-housing in two sexes.
Figure 8. Sex difference in the concentration of certain BAs in the liver, serum, and cecal contents. &p < .05, &&p < .01 compared with female group. n = 5–10 mice per group.
Sex-specific correlation between BAs and gut microbiota
The correlation analysis between the top 20 families of the gut microbiome and individual BA in the cecum was performed in each sex (Figure S11). In male mice, Erysipelotrichaceae and Staphylococcaceae, which are under Firmicutes phylum, were negatively correlated with 6-ketoLCA, GCA, 12-ketoLCA, TωMCA, ωMCA, GDCA, 7-HDCA, CA, and βMCA (Figure S9A). However, most of the above BAs were positively correlated with Lachnospiraceae and one unknown family under Saccharibacteria. Probiotic families Lactobacillaceae and Bifidobacteriaceae were clustered together with Lactobacillaceae positively correlated with THCA (Figure S11A). In female mice, Lactobacillaceae clustered separately with Bifidobacteriaceae and correlated with six BAs but not THCA. In addition, sex-specific correlation of Erysipelotrichaceae and Lachnospiraceae with BAs was observed (Figure S11B). These results revealed a sex-specific correlationship between the BAs and gut microbiota that was significantly altered by co-housing, which is indicative of the crosstalk between BAs and gut microbiota.
Discussion
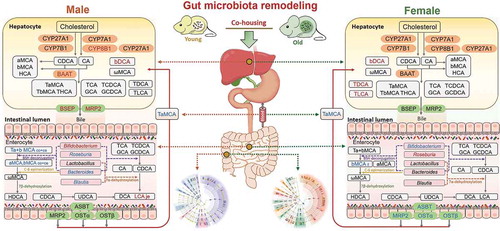
The aging-related inflammation and disordered metabolism are sex-specific and presumably associated with the dysregulation in gut microbiota and BA homeostasis.Citation35,Citation50-Citation53 The presented data establish a direct link between gut microbiota and aging-associated BA profile changes in multiple tissues of both sexes. We demonstrate that aging has a greater impact on changing hepatic BA profiles in females, but gut BA profiles in males. As summarized in , co-housing old mice with young mice strikingly changed gut microbiota composition in old mice and shifted BA profiles in multiple tissues to rescue aging-induced imbalanced BA homeostasis in a sex-specific manner. These findings suggest prevention and treatment of aging-associated disorders by targeting gut microbiota-regulated BA signaling should take sex into account.
Figure 9. Gut microbiota remodeling by co-housing reverses the dysregulation of systemic BA homeostasis induced by aging.
Gut microbiota plays essential roles in regulating host metabolism and inflammation, while its composition changes throughout the life span and is known to be associated with host sex.Citation51,Citation52 In the present study, among the top 10 differential genera during aging, four genera had opposite changes in two sexes. Norank_f_Erysipelotrichaceae was found to be either reduced or increased in male or female mice during aging. It is consistent with previous report of increased abundance of Erysipelotrichaceae in female mouse model of Alzheimer’s disease.Citation55 Meanwhile, Erysipelotrichaceae is also associated with the hyperlipidemia.Citation56,Citation57 Given the fact of higher rate of Alzheimer’s disease in females than males,Citation58 and the association of Alzheimer’s disease with dysregulated lipid metabolism,Citation59,Citation60 the increased abundance of norank_f_Erysipelotrichaceae in old female mice is a probable risk factor for occurrence of aging-related disorders in females. The bacteria belonging to Bifidobacterium genus have been widely used as probiotics.Citation61,Citation62 Our data showed the relative abundance of Bifidobacterium genus was reduced in male but increased in female mice during aging, which may account for the physiological disparity between males and females. Consistent evidence of reduced Bifidobacterium genus has been reported in old male mice and humans.Citation5,Citation41,Citation63,Citation64 In addition, Roseburia was increased or reduced in male and female mice during aging, respectively. Many species of Roseburia contain BSHs and can produce health-promoting butyrate. The alteration of Roseburia with age in male mice is consistent with previous report in miceCitation41 but was contrary to the findings in humans, which showed reduced relative abundance in centenarians.Citation30 It should be taken into account that the changes of the composition of gut microbiota vary in different studies during aging between mice and humans, which might be associated with the differences in anatomical location, diet, lifestyle, sex, and age. The divergent changes of microbiota profile during aging might lead to the disparity in the metabolism and immune response between the two sexes.
The biological functions of BAs are diversified which are associated with their concentrations and organ distributions.Citation48 Our data showed that total BA concentrations in liver reduced in female mice, whereas they remained relatively constant with age in male mice. In addition, total BA concentrations in serum increased in both female and male mice. However, Fu et al found total BA concentrations in liver of both sexes and in serum of male mice remained relatively constant from 3 to 27 months, only total BA in serum of female mice significantly increased with age.Citation10 This inconsistent finding might be due to the types of BAs detected in different studies. We quantified 39 BAs in our current study, while only 20 of them were measured in previous report, and several BAs with high proportions, such as HCA and 7-DHCA, were not included.Citation10 Secondary BAs, DCA and LCA, which are generated by gut microbiota, tended to be reduced in the serum and liver of old female mice compared with young female mice. Differing with the changes in females, old male mice had 15-fold induction of hepatic DCA level. These two BAs are not only agonists for FXR but also can activate TGR5 signaling to improve metabolism.Citation65–Citation67 However, excessive DCA produces reactive oxygen species that cause DNA damage and senescence-associated secretory phenotype, which facilitate high fat diet-induced hepatocellular carcinoma growth.Citation68 Therefore, reduced levels of DCA and LCA in old female mice might lead to less activation of FXR and TGR5 signaling which affect metabolism. On the other hand, elevated hepatic DCA in the liver of male mice may induce inflammatory signaling that are associated with the development of cancer, leading to the sex difference in the incidence of liver diseases.Citation68–Citation71 These results suggest the potential difference of the activation of BA receptors and DCA induced toxicity between the two sexes which could lead to different immune response and metabolism during aging. In addition, it is well known that the secondary BA concentration in feces is related to the incidence of colonic cancer.Citation72 The concentration of the major secondary fecal BAs, DCA and LCA, increased with aging and was significantly higher in elderly subjects compared to young adults.Citation73 In line with the fact of age-related incidence rates of colon cancer are higher for men than women,Citation74 our results also showed that aging increased DCA (4.6-fold increase) and LCA (2.7-fold increase) in the colon of male mice but not female mice.
A very intriguing finding in the present study was that aging increased the ratio of primary to secondary BAs while co-housing reduced it in the liver, serum, and all four intestinal segments of both sexes. Gut microbiota has essential effects on secondary BAs production. Blautia and Bacteroides genera, which include many species that convert primary BAs to secondary BAs by 7α-dehydroxylation,Citation13,Citation44,Citation46 were increased in old male mice and reduced after co-housing. Though there are many genera that can regulate secondary BAs production, Blautia and Bacteroides were markedly influenced by age and gut microbiota remodeling in a sex-dependent manner, suggesting their divergent roles in regulating host health in two sexes which need further study.
The beneficial or detrimental effects of BAs are also associated with the extent of hydrophobicity or hydrophilicity. Previous study reports that total BAs in serum and liver became more hydrophilic with aging in both male and female mice.Citation10 In the present study, we also found increased ratios of conjugated to unconjugated BAs in old mice, leading to increased hydrophilicity of BAs in the liver and serum, which was consistent with the previous finding. Increased hydrophobicity of BAs in tissues can cause cytotoxicity and produce reactive oxygen species, resulting in DNA damage, apoptosis, and necrosis.Citation75 Since the mRNA level of hepatic Baat, which involved in BAs conjugation, was reduced in old mice, it is possible that gut microbiota alters the BA composition during aging in order to increase the hydrophilicity of BAs, which might indicate an initiated protective mechanism to alter BA composition and compensate for disease susceptibility during aging. In the present study, the ratios of CDCA/TCDCA and CDCA/GCDCA were significantly increased in cecum contents of old mice and were decreased after co-housing. BSHs are a set of bacteria-derived enzymes for hydrolysis of conjugated BAs with different substrates and activities in bacteria.Citation21 Previous study indicated that BSH-T3 showed the highest enzyme activity when the substrates were TCDCA and GCDCA, followed by BSH-T1 and BSH-T4 based on human gut microbiome.Citation21 Although BSH-T3 showed the highest enzyme activity, it was only found in Lactobacillus, which contained 0.15% of the total relative abundance of BSHs. BSH-T1, with the relative abundance of 38.03%, was found in Blautia, Roseburia, and Ruminococcus_1. In addition, BSH-T4 was found in Bifidobacterium with 2.74% of the total relative abundance of BSHs.Citation21 Interestingly, age and co-housing induced abundance changes of Blautia and Roseburia in male mice as well as Bifidobacterium in female mice were consistent with the ratio changes of CDCA/TCDCA and/or CDCA/GCDCA, indicating these genera may play important roles in aging-associated BA alteration sex-specifically. However, BSHs containing bacteria that have high deconjugation activity toward MCA should be identified in future.
Interesting findings from the present study show both aging and co-housing significantly affect the concentration of MCAs in a tissue- and sex-dependent manner. The unconjugated and conjugated MCAs are unique BAs that only exist in rodents and not in humans. They play an important role as the signal molecules in regulating FXR signaling pathway which in turn affects host metabolism.Citation76,Citation77 In the liver, aging tended to reduce the concentration of αMCA and ωMCA in both sexes, while co-housing tended to increase ωMCA level in male mice. In the serum, old mice had increased trend of TαMCA, which is a more potent FXR antagonist than TβMCA, while co-housing reduced TαMCA levels. This change happened in both male and female mice suggesting the potent deactivation of FXR signaling in peripheral organs. However, previous study showed increased TαMCA with age in the serum was only found in female mice, not in male mice, which is not consistent with our findings.Citation10 It is worth noting that MCAs shifted more markedly in the intestine, especially in male mice. In the cecum, Tα+βMCA (13-fold increase), TωMCA (9-fold increase), αMCA (8-fold increase), βMCA (9-fold increase), and ωMCA (4-fold increase) were increased in old male mice. Similarly, in the colon, Tα+βMCA (11-fold increase), αMCA (5-fold increase), and βMCA (8-fold increase) were increased in old male mice while co-housing normalized all of them. Compared with the multiply changes of MCAs in male mice, it is interesting to note that only ileal βMCA was shifted by aging in female mice. These findings clearly suggest the divergent effects of age and gut microbiota remodeling on regulating BA profiles in two sexes. Because about 95% BAs are efficiently taken up in the ileal segment and active uptake of conjugated BAs is mediated by ileal ASBT,Citation78,Citation79 the elevated Tα+βMCA in the cecum and colon of old male mice might due to reduced ileal uptake that occurred during aging. Since Tα+βMCA are FXR antagonists, the increased level may have led to reduced intestinal FXR activity. Deactivation of hepatic FXR can cause liver cancer,Citation27,Citation80,Citation81 but deactivation of intestinal FXR may have some benefits. It has been shown that intestinal FXR deficient mice are resistant to diet-induced steatohepatitis, obesity, and insulin resistance.Citation15,Citation82 Therefore, old mice may have elevated intestinal Tα+βMCA levels to present beneficial effects while co-housed mice do not need to maintain high Tα+βMCA levels.
The sex differences in the concentrations of individual BAs shown in the present study provide important evidence that BAs may function as markers for longevity. Females have a longer life expectancy than males in many species, including humans.Citation32,Citation83,Citation84 In our study, both hepatic and serum concentrations of TCDCA, TαMCA, TUDCA were higher in female mice. The long-lived lit/lit mice were shown to have increased CDCA and UDCA in serum.Citation85 Therefore, the higher hepatic and serum concentrations of TCDCA and TUDCA might correlate with the tendency of increased longevity in female mice.
Although co-housing is increasingly adopted for investigating the role of gut microbiota,Citation86,Citation87 co-housing of mice for over 6 weeks can result in some extents of chronic stress and behavior dysfunction, but not present after 2 weeks of co-housing.Citation88 In our current study, to avoid unintended chronic stress or behavior change induced by co-housing, we adopted an intermittent co-housing strategy for gut microbiota remodeling. According to previous report, chronic stress can reduce body weight and disturb gut microbiota, with increased levels of inflammation promoting operational taxonomic units related to Helicobacter, Peptostreptococcaceae, Streptococcus, and Enterococcus faecalis.Citation89 Our data showed the body weight gain of co-housed old mice was comparable with their counterparts in either male or female mice during 10 weeks intermittent co-housing experiment. In addition, none of the stress-related bacteria was increased in the co-housed old mice, suggesting the intermittent co-housing did not induce obviously chronic stress in our current study. Even though, it should be cautious to interpret the results obtained from co-housing experiment, and more considerations should be taken in further study to clearly differentiate the impacts contributed by gut microbiota itself, or jointly with stress.
The current study provided a comprehensive description of the age-related changes of gut microbiota and BA profiles in male and female C57BL/6 mice. In addition, we found co-housing partially changed gut microbiota composition in a sex-specific manner. This might be due to the differential colonization and competitive ability of different gut bacteria, which leads to differential alteration of the relative abundance of each strain. In terms of the complicated crosstalks between gut microbiota and BAs, as well as the complex gut-liver axis in regulation of BA metabolism, our current results demonstrated that co-housing induced diversified changes of BAs either in absolute concentrations or relative composition sex-specifically.
In conclusion, aging is characterized by a series of disorders and susceptibility to various diseases with sexual disparity. Our current report reveals the interplay between gut microbiota and BA metabolism during aging, and more importantly, we demonstrate that gut microbiota remodeling can attenuate aging-related disorders, at least partially, through reverses the imbalanced BA homeostasis in a sex-specific manner. Our current findings highlight the potential of sex-specific strategy to prevent or treat aging-related disorders by targeting gut microbiota-regulated BA metabolism axis. Further investigations are warranted to elucidate the exact roles of altered BAs during aging in different sexes.
Methods and materials
Mice
Male and female C57BL/6 J mice of 24-month-old were provided by Laboratory Animal Center of Xiamen University (Xiamen, China). Four-week-old C57BL/6 J mice of both sexes were provided by Shanghai Laboratory Animal Center (Shanghai, China). All mice were housed in a 12-hour light (7 AM to 7 PM) and 12-hour dark (7 PM to 7 AM) cycle, with free access to water and chow diet. The experiments were conducted under the Guidelines for Animal Experiment of Shanghai University of Traditional Chinese Medicine and the protocol was approved by the institutional Animal Ethics Committee.
Co-housing experiments
An intermittent co-housing experiment was performed by consolidating 24-month-old old mice with 1-month-old young mice of the same sex for 10 weeks according to the reference with some modification.Citation88 In detail, the first round of co-housing lasted for 2 weeks and followed by 2 weeks interval of separation. Then, the second round of co-housing lasted for 4 weeks and followed by 2 weeks of separation. The experimental design is shown in ). In total, there were six groups including M_Y (young male), M_O (old male), M_O_CoH (co-housed old male), F_Y (young female), F_O (old female), and F_O_CoH (co-housed old female). At the end of the experiment, overnight fasted mice were sacrificed after anesthesia with 1% pentobarbital sodium solution by intraperitoneal injection. Samples were collected and immediately frozen at −80°C for further analysis.
Histological evaluation on the degree of hepatic lymphocyte infiltration
Liver tissues were fixed with 10% neutral formalin for 24 hours, embedded in paraffin, stained with hematoxylin-eosin staining (H&E) and sections were observed for the degree of hepatic lymphocyte infiltration under the light microscope. The degree of hepatic lymphocyte infiltration was evaluated according to a previous publication in a blinded way.Citation29 The criteria for scoring including 0 (absent), 1 (rare), 2 (mild), 3 (moderate), and 4 (severe).
Quantitative RT-PCR
Total RNA from ileum tissue was isolated using a RNeasy mini kit (#74104, QIAGEN, Germany) and total RNA of liver tissue was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized by the high Capacity cDNA Reverse Transcription Kit (#K1682, Thermo Fisher Scientific, USA). QRT-PCR was performed using SYBR Green (A25777, Thermo Fisher Scientific, USA). Gene expression was normalized to 18 s. The primers used are shown in supplementary table 1.
16 S rDNA sequencing
DNA samples were extracted from 50 to 100 mg cecal contents using E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, USA.). Qualified DNA samples were applied to amplification of 16 S rDNA V3-V4 region using the universal primers 338 F (ACTCCTACGGGAGGCAGCAG) and 806 R (GGACTACHVGGGTWTCTAAT). The sequencing was performed by the Illumina MiSeq PE300 system (Illumina, San Diego, USA) according to the standard protocols. Raw data files were demultiplexed, quality-filtered, and sequences whose overlap longer than 10 bp were merged using FLASH. The reads were clustered to OTUs with 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/) and chimeras were removed using UCHIME. The taxonomy of each sequence was analyzed by Ribosomal Database Project Classifier algorithm (http://rdp.cme.msu.edu/) against the 16 S rDNA database Silva (SSU123) using confidence threshold of 70%. The principal coordinates (PCoA) analysis based on unweighted_unifrac was conducted to reflect community similarity and overall difference of gut microbiota in each group, the difference between groups was analyzed by Adonis test.
BA extraction and quantification
Chemicals
All of the 39 bile acids standards including taurohyocholic acid (THCA), hyocholic acid (HCA), ω-muricholic acid (ωMCA), tauro-ω-muricholic acid (TωMCA), tauro-α muricholic acid (TαMCA), tauro-β-muricholic acid (TβMCA), tauroursodeoxycholic acid (TUDCA), glycoursodeoxycholic acid (GUDCA), glycohyodeoxycholic acid (GHDCA), taurohyodeoxycholi acid (THDCA), taurocholic acid (TCA), glycoursodeoxycholic acid (GCA), 12-dehydrocholic acid (12-DHCA), β-muricholic acid (βMCA), α-muricholic acid (αMCA), 7-dehydrocholic acid (7-DHCA), 3-dehydrocholic acid (3-DHCA), taurochenodeoxycholic acid (TCDCA), 3β-cholic acid (βCA), taurodeoxycholic acid (TDCA), glycochenodeoxycholic acid (GCDCA), glycol deoxycholic acid (GDCA), ursodeoxycholic acid (UDCA), hyodeoxycholic acid (HDCA), cholic acid (CA), ursocholic acid (UCA), 23-nordeoxycholic acid (NorDCA), norcholic acid (NorCA), allocholic acid (ACA), 3β-chenodeoxycholic acid (βCDCA), taurolithocholicacid (TLCA), 3β deoxycholic acid (βDCA), glycolithocholic acid (GLCA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), 6-ketolithocholic acid (6-ketoLCA), 7-ketolithocholic acid (7-ketoLCA), 12-ketolithocholic acid (12-ketoLCA), lithocholic acid (LCA) were purchased from Steraloids Inc. (Newport, RI) and TRC Chemicals (Toronto, ON, Canada), and 9 stable isotope-labeled standards were obtained from C/D/N Isotopes Inc. (Quebec, Canada) and Steraloids Inc. (Newport, RI). The standards and stable isotope-labeled standards were accurately weighed and prepared in methanol at a concentration of 5.0 mM (stock solution). Further dilution was performed to obtain a series of calibration concentration of 2000, 400, 160, 32, 12.8, 2.5, or 1 nM with methanol/water (50/50, v/v). Internal Standard (IS) concentrations were kept constant at all the calibration points at 100 nM for GCA-d4, TCA-d4, TCDCA-d9, UDCA-d4, CA-d4, GCDCA-d4, GDCA-d4, DCA-d4, and 200 nM for LCA-d4.
Methanol (Optima LC-MS), acetonitrile (Optima LC-MS), and formic acid (Optima LC-MS) were purchased from Thermo Fisher Scientific (Fair Lawn, NJ). Ultrapure water was produced by a Mill-Q Reference system equipped with an LC-MS Pak filter (Millipore, Billerica, MA).
Sample preparation
Quantitative analysis of BAs was conducted according to the previous publications.Citation90,Citation91 Briefly, each 20 µL of serum or standard solution was spiked with 180 µL of acetonitrile:methanol = 80:20 containing 9 internal standards (100 µl IS to acetonitrile:methanol = 80:20) and the extraction of bile acids was conducted at a laboratory shaker at 10°C and 1,500 rpm for 15 min. After centrifugation, the supernatant (170 µl) was transferred to a micro-centrifuge tube for lyophilization using a freeze dryer system (Labconco, Kansas City, MO). The residue was reconstituted with 1:1 (v/v) mobile phase B (30 µl, acetonitrile/methanol = 80:20, v/v) and mobile phase A (30 µl, water), and centrifuged at 13,500 g and 4°C for 20 min. The supernatant was transferred to a 96-well plate for LC-MS analysis.
Each 100 mg liver tissue sample was homogenized with 100 µL of 50% methanol using a Bullet Blender Tissue Homogenizer (Next Advance, Inc., Averill Park, NY). An aliquot of 150 µL of acetonitrile containing 9 internal standards was added and the second step extraction was performed using the homogenizer. After centrifugation, the supernatant was divided into two aliquots (200 µL and 10 µL) and transferred to a microcentrifuge tube for lyophilization. The residue was reconstituted in 1:1 (v/v) mobile phase B (acetonitrile/methanol = 95:5, v/v) and mobile phase A (water with formic acid, pH = 3.25), and centrifuged at 13,500 g and 4°C for 20 min. The supernatant was transferred to a 96-well plate for LC-MS analysis.
Each 100 mg Intestinal contents (jejunum, ileum, cecum, colon) sample were homogenized with 500 µL of ice-cold water. The mixture was vortexed for 4 min and then centrifuged at 13,200 rpm for 10 min at 4°C. A 300 µL aliquot of supernatant was transferred to a 2-ml tube, and the pellets were further extracted with ice-cold methanol using the same protocol. Another 300 µL aliquot of supernatant was added to the same tube as the initial aliquot, and 10 µL of IS (p-chlorophenylalanine in water, 5 g/ml) was added. The extraction was vortexed for 30 s and centrifuged at 13,000 rpm for 20 min. The supernatant was transferred to a 96-well plate for LC-MS analysis.
Instrumentation
An ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS) system (ACQUITY UPLC-Xevo TQ-S, Waters Corp., Milford, MA) was used to quantitate 39 bile acids in the mouse liver, serum, and intestine samples. All chromatographic separations were performed with ACQUITY UPLC BEH C18 1.7 µM VanGuard pre-column (2.1 × 5 mm) and ACQUITY UPLC BEH C18 1.7 µM analytical column (2.1 × 100 mm). The mobile phase consisted of 10 mM ammonium acetate adjusted to pH 3.25 using formic acid (mobile phase A) and acetonitrile/methanol (mobile phase B). The flow rate was 0.45 mL/min with the following mobile phase gradient: 0–1 min (5% B), 1–5 min (5–25% B), 5–15.5 min (25–40% B), 15.5–17.5 min (40–95% B), 17.5–19 min (95% B), 19–19.5 min (95–5% B), and 19.6–21 min (5% B). The column was maintained at 45°C and the injection volume of all samples was 5 μL. The mass spectrometer was operated with source and desolvation temperatures set at 150°C and 550°C. Bile acids were detected in the negative mode. The mass spectrometer was operated in negative ion mode with a 1.2kV capillary voltage. The source and desolvation gas temperature were 150°C and 550°C, respectively. The data were collected with multiple reaction monitor (MRM), and the cone and collision energy for each BA used the optimized settings from QuanOptimize application manager (Waters), according to the previous publications.
Data analysis
The raw data were processed using the TargetLynx application manager (Waters Corp., Milford, MA) to obtain calibration equations and the measured concentration of each bile acid in the samples.
RNA sequencing analysis
Hepatic total RNA was extracted using RNeasy mini kit (Qiagen, Germany), paired-end libraries were synthesized by using the TruSeq™ RNA Sample Preparation Kit (Illumina, USA) following TruSeq™ RNA Sample Preparation Guide. Purified libraries were quantified by Qubit® 2.0 Fluorometer (Life Technologies, USA) and validated by Agilent 2100 bioanalyzer (Agilent Technologies, USA) to confirm the insert size and calculate the mole concentration. Cluster was generated by cBot with the library diluted to 10 pM and then were sequenced on the Illumina HiSeq (Illumina, USA). The raw reads were filtered by Seqtk before mapping to genome using Tophat (version: 2.0.9).Citation92 The fragments of genes were counted using HTSeq followed by TMM (trimmed mean of M values) normalization.Citation93 Significant differential expressed genes were identified as those with a False Discovery Rate value above the threshold (Q < 0.05) and fold-change >2 using edgeR software.94 RNAseq data can be accessed in NCBI and the accession number is PRJNA611506.
Correlation analysis between bacterial taxonomy and BAs
Correlation between the top 20 families and bile acids was estimated by Spearman’s correlation coefficient analysis, which was visualized in heatmap including positive (red) or negative (blue) relationship. The significant correction was performed with the criteria of *p < .05, **p < .01.
Statistical analysis
Data are shown as means ± SEM unless otherwise noted. All the bar plots in this study were generated with Prism 8.0 (GraphPad, La Jolla, CA, USA). Differences between groups were calculated by Mann–Whitney U test or Kruskal–Wallis H test using SPSS 24.0 (IBM, SPSS, USA). In addition, the p value was adjusted using Benjamini-Hochberg to control the multiple testing false discovery rate in the analysis of bile acids and the differential bacteria. p < .05 was considered statistically significant.
Author contributions
Junli Ma took the responsibility for the animal experiments, data analysis, and manuscript writing; Ying Hong helped animal experiments and data analysis; Ningning Zheng contributed to analysis of microbiome data; Guoxiang Xie was responsible for performing targeted metabolomics on bile acids; Yuanzhi Lyu analyzed the RNAseq data; Zhenzhen Huang and Wenbin Wu contributed to the animal experiment and sample collection; Gaosong Wu and Yu Gu contributed to data analysis of bile acid profile; Chuchu Xi and Linlin Chen helped the animal experiments and sample processing; Yue Li, Xin Tao, and Jing Zhong helped the data analysis and manuscript writing; Lin Yuan, Min Lin, and Xiong Lu were responsible for the HE staining and histological analysis of liver tissue; Weidong zhang helped in project design; Wei Jia participated in the design of this study and bile acids data analysis; Lili Sheng was responsible for the data analysis and manuscript writing; Houkai Li supervised the project and revised the manuscript.
Supplemental Material
Download Zip (2.3 MB)Disclosure statement
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. PMID:24833586. doi:https://doi.org/10.1093/gerona/glu057.
- Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8(1):18–30. PMID:18692159. doi:https://doi.org/10.1016/j.arr.2008.07.002.
- Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586. PMID:29686666. doi:https://doi.org/10.3389/fimmu.2018.00586.
- Belikov AV. Age-related diseases as vicious cycles. Ageing Res Rev. 2019;49:11–26. PMID:30458244. doi:https://doi.org/10.1016/j.arr.2018.11.002.
- Vaiserman AM, Koliada AK, Marotta F. Gut microbiota: a player in aging and a target for anti-aging intervention. Ageing Res Rev. 2017;35:36–45. PMID:28109835. doi:https://doi.org/10.1016/j.arr.2017.01.001.
- Rehman T. Role of the gut microbiota in age-related chronic inflammation. Endocr Metab Immune Disord Drug Targets. 2012;12(4):361–367. PMID:23017185. doi:https://doi.org/10.2174/187153012803832620.
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21(4):455–66 e4. PMID:28407483. doi:https://doi.org/10.1016/j.chom.2017.03.002.
- Bodogai M, O’Connell J, Kim K, Kim Y, Moritoh K, Chen C, Gusev F, Vaughan K, Shulzhenko N, Mattison JA, et al. Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Sci Transl Med. 2018;10(467):eaat4271. PMID:30429354. doi:https://doi.org/10.1126/scitranslmed.aat4271.
- Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, Savelkoul HF, De Jonge MI, Boekschoten MV, Smidt H, Faas MM., et al. Aged gut microbiota contributes to systemical inflammaging after transfer to germ-free mice. Front Immunol. 2017;8:1385. PMID:29163474. doi:https://doi.org/10.3389/fimmu.2017.01385.
- Fu ZD, Csanaky IL, Klaassen CD. Gender-divergent profile of bile acid homeostasis during aging of mice. PLoS One. 2012;7(3):e32551. PMID:22403674. doi:https://doi.org/10.1371/journal.pone.0032551.
- Barcena C, Valdes-Mas R, Mayoral P, Garabaya C, Durand S, Rodriguez F, Fernández-García MT, Salazar N, Nogacka AM, Garatachea N, et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat Med. 2019;25(8):1234–1242. PMID:31332389. doi:https://doi.org/10.1038/s41591-019-0504-5.
- Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24(1):41–50. PMID:27320064. doi:https://doi.org/10.1016/j.cmet.2016.05.005.
- Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15(2):111–128. PMID:29018272. doi:https://doi.org/10.1038/nrgastro.2017.119.
- Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–3925. PMID:16473946. doi:https://doi.org/10.1073/pnas.0509592103.
- Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4(1):2384. PMID:24064762. doi:https://doi.org/10.1038/ncomms3384.
- Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, Greiner TU, Perkins R, Bäckhed F. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(3):429–437. PMID:26740296. doi:https://doi.org/10.1136/gutjnl-2015-310283.
- Li T, Chiang JY. Bile acids as metabolic regulators. Curr Opin Gastroenterol. 2015;31(2):159–165. PMID:25584736. doi:https://doi.org/10.1097/mog.0000000000000156.
- Wan Y-JY, Sheng L. Regulation of bile acid receptor activity. Liver Res. 2018;2(4):180–185. doi:https://doi.org/10.1016/j.livres.2018.09.008.
- Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89(1):147–191. PMID:19126757. doi:https://doi.org/10.1152/physrev.00010.2008.
- Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A. 2008;105(36):13580–13585. PMID:18757757. doi:https://doi.org/10.1073/pnas.0804437105.
- Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, Kalavagunta PK, Liao J, Jin L, Shang J, et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome. 2019;7(1):9. PMID:30674356. doi:https://doi.org/10.1186/s40168-019-0628-3.
- Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. PMID:22699609. doi:https://doi.org/10.1038/nature11234.
- Biagi E, Franceschi C, Rampelli S, Severgnini M, Ostan R, Turroni S, Consolandi C, Quercia S, Scurti M, Monti D, et al. Gut microbiota and extreme longevity. Curr Biol. 2016;26(11):1480–1485. PMID:27185560. doi:https://doi.org/10.1016/j.cub.2016.04.016.
- Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q, Geng B, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14. PMID:28143587. doi:https://doi.org/10.1186/s40168-016-0222-x.
- Canfora EE, Meex RCR, Venema K, Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol. 2019;15(5):261–273. PMID:30670819. doi:https://doi.org/10.1038/s41574-019-0156-z.
- Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Science. 2018;362(6416):776–780. PMID:30442802. doi:https://doi.org/10.1126/science.aau5812.
- Sheng L, Jena PK, Hu Y, Liu HX, Nagar N, Kalanetra KM, French SW, French SW, Mills DA, Wan YJY, et al. Hepatic inflammation caused by dysregulated bile acid synthesis is reversible by butyrate supplementation. J Pathol. 2017;243(4):431–441. PMID:28892150. doi:https://doi.org/10.1002/path.4983.
- Sheng L, Jena PK, Liu HX, Hu Y, Nagar N, Bronner DN, Settles ML, Bäumler AJ, Wan YJ, et al. Obesity treatment by epigallocatechin-3-gallate-regulated bile acid signaling and its enriched akkermansia muciniphila. Faseb J. 2018;fj201800370R. PMID:29882708. doi:https://doi.org/10.1096/fj.201800370R.
- Jena PK, Sheng L, Nagar N, Wu C, Barile D, Mills DA, Wan YJY. Synbiotics Bifidobacterium infantis and milk oligosaccharides are effective in reversing cancer-prone nonalcoholic steatohepatitis using western diet-fed FXR knockout mouse models. J Nutr Biochem. 2018;57:246–254. PMID:29800811. doi:https://doi.org/10.1016/j.jnutbio.2018.04.007.
- Bárcena C, Valdés-Mas R, Mayoral P, Garabaya C, Durand S, Rodríguez F, Fernández-García MT, Salazar N, Nogacka AM, Garatachea N, et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat Med. 2019;25(8):1234–1242. PMID:31332389. doi:https://doi.org/10.1038/s41591-019-0504-5.
- Geer EB, Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med. 2009;6(Suppl 1):60–75. PMID:19318219. doi:https://doi.org/10.1016/j.genm.2009.02.002.
- Wingard DL. The sex differential in morbidity, mortality, and lifestyle. Annu Rev Public Health. 1984;5(1):433–458. PMID:6372818. doi:https://doi.org/10.1146/annurev.pu.05.050184.002245.
- Islami F, Miller KD, Siegel RL, Fedewa SA, Ward EM, Jemal A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J Clin. 2017;67(4):273–289. PMID:28586094. doi:https://doi.org/10.3322/caac.21402.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. PMID:21296855. doi:https://doi.org/10.3322/caac.20107.
- Sheng L, Jena PK, Liu HX, Kalanetra KM, Gonzalez FJ, French SW, Krishnan VV, Mills DA, Wan YJY. Gender differences in bile acids and microbiota in relationship with gender dissimilarity in steatosis induced by diet and FXR inactivation. Sci Rep. 2017;7(1):1748. PMID:28496104. doi:https://doi.org/10.1038/s41598-017-01576-9.
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–590. PMID:30046148. doi:https://doi.org/10.1038/s41574-018-0059-4.
- Bohnsack JF, Brown EJ. The role of the spleen in resistance to infection. Annu Rev Med. 1986;37(1):49–59. PMID:3518612. doi:https://doi.org/10.1146/annurev.me.37.020186.000405.
- Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J Clin Invest. 2011;121(8):3220–3232. PMID:21747170. doi:https://doi.org/10.1172/jci41514.
- Gadaleta RM, Cariello M, Sabba C, Moschetta A. Tissue-specific actions of FXR in metabolism and cancer. Biochim Biophys Acta. 2015;1851(1):30–39. PMID:25139561. doi:https://doi.org/10.1016/j.bbalip.2014.08.005.
- Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24(1):41–50. PMID:27320064. doi:https://doi.org/10.1016/j.cmet.2016.05.005.
- van der Lugt B, Rusli F, Lute C, Lamprakis A, Salazar E, Boekschoten MV, Hooiveld GJ, Müller M, Vervoort J, Kersten S, et al. Integrative analysis of gut microbiota composition, host colonic gene expression and intraluminal metabolites in aging C57BL/6J mice. Aging (Albany NY). 2018;10(5):930–950. PMID:29769431. doi:https://doi.org/10.18632/aging.101439.
- Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. PMID:16299351. doi:https://doi.org/10.1194/jlr.R500013-JLR200.
- Lepercq P, Gerard P, Beguet F, Raibaud P, Grill JP, Relano P, Cayuela C, Juste C, et al. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol Lett. 2004;235(1):65–72. PMID:15158263. doi:https://doi.org/10.1016/j.femsle.2004.04.011.
- Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58(5):949–955. PMID:23333527. doi:https://doi.org/10.1016/j.jhep.2013.01.003.
- Sanchez B. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis: a role for bifidobacteria and lactobacilli? Nat Rev Gastroenterol Hepatol. 2018;15(4):205. PMID:29512648. doi:https://doi.org/10.1038/nrgastro.2018.23.
- Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes. 2013;4(5):382–387. PMID:23851335. doi:https://doi.org/10.4161/gmic.25723.
- Gerard P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens. 2013;3(1):14–24. PMID:25437605. doi:https://doi.org/10.3390/pathogens3010014.
- de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17(5):657–669. PMID:23602448. doi:https://doi.org/10.1016/j.cmet.2013.03.013.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. PMID:16199517. doi:https://doi.org/10.1073/pnas.0506580102.
- de la Cuesta-zuluaga J, Kelley ST, Chen Y, Escobar JS, Mueller NT, Ley RE, McDonald D, Huang S, Swafford AD, Knight R, et al. Age- and sex-dependent patterns of gut microbial diversity in human adults. mSystems. 2019;4(4). PMID:31098397. doi:https://doi.org/10.1128/mSystems.00261-19.
- Rizzetto L, Fava F, Tuohy KM, Selmi C. Connecting the immune system, systemic chronic inflammation and the gut microbiome: the role of sex. J Autoimmun. 2018;92:12–34. PMID:29861127. doi:https://doi.org/10.1016/j.jaut.2018.05.008.
- Org E, Mehrabian M, Parks BW, Shipkova P, Liu X, Drake TA, Lusis AJ, et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes. 2016;7(4):313–322. PMID:27355107. doi:https://doi.org/10.1080/19490976.2016.1203502.
- Morris A. Microbiota drives sex-specific differences. Nat Rev Endocrinol. 2018;15(1):4. PMID:30425340. doi:https://doi.org/10.1038/s41574-018-0127-9.
- Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30(3):332–338. PMID:24625896. doi:https://doi.org/10.1097/MOG.0000000000000057.
- Bauerl C, Collado MC, Diaz CA, Vina J, Perez MG. Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer’s disease during lifespan. Lett Appl Microbiol. 2018;66(6):464–471. PMID:29575030. doi:https://doi.org/10.1111/lam.12882.
- Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140(3):976–986. PMID:21129376. doi:https://doi.org/10.1053/j.gastro.2010.11.049.
- Martinez I, Wallace G, Zhang C, Legge R, Benson AK, Carr TP, Moriyama EN, Walter J. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl Environ Microbiol. 2009;75(12):4175–4184. PMID:19411417. doi:https://doi.org/10.1128/aem.00380-09.
- Munro CA. Sex differences in Alzheimer’s disease risk: are we looking at the wrong hormones? Int Psychogeriatr. 2014;26(10):1579–1584. PMID:25100433. doi:https://doi.org/10.1017/s1041610214001549.
- Wong MW, Braidy N, Poljak A, Pickford R, Thambisetty M, Sachdev PS. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimers Dement. 2017;13(7):810–827. PMID:28242299. doi:https://doi.org/10.1016/j.jalz.2017.01.008.
- Jia W, Rajani C, Kaddurah-Daouk R, Li H. Expert insights: the potential role of the gut microbiome-bile acid-brain axis in the development and progression of Alzheimer’s disease and hepatic encephalopathy. Med Res Rev. 2019. PMID:31808182. doi:https://doi.org/10.1002/med.21653.
- Messaoudi M, Violle N, Bisson JF, Desor D, Javelot H, Rougeot C. Beneficial psychological effects of a probiotic formulation (Lactobacillus helveticus R0052 and bifidobacterium longum R0175) in healthy human volunteers. Gut Microbes. 2011;2(4):256–261. PMID:21983070. doi:https://doi.org/10.4161/gmic.2.4.16108.
- De Wolfe TJ, Eggers S, Barker AK, Kates AE, Dill-McFarland KA, Suen G, Safdar N. Oral probiotic combination of lactobacillus and bifidobacterium alters the gastrointestinal microbiota during antibiotic treatment for clostridium difficile infection. PLoS One. 2018;13(9):e0204253. PMID:30265691. doi:https://doi.org/10.1371/journal.pone.0204253.
- Langille MG, Meehan CJ, Koenig JE, Dhanani AS, Rose RA, Howlett SE, Beiko RG. Microbial shifts in the aging mouse gut. Microbiome. 2014;2(1):50. PMID:25520805. doi:https://doi.org/10.1186/s40168-014-0050-9.
- Woodmansey EJ. Intestinal bacteria and ageing. J Appl Microbiol. 2007;102(5):1178–1186. PMID:17448153. doi:https://doi.org/10.1111/j.1365-2672.2007.03400.x.
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–9440. PMID:12524422. doi:https://doi.org/10.1074/jbc.M209706200.
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Mangelsdorf DJ, Shan B, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362–1365. PMID:10334992. doi:https://doi.org/10.1126/science.284.5418.1362.
- Song P, Rockwell CE, Cui JY, Klaassen CD. Individual bile acids have differential effects on bile acid signaling in mice. Toxicol Appl Pharmacol. 2015;283(1):57–64. PMID:25582706. doi:https://doi.org/10.1016/j.taap.2014.12.005.
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. PMID:23803760. doi:https://doi.org/10.1038/nature12347.
- Gupta S, Natarajan R, Payne SG, Studer EJ, Spiegel S, Dent P, Hylemon PB. Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J Biol Chem. 2004;279(7):5821–5828. PMID:14660582. doi:https://doi.org/10.1074/jbc.M310979200.
- Qiao L, Studer E, Leach K, McKinstry R, Gupta S, Decker R, Kukreja R, Valerie K, Nagarkatti P, Deiry WE, et al. Deoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosis. Mol Biol Cell. 2001;12(9):2629–2645. PMID:11553704. doi:https://doi.org/10.1091/mbc.12.9.2629.
- Xie G, Wang X, Huang F, Zhao A, Chen W, Yan J, Zhang Y, Lei S, Ge K, Zheng X, et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer. 2016;139(8):1764–1775. PMID:27273788. doi:https://doi.org/10.1002/ijc.30219.
- Hill MJ. Bile, bacteria and bowel cancer. Gut. 1983;24(10):871–875. PMID:6618266. doi:https://doi.org/10.1136/gut.24.10.871.
- Nagengast FM, van der Werf SD, Lamers HL, Hectors MP, Buys WC, van Tongeren JM. Influence of age, intestinal transit time, and dietary composition on fecal bile acid profiles in healthy subjects. Dig Dis Sci. 1988;33(6):673–678. PMID:3371139. doi:https://doi.org/10.1007/bf01540429.
- Purim O, Gordon N, Brenner B. Cancer of the colon and rectum: potential effects of sex-age interactions on incidence and outcome. Med Sci Monit. 2013;19:203–209. PMID:23511310. doi:https://doi.org/10.12659/MSM.883842.
- Yui S, Kanamoto R, Saeki T. Biphasic regulation of cell death and survival by hydrophobic bile acids in HCT116 cells. Nutr Cancer. 2009;61(3):374–380. PMID:19373611. doi:https://doi.org/10.1080/01635580802582744.
- Hu X, Bonde Y, Eggertsen G, Rudling M. Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J Intern Med. 2014;275(1):27–38. PMID:24118394. doi:https://doi.org/10.1111/joim.12140.
- Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(2):225–235. PMID:23395169. doi:https://doi.org/10.1016/j.cmet.2013.01.003.
- Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277(6):3801–3804. PMID:11733542. doi:https://doi.org/10.1074/jbc.R100057200.
- Martinez-Augustin O, Sanchez de Medina F. Intestinal bile acid physiology and pathophysiology. World J Gastroenterol. 2008;14(37):5630–5640. PMID:18837078. doi:https://doi.org/10.3748/wjg.14.5630.
- Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67(3):863–867. PMID:17283114. doi:https://doi.org/10.1158/0008-5472.CAN-06-1078.
- Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinog. 2007;28(5):940–946. PMID:17183066. doi:https://doi.org/10.1093/carcin/bgl249.
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang -Z-Z, Takahashi S, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125(1):386–402. PMID:25500885. doi:https://doi.org/10.1172/JCI76738.
- Grut M. Why do women live longer than men? Eur J Epidemiol. 1998;14(3):311. PMID:9663525. doi:https://doi.org/10.1023/a:1007431627515.
- Tower J. Sex-specific gene expression and life span regulation. Trends Endocrinol Metab. 2017;28(10):735–747. PMID:28780002. doi:https://doi.org/10.1016/j.tem.2017.07.002.
- Dai ZC. AIDS prevention and control in China. Chin Med J. 1991;104(10):795–798. PMID:1752138
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21(4):455–66.e4. PMID:28407483. doi:https://doi.org/10.1016/j.chom.2017.03.002.
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. PMID:22297845. doi:https://doi.org/10.1038/nature10809.
- Yang H, Jung S, Seo J, Khalid A, Yoo JS, Park J, Kim S, Moon J, Lee S-T, Jung K-H, et al. Altered behavior and neural activity in conspecific cagemates co-housed with mouse models of brain disorders. Physiol Behav. 2016;163:167–176. PMID:27211331. doi:https://doi.org/10.1016/j.physbeh.2016.05.031.
- Gao X, Cao Q, Cheng Y, Zhao D, Wang Z, Yang H, Wu Q, You L, Wang Y, Lin Y, et al. Chronic stress promotes colitis by disturbing the gut microbiota and triggering immune system response. Proc Natl Acad Sci U S A. 2018;115(13):E2960–E9. PMID:29531080. doi:https://doi.org/10.1073/pnas.1720696115.
- Xie G, Wang Y, Wang X, Zhao A, Chen T, Ni Y, Wong L, Zhang H, Zhang J, Liu C, et al. Profiling of serum bile acids in a healthy Chinese population using UPLC-MS/MS. J Proteome Res. 2015;14(2):850–859. PMID:25581415. doi:https://doi.org/10.1021/pr500920q.
- Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, Chen H, Zhao X, Zhang S, Zhou Z, et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 2013;27(9):3583–3593. PMID:23709616. doi:https://doi.org/10.1096/fj.13-231860.
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinf. 2009;25(9):1105–1111. PMID:19289445. doi:https://doi.org/10.1093/bioinformatics/btp120.
- Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinf. 2015;31(2):166–169. PMID:25260700. doi:https://doi.org/10.1093/bioinformatics/btu638.
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinf. 2010;26(1):139–140. PMID:19910308. doi:https://doi.org/10.1093/bioinformatics/btp616.