
ABSTRACT
We hypothesized that the antimicrobial peptide cathelicidin has a physiological role in regulating gut inflammatory homeostasis. We determined that cathelicidin synergizes with LPS to facilitate its internalization and signaling via endosomic TLR4 in colonic epithelium, evoking synthesis of the human neutrophil chemoattractant, CXCL8 (or murine homolog, CXCL1). Interaction of cathelicidin with LPS in the control of CXCL8/CXCL1 synthesis was assessed in human colon epithelial cells, murine colonoids and cathelicidin-null mice (Camp−/-). Mechanistically, human cathelicidin (LL-37), as an extracellular complex with LPS, interacted with lipid raft-associated GM1 gangliosides to internalize and activate intracellular TLR4. Two signaling pathways converged on CXCL8/CXCL1 production: (1) a p38MAPK-dependent pathway regulated by Src-EGFR kinases; and, (2) a p38MAPK-independent, NF-κB-dependent pathway, regulated by MEK1/2-MAPK. Increased cathelicidin-dependent CXCL8 secretion in the colonic mucosa activated human blood-derived neutrophils. These cathelicidin effects occurred in vitro at concentrations well below those needed for microbicidal function. The important immunomodulatory role of cathelicidins was evident in cathelicidin-null/Camp−/- mice, which had diminished colonic CXCL1 secretion, decreased neutrophil recruitment-activation and reduced bacterial clearance when challenged with the colitis-inducing murine pathogen, Citrobacter rodentium. We conclude that in addition to its known microbicidal action, cathelicidin has a unique pathogen-sensing role, facilitating LPS-mediated intestinal responses, including the production of CXCL8/CXCL1 that would contribute to an integrated tissue response to recruit neutrophils during colitis.
Introduction
Cathelicidins are small cationic peptides produced by epithelial cells, macrophages and polymorphonuclear leukocytes.Citation1,Citation2 A single cathelicidin gene is present in humans (cathelicidin antimicrobial peptide, CAMP), which yields a 37 amino acid peptide (leucine-leucine, LL-37) generated by extracellular cleavage of the C-terminus.Citation3 The murine counterpart is cathelicidin-related-antimicrobial-peptide (CRAMP), encoded by the gene Camp (formerly Cnlp).Citation4 LL-37 and CRAMP are considered homologous; they share similar structure (α-helical and net charge of +6) and antimicrobial capabilities and display interspecies (humans – mice) functions.Citation5,Citation8 Whereas cathelicidins are considered broad antibacterials against Gram-positive and Gram-negative bacteria,Citation5,Citation9,Citation11 the relevance of such activity in vivo is controversial. These peptides have weak killing activity under physiological conditions, including high salt concentrations or in the presence of sugars.Citation12,Citation13 Cathelicidin’s bacteriocidal activity is inhibited by bacterial surface modifications such as Salmonella spp lipid A acylation.Citation14
Apart from their direct bacteriocidal activities, cathelicidins were shown to modulate inflammatory responses, primarily in hematopoietic cells.Citation2 Cathelicidins enhance chemokine expression (CXCL1, CXCL8, CCL2, CCL5 and CCL10) by monocytes and reduce TNF-α production by macrophages challenged with lipopolysaccharide (LPS), lipotechoic acid (LTA) or lipoarabinomannan.Citation5,Citation15,Citation17 It has been proposed that cathelicidins recruit monocytes, neutrophils and eosinophils,Citation18,Citation20 possibly by activating chemotactic formyl peptide receptor 2 (FPR2).Citation19,Citation21 Although cathelicidins are produced by intestinal epithelial cells, potentially to ward off bacteria, a direct effect of cathelicidins on the gut epithelium and in the context of enteric infectious bacterial disease is unknown.
During infections caused by Gram-negative bacterial pathogens (e.g., Salmonella typhimurium, Escherichia coli), LPS, via Toll-like receptor 4 (TLR4) signaling, can provoke release of inflammatory cytokines.Citation22 In a murine infectious colitis model, TLR4 signaling induced by Citrobacter rodentium, a natural attaching and effacing pathogen, promotes the recruitment of neutrophils into colonic tissue.Citation23 Neutrophils have an early innate defense role in the colon controlling the bacterial burden,Citation24,Citation25 in part by regulating fecal shedding and dissemination of pathogens to extra-intestinal sites (mesenteric lymph nodes and liver).Citation26,Citation27 In humans, neutrophil migration is largely regulated by the interaction of chemokine CXCL8 with CXCR1/2 receptors. In mice, two functional homologs of CXCL8, CXCL1 (keratinocyte chemoattractant (KC)) and CXCL2 (macrophage inflammatory protein-2 (MIP-2)), are regarded as early-phase neutrophil chemokines.Citation28 Of note, mice deficient in CXCL1 have reduced neutrophil recruitment to the colon following infection with C. rodentium.Citation29 The unresolved conundrum is how the colonic epithelium can respond to invasion by Gram-negative enteric pathogens, while maintaining tolerance to LPS continuously released from commensal bacteria.Citation30,Citation31 In this work, we explored the hypothesis that cathelicidin produced in response to pathogens invading colonic mucosa has a protective role in the setting of C. rodentium induced colitis. Our postulate was prompted by the observation that cathelicidin-deficient mice (Camp−/-), have an exacerbated diarrhea when challenged with either a colitis-inducing chemical or infectious agent (e.g. Clostridium difficile).Citation32,Citation33 Here we examined signal transduction pathways and other mechanisms whereby cathelicidin, in concert with LPS, can upregulate the neutrophil chemokine, CXCL8/CXCL1, contributing to the influx of neutrophils during infectious colitis.
Results
Cathelicidins contributed to neutrophil recruitment and pathogen clearance in infectious colitis
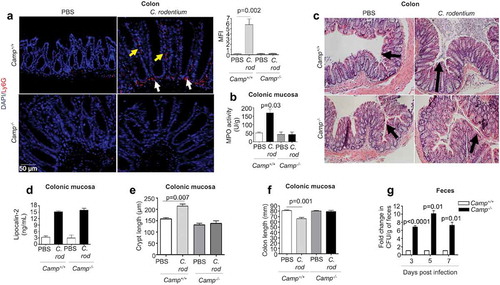
Neutrophils are necessary for controlling C. rodentium in mice; for example, mice rendered neutropenic due to anti-Gr-1 antibody treatment or depletion of CXCR2 have higher colonic bacterial burdens compared to wildtype mice.Citation27,Citation29 In the present study, cathelicidin-null (Camp−/-) mice had reduced neutrophil infiltration into the distal colon compared to Camp+/+ mice at peak C. rodentium infection (7 d post-infection; pi), as determined by immunofluorescence () and myeloperoxidase (MPO) activity (). Further, whereas infected Camp+/+ and Camp−/- mice had comparable histopathology (), expression of the cell damage marker lipocalin 2 () and reductions in cecum weight (Fig S1B), only infected Camp+/+ mice displayed increased crypt hyperplasia () and decreased colon length () as prominent hallmarks of colitis. Bacterial burden was also affected by the absence of endogenous cathelicidins. Fecal shedding of C. rodentium was ~8-fold greater in Camp−/- mice compared to Camp+/+ mice at 3, 5 and 7 d pi (). Further, C. rodentium was isolated from the spleen and liver of all Camp−/- mice but only in 50% of Camp+/+ mice (both for spleen and liver) (Fig S1C). Lack of cathelicidins in Camp−/- mice impacted not only the number of neutrophils infiltrating the colon but also their function. Neutrophils from Camp+/+ mice or a 1:1 mix of neutrophils from Camp+/+ and Camp−/- mice were more effective at in vitro killing of C. rodentium compared to Camp−/- neutrophils (at 30 min) (Fig S1A). Thus, cathelicidins can contribute to the influx of neutrophils and the clearance of C. rodentium.
Figure 1. Neutrophil activation, histopathology and fecal shedding in Camp+/+ and Camp−/- mice infected by Citrobacter rodentium. C57BL/6 Camp+/+ and Camp−/- mice were orally infected with C. rodentium (1 x 108 CFU in 200 μL of PBS) for 7 d. (a) Immunofluorescence neutrophil staining of murine colons with anti-Ly6 G antibody (5 μg/mL). Neutrophils were widely distributed in colon (lumen, crypt and lamina propria) although this representative image only shows neutrophils along crypts (yellow arrows) and in lamina propria (white arrows). Fluorescence was calculated using ImageJ 1.50i software and represented mean fluorescence intensity (MFI). (b) MPO activity in murine colonic mucosa after 7 d pi with C. rodentium. Data are represented as absolute values (MPO activity (U/g)) with respect to Camp+/+ PBS control. (c) H&E microphotography of murine colons. Black arrows denote crypt length. (d-f) Histograms representing absolute values of lipocalin-2 (ng/mL) secretions in feces (d), crypt length (e) and colon length (f) in control and infected mice. (g) For C. rodentium burden, fresh fecal pellets (1/per mouse) were collected at 3, 5 and 7 d pi (n = 4 mice/group) and serially diluted in sterile PBS, plated on McConkey agar and counted to obtain bacterial CFU/g. Data were normalized to infected Camp+/+ mice and represented as fold change in CFU/g of feces. One fold change represents ~105, 106 and 107 CFU/g for 3, 5 and 7 d pi, respectively. Data are shown as means ± SEM (n = 4 mice/group). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparison or two-tailed Student’s t-test for two groups) was considered significant.
Cathelicidins synergized with LPS to stimulate murine CXCL1 or human CXCL8 chemokines in colon
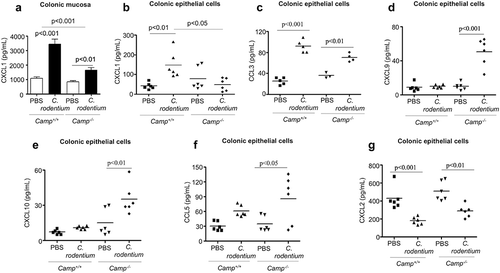
Based on in vitro studies, cathelicidins may influence migration of neutrophils to inflamed colons by chemoattraction, either directly via activation of FPR2 or indirectly by inducing pro-inflammatory cytokines (e.g., IL-1β, TNF-α, CXCL1).Citation17,Citation19,Citation34 Whereas fpr2−/- and wildtype mice have similar colonic neutrophil recruitment during C. rodentium infection,Citation35 excluding an impact of cathelicidins on FPR2 activation, the role of cathelicidins in inducing the production of chemoattractants has been less explored. In our studies, we observed increased CXCL1 secretion in colons of Camp+/+ mice compared to Camp−/- mice at the peak of C. rodentium infection (7 d pi) (). Other pro inflammatory cytokines, IL-1β (Fig S1D) and TNF-α (Fig S1E) were similarly increased in the colon after infection of Camp+/+ and Camp−/- mice. To determine the source of this differential expression of CXCL1, we assessed CXCL1 secretions in CD326+CD45− epithelial cells and CD45+ CD326− leukocytes isolated from colons of mice infected with C. rodentium (7 d pi). CXCL1 secretions were increased in colonic epithelial cells () and leukocytes from the lamina propria (Fig S1 F) of infected mice compared to uninfected controls; however, only in colonic epithelium was the amount of CXCL1 statistically significantly higher in infected Camp+/+ mice compared to Camp−/- (). Thus, whereas baseline constitutive Camp peptide expression was higher (~4-fold) in lamina propria leukocytes compared to colonic epithelium in Camp+/+ mice (Fig S1 G), we decided to assess if the colonic epithelium, an early sensor of enteric pathogens (S. typhimurium and E. coli) and producer of cathelicidin,Citation36,Citation38 had a particular role in producing chemokines via the synthesis of cathelicidin.Citation39 We determined that in addition to CXCL1 (), colonic epithelial cells from Camp+/+ mice infected with C. rodentium produced higher amounts of the related chemokine, CCL3 (). In contrast, the production of CXCL9, CXCL10 and CCL5 was increased in colonic epithelium from infected Camp−/- mice ((-f), respectively). Kinetics of the other neutrophil chemoattractant, CXCL2, did not differ between colonic epithelium from infected Camp+/+ and Camp−/- mice (). Thus, in addition to immune and stromal cells producing chemotactic factors and initiating inflammation (i.e. neutrophil recruitment), colonic epithelial cells particularly increase CXCL1 via cathelicidin.
Figure 2. Determination of chemokine production in colonic mucosa and primary colonic epithelial cells in Camp+/+ and Camp−/- mice challenged by Citrobacter rodentium. Camp+/+ and Camp−/- mice were infected with C. rodentium (1 x 108 CFU in 200 μL of PBS) for 7 d. (a) CXCL1 secretions in distal colonic mucosa of infected Camp+/+ and Camp−/- mice (ELISA). Chemokine production of CXCL1 (b), CCL3 (c) CXCL9 (d), CXCL10 (e), CCL5 (f) and CXCL2 (g) in isolated primary CD326+CD45− colonic epithelial cells (multiplex bead-based assay). Data are shown as mean ± SEM (n= 3–6/group). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparisons or two-tailed Student’s t-test for two groups) was considered significant.
Given the importance of TLR4 signaling and chemokine synthesis in neutrophil recruitment during enteric bacterial infections,Citation23,Citation27,Citation29 we hypothesized that a combination of cathelicidin and LPS (LPS+LL-37) could synergize to regulate CXCL1 (murine) or CXCL8 (human). Single addition of LPS, LL-37 or a scambled LL-37 peptide, did not increase secretion of CXCL8 by human colon-derived epithelial cell lines. Remarkably, a combination of LPS+LL-37 given simultaneously increased CXCL8 secretion in 2 colonic epithelial cell lines: HT29 (early (4 h) () and later (16 h) (Fig S2A)) and T84 cells (where only apical secretion was observed; ). Synergistic increases in CXCL8 secretion were observed in HT29 cells with heat-killed S. typhimurium and LL-37 (Fig S2B). E. coli (O111:B4)-LPS also had similar synergistic synthesis of CXCL8 when combined with LL-37 (colonic HT29 cells at 4 h; data not shown).
Figure 3. CXCL1/CXCL8 synthesis in colonic epithelium stimulated with cathelicidins and LPS or Salmonella typhimurium infection. (a-b) CXCL8 were determined by ELISA in cell supernatants from human colonic HT29 epithelial cells (a) and human colonic T84 epithelial cells (b). Challenge consisted of LL-37 (10 μg/mL) and LPS (1 μg/mL), either alone or in combination for 4 h. (c-d) CXCL1 protein expression determined by ELISAs in homogenized colons of Camp−/- mice (n = 3–5 mice/per group) (c) and colonoids from Camp+/+ mice (d) treated with LPS (1 μg/g or 1 μg/mL), LL-37 (1 μg/g or 10 μg/mL) or scrambled-sequence peptide sLL-37 (10 μg/mL; only in mouse colonoids), either alone or in combination (intraperitoneally for 3 h in (C) and ex vivo for 4 h in (D)). (e-f) HT29 cells either transfected with sham plasmid/knocked-down for LL-37 (ShLL-37) or untransfected were challenged with S. typhimurium (MoI 10:1; 2 × 107 CFU) for variable time points (up to 8 h). Bar graphs are representing LL-37 secretions (1 fold ~250 pg/mL) (e) and CXCL8 secretions (f) in cell supernatants. Graphs are representative of three independent experiments. Data are shown as mean ± SEM (n = 3 independent experiments done in triplicate, unless mentioned otherwise). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparison or two-tailed Student’s t-test for two groups) was considered significant. ND = “not detected”.
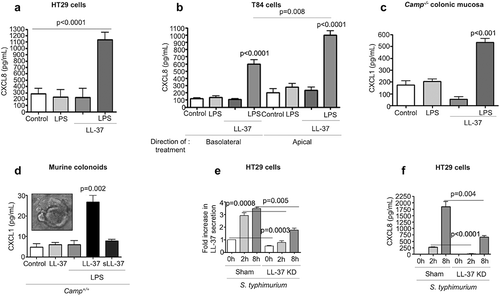
Due to interspecies functionality and reported immunological similarities between CRAMP and LL-37,Citation5,Citation19,Citation34 we tested whether LL-37 synergized with LPS to induce chemokines in murine colons. Intraperitoneal injection with LPS and synthetic LL-37, but neither LPS nor LL-37 alone, increased CXCL1 secretion in colons of Camp−/- mice (). However, neither LPS or LL-37 (alone or in combination) increased neutrophil MPO activity above baseline (PBS only control) in colons (Fig S2 C). LPS+LL-37 increased CXCL1 in colonoids developed from Camp+/+ mice (4 h) (), whereas LPS combined with scrambled LL-37 (only ()) had no such effect on CXCL1 (-d). Synergy between Gram-negative bacteria and cathelicidins was evident in HT29 cells knocked-down (KD) in endogenous cathelicidin (ShLL-37) (). ShLL-37 KD cells had reduced secretion of CXCL8 when exposed to S. typhimurium compared to sham-transfected cells at early (2 h) and later (8 h) () time points, with no cytotoxic side effects (Fig S2D).
Such synergistic CXCL8 synthesis with LPS+cathelicidins was not observed in leukocytes; in contrast, a decrease in TNF-α secretion was detected in phorbol 12-myristate-13-acetate (PMA) differentiated THP-1 cells treated with LPS+LL-37 (Fig S2E). Thus, LL-37 (and CRAMP) synergized with LPS or LPS+ bacteria to promote substantial production of neutrophilic chemoattractants, CXCL8 or CXCL1, from human and murine colonic epithelia, respectively.
LPS and LL-37 through extracellular interaction induced CXCL8 secretion via TLR4
Cathelicidins modulate colonic epithelial TLR4 in the presence of LPS, Salmonella spp. or E. coli.Citation40,Citation41 Herein, there was a key role of colonic epithelial TLR4 for cathelicidin-induced CXCL8 secretion. Inhibition of TLR4 with LPS-RS blocked CXCL8 mRNA and protein synthesis in HT29 cells challenged with LPS+LL-37 (-b). Further, TLR4 KD (ShTLR4) HT29 cells had diminished CXCL8 production when stimulated with LL-37+LPS compared to sham-transfected controls (c). The TLR4 co-adaptor protein MD-2 was not required for the production of CXCL8, as determined by a selective pharmacological MD-2 antagonist, L48H37 (Fig S3A). Secretion of the TLR4 accessory LPS-binding protein (LBP) was negligible in HT29 cells, irrespective of treatment (data below detection) and therefore could not account for the action of LPS/cathelicidin complex. When testing other TLR ligands and production of chemokines by HT29 cells, LL-37 reduced TLR9 agonist oligodinucleotide (ODN) mediated CXCL8 secretion but had no effect (synergistic or antagonistic) with the TLR5 agonist flagellin (Fig S2A). We concluded that the combined action of LPS+cathelicidin was due to TLR4 activation.
Figure 4. TLR4 dependent CXCL8 synthesis in colonic epithelial cells stimulated with cathelicidins and LPS. (a-d) HT29 cells used were either normal (A-B and D) or sham transfected/knocked-down in TLR4 (c). (a-c) Cells were pre-treated with LPS-RS (5 μg/mL) (or not; in C) followed by LPS (1 μg/mL) and LL-37 (10 μg/mL) alone or in combination. (d) Cells were treated by a mix of either LPS (1 μg/mL) or LL-37 (10 μg/mL) (1 h, 37°C) followed by substitution with polymyxin B (Poly B; 10 μg/mL) or LPS (1 μg/mL)+polymyxin B (Poly B; 10 μg/mL)/bovine serum albumin (BSA; 10 μg/mL) (1 h, 37°C) followed by LL-37 (10 μg/mL). Polymyxin B alone and BSA were used as negative controls. (a) CXCL8 mRNA levels were assessed by qPCR. GAPDH was used as housekeeping control. (b-d) CXCL8 secretion was quantified by ELISA after 4 h. Data are shown as means ± SEM (n = 3 independent experiments done in triplicate). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparison or two-tailed Student’s t-test for two groups) was considered significant.
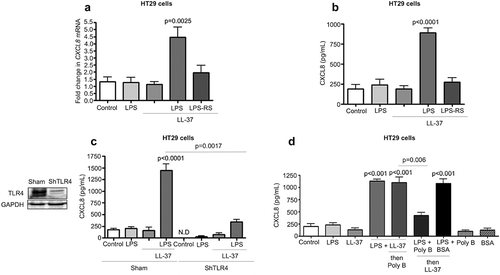
The next step was to determine if LPS bound to LL-37 to form a non-covalent complex prior to interacting with its colonic epithelium target. To this end, we first treated LPS with polymyxin B, a cyclic cationic polypeptide that binds to and neutralizes LPS. Prior treatment of LPS with polymyxin B prevented its ability to work in combination with cathelicidin to stimulate CXCL8 production by HT29 cells (). Conversely, if LPS and cathelicidin were first allowed to interact, followed by the addition of polymyxin B, the combined action of LPS+cathelicidin to enhance CXCL8 production in HT29 cells was not affected (). These data implicated the extracellular formation of a polymyxin B resistant complex between LPS and LL37 to stimulate CXCL8 synthesis.
LL-37 promoted CXCL8 synthesis via monosialotetrahexosylganglioside (GM1)-lipid raft mediated LPS uptake and intracellular TLR4 signaling
We inferred that LL-37 interacted with LPS prior to its activation of TLR4 to induce CXCL8 production. Therefore, we sought to determine how this complex reached TLR4, which can have an intracellular location for its action.Citation42,Citation43 We speculated that an LPS-cathelicidin complex may be internalized, as cathelicidins can act as a cell-penetrating peptide carrying cargo into cells.Citation44 Additionally, the sequence of cathelicidin has a distant relationship to the cholera toxin B-subunit (CTB), known to internalize the A subunit of cholera toxin via ganglioside GM1 cell surface binding.Citation45,Citation46 We, therefore, hypothesized that LL-37+LPS complexes interact with lipid rafts and associated membrane GM1 gangliosides to internalize into epithelia and promote intracellular TLR4 signaling. For this, we determined intracellular TLR4 expression in HT29 cells was ~3-fold higher than cell surface expression (Fig S3B). Surface staining of TLR4 diminished in HT29 cells after challenge with LPS+LL-37, but not with LPS or LL-37 alone (). Furthermore, Alexa488-conjugated-LPS in combination with LL-37 (), but not on its own or together with a scrambled LL-37 (Fig S3 C), was time-dependently internalized into HT29 cells. Internalized LPS was visualized within cells at higher magnification using a membrane marker (c). Addition of exogenous GM1 (100 μg/mL) to compete with LL-37+LPS for lipid rafts, reduced CXCL8 secretion (). Moreover, chemical lipid raft disruption with a mixture of methyl-β-cyclodextrin (500 μM) and mevinolin (250 ng/mL) (MM) abolished the ability of LPS+LL-37 to induce CXCL8 secretion () and internalization of Alexa488-conjugated-LPS (f).Citation47 Other endocytic pathways, including dynamin GTPase-dependent or actin-dependent processes assessed by pharmacological inhibition with dynamin inhibitor (D15) or actin assembly inhibitor cytochalasin D (CytoD) respectively, were not involved in CXCL8 synthesis () or uptake of Alexa488-conjugated-LPS by HT29 cells (only cytochalasin D; f). In regards to cholera toxin, unlike LL-37, CTB did not synergize with LPS to enhance CXCL8 synthesis by HT29 cells (Fig S3D). Previous studies have shown that CTB facilitates the internalization of LPS and thus, activation of caspase-11 in murine macrophages.Citation48 However, we observed no caspase-11 cleavage (activation) in primary colonic epithelium (i.e. murine Camp−/- colonoids) after LPS and LL-37 treatments, either alone or in combination (up to 2 h) (Fig S3E). In conclusion, LPS+LL-37 synergism in promoting CXCL8 production by colonic epithelial cells was dependent on extracellular binding with GM1-containing lipid rafts and intracellular TLR4 signaling.
Figure 5. LL-37-mediated GM1-lipid raft dependent LPS uptake regulates CXCL8 synthesis via intracellular TLR4 signaling in colonic epithelial cells. (a) Fluorescent images depicting TLR4 localization in non-permeabilized HT29 cells after LPS (1 μg/mL) ± LL-37 (10 μg/mL) for 4 h (anti-TLR4 antibody; yellow). A bar graph representing quantification of TLR4 fluorescence as mean fluorescence intensity (MFI). (b-c) HT29 cells were challenged with a combination of Alexa488-conjugated-LPS (1 μg/mL) and LL-37 (10 μg/mL) for variable intervals (up to 4 h) followed by quantification of fluorescence (b) or combined with wheat germ agglutinin (WGA) as a membrane marker (for 4 h) (c). (d) CXCL8 was quantified by ELISA in supernatants from HT29 cells treated with LPS and LL-37 ± exogenous monosialotetrahexosylganglioside (GM1; 100 μg/mL). GM3 (100 μg/mL) was used as a negative control. (e-f) HT29 cells were pre-treated with a mix of lipid raft inhibitors (MM: methyl-β-cyclodextrin (500 μM) and mevinolin (250 ng/mL)), or endocytosis inhibitors cytochalasin D (CytoD; 2 μM), or dynamin-dependent endocytosis inhibitor (D15; 10 μM; only for (e)) for 1 h, followed by stimulation with nonconjugated LPS (1 μg/mL) (E) or Alexa488-conjugated-LPS (1 μg/mL) (f) and LL-37 (10 μg/mL), either alone or in combination for 4 h. (E) CXCL8 protein secretion was quantified using ELISA. (b and f) LPS uptake was assessed using immunocytochemistry and quantified using ImageJ 1.50i software. Fluorescence was calculated and presented as mean fluorescence intensity (MFI) for three independent experiments. Data are shown as means ± SEM (n = 3 independent experiments, done in triplicate). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparisons or two-tailed Student’s t test for two groups) were considered statistically significant.
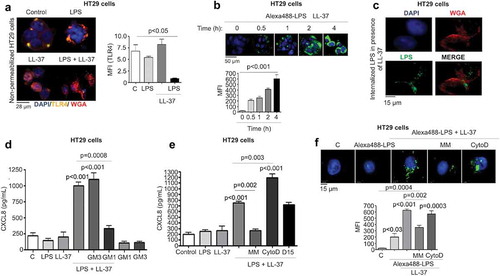
LL-37/LPS complex initiated p38MAPK signaling to promote CXCL8 synthesis
Intracellular signaling for CXCL8 synthesis in colonic epithelium involves protein tyrosine kinases and mitogen-activated protein kinases (MAPKs),Citation49 although the detailed interactive pathway is elusive. Here, LPS+LL-37 resulted in a time-dependent increase in phosphorylation of p38MAPK (up to 4 h) (). Moreover, p38MAPK inhibition by SB203580 blocked LPS+LL-37 induced CXCL8 mRNA and protein synthesis in HT29 cells (-c). Next, we focussed on how TLR4 facilitates p38MAPK phosphorylation in LPS+LL-37 induced CXCL8 secretion. Of interest was the sarcoma family kinase (Src), as it is linked to TLR4 and EGFR in human mammary epithelial cells in LPS-induced lethality.Citation50 There was a trend toward increased phosphorylation activation of EGFR by LPS+LL-37 that was reduced upon inhibition of EGFR (by AG1478), as well as Src (by PP1) (data shown for 2 h; ). Further, LPS+LL-37 induced p38MAPK activation was stronger than effects promoted by either stimuli alone (data shown for 2 h; ). Of significance, inhibitors of the EGFR (AG1478), Src (PP1) and TLR4 (LPS-RS) all reduced phosphorylation of p38MAPK (); furthermore, all three inhibitors attenuated CXCL8 secretion in HT29 cells stimulated by LPS+LL-37 (f-g). Pharmacological inhibition of other membrane-based receptors that reportedly can bind LL-37, including formyl peptide receptor-like 1 (FPRL-1), P2X purinoceptor 7 (P2X7) and the EGFR2/ErbB2 that can also be cross-activated by matrix metalloproteinases (MMP),Citation51 did not affect the synergistic action of LPS+LL37 to stimulate CXCL8 secretion (Fig S4A). Taken together, we inferred that synthesis of CXCL8 in colonic epithelium by LPS+LL-37 relied on the TLR4 mediated activation of p38MAPK signaling in concert with activation of EGFR and Src kinase, which appeared to be upstream of p38MAPKinase.
Figure 6. Src-EGFR kinase-mediated p38MAPK signaling promotes CXCL8 synthesis in colonic epithelial cells stimulated by cathelicidins and LPS. (a) Activation of p38MAPK in HT29 cells stimulated by LPS+LL-37 was detected by western blotting with antibodies against phosphorylated p38MAPK. Total loading was confirmed after immunoblotting with p38MAPK. (b-g) HT29 cells were pre-treated with SB203580 (10 μM) (b-c), AG1478 (1 μM) (d-f), the Src kinases inhibitor PP1 (1 μM) (D-E and G) or LPS-RS (5 μg/mL) (e) for 1 h, followed by LPS (1 μg/mL) and LL-37 (10 μg/mL) treatment. CXCL8 mRNA synthesis was quantified by qPCR after 2 h (n = 4) (b) or CXCL8 protein secretion (n = 4) by ELISA after 4 h (c). (d) Activation of EGFR was assessed after 2 h using western blotting for phosphorylated EGFR. Normalization was done with total EGFR. (E) Activation of p38MAPK was assessed after 2 h using western blotting for phosphorylated p38MAPK. p38MAPK was blotted as housekeeping control. (f-g) Total CXCL8 secretions were quantified using ELISA. Data are shown as means ± SEM (n = 3 independent experiments done in triplicate, unless mentioned otherwise in respective sub-figure). P < .05 (two-tailed Student’s t-test for two groups) was considered significant.
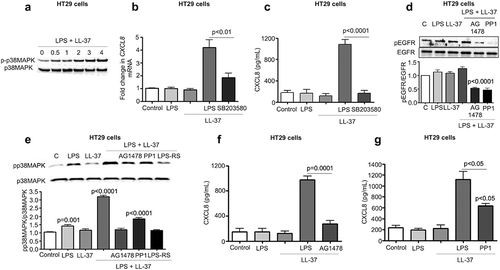
MEK1/2 mediated NF-κB activation contributed to colonic CXCL8 production induced by LL-37/LPS
Activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is essential for CXCL8 synthesis in airway and cervical epithelial cells.Citation52 In agreement with these findings, LPS+LL-37 induced a time-dependent phosphorylation of the p65-NF-κB subunit in HT29 cells (Fig S4B). Pharmacological inhibition of NF-κB-activating IκB kinase β (IKK) complex by PS1145 blocked the upregulation of CXCL8 mRNA and protein secretion in HT29 cells (-b). We thus assessed the ability of LL-37+ LPS to stimulate NF-κB promoter elements upstream of CXCL8 in HT29 cells using luciferase reporter constructs.Citation53 Luciferase reporter activity was abolished upon mutation of the NF-κB promoter site, whereas it remained active after mutations of the activator protein (AP)-1 or nuclear factor for IL-6 expression (NFIL-6) sites (c). This LPS+LL-37-mediated NF-κB p65 subunit phosphorylation was partially and significantly downregulated in TLR4 KD cells ().
Figure 7. MEK1/2 kinase-dependent NF-κB activation is required for CXCL8 synthesis induced by cathelicidins and LPS in colonic epithelial cells. (a-b) HT29 cells were pre-treated with the NF-κB inhibitor PS1145 (1 μM; 1 h) followed by LPS (1 μg/mL) and LL-37 (10 μg/mL). CXCL8 gene synthesis was quantified by qPCR after 2 h (n = 4) (a) and protein secretion by ELISA after 4 h (b). (c) HT29 cells were transfected with 174 bp wild type (FL) or mutant CXCL8 promoter construct (containing individual mutated sites for either NF-κB or AP-1 or NF-IL-6) upstream of the luciferase gene, followed by stimulation with LPS (1 μg/mL) and LL-37 (10 μg/mL) for 4 h. Data are represented as fold change in relative light units (RLU), normalized to renilla luciferase transfection control. IL-1β (10 ng/mL) was used as positive control. (d-e) HT29 cells sham transfected/knocked down for TLR4 (ShTLR4) (d) or treated with inhibitors for Src kinases (PP1; 1 μM), EGFR kinase (AG1478; 1 μM) and p38MAPK (SB203580; 2 μM) for 1 h (e) were stimulated by LPS+LL-37, either alone or in combination for 2 h. p65 phosphorylation was assessed by western blotting with specific antibodies. (f-g) HT29 cells normal and/or inhibited for ERK1/2 (PD98059; 20 μM) (only (g)) were stimulated by LPS+LL-37, either alone or in combination for 2 h. ERK1/2 (f) or p65 phosphorylation (G) was assessed by western blotting with specific antibodies. Total loading was confirmed after blotting for GAPDH. Data are shown as means ± SEM (n = 3 independent experiments done in triplicate). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparison or two-tailed Student’s t-test for two groups) were considered significant.
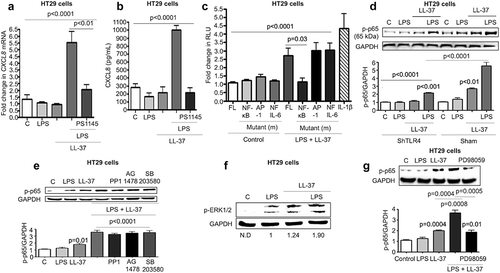
Inhibition of Src, EGFR kinase and p38MAPK had no effect on LL-37+ LPS-induced phosphorylation of NF-κB/p65 (). In search of an alternate signal pathway(s) to regulate NF-κB, we determined LPS+LL-37 enhanced phosphorylation-activation of extracellular signal-regulated kinase 1/2 (ERK1/2), downstream of mitogen-activated protein kinase (MEK) 1/2 (f). Moreover, pharmacological inhibition of MEK1/2 by PD98059 blocked phosphorylation-transactivation of the NF-κB subunit p65 (g) in HT29 cells exposed to LPS+LL-37 with a concomitant reduction in CXCL8 secretion (Fig S4 C). Similarly, phosphorylation-transactivation of the NF-κB subunit p65 was observed in LPS-challenged bone marrow-derived macrophages from Camp+/+, but not Camp−/- mice (Fig S4D). The proposed signaling pathways were verified in colons of C. rodentium-infected mice, where increased phosphorylation-activation of EGFR (a component of p38MAPK signaling axis) and NF-κB (p65) occurred in Camp+/+ compared to Camp−/- mice (Fig S4E). Thus, synergistic CXCL8 synthesis in colonic epithelium stimulated by LPS+LL37 was contingent upon two signals: p38MAPK activation (regulated by Src and EGFR kinases) and p38MAPK-independent NF-κB activation, downstream of TLR4 and MEK1/2 MAPK.
Secreted colonic CXCL8 activated human blood-derived neutrophils
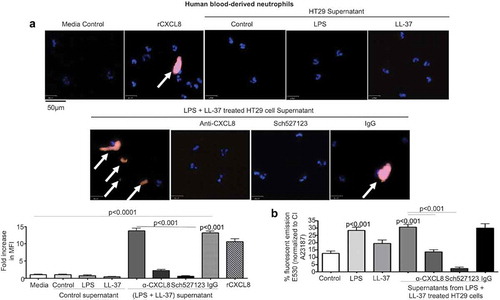
CXCL8 is a potent chemokine that activates a Gq-protein coupled CXCR1/2 receptor expressed on the surface of neutrophils.Citation54 This action promotes calcium mobilization in neutrophils which kills microbial pathogens.Citation55,Citation56 To determine the biological significance of CXCL8 secreted by colonic epithelium when stimulated with LPS+LL-37, we assessed neutrophil responses. Secreted products released by HT29 cells in response to LPS+LL-37 promoted the secretion of neutrophil elastase () and increased calcium flux () in naïve blood-derived human neutrophils. This response was mostly attributed to CXCL8, since an anti-CXCL8 antibody and a CXCL8 receptor antagonist blocked both events (-b). Taken together, LPS+LL-37 enhanced colonic epithelial cell release of bioactive CXCL8 at levels capable of activating neutrophils.
Figure 8. Secreted CXCL8 from colonic epithelium stimulated by cathelicidins and LPS induces calcium flux and activation of neutrophils. (a-b) Human neutrophils (1 x 104/well in an 8-well chamber) were incubated at 37° C for 1 h alone (a) or pre-incubated with Fluo4-NW dye (1 x 106 cells, 45 m, RT) (b). Neutrophils were exposed to supernatants from HT29 cells unstimulated (control) or stimulated with LPS (1 μg/mL) and LL-37 (10 μg/mL), either alone or in combination, for 4 h ± anti-CXCL8 antibody (1 μg/mL) or CXCR1/2 inhibitor SCH527123 (20 μM). rCXCL8 was used as positive control. IgG was used as an isotype control. (A) Neutrophil activation was assessed by neutrophil elastase secretion (depicted by white arrows) using immunocytochemistry with anti-neutrophil elastase antibody (5 μg/mL). Data are represented as fold increase in MFI normalized to respective control, for three independent experiments. (B) Data are represented as percentage fluorescence emission at 530 nm of positive control calcium ionophore A23187 (CI A23187). Data are shown as means ± SEM (n = 3 independent experiments done in triplicate). P < .05 (one-way ANOVA post hoc Bonferroni correction for multiple group comparison or two-tailed Student’s t-test for two groups) was considered significant.
Discussion
This study demonstrates that endogenous cathelicidin/LL-37, beyond its bactericidal activities when secreted by hematopoietic and non-hematopoietic cells, synergizes with LPS by promoting LPS internalization into colonic epithelium via lipid rafts. The LPS then initiates intracellular TLR4 signaling which in turn elevates the production of neutrophil chemoattractants (human CXCL8 or murine CXCL1). This mechanism seems key in infectious colitis. Studies on cathelicidin-null (Camp−/-) mice had shown endogenous cathelicidin is needed for controlling C. rodentium infection, mostly attributed to antimicrobial actions.Citation57,Citation58 We confirmed that the elimination of C. rodentium was impaired in the absence of cathelicidin; however, we propose a hitherto unrecognized “pathogen sensing” mechanism of cathelicidin at the colonic epithelium (). Cathelicidin from colonic epithelium in synergy with Gram-negative bacteria/LPS could initiate and/or contribute to the chemotaxis of neutrophils, either directly via known receptors (e.g., FPR2) or indirectly by releasing immune effectors (e.g., CXCL1) upon communication with local stromal or immune cells.Citation2,Citation34,Citation59 Thus, epithelial production of cathelicidins and chemoattractants could act in alliance with other gut cells (e.g.resident or recruited leucocytes) to recruit neutrophils following infection to restrict the pathogen (e.g. C. rodentium) with the bystander effect of collateral inflammatory tissue damage.Citation60 Indeed, we observed Camp+/+ neutrophils are better at the direct killing of C. rodentium in vitro than Camp−/- neutrophils. In agreement, CXCL1 expression in intestinal stromal cells correlates with early neutrophil recruitment into the colonic mucosa which could migrate into the lumen to eliminate IgG opsonized virulent C. rodentium.Citation61,Citation62 Since leukocytes are major sources of cathelicidin, future studies should employ bone marrow chimeric mice (Camp+/+ <-> Camp−/-)Citation63 to determine the relative contribution of epithelial-derived verses leucocyte-derived cathelicidin and associated CXCL1 in neutrophil recruitment into the colon.
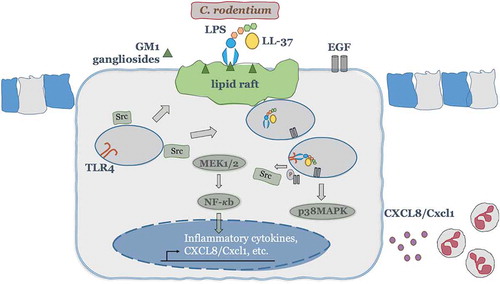
Figure 9. A theoretical scheme of signaling mechanisms elicited by cathelicidins in synergy with LPS to promote CXCL8 secretion in colonic epithelium and subsequent neutrophil recruitment/activation. In colonic epithelial cells, LL-37 physically binds and facilitates LPS uptake via interaction with GM1 (monosialotetrahexosylganglioside) in lipid rafts, where EGFR and Src kinases reside. Intracellularly, LPS interacts with TLR4 to promote activation of two signaling axes: one p38MAPK dependent, which relies on Src and EGFR kinases cross-talk and subsequent p38MAPK activation; second NF-κB dependent, which signals via MEK1/2 but independent of Src, EGFR and p38MAP kinases. Both signaling pathways together promote colonic CXCL8 protein synthesis and secretion. The induced colonic CXCL8 chemokine regulates intestinal defenses via neutrophil recruitment/activation.
The cathelicidin-dependent synergistic increase in CXCL1 synthesis upon C. rodentium infection was mostly restricted to CD326+CD45− colonic epithelium although baseline Camp expression was higher in CD45+CD326− leukocytes located in the lamina propria. In fact, the effect of LPS+LL-37 inducing CXCL8 in the epithelium differed from immunosuppressive effects of LPS+LL-37 on macrophages and in a sepsis model.Citation2,Citation64,Citation66 In this regard, TNF-α secretion was reduced following LPS+LL-37 treatment of PMA-differentiated human monocytes (THP-1), and CXCL8 synthesis was unaffected. Such disparate effects of cathelicidin in hematopoietic vs. non-hematopoietic cells could be attributed to the pleiotropic nature of cathelicidins acting via multiple cell surface (EGFR, P2X7 and FPR2) and cytoplasmic (TLR9 and GAPDH) receptors.Citation67 Additionally, we observed systemic (ip.) LPS+LL-37 delivery in Camp−/- mice did not promote neutrophil recruitment/activity into the colon. Neutrophil recruitment is a muti-factorial process regulated by a localized gradient of chemokines factors, which include CXCL1 but also others such as C5a complementary protein and leukotrienes.Citation68 Indeed, Camp−/- mice showed a reduced capacity to produce specific chemokines (CXCL1 and CCL3), whereas others (CXCL9 and CXCL10) were increased in Camp−/- mice, likely as a compensatory event. Thus, defective gut defense in cathelicidin-null mice was, at least in part, attributed to a reduced CXCL1-stimulated influx of neutrophils during infectious colitis and, LPS+LL-37 can drive epithelial chemokine responses at concentrations of LL-37 that were below microbicidal or cytotoxic.Citation69
The synergistic effect on colonic CXCL8 synthesis by LPS/LL-37 complex seems to specifically involve signaling by TLR4. The ability of cathelicidin to promote internalization of LPS was consistent with reports that cathelicidin aggregated with DNA, possibly due to the positive charges in the cathelicidin sequence, to promote internalization of DNA into endosomal compartments wherein it triggered TLR9 signaling.Citation70 However, LL-37 either reduced or did not enhance colonic CXCL8 production when elicited by oligodinucleotide (TLR9 activation) and flagellin (TLR5 ligand), respectively. Similar differential responses of cathelicidins to TLR ligands have been reported. In human bronchial epithelial cells, LL-37 increased CXCL8 secretion in the presence of the TLR1/2 ligand PAM3 CSK4,Citation71 but decreased the impact of double-stranded RNAs (TLR3 ligand).Citation71,Citation72 Also, CXCL8 mRNA synthesis was reduced by LL-37+ oligodinucelotide in colonic epithelial cells.Citation40 Furthermore, in murine macrophages, LPS-containing outer membrane vesicles shed by C. rodentium were endocytosed, which then activated caspase-11 and mediated cell death.Citation73 We did not observe caspase-11 activation and/or cytotoxicity in colonic epithelium after LPS+LL-37 treatment, suggesting LL-37 is particularly involved in signaling via endosomal TLR4 and secreting CXCL1 chemokine in colonic epithelium. It is noted that CXCL8 synthesis was still partially evident in TLR4 knockdown HT29 cells which may reflect residual TLR4 expression in knockdown cells and/or involvement of other pattern-recognition-receptors (PRR). However, future studies should be pursued in TLR4 and other PRR deficient mice to fully decipher the roles of TLRs in LPS and LL-37 synergism.
Both LPS trafficking and intracellular availability of LPS+LL-37 were crucial to enhance TLR4 signaling for CXCL8 secretion as confirmed in intestinal epithelial cells with different functional characteristics (HT29, T84), wildtype and Camp−/- mice, and ex vivo murine colonoids. The surface and intracellular compartments of colonic epithelium express TLR4 but intracellular TLR4 was key for LPS+LL-37-driven chemokine synthesis.Citation42 This intracellular TLR4 preponderance in the intestinal epithelium could be strategic in avoiding unwarranted inflammation.Citation74 Co-factors of TLR4, such as MD-2 and LBP, were not involved in LPS+LL-37 induced CXCL8 secretion. Expression of MD-2 was negligible in intestinal epithelium and only overexpression of MD-2 in HT29 cells promoted CXCL8 secretion upon LPS challenge.Citation75,Citation77 Similarly, exogenous LBP failed to induce CXCL8 secretion in methotrexate differentiated HT29 cells challenged with LPS.Citation78 Increased CXCL8 secretion did not occur in the presence of polymyxin B and LPS, perhaps due to the anti-endotoxin activity of polymyxin B. Moreover, cathelicidin or LPS acting on their own did not enhance colonic CXCL8; for instance, synthesis of CXCL8 in colonic epithelium (either mRNA transcription or protein translation) via phosphorylation of NF-κB (p65) did not occur with the addition of LPS alone (for up to 4 h). In agreement, only high concentrations of LPS (1 μg/mL for 18 h) increased CXCL8 production in previous studies with HT29 cells.Citation79 Further, ip. administration of LPS did not affect colonic CXCL1 synthesis, whereas LL-37 alone reduced CXCL1 levels in Camp−/- mice. It is likely that LPS does not increase CXCL1 in Camp−/- mice due to the absence of cathelicidin that is required to act together with LPS. In the case of LL-37 acting alone, perhaps lower concentrations of the peptide (<20 μg/mL) may induce expression of Toll interacting-protein (Tollip, a negative regulator of TLRs), thereby reducing basal CXCL1 synthesis (unpublished data). This hypothesis remains to be tested. Therefore, LPS+LL-37 physically interacted with TLR4 to promote colonic CXCL8 secretion, independent of MD2 and LBP adaptors. This multi-checkpoint strategy in the gut might prevent unnecessary inflammation in the presence of commensals and activate the colonic epithelium (CXCL8 synthesis) only during an infectious challenge, when a Gram-negative pathogen (or its LPS) is available and can form complexes with cathelicidins.
Mechanistically, we inferred a physical extracellular interaction between LL-37 and LPS, followed by internalization of an LPS+LL-37 complex via GM1-containing lipid rafts. Precise sites of interaction between LPS and LL-37, as for the interaction between cathelicidin and self-DNA,Citation70 remain to be determined. The interaction between LL-37 and other negatively charged ligands like LPS and DNA may well involve multiple lysine (K)/arginine (R) residues in LL-37. Positive charges on cathelicidins in proximity to negatively charged LPS would enable embedding of LL-37 hydrophobic helices into LPS micelles.Citation80,Citation81 Of the 10 positive lysine/arginine side chains in LL-37, three are situated immediately upstream of a negatively charged aspartic or glutamic acid, whereas four are present as dibasic clusters (FFR140K141 S; FK151R152IVQRI). The two C-terminal LL-37 basic residues are spaced by four amino acids (LR162NLVPR167TES), one of which is a proline that would generate a ‘twist’ between the two arginines, thereby limiting ligand interaction. Thus, if the positive side-chains in LL-37 were involved in an LPS or DNA interaction, the most likely sequences would be FFR140K141 S and FK151R152IVQRI. Of note, both of these sequences, but not the C-terminal arginines of LL-37 (LR162 NLVPR167TES), would also be present in the cathelicidin-derived inflammatory peptide, FA-29.Citation82 In principle, both LL-37 and FA-29 could synergize with LPS to drive CXCL8/cytokine synthesis via TLR4. Our continuing work is aimed at testing these hypotheses to identify the precise site whereby LPS interacts with LL-37 and to determine if FA-29, like cathelicidin, can synergize with LPS to drive cytokine production. Based on our data, the LPS+LL-37 complex gained access to intracellular TLR4 via a GM1/lipid raft mechanism. In support of this finding, lipid rafts were necessary for LPS signaling via intracellular TLR4 in murine small intestinal crypt (m-ICcl2) cells and lipid rafts were involved in ability of LL-37 to promote endocytosis of LPS in lung epithelial cells.Citation47,Citation83 Further work identifying the nature of the LL-37/LPS interaction should clarify how this complex interacts with lipid raft-containing ganglioside GM1. Interestingly, ganglioside GM1-mediated lipid rafts are key in the internalization of cholera toxin A/B. However, whereas cholera toxin promoted caspase-11 activation in murine macrophages by increasing levels of LPS in the cytoplasm,Citation48 LL-37 likely increases endosomal LPS trafficking in colonic epithelium as showed by insufficient caspase-11 activation. Indeed, cholera toxin and LPS did not synergistically enhance CXCL8 synthesis in colonic epithelium perhaps, due to inefficient activation of endosomal TLR4 as compared to the LPS+LL-37 complex.
Two signal pathways converged on the regulation of CXCL8 production by LPS+LL-37 in the colonic epithelium (). First, LPS+LL-37 promoted CXCL8 synthesis via p38MAPK activation, dependent on Src and EGF receptor-kinase activity. Interestingly, p38MAPK extended the CXCL8 mRNA half-life in HT29 cells stimulated with TNF-α,Citation84 whereas EGFR kinase stabilized amphiregulin mRNA in keratinocytes upon exposure to ultraviolet B radiation.Citation85 Thus, our reported Src-EGFR-p38MAPK signaling could be a cathelicidin downstream effect in the regulation of colonic CXCL8 synthesis, aiding in mRNA stabilization. Second, CXCL8 synthesis was dependent on MEK1/2 and NF-κB (p65) phosphorylation/activation. Likewise, LL-37 promoted NF-κB dependent CXCL8 secretion in human monocytes.Citation16 Further, we determined Camp+/+ BMMs had higher NF-κB activation than Camp−/- BMMs upon LPS challenge. This contrasted with previous studies where exogenous LL-37 inhibited LPS-induced nuclear translocation of NF-κB in leukocytes.Citation65 It is recognized that endogenous and exogenous cathelicidins could signal differentially on various cell types. Exogenous CRAMP diminished LPS-induced NF-κB activation in macrophages, whereas endogenous CRAMP promoted NF-κB activation.Citation86 The outcome of cathelicidin-bacterial ligand(s) interaction appeared to be dependent upon the nature of the bacterial molecule, microenvironmental conditions, the cell and the receptor (sub)types it expressed.
In summary, we describe cathelicidin-mediated “pathogen sensing” in the colonic epithelium via ganglioside GM1/lipid rafts mediated LPS internalization and sequential cross-talk with intracellular TLR4. Such host–pathogen interaction at the colonic mucosa resulted in the production of the neutrophil chemoattractant CXCL8 (humans)/CXCL1 (mice) and pathogen clearance, representing a novel endogenous innate cathelicidin defense in the setting of infectious colitis.
Methods
Ethics statement
All studies in mice were conducted following regulations specified by the Canadian Guidelines for Animal Welfare (CGAW) and were approved by the University of Calgary Health Sciences Animal Care Committee (AC16-0092).
Salmonella typhimurium
S. typhimurium (clinical strain LT2/ATCC 700720), provided by Dr. J. De Buck Univ. Calgary, was grown in Luria-Bertani (LB) Miller broth (IBI Scientific) (18 h, 37°C at 225 rpm) and streaked out on an LB agar plate.
Murine models and tissue processing for cytokine analysis
Male 8-wk-old wild-type Camp+/+ and cathelicidin-null Camp−/- C57BL/6 mice (B6.129X1-CamptmCitation1Rlg/J; The Jackson Laboratory) were littermates and were co-housed for several generations in a pathogen-free environment (Univ. Calgary). For systemic LPS challenge, Camp−/- mice were injected i.p. either with phosphate-buffered saline (PBS, Gibco, Life Technologies; 1x) as vehicle or with LPS derived from S. typhimurium (L6143; Sigma-Aldrich) and synthetic LL-37 amide trifluoroacetate salt peptide (>98.6% purity, H-6224.0005; Bachem) administered either alone or in combination (both at 1 μg/g). Mice were humanely euthanized 3 h post-challenge to collect colon samples. Distal colon was sampled, weighed, and suspended in either hexadecyltrimethylammonium bromide (HTAB) buffer (50 mg tissue wet-weight/mL) for myeloperoxidase (MPO) activity or, in sterile-PBS containing protease inhibitor cocktail (50 mg tissue wet-weight/mL) for ELISAs.Citation87 After tissue homogenization, IL-1β, TNF-α and CXCL1 cytokines were quantified by quantitative ELISA (DY401, DY410 and DY453, respectively; R&D Systems). MPO activity was determined as described and is represented as units (U)/g.Citation88 Data are presented as absolute values normalized to tissue wet weight.
For the infectious colitis model, C. rodentium (DBS-100) was cultured on McConkey agar plates (16 h, 37°C) and single colonies sub-cultured in LB broth (5 mL, 16 h, 37°C) without shaking. Mice were gavaged orally with 200 μL PBS (1x) or C. rodentium (~1 × 108 CFU in 200 μL), a reported dose for establishing infection in C57BL/6 mice,Citation89,Citation90 and humanely euthanized 7 d pi. Distal colon was processed for MPO activity determination and ELISA as discussed above or in denaturing cell extraction buffer (DCEB; FNN0091; Thermo Fischer Scientific) containing protease inhibitor cocktail (50 mg/mL) for western blotting. MPO activity was determined and represented as units (U)/g.Citation87 For CXCL1 and lipocalin-2 (DY1857; R&D Systems), ELISA was performed as discussed above.
Chemokine content in acutely isolated intact colonic epithelial cells was assessed by multiplex bead-based assay (Chemokine Discovery MD31, Eve Technologies). Colonic epithelial cells were recovered as described.Citation91 Briefly, entire colons were aseptically removed from PBS control and C. rodentium infected Camp+/+ and Camp−/- mice and rinsed with cold PBS (1X). Colons were cut-open longitudinally, sectioned (2-cm pieces) and suspended into isolation buffer containing EDTA (5 mM; 15575020, ThermoFisher Scientific) and DTT (0.324 M; D0632, Sigma-Aldrich) in 10 mL PBS. Isolated cells in suspension were incubated (37°C, 20 min on a rocker-shaker), passed through a cell strainer (70 μm; 10199656, VWR), washed with cold PBS (3X) and processed for downstream applications. For isolation of lamina propria cells, colon pieces devoid of epithelial cells were transferred into RPMI media, minced into (~5 mm) pieces and incubated with Liberase (100 units of activity/mL; containing collagenases I and II and Thermolysin; 5401119001; Sigma-Aldrich) (37°C in rocker-shaker, 1 h). The Liberase digestion process was repeated twice and lamina propria cells containing supernatant were collected in-between digestions. These cells were washed with cold PBS and processed for downstream applications. Purity of primary colonic epithelial and lamina propria cell populations was determined as CD326+ (EpCAM+; 563477, BDbioscience) CD45− (103139, BDbioscience) and CD326− CD45+ cells, respectively, using flow cytometry (~85–90%; data not shown). Data for multiplex chemokine assay were normalized to the total amount of protein (pg/mL).
To measure CFU/g of C. rodentium in tissue samples of liver and spleen, these organs were homogenized in PBS (50 mg/mL). Lysates were then plated on MacConkey agar plates at various dilutions and C. rodentium colonies quantified. To determine fecal shedding of C. rodentium, fresh fecal pellets (1 per mouse) were obtained aseptically from infected mice at 3, 5 and 7 d pi, and cultured on McConkey agar plates at various dilutions. Bacterial colonies were counted and data were represented as fold increase in CFU/g normalized to Camp+/+ infected mice. A qPCR validation was done using C. rodentium specific primers against espB (extracellularly secreted protein B) gene (forward primer: 5′-ATGCCGCAGATGAGACAGTTG-3′ and reverse primer: 5′-CGTCAGCAGCCTTTTCAGCTA-3′) (data not shown).Citation92
Murine colonoids
Mini 3D-gut colonoids were developed from murine colonic crypts as described.Citation93 Upon isolation, crypts were cultured on matrigel matrix for 10 d until colonoid morphology was apparent. After treatment(s), cell lysates were collected in DCEB containing protease inhibitor cocktail for western blotting, and supernatants were collected for quantification of CXCL1 by ELISAs.
Isolation of murine bone marrow macrophages
For isolation and culture of bone marrow macrophages (BMMs),Citation94 femoral and tibial bones were aseptically removed from Camp+/+ and Camp−/- mice. Upon isolation, bone marrow monocytes were cultured (6 d) in Roswell Park Memorial Institute 1640 medium (RPMI; Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS; Benchmark Gemini Bio-Products), 2 mM l-glutamine, 50 μM 2-mercaptoethanol, 10 mM HEPES buffer (pH 7.4), 1% penicillin (100 U ml−1)/streptomycin (100 μg ml−1; HyClone Thermo, Fisher Scientific) and 10% conditioned media from L929 cells (as a source of macrophage colony-stimulating factor required for macrophage lineage differentiation). For experiments, BMMs cultured in RPMI without FBS and antibiotics were challenged with LPS (1 μg/mL) for 1 h (for immunoblotting). Cell lysate proteins were isolated using DCEB buffer and assessed for phospho-NF-κB p65-Ser536 (3033; Cell Signaling Technology) activation. GAPDH was used as housekeeping control.
Isolation of neutrophils from murine bone marrows
Bone marrows from Camp+/+ and Camp−/- mice were isolated as described above. In brief, bone marrows were laid on the top of a three-layered Percoll gradient (i.e., 72%, 64% and 52%) (17089102; Amersham Bioscience) and centrifuged (650 x g, 4°C, 30 min, zero de-acceleration). Neutrophils were collected from the layer between 72% and 64% Percoll, washed with PBS (1X) and resuspended in RPMI media at a concentration of 1 × 107 cells/mL.
In vitro killing assay of Citrobacter rodentium by murine neutrophils
Murine bone marrow neutrophils from Camp+/+ and Camp−/- mice were isolated and purified using Percoll gradient as discussed above. Neutrophils were resuspended as individual genotypes (Camp+/+ or Camp−/-) as well as in a 1:1 mixture (50% Camp+/+ neutrophils and 50% Camp−/- neutrophils) in RPMI media without serum and antibiotic. Neutrophils (1 x 105 cells/mL) were incubated with bioluminescent C. rodentium (DBS100; MoI 1; 1 × 105 CFU) (30 min at 37°C), followed by assessment of C. rodentium luminescence relative to growth in LB media. C. rodentium growth in RPMI media was used as control. Data were represented as “% C. rodentium killing”’.
Cell culture
The human colon adenocarcinoma-derived HT29 and T84 epithelial cell lines, and THP-1 human monocytic cells (provided by Dr. K. Chadee, Univ. of Calgary) were cultured in Dulbecco’s Modified Eagle’s Media (DMEM; Gibson, Life Technologies) and RPMI-1640, respectively, supplemented with 10% fetal bovine serum (FBS; Benchmark Gemini Bio-Products), 1% penicillin (100 U ml−1)/streptomycin (100 μg ml−1; HyClone Thermo, Fisher Scientific) in a humidified environment with 5% CO2. For experiments, cells were cultured either in DMEM (for colonic cells) or RPMI (for THP-1) without FBS or antibiotics.
HT29 cells were stimulated with S. typhimurium, at a multiplicity of infection (MoI) of 10:1 (~2 × 107 CFU; OD600 (1) = ~0.8 × 108 CFU/mL) for variable intervals, as indicated in the figures, heat-inactivated S. typhimurium (2 x 108 CFU equivalent; 15 mins at 70°C), LPS from S. typhimurium (L6143, Sigma-Aldrich), LPS from E.coli O111:B4 (437627, Sigma-Aldrich), Alexa Fluor® 488-conjugated LPS from S. minnesota (L-23356; Molecular Probes), flagellin (SRP8029; Sigma-Aldrich), ODN (tlrl-2395; Invivogen) or CTB (C9903; Sigma-Aldrich). Concentrations of LL-37 (10 μg/mL) and LPS (1 μg/mL) (Fig S5A-D) and the time-point (4 h) examined (Fig S5E) were based on our previous studies of CXCL8 kinetics. Cytotoxicity induced by S. typhimurium (~2 × 107 CFU) was verified with a PierceTM LDH Cytotoxicity Assay Kit (88953; ThermoFisher Scientific). Effects of LPS on colonic cells were further studied using a TLR4-competitive inhibitor of LPS isolated from Rhodobacter sphaeroides (LPS-RS, tlrl-prslps; Invivogen) and polymyxin B sulfate salt (1405–20-5; Sigma-Aldrich). These treatments were combined with synthetic LL-37 or LL-37 scrambled peptide (63708; Ana Spec). Absence of cytotoxic effects of LPS (10 ng/mL to 5 μg/mL) and LL-37 (1 to 30 μg/mL) was confirmed using a PierceTM LDH Cytotoxicity Assay Kit.
THP-1 human monocytic cells were differentiated with PMA (P8139; Sigma-Aldrich, 20 ng/mL) for 3 d followed by simultaneous treatment with LPS (1 μg/mL) and LL-37 (10 μg/mL),Citation95 either alone or in combination, for 4 h. Cell supernatants were then collected and CXCL8 (DY208; R&D Systems) and TNF-α (DY210; R&D Systems) cytokine secretions were quantified as absolute values (pg/mL) using ELISA.
Signal transduction pathways
Intracellular signaling was investigated by pharmacologically blocking human EGFR 2/ErbB2 (TAK 165, 366017–09-6; Tocris Bioscience), FPRL1 (WRW4, 2262; Tocris Bioscience), MMP (GM6001, 2983; Tocris Bioscience), P2X7 (A740003, 3701; Tocris Bioscience), EGFR-kinase (AG1478 hydrochloride, 1276; Tocris Bioscience), Src kinase (PP1, Calbiochem), MEK1/2 (PD98059, 9900; Cell Signaling Technology and U0126, 1144; Tocris Bioscience), p38 MAPK (SB203580, 1202; Tocris Bioscience), MD-2 (L48H37, SML1443; Sigma-Aldrich) and NF-κB activating kinase, i.e. IKKβ (PS-1145; Cayman Chemical). Endocytic processes were investigated by inhibiting endocytosis (D15, 2334; Tocris Bioscience), actin polymerization (Cytochalasin D, C8273; Sigma-Aldrich) and lipid raft formation (mevinolin, M2147, HMG-CoA reductase inhibitor; Sigma-Aldrich and methyl-β-cyclodextrin, 332615, cholesterol solubilizing agent; Sigma-Aldrich). Cells were pre-treated (or not) for 1 h with inhibitors at concentrations either based on their IC50 values as recommended by manufacturer(s) or obtained from previous studies and maintained in culture medium without FBS or antibiotics.Citation40,Citation47,Citation96
Activation/phosphorylation of signal components (p38MAPK, ERK1/2 and NF-κB) was monitored by western blot analysis. Total proteins from cell lysates were isolated using DCEB buffer with a protease inhibitor cocktail. Extracted proteins were blotted using specific primary antibodies to detect phospho-p38 MAPK-Thr180/Tyr182 (4511; Cell Signaling Technology), p38 MAPK (9212; Cell Signaling Technology), phospho-ERK1/2 (4370; Cell Signaling Technology), phospho-NF-κB p65-Ser536, anti-caspase-11 (ab22684; Abcam) and human GAPDH-6C5 (1001; Calbiochem). Horseradish-peroxidase-conjugate (HRP) goat anti-mouse IgG (H + L) (115–035-146; Jackson ImmunoResearch) or HRP goat anti-rabbit IgG (H + L) (115–035-144; Jackson ImmunoResearch) was used as secondary antibodies and developed using the Clarity Western ECL Detection System (BioRad). Image capture and densitometric analyses were performed with ChemiDoc MP Imaging system and ImageLab 4.0.1 software (BioRad), respectively. Normalization was done with reference to either GAPDH (housekeeping protein) or respective total protein (p38MAPK). Results were reported as mean fold change of target expression in stimulated groups, compared to unstimulated control group.
Detection of CXCL8 protein secretion and gene transcription of CXCL8 and Camp
Secretion of human CXCL8 from epithelial cells was quantified using ELISA (DY208; R&D Systems). Transcription of human CXCL8 and murine Camp mRNA was quantified by quantitative real-time polymerase chain reaction (qPCR) using pre-designed primers (RTCitation2 qPCR Primer Assay, Qiagen) specific for human CXCL8 (PPH00568A; NM_000584.3), murine Camp (PPM25023A; NM_009921), human GAPDH (PPH00150 F; NM_002046.5) and murine GAPDH (PPM02946E; NM_008084) with verified specificity and efficiency (>95%) to ensure amplification of a single product of the correct size, as indicated in MIQE guidelines.Citation97 Target gene mRNA values were corrected relative to the normalizer, GAPDH. Data were analyzed using the 2−ΔΔCT method and reported as mean fold change of target transcript levels in stimulated groups versus untreated control group or CD45+ CD326− leukocytes vs. CD326+CD45− colonic epithelium for murine Camp.
LPS uptake in colonic epithelium
HT29 cells were treated with Alexa488-conjugated-LPS ± LL-37 or scrambled LL-37 peptides, fixed with 4% PFA in the dark (15 min, RT), and counterstained for nuclei with 4ʹ, 6-diamidino-2- phenylindole (DAPI, 62247; ThermoFisher Scientific) (1:1,000, 30 min, RT). Slides were then examined using a wide-field immunofluorescence microscope (IX71Olympus). Assessment of Alexa488-conjugated-LPS uptake by colonic cells was performed using ImageJ 1.50i software (© National Institute of Health). Fluorescence intensity was calculated in randomly selected fields of view (five/replicate), either with a cluster of cells (for time-dependent LPS uptake assay) or single cells (for LPS uptake in presence of inhibitors) per replicate, for a total of three replicates/experiment. Data were reported as MFI for three independent experiments.
TLR4 localization in colonic epithelium
HT29 cells (treated with LPS ± LL-37 or not) were either permeabilized (for intracellular TLR4) or not (surface TLR4), followed by analysis using immunofluorescence or flow cytometry. For immunofluorescence, cells were fixed as discussed above and permeabilized (or not) using 0.1% Triton X-100 (T8787; Sigma-Aldrich). Cells were then incubated with anti-TLR4 primary antibody (ab89455; Abcam) (1:100, O/N, 4°C) followed by cyanine 3-conjugated (Cy3) donkey anti-mouse IgG (H + L) (715–165-150; Jackson ImmunoResearch) secondary antibody (1:200, 1 h, RT). Cells were counterstained for nuclei with DAPI (1:1000, 30 min, RT) and/or Alexa647-conjugated wheat germ agglutinin (WGA; W32466; ThermoFisher Scientific) (1:500, 30 min, RT), which binds membrane expressing N-acetyl-D-glucosamine and sialic acid. Slides were examined using a wide-field immunofluorescence microscope (IX71Olympus). Data were reported as MFI for three independent experiments. For fluorescence-activated cell sorting (FACS) analysis, HT29 cells permeabilized (or not) were incubated either with PE-labeled anti-TLR4-CD284 (12–9917-41; eBioscience) or PE-labeled isotype mouse IgG (12–4724-41; eBioscience) (0.5 μg/mL, 30 min, 4°C). Cells were then analyzed using FACS (BDTM LSR II; BD-Bioscience). Data were obtained as mean fluorescence intensity (MFI) and represented as TLR4 expression normalized to surface expression.
TLR4 and LL-37 knock-down in colonic epithelial cells
A short hairpin (Sh)-TLR4 or (Sh)-LL-37 pGFP-V-RS plasmid vector or non-effective scrambled shRNA construct (sham) (TG320555 and TG314213; Origene) was transfected into confluent HT29 cells using Cell Line Nucleofector® Kit V (VCA-1003; Lonza). Transfected cells were selected for puromycin resistance (30 μg/mL). Knockdown efficiency was ~80-90 and 70–80% for TLR4 and LL-37, respectively, as assessed either through relative intensity quantification of TLR4 protein bands (ab13867; Abcam) or LL-37 ELISA (HK321; Hycult Biotechnology). Cells were maintained by constantly culturing them in DMEM media supplemented with puromycin (15 μg/mL).
Luciferase CXCL8 promoter transfection in colonic epithelium
A pGL3 basic plasmid containing 174 bp CXCL8 promoter construct (−166 to +8 relative to transcriptional start site) upstream of a firefly luciferase gene and a pRL null plasmid constitutively expressing renilla luciferase gene were utilized (provided by Dr. D. Proud, Univ. Calgary). The CXCL8 promoter construct contained either intact (174 bp full length; FL) or individually mutated sites for NF-κB, activator protein (AP)-1 and nuclear factor for IL-6 expression (NFIL-6) (site-directed mutagenesis; denoted as mNF-κB, mAP-1 and mNFIL-6, respectively). HT29 cells were co-transfected with the construct (1 μg) and pRL (0.1 μg) in DMEM media (FBS and antibiotic free) using TransIT transfection reagent (MIR5405; Mirus Bio LLC). pRL null plasmid was used as transfection control. Recombinant human interleukin-1β (IL-1β; 10 ng/mL, 8900; Cell Signaling Technology) was used as a positive control for CXCL8 promoter assay. Results were quantified using a dual-luciferase reporter assay (PR-E1910; Promega). Data were obtained as fold change in relative light units (RLU) of firefly luciferase activity with respect to control group, for three independent experiments.
Histological assessment and immunofluorescence in murine colon
Colonic tissues fixed in 10% neutral-buffered formalin and embedded in paraffin were stained with hematoxylin-eosin (H&E). For immunofluorescence, sections were deparaffinized, blocked in PBS-Tw containing 10% donkey serum (017–000-021; Jackson ImmunoResearch), 1% bovine serum albumin (BSA, 9048–46-8; Amresco), and 0.3 M glycine (1 h, RT), and incubated with primary Alexa647 tagged-anti-lymphocyte antigen 6 complex locus G6D (Ly6 G) antibody (5 μg/mL; MAB91671; R&D Systems) diluted in PBS (16 h, 4ºC). Slides were counterstained with DAPI and examined using IX71Olympus microscope. Integrated fluorescence intensity per mouse was calculated using ImageJ 1.50i software in five randomly selected fields of view and data were reported as MFI from n = 4 mice.
Neutrophil activation
Human neutrophils from healthy donors were isolated using Lympholyte®-poly solution (CL5070; Cedarlane) and stimulated with supernatants (500 μL) harvested from HT29 cells, either untreated (control; serum-free DMEM media) or treated with LPS ± LL-37 (for 4 h). Immunofluorescence analysis was performed as described above, using an anti-neutrophil elastase antibody (5 μg/mL; MAB91671; R&D Systems) and secondary Alexa549-conjugated donkey anti-mouse IgG antibody. For neutrophil elastase secretion quantification, fluorescence intensity was calculated using ImageJ 1.50i software. Average fluorescence intensity per replicate was quantified in five randomly selected fields of view and represented as fold increase in MFI.
Calcium flux assay
Neutrophils were isolated as discussed above. For calcium flux assay, neutrophils were incubated with Fluo4-no wash calcium dye (Fluo4 NW, F1242; Invitrogen) (1 mL; 45 m, RT). Then, neutrophils were washed with PBS and resuspended in calcium-magnesium-containing HBSS. Neutrophils (1 x 106) were transferred into a cuvette and stimulated with supernatants (100 μL) harvested from either untreated (control; serum-free DMEM media) or LPS and/or LL-37 treated colonic epithelial HT-29 cells. To confirm the role of CXCL8, either supernatants from LPS-LL37 treated HT29 cells were blocked (or not) with anti-CXCL8 antibody (1 μg/mL, MAB208; R&D Systems) or mouse IgG1 isotype control (1 μg/mL, 5415 S; Cell Signaling Technology) (4°C; 1 h) or with CXC receptor1/2 (CXCR1/2) inhibitor SCH 527123 (20 μM, A3802; APExBIO), prior to stimulation of neutrophils. Recombinant human CXCL8 (200 ng/mL, 208-IL-010; R&D Systems) was used as a positive control. For calcium flux assay, calcium signals were monitored at an excitation wavelength of 480 nm and an emission wavelength of 530 nm, recorded with the Aminco Bowan series II fluorimeter and the AB2 software (Thermo Fisher Scientific). Data were expressed as a percentage of the emission fluorescence normalized to maximum fluorescence caused by 2.5 μM calcium ionophore A23187. All data recorded were within the range of fluorescence obtained with calcium ionophore A23187.
Statistical analyses
Analytical data represented as histograms were recorded as mean values with bars representing standard errors of the mean (SEM) from a minimum of three independent experiments, with data obtained in triplicates, unless otherwise mentioned. Normality was assessed using D’Agostino & Pearson omnibus normality or Shapiro-Wilk (Royston) tests. All comparisons were performed using two-sided unpaired Student’s t-test, one-way analysis of variance (ANOVA) with a post hoc Bonferroni correction for multiple group comparisons or one-tailed z-test for comparing fractions. A P- value was assigned to each group with reference to control group, unless shown specifically on the graph. A P value of <0.05 was considered significant. All statistical analysis was performed with Graph Pad Prism software (Graph Pad 5.0).
Author contributions
RH performed the in vitro and in vivo experiments, acquisition, analysis of data, and prepared the figures. AB conducted mice infection, flow cytometry studies in epithelial and leukocytes and contributed in manuscript editing. GB participated in mice infection, PCR experiments and manuscript editing. FL participated in performing colonoid experiments. HJ assisted in the colonoid experiments. DM participated in manuscript review and editing. MDH participated in conceiving the experiments and edited the manuscript, contributed reagents, materials, and analysis tools. RH and EC conceived this research, designed the experiments, and wrote the manuscript. All authors reviewed and approved the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (1.9 MB)Acknowledgments
The authors thank D Proud (Univ. Calgary) for assisting in the experimental design and providing plasmid constructs for CXCL8 and, J Kastelic (Univ. Calgary) for editing this manuscript. Immunofluorescence studies were conducted in the Live Cell Imaging Facility, Snyder Institute, University of Calgary.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kosciuczuk EM, Lisowski P, Jarczak J, Strzalkowska N, Jozwik A, Horbanczuk J, Krzyzewski J, Zwierzchowski L, Bagnicka E. Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep. 2012;39:10957–24. doi:10.1007/s11033-012-1997-x.
- Hancock RE, Haney EF, Gill EE. The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol. 2016;16:321–334. doi:10.1038/nri.2016.29.
- Reinholz M, Ruzicka T, Schauber J. Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease. Ann Dermatol. 2012;24:126–135. doi:10.5021/ad.2012.24.2.126.
- Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414(6862):454–457. doi: 10.1038/35106587.
- Coorens M, Scheenstra MR, Veldhuizen EJ, Haagsman HP. Interspecies cathelicidin comparison reveals divergence in antimicrobial activity, TLR modulation, chemokine induction and regulation of phagocytosis. Sci Rep. 2017;7(1):40874. doi: 10.1038/srep40874.
- Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005; 7(2): 179–196. http://www.ncbi.nlm.nih.gov/pubmed/16053249
- Barlow PG, Svoboda P, Mackellar A, Nash AA, York IA, Pohl J, Davidson DJ, Donis RO, Kovats S. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One. 2011;6(10):e25333. doi: 10.1371/journal.pone.0025333.
- Alt JA, Qin X, Pulsipher A, Orb Q, Orlandi RR, Zhang J, Schults A, Jia W, Presson AP, Prestwich GD, et al. Topical cathelicidin (LL-37) an innate immune peptide induces acute olfactory epithelium inflammation in a mouse model. Int Forum Allergy Rhinol. 2015;5(12):1141–1150. doi:10.1002/alr.21634.
- Kao C, Lin X, Yi G, Zhang Y, Rowe-Magnus DA, Bush K Cathelicidin antimicrobial peptides with reduced activation of Toll-like receptor signaling have potent bactericidal activity against colistin-resistant bacteria. mBio 2016; 7. doi: 10.1128/mBio.01418-16.
- Scheenstra MR, van den Belt M, Tjeerdsma-van Bokhoven JLM, Schneider VAF, Ordonez SR, van Dijk A, Veldhuizen EJA, Haagsman HP. Cathelicidins PMAP-36, LL-37 and CATH-2 are similar peptides with different modes of action. Sci Rep. 2019;9:4780. 10.1038/s41598-019-41246-6
- Bommineni YR, Dai H, Gong YX, Soulages JL, Fernando SC, Desilva U, Prakash O, Zhang G. Fowlicidin-3 is an alpha-helical cationic host defense peptide with potent antibacterial and lipopolysaccharide-neutralizing activities. The FEBS Journal. 2007;274:418–428. doi:10.1111/j.1742-4658.2006.05589.x.
- Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi:10.1016/s0092-8674(00)81099-5.
- Scott A, Weldon S, Buchanan PJ, Schock B, Ernst RK, McAuley DF, Tunney MM, Irwin CR, Elborn JS, Taggart CC. Evaluation of the ability of LL-37 to neutralise LPS in vitro and ex vivo. PLoS One. 2011;6:e26525. doi:10.1371/journal.pone.0026525.
- Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell. 1998;95:189–198. doi:10.1016/s0092-8674(00)81750-x.
- Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. 2004;172:3758–3765. doi:10.4049/jimmunol.172.6.3758.
- Mookherjee N, Hamill P, Gardy J, Blimkie D, Falsafi R, Chikatamarla A, Arenillas DJ, Doria S, Kollmann TR, Hancock RE Systems biology evaluation of immune responses induced by human host defence peptide LL-37 in mononuclear cells. Mol Biosyst 2009; 5:483–496. doi: 10.1039/b813787k.
- Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. 2002;169:3883–3891. doi:10.4049/jimmunol.169.7.3883.
- Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol. 2006;140:103–112. doi:10.1159/000092305.
- Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–6265. doi:10.4049/jimmunol.174.10.6257.
- Beaumont PE, McHugh B, Gwyer Findlay E, Mackellar A, Mackenzie KJ, Gallo RL, Govan JR, Simpson AJ, Davidson DJ. Cathelicidin host defence peptide augments clearance of pulmonary Pseudomonas aeruginosa infection by its influence on neutrophil function in vivo. PLoS One. 2014;9:e99029. doi:10.1371/journal.pone.0099029.
- Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol. 2003;171:6690–6696. doi:10.4049/jimmunol.171.12.6690.
- Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167:1609–1616. doi:10.4049/jimmunol.167.3.1609.
- Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. 2006;74:2522–2536. doi:10.1128/IAI.74.5.2522-2536.2006.
- Mumy KL, McCormick BA. The role of neutrophils in the event of intestinal inflammation. Curr Opin Pharmacol. 2009;9:697–701. doi:10.1016/j.coph.2009.10.004.
- Cheminay C, Chakravortty D, Hensel M. Role of neutrophils in murine salmonellosis. Infect Immun. 2004;72:468–477. doi:10.1128/IAI.72.1.468-477.2004.
- Maier L, Diard M, Sellin ME, Chouffane ES, Trautwein-Weidner K, Periaswamy B, Slack E, Dolowschiak T, Stecher B, Loverdo C, et al. Granulocytes impose a tight bottleneck upon the gut luminal pathogen population during Salmonella typhimurium colitis. PLoS Pathog. 2014;10:e1004557. doi:10.1371/journal.ppat.1004557.
- Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol. 2007;179:566–577. doi:10.4049/jimmunol.179.1.566.
- De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:4930–4937. doi:10.1182/blood-2013-02-486217.
- Spehlmann ME, Dann SM, Hruz P, Hanson E, McCole DF, Eckmann L. CXCR2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J Immunol. 2009;183:3332–3343. doi:10.4049/jimmunol.0900600.
- Im E, Riegler FM, Pothoulakis C, Rhee SH. Elevated lipopolysaccharide in the colon evokes intestinal inflammation, aggravated in immune modulator-impaired mice. Am J Physiol Gastrointest Liver Physiol. 2012;303:G490–7. doi:10.1152/ajpgi.00120.2012.
- Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–1070. doi:10.1053/j.gastro.2004.01.007.
- Koon HW, Shih DQ, Chen J, Bakirtzi K, Hing TC, Law I, Ho S, Ichikawa R, Zhao D, Xu H, et al. Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology. 2011;141:1852–63 e1-3. doi:10.1053/j.gastro.2011.06.079.
- Hing TC, Ho S, Shih DQ, Ichikawa R, Cheng M, Chen J, Chen X, Law I, Najarian R, Kelly CP, et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut. 2013;62:1295–1305. doi:10.1136/gutjnl-2012-302180.
- De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi:10.1084/jem.192.7.1069.
- Sharba S, Venkatakrishnan V, Padra M, Winther M, Gabl M, Sundqvist M, Wang J, Forsman H, Linden SK. Formyl peptide receptor 2 orchestrates mucosal protection against Citrobacter rodentium infection. Virulence. 2019;10:610–624. doi:10.1080/21505594.2019.1635417.
- Bruno VM, Hannemann S, Lara-Tejero M, Flavell RA, Kleinstein SH, Galan JE. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 2009;5:e1000538. doi:10.1371/journal.ppat.1000538.
- Moxley RA, Smith DR. Attaching-effacing Escherichia coli infections in cattle. Vet Clin North Am Food Anim Pract. 2010;26:29–56. table of contents. doi:10.1016/j.cvfa.2009.10.011.
- Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun. 2002;70:953–963. doi:10.1128/IAI.70.2.953-963.2002.
- Zhang J, Jiao Y, Hou S, Tian T, Yuan Q, Hao H, Wu Z, Bao X. S100A4 contributes to colitis development by increasing the adherence of Citrobacter rodentium in intestinal epithelial cells. Sci Rep. 2017;7:12099. 10.1038/s41598-017-12256-z
- Marin M, Holani R, Shah CB, Odeon A, Cobo ER. Cathelicidin modulates synthesis of Toll-like receptors (TLRs) 4 and 9 in colonic epithelium. Mol Immunol. 2017;91:249–258. doi:10.1016/j.molimm.2017.09.011.
- Marin M, Holani R, Blyth GAD, Drouin D, Odeon A, Cobo ER. Human cathelicidin improves colonic epithelial defenses against Salmonella Typhimurium by modulating bacterial invasion, TLR4 and pro-inflammatory cytokines. Cell Tissue Res. 2019;376(3):433–442. 10.1007/s00441-018-02984-7
- Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, Barton GM. A map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity. 2018;49(3):560–75 e6. doi: 10.1016/j.immuni.2018.07.016.
- Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. 2002;195(5):559–570. doi:10.1084/jem.20011788.
- Seil M, Nagant C, Dehaye J-P, Vandenbranden M, Lensink MF. Spotlight on human LL-37, an immunomodulatory peptide with promising cell-penetrating properties. Pharmaceuticals (Basel). 2010;3(11):3435–3460. doi:10.3390/ph3113435.
- Hollenberg MD, Fishman PH, Bennett V, Cuatrecasas P. Cholera toxin and cell growth: role of membrane gangliosides. Proc Natl Acad Sci U S A. 1974;71(10):4224–4228. doi:10.1073/pnas.71.10.4224.
- Bennett V, Craig S, Hollenberg MD, O’Keefe E, Sahyoun N, Cuatrecasas P. Structure and function of cholera toxin and hormone receptors. J Supramol Struct. 1976;4(1):99–120. doi: 10.1002/jss.400040110.
- Shaykhiev R, Sierigk J, Herr C, Krasteva G, Kummer W, Bals R. The antimicrobial peptide cathelicidin enhances activation of lung epithelial cells by LPS. Faseb J. 2010;24(12):4756–4766. doi: 10.1096/fj.09-151332.
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi: 10.1126/science.1240988.
- Toumi F, Neunlist M, Denis MG, Oreshkova T, Laboisse CL, Galmiche J-P, Jarry A. Vasoactive intestinal peptide induces IL-8 production in human colonic epithelial cells via MAP kinase-dependent and PKA-independent pathways. Biochem Biophys Res Commun. 2004;317(1):187–191. doi: 10.1016/j.bbrc.2004.03.033.
- De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci U S A. 2015;112(31):9680–9685. doi: 10.1073/pnas.1511794112.
- Verjans E-T, Zels S, Luyten W, Landuyt B, Schoofs L. Molecular mechanisms of LL-37-induced receptor activation: an overview. Peptides. 2016;85:16–26. doi:10.1016/j.peptides.2016.09.002.
- Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol. 1994;56(5):554–558. doi:10.1002/jlb.56.5.554.
- Wiehler S, Proud D. Interleukin-17A modulates human airway epithelial responses to human rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L505–L15. doi: 10.1152/ajplung.00066.2007.
- Wu D, LaRosa GJ, Simon MI. G protein-coupled signal transduction pathways for interleukin-8. Science. 1993;261(5117):101–103. doi:10.1126/science.8316840.
- Gupta AK, Giaglis S, Hasler P, Hahn S, Palaniyar N. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS One. 2014;9(5):e97088. doi: 10.1371/journal.pone.0097088.
- Brinkmann V. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385.
- Mann EA, Harmel-Laws E, Cohen MB, Steinbrecher KA. Guanylate cyclase C limits systemic dissemination of a murine enteric pathogen. BMC Gastroenterol. 2013;13(1):135. doi: 10.1186/1471-230X-13-135.
- Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. 2005;174(8):4901–4907. doi:10.4049/jimmunol.174.8.4901.
- Szabady RL, McCormick BA. Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front Immunol. 2013;4:220. doi:10.3389/fimmu.2013.00220.
- Thorsvik S, Bakke I, van Beelen Granlund A, Royset ES, Damas JK, Ostvik AE, Sandvik AK. Expression of neutrophil gelatinase-associated lipocalin (NGAL) in the gut in Crohn’s disease. Cell and Tissue Research. 2018;374(2):339–348. 10.1007/s00441-018-2860-8
- Lee Y-S, Yang H, Yang J-Y, Kim Y, Lee S-H, Kim JH, Jang YJ, Vallance BA, Kweon M-N, Bäumler AJ. Interleukin-1 (IL-1) signaling in intestinal stromal cells controls KC/CXCL1 secretion, which correlates with recruitment of IL-22-secreting neutrophils at early stages of Citrobacter rodentium infection. Infect Immun. 2015;83(8):3257–3267. doi: 10.1128/IAI.00670-15.
- Kamada N, Sakamoto K, Seo S-U, Zeng M, Kim Y-G, Cascalho M, Vallance BA, Puente JL, Nunez G. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe. 2015;17(5):617–627. doi: 10.1016/j.chom.2015.04.001.
- Kovach MA, Ballinger MN, Newstead MW, Zeng X, Bhan U, Yu F-S, Moore BB, Gallo RL, Standiford TJ. Cathelicidin-related antimicrobial peptide is required for effective lung mucosal immunity in Gram-negative bacterial pneumonia. J Immunol. 2012;189(1):304–311. doi: 10.4049/jimmunol.1103196.
- Coorens M, Schneider VAF, de Groot AM, van Dijk A, Meijerink M, Wells JM, Scheenstra MR, Veldhuizen EJA, Haagsman HP. Cathelicidins inhibit Escherichia coli –induced TLR2 and TLR4 activation in a viability-dependent manner. J Immunol. 2017;199(4):1418–1428. doi: 10.4049/jimmunol.1602164.
- Mookherjee N, Brown KL, Bowdish DM, Doria S, Falsafi R, Hokamp K, Roche FM, Mu R, Doho GH, Pistolic J, et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J Immunol. 2006;176(4):2455–2464.doi:10.4049/jimmunol.176.4.2455.
- Cirioni O, Giacometti A, Ghiselli R, Bergnach C, Orlando F, Silvestri C, Mocchegiani F, Licci A, Skerlavaj B, Rocchi M, et al. LL-37 protects rats against lethal sepsis caused by gram-negative bacteria. Antimicrob Agents Chemother. 2006;50(5):1672–1679. doi:10.1128/AAC.50.5.1672-1679.2006.
- Hemshekhar M, Choi KYG, Mookherjee N. Host defense peptide LL-37-mediated chemoattractant properties, but not anti-inflammatory cytokine IL-1RA production, is selectively controlled by Cdc42 Rho GTPase via G protein-coupled receptors and JNK mitogen-activated protein kinase. Front Immunol. 2018;9:1871. doi:10.3389/fimmu.2018.01871.
- De Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16(6):378–391. doi: 10.1038/nri.2016.49.
- Sakoulas G, Kumaraswamy M, Kousha A, Nizet V Interaction of antibiotics with innate host defense factors against Salmonella enterica serotype newport. mSphere 2017; 2. doi: 10.1128/mSphere.00410-17
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–569. doi:10.1038/nature06116.
- Filewod NC, Pistolic J, Hancock RE. Low concentrations of LL-37 alter IL-8 production by keratinocytes and bronchial epithelial cells in response to proinflammatory stimuli. FEMS Immunol Med Microbiol. 2009;56(3):233–240. doi: 10.1111/j.1574-695X.2009.00571.x.
- Lai Y, Adhikarakunnathu S, Bhardwaj K, Ranjith-Kumar CT, Wen Y, Jordan JL, Wu LH, Dragnea B, San Mateo L, Kao CC. LL37 and cationic peptides enhance TLR3 signaling by viral double-stranded RNAs. PLoS One. 2011;6(10):e26632. doi: 10.1371/journal.pone.0026632.
- Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VK. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. 2016;165(5):1106–1119. doi: 10.1016/j.cell.2016.04.015.
- Meng D, Zhu W, Shi HN, Lu L, Wijendran V, Xu W, Walker WA. Toll-like receptor-4 in human and mouse colonic epithelium is developmentally regulated: a possible role in necrotizing enterocolitis. Pediatr Res. 2015;77(3):416–424. doi: 10.1038/pr.2014.207.
- Abreu MT, Arnold ET, Thomas LS, Gonsky R, Zhou Y, Hu B, Arditi M. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem. 2002;277(23):20431–20437. doi: 10.1074/jbc.M110333200.
- Vamadevan AS, Fukata M, Arnold ET, Thomas LS, Hsu D, Abreu MT. Regulation of toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis. Innate Immunity. 2010;16(2):93–103. doi: 10.1177/1753425909339231.
- Grondin V, Seksik P, Dumont S, Thomas G, Trugnan G, Flejou JF, Masliah J, Wendum D, Bachelet M. Regulation of colon cancer cell proliferation and migration by MD-2 activity. Innate Immunity. 2011;17(4):414–422. doi: 10.1177/1753425910375583.
- Bocker U, Yezerskyy O, Feick P, Manigold T, Panja A, Kalina U, Herweck F, Rossol S, Singer MV. Responsiveness of intestinal epithelial cell lines to lipopolysaccharide is correlated with toll-like receptor 4 but not toll-like receptor 2 or CD14 expression. Int J Colorectal Dis. 2003;18(1):25–32. doi: 10.1007/s00384-002-0415-6.
- Eckmann L, Jung HC, Schurer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF. Differential cytokine expression by human intestinal epithelial cell lines: regulated expression of interleukin 8. Gastroenterology. 1993;105(6):1689–1697. doi: 10.1016/0016-5085(93)91064-O.
- Bociek K, Ferluga S, Mardirossian M, Benincasa M, Tossi A, Gennaro R, Scocchi M. Lipopolysaccharide phosphorylation by the WaaY Kinase affects the susceptibility of Escherichia coli to the human antimicrobial peptide LL-37. J Biol Chem. 2015;290(32):19933–19941. doi: 10.1074/jbc.M114.634758.
- Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. 2008;283(47):32637–32643. doi: 10.1074/jbc.M805533200.
- Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13(8):975–980. doi:10.1038/nm1616.
- Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. 2003;198(8):1225–1235. doi: 10.1084/jem.20022194.
- Jijon HB, Panenka WJ, Madsen KL, Parsons HG. MAP kinases contribute to IL-8 secretion by intestinal epithelial cells via a posttranscriptional mechanism. Am J Physiol Cell Physiol. 2002;283(1):C31–41. doi: 10.1152/ajpcell.00113.2001.
- Nakayama H, Fukuda S, Matsushita N, Nishida-Fukuda H, Inoue H, Shirakata Y, Hashimoto K, Higashiyama S. Human antigen R-mediated mRNA stabilization is required for ultraviolet B-induced autoinduction of amphiregulin in keratinocytes. J Biol Chem. 2013;288(15):10338–10348. doi: 10.1074/jbc.M112.417527.
- Pinheiro da Silva F, Gallo RL, Nizet V. Differing effects of exogenous or endogenous cathelicidin on macrophage toll-like receptor signaling. Immunol Cell Biol. 2009;87(6):496–500. doi: 10.1038/icb.2009.19.
- Kishida K, Kohyama M, Kurashima Y, Kogure Y, Wang J, Hirayasu K, Suenaga T, Kiyono H, Kunisawa J, Arase H. Negative regulation of DSS-induced experimental colitis by PILRα. Int Immunol. 2015;27(6):307–314. doi: 10.1093/intimm/dxv004.
- Dann SM, Eckmann L. Innate immune defenses in the intestinal tract. Curr Opin Gastroenterol. 2007;23(2):115–120. doi: 10.1097/MOG.0b013e32803cadf4.
- Vong L, Pinnell LJ, Maattanen P, Yeung CW, Lurz E, Sherman PM. Selective enrichment of commensal gut bacteria protects against Citrobacter rodentium induced colitis. Am J Physiol Gastrointest Liver Physiol. 2015;309(3):G181–92. doi: 10.1152/ajpgi.00053.2015.
- Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, Wallace JL, Hollenberg MD, Vergnolle N. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci U S A. 2005;102(23):8363–8368. doi: 10.1073/pnas.0409535102.
- Pan D, Das A, Liu D, Veazey RS, Pahar B, Shoukry NH. Isolation and characterization of intestinal epithelial cells from normal and SIV-infected rhesus macaques. PLoS One. 2012;7(1):e30247. doi: 10.1371/journal.pone.0030247.
- Sagaidak S, Taibi A, Wen B, Comelli EM. Development of a real-time PCR assay for quantification of Citrobacter rodentium. J Microbiol Methods. 2016;126:76–77. doi:10.1016/j.mimet.2016.05.008.
- Fernando EH, Dicay M, Stahl M, Gordon MH, Vegso A, Baggio C, Alston L, Lopes F, Baker K, Hirota S, et al. A simple, cost-effective method for generating murine colonic 3D enteroids and 2D monolayers for studies of primary epithelial cell function. Am J Physiol Gastrointest Liver Physiol. 2017;313(5):G467–G75. doi:10.1152/ajpgi.00152.2017.
- Weischenfeldt J, Porse B Bone Marrow-Derived Macrophages (BMM): isolation and applications. CSH Protoc 2008; 2008:pdb prot5080. doi: 10.1101/pdb.prot5080
- Starr T, Bauler TJ, Malik-Kale P, Steele-Mortimer O, Ko DC. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella typhimurium. PLoS One. 2018;13(3):e0193601. doi: 10.1371/journal.pone.0193601.
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271(2):695–701. doi: 10.1074/jbc.271.2.695.
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi:10.1373/clinchem.2008.112797.