
ABSTRACT
Infant formula feeding, compared with human milk, has been associated with development of a distinct infant gut microbiome, but no previous study has examined effects of formula with added sugars. This work examined differences in gut microbiota among 91 Hispanic infants who consumed human milk [at breast (BB) vs. pumped in bottle (BP)] and 2 kinds of infant formula [(traditional lactose-based (TF) vs. lactose-reduced with added sugar (ASF)]. At 1 and 6 months, infant stool was collected to characterize gut microbiota. At 6 months, mothers completed 24-hour dietary recalls and questionnaires to determine infant consumption of human milk (BB vs. BP) or formula (TF vs. ASF). Linear regression models were used to determine associations of milk consumption type and microbial features at 6 months. Infants in the formula groups exhibited a significantly more ‘mature’ microbiome than infants in the human milk groups with the most pronounced differences observed between the ASF vs. BB groups. In the ASF group, we observed reduced log-normalized abundance of Bifidobacteriaceae (TF-BB Mean Difference = −0.71, ASF-BB Mean Difference = −1.10), and increased abundance of Lachnospiraceae (TF-BB Mean Difference = +0.89, ASF-BB Mean Difference = +1.20). We also observed a higher Community Phenotype Index of propionate, most likely produced by Lachnospiraceae, in the ASF group (TF-BB Mean Difference = +0.27, ASF-BB Mean Difference = +0.36). This study provides the first evidence that consumption of infant formula with added sugar may have a stronger association than birth delivery mode, infant caloric intake, and maternal BMI on the infant’s microbiome at 6 months of age.
Introduction
Factors that impact the composition of the infant gut microbiome include delivery mode,Citation1 gestational age,Citation2,Citation3 genetics,Citation4 antibiotic use, maternal diet,Citation5,Citation6 and infant feeding type (e.g., human milk vs. formula).Citation7,Citation8 While more recent studies suggest that infant feeding practices may be the largest driver of gut microbial development, earlier studies have compared human milk feeding versus formula feeding in general without considering the effects of infant formulas that contain added sugars.
Despite well-known differences in the gut microbiota between human milk-fed and formula-fed infants, the impact of early introduction of added sugars in formula on the development of the infant gut microbiota has not been previously examined. Formulas with added sugar are labeled as “gentle” and marketed to improve colic or “fussiness” in infants by removal of lactose.Citation9,Citation10 However, these “gentle” formulas are distinct from other traditional formulas because they contain lower concentrations of lactose (and therefore galactose as well), often displaced by added sugar in the form of corn syrup solids. Infants who are exposed to lactose-reduced formula with added sugar can conceivably have an altered gut microbiome, as has been revealed in animal studies that examined the microbiome of juvenile rodents.Citation11 To our knowledge, no human studies have examined the influence of lactose-reduced formula with added sugar on gut microbiota of infants. Therefore, the primary aim of this work was to examine differences in gut microbiota of infants that consumed: (1) human milk directly from breast and no formula; (2) pumped human milk delivered to the infant from a bottle and no formula; (3) traditional lactose-based formula; and (4) lactose-reduced formula with added sugar.
Results
General characteristics of infant cohort
This study included data from 91 Hispanic infants from the Los Angeles area. Infants were grouped by milk consumption type at 6 months of age: those who consumed human milk directly from breast and no formula (BB, n = 14), those who consumed pumped human milk and no formula (BP; n = 19), those who consumed a traditional formula (TF; n = 30) and those who consumed a lactose-reduced formula with added corn syrup solids formula (ASF; n = 29). Information regarding macronutrient composition and average macronutrient intake of infants by feeding type are provided in Supplemental . Briefly, the lactose-reduced formula with added sugar contains more glucose and maltose, has a higher glycemic index, and contains more added sugar (as measured by available carbohydrates) vs. traditional lactose-based formula and human milk. provides a summary of participants’ demographics, anthropometrics, body composition at 6 months of age by feeding type. Statistically significant differences between feeding-type groups were observed for mother’s BMI so this was included as a covariate in the analysis ().
Table 1. General characteristics of 6-month old infants by milk consumption.
Differences in diversity of gut microbiota at 6 months of infant age
At 6 months of age, there was no statistically significant difference in alpha diversity between milk consumption type as measured by Shannon’s index, Simpson’s index, richness, evenness, and total sequence reads per samples (). To examine the beta diversity of the infants’ microbiome, we applied multidimensional scaling of the ASVs using DEICODE Citation12 and examined the first three axes which explained ~100% of the variation. At 6 months of infant age, there was a significant difference in the beta diversity in the gut microbiota between human milk and formula (R2 = 0.11, p value = 0.002) but not between the two formula types (R2 = 0.033, p value = 0.1518) or between the two modes of breast delivery (R2 = 0.00378, p value = 0.92). Milk consumption type had a larger effect (Beta = 0.11, p value = 0.02) on the composition of the microbiome than birth delivery mode, maternal BMI and infant caloric intake ().
Figure 1. Multidimensional scaling (MDS) of 16S rRNA gene sequences colored by milk consumption type at 6 months. There is statistically significant separation between formula-fed infants and breastfed infants for both MDS axis 1 and axis 2, p values computed by independent ANOVA tests of each axis (a). Milk consumption type has a larger effect on the first MDS axis than delivery mode and infant weight (b). The phylum Proteobacteria has the highest mean relative abundance in the breast-fed (BB) group while Bacteroidetes has the highest mean relative abundance in the two formula groups (TF and ASF) (c). The family Enterobacteriaceae has the highest mean relative abundance in the BB group while Bacteroidaceae has the highest mean relative abundance in TF and ASF groups (d).
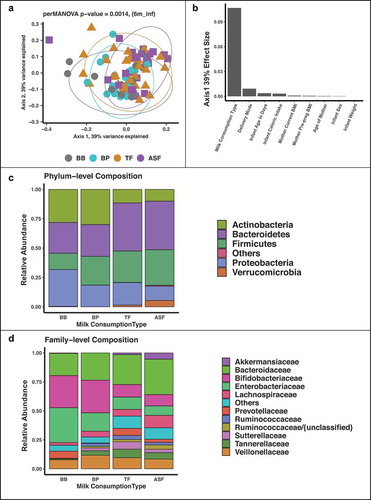
Table 2. Average diversity across milk consumption type.
Differences in abundances of gut microbiota at 6 months of infant age
We identified microbes that were differentially enriched or depleted in the four infant feeding groups. Using LEfSe discriminant analysis, we observed taxonomic groups in which pairwise differences in the milk consumption groups were discriminant among the groups (). We further examined the differences in abundances of each microbe using linear regression models adjusting for covariates (Supplemental Table 2–5). In particular, we found that relative to human milk (BB and BP), infants who consumed formula (TF and ASF) had a lower relative abundance of the family Bifidobacteriaceae (TF-BB Mean Difference = −0.71, HSD p value > 0.05; ASF-BB Mean Difference = −1.10, HSD p value = 0.002; TF-BP Mean Difference = −0.49, HSD p value > 0.05; ASF-BP Mean Difference = −0.83, HSD p value = 0.01). The depletion of Bifidobacteriaceae relative to the human milk groups was greater in the ASF than the depletion when compared to the TF group. Also, we found that formula-fed infants had significantly increased abundances of ASVs assigned to the family Lachnospiraceae (TF-BB Mean Difference = +0.89, HSD p value = 0.001; ASF-BB Mean Difference = +1.20, HSD p value = 3.3 × 10−6; TF-BP Mean Difference = −0.23, HSD p value > 0.05, ASF-BP Mean Difference = +0.58, HSD p value = 0.03). This relative enrichment was greater in ASF than TF. We also found that when compared to the group consuming traditional formula (TF), there was a significant increase in the family Acidaminococcaceae in the group of infants consuming lactose-reduced formula with added corn syrup solids (ASF-TF Difference = +0.71, HSD p value = 0.03) (, ). These findings were independent of infant’s sex, age in days, infant weight and abundance of the respective gut microbe at 1 month and also independent of mother’s age, current BMI, prepregnancy BMI, and delivery mode.
Figure 2. Linear discriminant analysis shows the microbial taxonomic groups that are enriched and depleted in infants consuming formula versus infants whose sole milk source is human milk. When comparing the gut microbiome of infants in the who consume expressed human milk in a bottle (BP) to infants who consume traditional formula (TF) (a), differences are apparent, but more differences result from the comparison of the gut microbiomes of infants whose only source of milk is through breastfeeding (BB) to those in the TF group (b). Even more microbes are revealed to be discriminant when we compare BP group and group consuming lactose-reduced formula with added-sugar (ASF) (c) and the BB group and ASF (d).
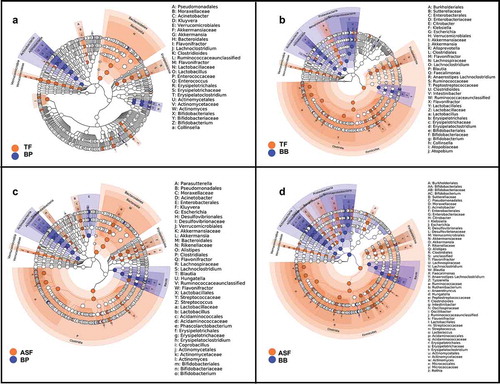
Figure 3. Log-normalized relative abundances of taxonomic families that are significantly different among the milk consumption types. At 6 months, microbes belonging to Bifidobacteriaceae are enriched in breastfed infants compared to formula fed infants with the infants who consume lactose-reduced formula with added corn syrup solids (ASF) having the lowest abundance (a). Enterobacteriaceae was also enriched in breastfed infants compared to formula fed infants but it was also significantly lower in the group who consume some human milk from a bottle (b). Further, microbes belonging to the families Lachnospiraceae are enriched in formula fed infants compared to breastfed infants with the infants in the ASF group having the highest abundance (c). Acidaminococcaceae was significantly higher in the infants who consume lactose-reduced formula with added corn syrup solids (ASF) (d).
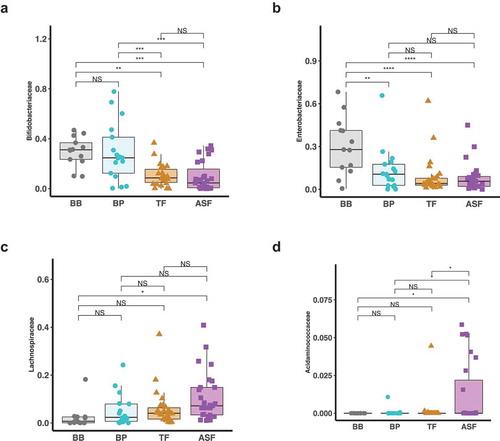
Table 3. Taxonomic families determined to be significantly different among milk consumption type groups.
Differences in predicted metabolic functions and phenotypes at 6 months
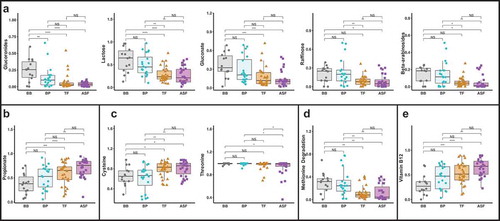
Finally, to examine differences in the functional potential of gut bacteria by milk consumption type, we applied the phenotype profiling approach using the obtained taxa relative abundances to quantify the fractional representation of predicted metabolic features (phenotypes) in the analyzed microbiome samples. In this analysis, we extended the approach previously described for B vitaminsCitation13,Citation14 toward a tentative prediction of requirements and utilization capabilities for major nutrients (amino acid and carbohydrates) as well as a potential to produce major short chain fatty acids (SCFAs). The details of our computational approach, which is based on in silico pathway reconstructionCitation15-Citation17 and is fundamentally similar to a broadly utilized analysis of pathway abundance (as implemented via combination of PICRUSt and MinPathCitation18,Citation19 (are outlined in Methods, and the results are summarized in Supplemental Table 6. We have identified significant differences among the distinct milk consumption groups in predicted metabolic capabilities of their respective microbial communities pertaining to a subset of 25 metabolites spanning: (i) utilization of 19 mono- and oligosaccharides (ii) biosynthesis of cysteine, threonine and B12; (iii) lysine and methionine degradation; and (iv) propionate production ().
Table 4. Predicted community metabolic phenotypes determined to be significantly different among milk consumption type groups.
Among such discriminating metabolic capabilities quantified by Community Phenotype Indices (CPI, on the scale from 0 to 100% fractional representation) is lactose utilization showing a two-fold elevated CPI value in breastfed groups (BB and BP) compared to formula-fed groups (TF and ASF). The predicted utilization capabilities for glucuronides, gluconate, beta-arabinosides, and raffinose, were also elevated two- to five-fold in both human milk fed groups. CPI for methionine degradation was 2.5-fold higher in both human milk fed groups, whereas the biosynthetic propensity for cysteine and cobalamin (vitamin B12) biosynthesis were decreased 1.5- to 2-fold in the human milk fed groups. Predicted propionate production capability appeared to be ~1.5- to 2-fold higher in the formula-fed groups, but was also elevated in babies who consumed pumped human milk from a bottle ().
Figure 4. CPIs of predicted functions that are significantly different among the four milk consumption types. Gut microbial profiles derived from 16S rRNA sequencing of stool samples from 6 month old infants were used to predict functional phenotypes of the gut microbes. There were significant differences in carbohydrate utilization (a), short-chain fatty acid (SCFA) production (b), amino acid biosynthesis and degradation (C and D) and vitamin biosynthesis (e).
Discussion
This study provides the first evidence that infant formula with added sugar compared to traditional formula and human milk may have a stronger association than birth delivery mode, infant caloric, and maternal BMI on the infant’s microbiome. Differences in the infant microbiome were observed between the various feeding groups examined in this study, with formula-fed infants exhibiting a distinct microbiome with characteristics of premature maturation that is even more pronounced in infants fed formula with added sugars (ASF). The most distinct changes occurred in specific microbe families associated with the consumption of a Western diet, characterized by high-fat and high-carbohydrate contentCitation4-Citation6,Citation8.
Microbes belonging to the family Lachnospiraceae (part of the phylum Firmicutes) were found to be elevated in infants fed formula, with the highest abundance in those who consumed ASF. We found that CPI for propionate production was increased in the formula-fed groups compared to infants who consumed expressed or pumped human milk. The family Lachnospiraceae is known to contain genes that contribute to the CPI of predicted propionate production,Citation20 and it has been shown that increased abundance of Lachnospiraceae is associated with diets that are high in carbohydrates.Citation21 Members of the family closely related to Lachnospiraceae are frequently found in the rumen of cows, goats and other ruminants,Citation22 and are linked to obesity related conditionsCitation23 with the genus Blautia (a member of the Lachnospiraceae family) having been shown to be associated with obesity in Hispanic children.Citation24,Citation25 It follows that infants who consume either traditional lactose-based formula or lactose-reduced formula (which contains partially hydolyzed milk and whey protein concentrate solids) would have an increased abundance of microbes belonging to the Lachnospiraceae family.
A depletion of microbes belonging to Bifidobacteriaceae (a member of the phylum Actinobacteria) was observed in infants who consumed any formula, with greater decreases in ASF group. The gut microbiome of infants who consume human milk need to contain members of Bifidobacteriaceae, since specifically the species B. longum subsp. infantis is able to fully metabolize human milk oligosaccharides, one of the most abundant components of human milk.Citation26 Also, we observed that CPI for lactose utilization, which is the most abundant milk sugar, was significantly higher in breastfed groups (BB and BP) compared to formula-fed groups (TF and ASF). Lactose utilization is one of the most common catabolic phenotype among Bifidobacterium spp., which could explain the increased predicted lactose utilization in the two groups consuming human milk and no formula.
In addition to the aforementioned microbes that were significantly different between groups of infants, we found that abundance of Acidaminococcaceae was higher in infants consuming lactose-reduced formula with added sugar compared to those who consume traditional formula. Currently, research examining the impact of diet on Acidaminococcaceae is sparse, with only a few studies in humans and in animal models. A member of the family Acidaminococcaceae, the genus Phascolarctobacterium, has been shown to be directly associated with the consumption of a Western diet, characterized by high fat and high carbohydrate content.Citation4-Citation8, Phascolarctobacterium has also been shown to be significantly higher in humans over the age of one compared to those less than one year oldCitation27
The importance of this study is bolstered by the inclusion of gut microbiota data at 1 month of age for use as baseline data, a timepoint in which a subset of the infants who are in the ASF and TF groups are exclusively breastfed. Controlling for microbiota at 1 month allowed for baseline shifts in the microbiome of the infants to be accounted for when examining differences by milk consumption type in the 6-month microbiome. Furthermore, the 1-month timepoint data was useful in demonstrating that these differences emerge with the introduction of infant formula. Additionally, we are able to distinguish between infants who consume human milk either directly from breast or from the bottle. This further division of the human milk consuming group allowed us to more confidently associate differential microbes with human milk as distinct from the act of breastfeeding. Our findings correspond with previous work that has shown infant outcomes differ in breastfed infants versus infants who consume human milk in a bottle.Citation28,Citation29
A limitation of the study is that the exact amount and type of milk consumed was not directly measured; however, formula type was assessed using maternal 24-hour dietary recalls. While it is likely that the type of formula consumed according to the dietary recall is indeed the primary formula type for the infant, we cannot be certain that the formula type consumed at the time of recall was consumed for a long time period. Also, while we were able to characterize the metabolic functions and phenotypes of the gut microbiome in this group of infants, this characterization is a prediction based on mapping of 16S data to the curated collection of metabolic phenotypes. It is important to note that this applied approach has a number of important limitations originating from both: (i) limited accuracy of phenotype projection from curated reference collection over 16S phylogenetic profiles; and (ii) the unknown extent of a predicted metabolic potential realization under specific conditions in the niche, a subject of complex regulation. Such functional predictions can be further refined by a combination with other -omics measurements and provide a starting point for focused experimental validation.Citation30 This study provides new evidence that infants consuming lactose-reduced formula high in glucose have a more mature microbiome than infants consuming traditional formula and human milk. Future studies are necessary to determine the long-term effects on the developing infant gut microbiome and on the growth and development of infants who consume the lactose-reduced formula for an extended period of time.
Methods
Participants
This research examined data from 91 Hispanic infant-mother pairs enrolled in our ongoing prospective study (known as Mother’s Milk funded through DK110793) with the aim of determining the impact of breastfeeding and dietary sugar intake during the first two years of life on adiposity, possibly mediated by alterations in the developing gut microbiome. The inclusion and exclusion criteria for the Mother’s Milk study were previously reported in detail in Berger et al.Citation31 Fecal samples to characterize the infants’ gut microbiome were collected at 1 month (to control for individual differences in microbiome) and 6 months of age, at which times infants’ length and weight were also measured. Mothers of the children completed several surveys to assess medical history and feeding behavior at 1 month and 6 months postpartum. Adjustment covariates extracted from these results included mother age at birth, pre-pregnancy BMI, mode of delivery, and mother current BMI as well as infant sex, age (days), and weight. To assess milk consumption type at 6 months of age, 24-hour diet recalls were performed in duplicate (first in person, second by telephone) for the infants by interviewing the mothers using the “multiple-pass” method and analyzed using the Nutrition Data System for Research software version 2014, developed by the Nutrition Coordinating Center, University of Minnesota. These dietary recalls allowed us to determine the milk consumption type and the frequency of milk consumption. Infants were divided into four groups based on feeding mode and type: (1) human milk directly from breast and no formula; (2) pumped human milk delivered from a bottle and no formula; (3) traditional lactose-based formula; and (4) lactose-reduced formula with added sugar as corn syrup solids.
Ethics
Written parental consent for inclusion in these studies were obtained prior to any testing procedure for participants under 18 years of age. The University of Southern California and Children’s Hospital Los Angeles Institutional Review Board approved that these studies were conducted in accordance with the Declaration of Helsinki.
Stool collection and DNA extraction/sequencing
Stool samples were collected using OMNIgene GUT collection kits (DNA Genotek, Ottawa, ON, CAN). Samples were stored at –80°C. 16S rRNA amplicon sequencing was used to characterize the microbiota. We prepared sample slurries according to the methods used by Flores et al.Citation32 and subsequently extracted DNA using the PowerSoil‐htp 96 well soil DNA isolation kit (Mo Bio Laboratories, cat. no. 12955‐4) as recommended by the Earth Microbiome Project.Citation33 Sequencing methods can be found in the Supplemental Material.
16S data processing
Sequence reads were demultiplexed with deML (https://grenaud.github.io/deML/). We used default parameters in DADA2Citation34(p2) (https://benjjneb.github.io/dada2/), available from QIIME2 software package for sequence read filtering, denoising, paired-read merging, removal of chimeras and obtaining of amplicon sequence variants (ASVs) with abundances. We assigned taxonomy to the obtained ASV sequences by blast with Ribosomal Database Project (RDP, version 11.5)Citation35 and NCBI 16S rRNA gene combined databases using multitaxonomy approach (MTA). Further details of MTA approach can be found in the Supplemental Material. To calculate beta-diversity of the obtained ASVs, we used DEICODE, an Aitchison distance matrix that is robust to sparsity.Citation12 ASVs were also collapsed into a total of 366 taxonomic groups (kingdom: n = 3; phylum: n = 13; class: n = 27; order: n = 40; family: n = 89; genus: n = 194; and species: n = 349). The raw 16S rRNA gene sequence reads used for this study are available on the NCBI Short Read Archive associated with the NCBI BioProject PRJNA589488.
Prediction of metabolic functions and phenotypes
Functional gene assignments and metabolic reconstructions were performed using the SEED database and Web tools that allow subsystem-based analysis of ~6,000 bacterial genomes, including a subset of 2,660 reference human gut microbial genomes representing 690 species.Citation36 The collection of curated metabolic subsystems includes (i) biosynthesis of essential nutrients (vitamins, amino acids), (ii) uptake and fermentation of carbohydrates including mono-, oligo-saccharides, sugar acids and alcohols, (iii) degradation of amino acids, and (iv) production of short chain fatty acids (SCFAs). Details on how 16S rRNA sequence reads were used to determine metabolic phenotypes from the metabolic reconstruction can be found in the Supplemental Methods. For each phenotype, we obtained a Community Phenotype Index (CPI) which represent a fractional representation (on a scale 0–100%) of microbial cells with a metabolic phenotype.
Statistical analysis
To examine differences in diversity of gut microbiota, we conducted a perMANOVA (permutation multivariate analysis of variance) of beta diversities using 10,000 permutations. Pairwise linear discriminant analysis was used to determine taxa that are differentially abundant based on milk consumption types using the LEfSe algorithm on the Galaxy web applicationCitation33,Citation34,37 with the default parameters. Further, taxa that were considered to be differentially abundant between the ASF and BB groups were examined using linear regression models to determine if differences in abundances were statistically significant. Equation (1) was used to determine differences in microbial features (log-normalizedCitation38 relative abundance of taxonomic groups and CPI of predicted metabolic phenotypes) by infant’s milk consumption type.
(1) microbialFeaturei, 6 month = milk consumption type + mother age + maternal pre-pregnancy BMI+ delivery mode (vaginal/c-section) + maternal BMI + infant sex + infant age in days + infant weight + microbialFeaturei,1-month +e
The p-value from an ANOVA comparing these models to a reduced model that excludes “milk consumption type” as a dependent variable was used to determine significance of milk consumption type in the model. For microbiota found to be significantly associated with milk consumption type, the pairwise differences in the four milk consumption type groups were examined using the Tukey’s Honestly Significant Difference (HSD) test as a post-hoc analysis. Confidence intervals (95%) for false discovery rates were calculated using the fdrci R packageCitation39 with 1,000 permutations. Tests in which both p value was less than 0.05 and upper level of confidence interval for false discovery rate was less than 0.25 were considered to be statistically significant. All statistical analyses were performed in the R statistical computing languageCitation40 (version 3.6.0) and figures were generated using the ggplot2 R package.Citation41 The code generated to conduct these statistical analyses can be found on GitHub using the link https://github.com/rbarner/infantMilkConsumption.
Authors contributions
RBJ conceptualized the research question, refined the research question, coordinated acquisition of data, analyzed and interpreted data, wrote the initial manuscript and has primary responsibility for the final content. PKB, JFP, and TLA refined the research question, coordinated acquisition of data, interpreted data, and revised the manuscript critically for important intellectual content. JM provided statistical input, interpreted data and revised the manuscript critically for important intellectual content. JF refined the research question and revised the manuscript critically for important intellectual content. SNI developed predicted metagenomic computational pipeline, calculated phenotypic metrics and revised the manuscript critically for important intellectual content. DAR and ALO produced predicted metabolic functions in the reference genomes and revised the manuscript critically for important intellectual content. LB and MIG conceptualized and designed the study, coordinated and supervised acquisition of data, and revised the manuscript critically for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Disclosure of potential conflicts of interest
Michael I. Goran is a scientific advisor for Yumi. The authors have no other conflicts of interest relevant to this article to disclose.
Supplemental Material
Download MS Word (146.1 KB)Acknowledgments
We would like to acknowledge the efforts of our clinical team, Carla Flores, Danielle Garcia, Emily Leibovitch, Sinthia Arcadia Rodriguez, Maggie Gutierrez, Rosa Rangel, Claudia Rios, and Elizabeth Campbell who conducted recruitment and study visits. Finally, we would also like to acknowledge the mothers and children, whose participation and commitment to this study made this research possible.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. PNAS. 2010;107(26):11971–14. doi:doi:10.1073/pnas.1002601107.
- Ardissone AN, de la Cruz DM, Davis-Richardson AG, Rechcigl KT, Li N, Drew JC, Murgas-Torrazza R, Sharma R, Hudak ML, Triplett EW. Meconium microbiome analysis identifies bacteria correlated with premature birth. Plos One. 2014;9(3):e90784. doi:doi:10.1371/journal.pone.0090784.
- Groer MW, Luciano AA, Dishaw LJ, Ashmeade TL, Miller E, Gilbert JA. Development of the preterm infant gut microbiome: a research priority. Microbiome. 2014;2(1):38. doi:doi:10.1186/2049-2618-2-38.
- Goodrich JK, Waters JL, Poole AC, Sutter J, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell J, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi:doi:10.1016/j.cell.2014.09.053.
- Chu DM, Antony KM, Ma J, Prince AL, Showalter L, Moller M, Aagaard KM. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016;8(1):77. doi:doi:10.1186/s13073-016-0330-z.
- Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, Blundell P, Alan Harris R, Frias AE, Grove KL. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat Commun. 2014;5(1):3889. doi:doi:10.1038/ncomms4889.
- Yasmin F, Tun HM, Konya TB, Guttman DS, Chari RS, Field CJ, Becker AB, Mandhane PJ, Turvey SE, Subbarao P, et al. Cesarean section, formula feeding, and infant antibiotic exposure: separate and combined impacts on gut microbial changes in later infancy. Front Pediatr. 2017;5:200. doi:doi:10.3389/FPED.2017.00200.
- Guaraldi F, Salvatori G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Front Cell Infect Microbiol. 2012:2. doi:doi:10.3389/fcimb.2012.00094.
- Rossen LM, Simon AE, Herrick KA. Types of infant formulas consumed in the United States. Clin Pediatr (Phila). 2016;55(3):278–285. doi:doi:10.1177/0009922815591881.
- Stang J, Hoss K, Story M. Health statements made in infant formula advertisements in pregnancy and early parenting magazines: a content analysis. ICAN. 2010;2(1):16–25. doi:doi:10.1177/1941406409359806.
- Noble EE, Hsu TM, Jones RB, Fodor AA, Goran MI, Kanoski SE. Early-life sugar consumption affects the rat microbiome independently of obesity. J Nutr. 2017;147(1):20-28. doi:10.3945/jn.116.23881.
- Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, Zengler K. A novel sparse compositional technique reveals microbial perturbations. mSystems. 2019;4(1):e00016–19. doi:doi:10.1128/mSystems.00016-19.
- Rodionov DA, Arzamasov AA, Khoroshkin MS, Iablokov SN, Leyn SA, Peterson SN, Novichkov PS, Osterman AL. Micronutrient requirements and sharing capabilities of the human gut microbiome. Front Microbiol. 2019:10. doi:doi:10.3389/fmicb.2019.01316.
- Sharma V, Rodionov DA, Leyn SA, Tran D, Iablokov SN, Ding H, Peterson DA, Osterman AL, Peterson SN. B-vitamin sharing promotes stability of gut microbial communities. Front Microbiol. 2019:10. doi:doi:10.3389/fmicb.2019.01485.
- Leyn SA, Maezato Y, Romine MF, Rodionov DA. Genomic reconstruction of carbohydrate utilization capacities in microbial-mat derived consortia. Front Microbiol. 2017:8. doi:doi:10.3389/fmicb.2017.01304.
- Arzamasov AA, van Sinderen D, Rodionov DA. Comparative genomics reveals the regulatory complexity of bifidobacterial arabinose and arabino-oligosaccharide utilization. Front Microbiol. 2018:9. doi:doi:10.3389/fmicb.2018.00776.
- Bouvier JT, Sernova NV, Ghasempur S, Rodionova IA, Vetting MW, Al-Obaidi NF, Almo SC, Gerlt JA, Rodionov DA. Novel metabolic pathways and regulons for hexuronate utilization in proteobacteria. J Bacteriol. 2019;201:2. doi:doi:10.1128/JB.00431-18.
- Ye Y, Doak TG, Parsimony A. Approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput Biol. 2009;5(8):e1000465. doi:doi:10.1371/journal.pcbi.1000465.
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi:doi:10.1038/nbt.2676.
- Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. doi:doi:10.1111/1462-2920.13589.
- Tidjani Alou M, Lagier J-C, Raoult D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Hum Microbiome J. 2016;1:3–11. doi:doi:10.1016/j.humic.2016.09.001.
- Kittelmann S, Seedorf H, Walters WA, Clemente JC, Knight R, Gordon JI, Janssen PH. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. Plos One. 2013;8(2):e47879. doi:doi:10.1371/journal.pone.0047879.
- Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–626. doi:doi:10.1038/nature11400.
- Nirmalkar K, Murugesan S, Pizano-Zárate ML, Villalobos-Flores L, García-González C, Morales-Hernández R, Nuñez-Hernández J, Hernández-Quiroz F, Romero-Figueroa M, Hernández-Guerrero C, et al. Gut microbiota and endothelial dysfunction markers in obese mexican children and adolescents. Nutrients. 2018;10(12):2009. doi:doi:10.3390/nu10122009.
- Murugesan S, Ulloa-Martínez M, Martínez-Rojano H, Galván-Rodríguez FM, Miranda-Brito C, Romano MC, Piña-Escobedo A, Pizano-Zárate ML, Hoyo-Vadillo C, García-Mena J, et al. Study of the diversity and short-chain fatty acids production by the bacterial community in overweight and obese Mexican children. Eur J Clin Microbiol Infect Dis. 2015;34(7):1337–1346. doi:doi:10.1007/s10096-015-2355-4.
- Bode L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology. 2012;22(9):1147–1162. doi:doi:10.1093/glycob/cws074.
- Wu F, Guo X, Zhang J, Zhang M, Ou Z, Peng Y. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp Ther Med. 2017;14(4):3122–3126. doi:doi:10.3892/etm.2017.4878.
- Azad MB, Vehling L, Chan D, Klopp A, Nickel NC, McGavock JM, Becker AB, Mandhane PJ, Turvey SE, Moraes TJ, et al. Infant feeding and weight gain: separating breast milk from breastfeeding and formula from food. Pediatrics. 2018;142(4):4. doi:doi:10.1542/peds.2018-1092.
- Klopp A, Vehling L, Becker AB, Subbarao P, Mandhane PJ, Turvey SE, Lefebvre DL, Sears MR, Azad MB, Daley D, et al. Modes of infant feeding and the risk of childhood asthma: a prospective birth cohort study. J Pediatr. 2017;190(192–199):e2. doi:doi:10.1016/j.jpeds.2017.07.012.
- Gehrig JL, Venkatesh S, Chang H-W, Hibberd MC, Kung VL, Cheng J, Chen RY, Subramanian S, Cowardin CA, Meier MF, et al. Effects of microbiota-directed foods in gnotobiotic animals and undernourished children. Science. 2019;365(6449):6449. doi:doi:10.1126/science.aau4732.
- Berger PK, Plows JF, Jones RB, Pollock NK, Alderete TL, Ryoo JH, Goran MI. Maternal blood pressure mediates the association between maternal obesity and infant weight gain in early postpartum. Pediatr Obes. 2019;14(11):e12560. doi:10.1111/ijpo.12560.
- Flores GE, Henley JB, Fierer N, Direct A. PCR approach to accelerate analyses of human-associated microbial communities. Plos One. 2012;7(9):e44563. doi:doi:10.1371/journal.pone.0044563.
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457–463. doi:doi:10.1038/nature24621.
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi:doi:10.1038/nmeth.3869.
- Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR. The RDP (ribosomal database project). Nucleic Acids Res. 1997;25(1):109–111. doi:10.1093/nar/25.1.109.
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014;42(D1):D206–D214. doi:doi:10.1093/nar/gkt1226.
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:doi:10.1186/gb-2011-12-6-r60.
- McCafferty J, Mühlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, Fodor AA. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. Isme J. 2013;7(11):2116–2125. doi:doi:10.1038/ismej.2013.106.
- Millstein J, Volfson D. Computationally efficient permutation-based confidence interval estimation for tail-area FDR. Front Genet. 2013;4:179. doi:doi:10.3389/fgene.2013.00179.
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing Vienna Austria. 2016; 0:{ISBN} 3-900051-07-0. doi:10.1038/sj.hdy.6800737.
- Wickham H. Ggplot2: elegant graphics for data analysis. New York (NY): Springer; 2016.