
ABSTRACT
Comensal Bacteroidota (Bacteroidota) and Enterobacteriacea are often linked to gut inflammation. However, the causes for variability of pro-inflammatory surface antigens that affect gut commensal/opportunistic dualism in Bacteroidota remain unclear. By using the classical lipopolysaccharide/O-antigen ‘rfb operon’ in Enterobacteriaceae as a surface antigen model (5-rfb-gene-cluster rfbABCDX), and a recent rfbA-typing strategy for strain classification, we characterized the integrity and conservancy of the entire rfb operon in Bacteroidota. Through exploratory analysis of complete genomes and metagenomes, we discovered that most Bacteroidota have the rfb operon fragmented into nonrandom patterns of gene-singlets and doublets/triplets, termed ‘rfb-gene-clusters’, or rfb-‘minioperons’ if predicted as transcriptional. To reflect global operon integrity, contiguity, duplication, and fragmentation principles, we propose a six-category (infra/supra-numerary) cataloging system and a Global Operon Profiling System for bacteria. Mechanistically, genomic sequence analyses revealed that operon fragmentation is driven by intra-operon insertions of predominantly Bacteroides-DNA (thetaiotaomicron/fragilis) and likely natural selection in gut-wall specific micro-niches or micropathologies. Bacteroides-insertions, also detected in other antigenic operons (fimbriae), but not in operons deemed essential (ribosomal), could explain why Bacteroidota have fewer KEGG-pathways despite large genomes. DNA insertions, overrepresenting DNA-exchange-avid (Bacteroides) species, impact our interpretation of functional metagenomics data by inflating by inflating gene-based pathway inference and by overestimating ‘extra-species’ abundance. Of disease relevance, Bacteroidota species isolated from cavitating/cavernous fistulous tract (CavFT) microlesions in Crohn’s Disease have supra-numerary fragmented operons, stimulate TNF-alpha from macrophages with low potency, and do not induce hyperacute peritonitis in mice compared to CavFT Enterobacteriaceae. The impact of ‘foreign-DNA’ insertions on pro-inflammatory operons, metagenomics, and commensalism/opportunism requires further studies to elucidate their potential for novel diagnostics and therapeutics, and to elucidate the role of co-existing pathobionts in Crohn’s disease microlesions.
Introduction
The causes for variability of surface pro-inflammatory antigens that affect gut commensal/opportunistic dualism within the phylum Bacteroidota in the gut and its micropathologies remain unclear.Citation1,Citation2 An operon is a unit of DNA that consists of a cluster of contiguous genes. Herein,Citation3–5 we proposed that antigenic variability in the phylum could be due to disrupted integrity patterns in antigenic operons. Among several antigenic operons, the production of the O-antigen (polysaccharide) component of lipopolysaccharides (LPS), widely present in gram-negative bacteria, is influential on inflammation but variable across bacteria. The phylum Bacteroidota (Bacteroidetes), composed primarily of gram-negative gut commensals,Citation6–10 is known to have several opportunistic pathogenic species (‘pathobionts’),Citation11–19 which for unclear reasons have commensal/pathogenic dualism. Concerningly, species from the phylum have been proposed as future probiotics because some strains modulate gut local immunity locallyCitation6,Citation20,Citation21 or influence the susceptibility to chronic extra-intestinal diseases (e.g., Parabacteroides goldsteinii attenuates obstructive pulmonary disease).Citation15
The precise role of Bacteroidota in inflammatory bowel diseases (IBD), namely Crohn’s disease (CD) and ulcerative colitis (UC), remains undefined.Citation22 Supporting a chronic pathogenic role in IBD complications, we recently discovered that the inflamed bowel of CD patients who required surgery to remove severe inflamed bowel segments have a unique type of gut wall cavitating micropathologies that we earlier termed as ‘cavernous fistulous micropathologies’ (CavFT;/cavftas/) resembling cavern formations. Citation23 Of notoriety, CavFTs harbor cultivable bacteria, namely Bacteroidota and Enterobacteriaceae (E. coli).Citation5,Citation24,Citation25 Focused on CavFT, genomic analyses of consecutive Parabacteroides distasonis (Bacteroidota) isolates from two unrelated patients that underwent surgery for CD indicated for the first time that a novel lineage might have become adapted to CavFTs, swapping large fragments of DNA with Bacteroides, been likely transmissible in the community.Citation25 To classify P. distasonis and other Bacteroidota, and to facilitate the orderly study of such commensal/pathogenic dualism in the phylum, we recently proposed the use of the LPS/O-Antigen gene rfbA for the genotyping and classification of Bacteroidota.Citation3
Compared to the highly conserved lipid A gene (lpxK; the lipid component in LPS), rfbA-typing studies in our laboratoryCitation3 suggested that the genes forming the rfb operon (synonym rmlCitation26) responsible for the O-antigen synthesis (saccharide component in LPS in E. coli could also be used to aid in the identification of features to differentiate probiotic from pathogenic strains in Bacteroidota.Citation3 In E. coli K12, the initial step of the O-antigen pathway involves a reversible transfer reaction facilitated by the enzyme rfbA (glucose-1-phosphate thymidyltransferase),Citation26 with subsequent stages mediated by the enzymes rfbB (dTDP-D-glucose-4,6-dehydratase), rfbC (dTDP-4-keto-6-deoxy-D-glucose-3,5-epimerase), and rfbD (dTDP-4-keto-L-rhamnose reductase), respectively.Citation26 Of genotyping relevance, rfbA-typing of P. distasonis as a model for Bacteroidota revealed that historical strains isolated from pathological sources belonged to one of four rfbA types (rfbA-Type I).Citation3
Previous studies have paid great attention to glycosyltransferases and heptosyltransferases and their role in LPS formation in Bacteroidota, but less attention has been placed on rfb genes.Citation27–29 In Campylobacter jejuni, the genetic lipooligosaccharides loci consist of 19 genes, encompassing 11 glycosyltransferases and one gene encoding lipid A acyltransferase. Notably, the genes rfbA and rfbB hold significant roles in the synthesis of lipooligosaccharides that incorporate dideoxyhexosamine.Citation30 To date, however, most mechanistic studies have focused on model organisms within Bacteroidota, for instance, B. thetaiotaomicron.Citation27–29 However, there are no studies comprehensively examining the genetic architecture of the rfb operon across various genera to determine if general extrapolations can be made across the phylum, or even across strains within a genus. Because polysaccharides can also be found in bacterial capsules, it is important to note that the interaction between capsular polysaccharide gene clusters do not appear to be involved in lipooligosaccharides biosynthesis, based on studies conducted with a strain of B. thetaiotaomicron.Citation29
Although lipid A in Bacteroidota is known to induce lower TLR4 inflammatory activation compared to E. coli,Citation6,Citation15,Citation21,Citation31 the genetic integrity and contiguity across rfb-genes in Bacteroidota operons remains poorly understood.Citation20 Herein, we conducted an expanded typing analysis across genes in all rfb operons seen in representative Bacteroidota using i) validated principles of rfb typing methods we implemented for rfbA,Citation3 ii) complete genomes and NCBI genome and metagenome (WGS) databases, iii) new genomes sequenced from CavFT in our laboratory for macrophage and peritonitis experiments, iv) the classical Enterobacteriaceae rfb operon contiguity as referent, and v) Parabacteroides and Alistipes as emerging pathogenic/probiotic modelsCitation1,Citation2 to identify potential genomic structural signatures that could be used to classify bacteria and guide mechanistic studies that could be impacting surface antigens favoring Bacteroidota switches from commensalism to pathobiont dualism in the gut and local micro-niches, such as CavFT in CD patients.
Results
The classic rfb operon is contiguous in Enterobacteriaceae
As a referent to illustrate the arrangement of rfb genes involved in the O-antigen synthesis operon in Enterobacteriaceae, we analyzed reference genomes from E. coli, Klebsiella variicola, and Salmonella enterica. A schematic of the LPS/O-antigen molecule is shown in . In E. coli K12, illustrates the contiguous arrangement of five genes which functionally enable the en bloc transcription of the rfb operon (rfbABCDX). Regardless of transcriptional orientation (positive/negative sense), other Enterobacteriaceae have the same contiguous arrangement of the rfb genes. Notably, while the length of rfb operons vary across this family, bacteria predominantly have at least four genes (rfbABCD), organized contiguously, with sporadic singly duplicated genes. Contiguity is so conserved within the family that the genus Salmonella, for instance, has species with operons composed of up to 15 rfb genes that run contiguously in the same DNA strand ( and Supplementary Figure S1a-b).
Figure 1. Operon integrity, fragmentation and diversification profiling and classification in Bacteroidota based on the number/presence of classical rfb genes ABCD and non-rfb genes. a) lipopolysaccharide (LPS) and O-antigen schematics. b) classical contiguity of rfb genes ABCD in the reference genome of E. coli K12 (rfbXABCD; XCADB; [rfb:5]). Horizontal lines represent the bacterial genome length, distribution of rfb genes/operons (shaded circles; darker = more genes) and rfb gene orientation (+, sense; –, antisense). nL: n of rfb clusters or gene singlet loci. Note orientations/duplications. c) classification of Bacteroidota based on rfb operon integrity. Six categories of operon arrangement & ‘rfb-gene-cluster’ fragmentation are depicted to summarize the observations of this study. For the purpose of operon classification, based on continuity vs. fragmentation, and diversification, the analysis summary is focused on the presence/absence of ‘expected contiguous rfb genes ABCD’ irrespective of other genetic elements required in any operon (e.g. transcribers, promoters, enhancers). Prediction analysis (see methods) indicates that such fragmented yet contiguous rfb-gene-clusters (minioperons) in Bacteroidota, including additional (non rfb) genes, have resulted in a diversified set of operons’ that are computationally-predictable functional transcriptional units involving the additional non-rfb gene. The location of non-rfb gene in the cluster of ‘diversified operons’ could be represented with an exclamation mark (!) for the locations of non-rfb gene in the cluster; wherein Alistipes finegoldii DSM17242 has non-rfb(!) genes within its operons rfb(BDCA!) indicates (B!1D!2C!3A)(----), and where (!Citation1=glycosyltransferase, !Citation2=acyltransferase and !Citation3= acetyltransferase). Supplementary Figure S1c illustrates in context rfb operon fragmentation and diversification for Alistipes, Bacteroides, Parabacteroides, Prevotella, Paraprevotella, Barnesiella, Tannerella, Odoribacter and Porphyromonas.
![Figure 1. Operon integrity, fragmentation and diversification profiling and classification in Bacteroidota based on the number/presence of classical rfb genes ABCD and non-rfb genes. a) lipopolysaccharide (LPS) and O-antigen schematics. b) classical contiguity of rfb genes ABCD in the reference genome of E. coli K12 (rfbXABCD; XCADB; [rfb:5]). Horizontal lines represent the bacterial genome length, distribution of rfb genes/operons (shaded circles; darker = more genes) and rfb gene orientation (+, sense; –, antisense). nL: n of rfb clusters or gene singlet loci. Note orientations/duplications. c) classification of Bacteroidota based on rfb operon integrity. Six categories of operon arrangement & ‘rfb-gene-cluster’ fragmentation are depicted to summarize the observations of this study. For the purpose of operon classification, based on continuity vs. fragmentation, and diversification, the analysis summary is focused on the presence/absence of ‘expected contiguous rfb genes ABCD’ irrespective of other genetic elements required in any operon (e.g. transcribers, promoters, enhancers). Prediction analysis (see methods) indicates that such fragmented yet contiguous rfb-gene-clusters (minioperons) in Bacteroidota, including additional (non rfb) genes, have resulted in a diversified set of operons’ that are computationally-predictable functional transcriptional units involving the additional non-rfb gene. The location of non-rfb gene in the cluster of ‘diversified operons’ could be represented with an exclamation mark (!) for the locations of non-rfb gene in the cluster; wherein Alistipes finegoldii DSM17242 has non-rfb(!) genes within its operons rfb(BDCA!) indicates (B!1D!2C!3A)(----), and where (!Citation1=glycosyltransferase, !Citation2=acyltransferase and !Citation3= acetyltransferase). Supplementary Figure S1c illustrates in context rfb operon fragmentation and diversification for Alistipes, Bacteroides, Parabacteroides, Prevotella, Paraprevotella, Barnesiella, Tannerella, Odoribacter and Porphyromonas.](/cms/asset/c00d1d87-da77-4ed1-b737-446c289fd80a/kgmi_a_2350150_f0001_oc.jpg)
The rfb operon in Bacteroidota is often fragmented into ‘rfb-gene-clusters’
The primary focus of this study was on the rfb genes present in the genome of the Bacteroidota phylum. To assess the spatial integrity of the rfb operon in Bacteroidota, we first examined the rfb gene profile (presence/absence) in available complete genomes. Analysis revealed that the rfb genes in Parabacteroides, Bacteroides, and Prevotella were not contiguous but fragmented and dispersed throughout the genome, as individual rfb genes (singlets), or as combinations of 2 or 3 rfb genes, herein referred to as duplet or triplet ‘rfb-gene-clusters’ (or minioperons). For descriptive purposes, rfb-singlets/doublets/triplets are generically and arbitrarily deemed as ‘rfb-gene-clusters’.
In contrast, Alistipes, Porphyromonas and Odoribacter have rfb operons composed of the same primordial genes as Enterobacteriaceae (rfbACDB), being intact and/or duplicated, supporting their genomic potential to be functionally capable of LPS-proinflammatory induction. Within the phylum, operon fragmentation products, 'rfb-gene-clusters', result in various combinations of rfb singlet genes and doublets/triplets with variable orientations (sense: +; antisense: –, Supplementary Figure S1c). With E. coli as referent, using four rfb genes (n = 4) as the median maximum number of contiguous genes observed among Bacteroidota in this study, herein we propose that rfb operons in Bacteroidota can be classified into at least five categories: i) Mono-operon (single contiguous operon, regardless of numbers of genes in cluster/locus), ii) Di-operon (duplicated operon), iii) fragmented normo-numerary operon (operon fragmented with 4 rfb clusters/loci, nL = 4), iv) fragmented infra-numerary operon (nL < 4 clusters/loci), and v) fragmented supra-numerary operon (nL > 4 clusters/loci; ), this includes bacteria such as Parabacteroides goldsteinii BFG-241, which possesses both a conserved rfb gene cluster (namely rfbBDCA(----)) and a supra-numerary operon integrity profile for fragmented rfb gene clusters comprising rfbCB(++), rfbA(+), rfbCD(++), rfbA(–), and rfbD(+), with nL = 6. Used alongside a baseline referent, this system of categorization and cataloging may be applied to any operon system.
Envisioning the potential combinatorial variability for the O-antigen via the theoretical pairwise combination of rfb genes in Bacteroidota, we next determined that mathematical permutations of rfb-gene-clusters could yield thousands of possible combinations unlikely to yield functional O-antigen polysaccharides (Supplemental Figure S2a), which proves technically challenging to verify experimentally for all bacteria by requiring innumerable growth conditions.
Organized, illustrates that the rfb operon in Enterobacteriaceae is contiguous, but for descriptive purposes in Bacteroidota, the operon patterns observed for the rfb operon in Bacteroidota are largely diversified across genera within the phylum. Overall, rfb operons can be either intact, duplicated, or fragmented into ‘rfb-gene-clusters’ or ‘minioperons’ creating diversification of combinatorial possibilities for the operon. In addition, the presence of other (non-rfb) genes interspersed in contiguity with the well-known rfbABCD genes, or with other rfb genes commonly seen in the phylum Proteobacteria (Pseudomonadota; e.g., rfbFGH) represents an additional arrangement of rfb operons in Bacteroidota which we referred to as ‘diversified rfb operons’ in which non-rfb genes are predictably transcribed together with rfb genes as functional transcriptional units.
rfb-gene-cluster patterns in Parabacteroides suggests rfb gene distancing mechanism
Since the study of potential operon fragmentation mechanisms could be better achieved using strains with fully sequenced genomes, we next conducted an arrangement analysis to determine if rfb operon fragmentation was common within any given species and if they followed statistically significant patterns among unrelated strains of the same species. Using P. distasonis as a model Bacteroidota species and strain ATCC8503 (peritonitis, USA/1935) as the historic referent species for the genus Parabacteroides, analyses revealed that P. distasonis have their rfb operons invariably fragmented (12/12, Fisher’s exact p < .00001), following unique patterns of gene combinations, duplications, or orientations that are significantly more likely to be supra-numerary than infra-numerary (Fisher’s exact p = .0001, ). These findings suggest that some species are more likely to gain rfb loci (P. distasonis), compared to others which could be more likely to lose rfb loci (Barnesiella viscericola).
Figure 2. The occurrence, patterns and inversions of rfb-gene-clusters in P. distasonis and B. thetaiotaomicron suggests mechanism of operon fragmentation in Bacteroidota. a) fragmentation of rfb operon in P. distasonis is in modern times supra-numerary compared to ATCC8503 strain from USA/1933. b) fragmentation has resulted in conserved singlet, doublet/triplet patterns in P. distasonis. c) heatmap clustering of P. distasonis strains based on rfb-gene-cluster shows distinct clades. Additional information is available in supplementary figure S2b. d) unique rfbFGC->rfbA rfb-gene-cluster distancing pattern (downward red/black arrows and solid circles) in B. thetaiotaomicron is also present in novel CavFT strains of P. distasonis from gut wall lesions in Crohn’s disease. Notice the orientation sense and patterns. e) SDS-PAGE of LPS extract analysis of E. coli and P. distasonis CavFT-46 shows that P. distasonis is unable to produce O-antigen polysaccharides in diverse growth conditions (Fisher’s exact p=0.079), the observed higher molecular weight band (approximately 130Kb) in P. distasonis suggests saccharide is of capsular origin, rather than lipopolysaccharide (see manuscript).
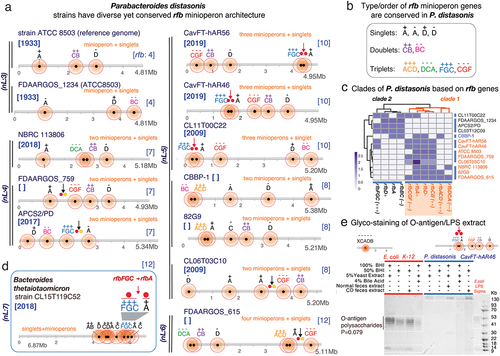
Fragmentation patterns were also highly conserved among P. distasonis. Specifically, rfbA and rfbD genes were present in rfb-gene-clusters or as singlets, whereas the genes rfbB, rfbC, rfbG, and rfbF were exclusively present only as doublet- or triplet- rfb-gene-cluster arrangements (). Based on such conservancy, P. distasonis strains belong to at least two distinctive phylogenetic clades (, Supplementary Figure S2b), suggesting that not all P. distasonis would be the same, and emphasizing the need to better classify Bacteroidota isolates for functional studies.
Mechanistically insightful, outside of this species we found that a unique combination and distancing of two rfb-gene-clusters (rfbFGC->rfbA; abbreviation for rfbFGC(+++) positive-sense triplet followed by a rfbA(+) positive-sense singlet), overrepresented in some P. distasonis strains (n = 6/12, Fisher’s exact p < .001, vs. numerous other possibilities), was also present in a B. thetaiotaomicron strain CL15T119C52 (), a phenomenon not previously reported in the literature.
Considering that B. thetaiotaomicron lacks LPS-polysaccharide formationCitation32 and has anti-inflammatory and immunomodulatory properties,Citation18,Citation19 the presence of such rfbFGC->rfbA fragmentation pattern in both P. distasonis/B thetaiotaomicron suggests a mechanism for operon fragmentation that could explain beneficial effects for both species/strains.Citation2 Remarkably, an inter-genus rfbFGC->rfbA pattern, exclusively present in P. distasonis strains of CavFT origin and CL11T00C22,Citation5 have conserved rfb–gene-cluster sequences but variable inter-rfb–gene-cluster rfbFGC->rfbA distances, suggesting that DNA insertion within the flanking rfb genes could be the reason for gene–gene separation in an ongoing permissible process of gene exchange between P. distasonis and B. thetaiotaomicron.
Of novelty, variable inter-rfb-gene-cluster orientations for the same rfbFGC->rfbA pattern in other P. distasonis genomes (APCS2/PD, FDAARGOS_615, and CL06T03C10) suggest the existence of conserved rfbFGC->rfbA inversions with a pivot point in the inter-rfb-gene-cluster rfbFGC->rfbA region. This observation indicates that such a potential gene-gene separation mechanism is highly conserved, yet variable. Furthermore, it offers an evolutionary explanation to the presence of unique arrangements across certain strains, lineages, or possible niches that could reflect favored co-habitation of both genera, genetic exchange, selection, and niche adaptation (notice the red vs. black arrows for rfbFGC->rfbA in ).
To elucidate the effect of rfb-gene-clusters on LPS/O-antigen production and structure, we opted to verify the presence/absence of the O-antigen polysaccharide in P. distasonis CavfT-hAR46, under five unique growth conditions, as a cultivable Bacteroidota model with 5 rfb-gene-cluster fragments (vs. E. coli with one rfb operon). Of functional importance, the classical rfb operon in E. coli effectively produced a typical O-antigen/polysaccharide as expected in all conditions, with repeated bands between 41 and 53 kb in SDS-PAGE ().Citation33,Citation34 However, as anticipated for a P. distasonis strain with supra-numerary rfb fragmentations, O-antigen polysaccharides were not detected in any of the conditions, indicating likely non-functionality (5/5 vs. 0/5, Fisher’s exact p = 0.0079). Based on recent studies using B. thetaiotaomicron and mutant variations lacking capsular polysaccharide genes,Citation32 the higher molecular weight band (around 130KD) in the case of P. distasonis may correspond to capsular saccharides rather than lipopolysaccharides (). Of interest, fragmentation of the rfb-operon was also observed in other genera within the Bacteroidota phylum, including Phocaeicola vulgatus and Phocaeicola dorei which are known to produce LPS (Supplementary Figure S1),Citation35,Citation36 further illustrating the need for additional studies to understand how the conservation or fragmentation of the rfb operon influences the structure of LPS across genera and strains.
The rfb operon in Alistipes is intact or duplicated suggesting benefit for survival
Contrasting Parabacteroides, the Alistipes genus primarily exhibits no operon fragmentation. Unique conserved organization and orientation was observed in all examined Alistipes species which have 4-gene contiguous rfb operons (BDCA or CADB; ). When fragmentation was present (A. shahii, A. dispar), Alistipes have rfb-gene-cluster doublets (rfbCA, DB, AC, GF) not seen in Parabacteroides, furthers supporting that genus-specific mechanisms of operon fragmentation or conservation vary across Bacteroidota. However, an exception was observed in Alistipes finegoldii DSM17242, Alistipes communis 6CPBBH3, and Alistipes onderdonkii subsp. vulgaris strain 3BBH6 where an additional (non-rfb) gene was present within continuous rfb genes in the operons. Like in the case of Alistipes finegoldii DSM17242 in rfbBDCA(----), non-rfb genes are present B!1D!2C!3D((----)-(!1=glycosyltransferase, !2=acyltransferase and !3= acetyltransferase, see for detail). Intriguingly, as a major difference within other Bacteroidota, most Alistipes species have duplicated operons (5/8 vs. 1/22, B. fragilis, Fisher’s exact p = 0.0018), indicating that the genus has genomic potential for enhanced O-antigen production and possibly pro-inflammatory LPS effects that could be necessary or beneficial for Alistipes adaptation/survival. Alternatively, operon duplication with minimal fragmentation suggests that i) fragmentation is non-sustainable or rather deleterious for Alistipes, and/or ii) its genetic mechanisms of operon maintenance allow for operon variability (duplication, inversions, sequence), but not for gene-operon separation, unlike Parabacteroides in which fragmentation predominates (12/12 vs. 2/8, A. shahii & A. dispar, Fisher’s exact p = 0.0049). No rfb singlets were observed in Alistipes.
Figure 3. The rfb operon in Alistipes is contiguous and duplicated suggesting evolutionary benefit. a) Alistipes has contiguous rfb operons with frequent duplication and less common incorporation of rfb-gene-cluster duplets. b) patterns of conserved rfb-gene-clusters in Alistipes differ from Parabacteroides. c) schematics of gene-gene rfb distances measured within and between rfb-gene-clusters. d) Parabacteroides and Bacteroides demonstrate the greatest variance in number of rfb operon fragments and gene-gene distances. Alistipes and enterobacteriaceae are similarly contiguous. Intergene distances for Parabacteroides, Bacteroides and Prevotella were greater than enterobacteriaceae (0.54 ± 0.84Mb, 0.49 ± 0.86Mb, 0.34 ± 0.32Mb, respectively, vs. 0.13 ± 0.46Mb, p < .001). Alistipes gene distribution is similar to enterobacteriaceae (0.17 ± 0.49Mb, p = .79). e) rfb-gene-cluster sequence homologies for Alistipes vs. Parabacteroides based on rfb-gene-cluster orientation between and within genera/species. Two tailed-T tests p < .01 **, p < .001 ***. f) alignment and g) phylogeny based on rfb operon sequences. Note that Alistipes clusters are driven by the sense/antisense orientation of the operons.
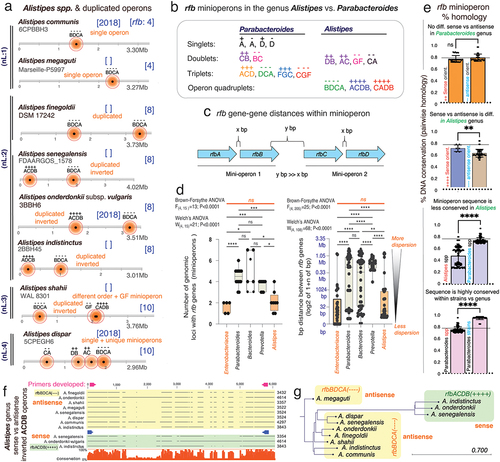
The rfb-gene-cluster types in Alistipes and Parabacteroides indicate that gene patterns and orientations are unique and vary, being potentially useful as signatures for lineage identification and classification (). When we quantified the magnitude of fragmentation (gene–gene distances in number of nucleotides) across genera, we found that Parabacteroides, Bacteroides and Prevotella had similarly more fragmentation on average than Enterobacteriaceae (p < .001, p = .0151, p = .010, respectively), but not Alistipes compared to Enterobacteriaceae (2.1 vs. 1.6, p = .89, ).
Most rfb singlets/rfb-gene-clusters are predicted to be transcribed as unique transcriptional units
To determine whether rfb singlets/rfb-gene-clusters could be transcribed as a simple unit or as a complex unit carrying other non-rfb genes, we used and compare three software tools that rely on predicted promoter and terminator sequences flanking the genes of interest to generate the list of genes that could be transcribed with the rfb singlets/rfb-gene-clusters. The software used included FGENESB Bacterial Operon GenePrediction(http://www.softberry.com/berry.phtml?topic=fgenesb&group=programs&subgroup=gfindb),Citation37 Operon-Mapper (https://biocomputo.ibt.unam.mx/operon_mapper/),Citation38 and BioCyc (https://biocyc.org/).Citation39 Since the efficiency of such tools predicting operon transcriptional units is slightly variable, we first tested the tools using the genome and operon data from E. coli K12. To achieve this, we extracted a 10,000 bp nucleotide DNA sequence from the upstream and downstream regions flanking the rfbABCDX operons for analysis with such operon transcription predictors. By doing this informatic analysis, we found that Operon-Mapper predicted that 13 genes would be transcribed as a unit together within the rfbABCDX operon, while FGENESB predicted 12 genes transcribed as a sole operon. As a major discrepancy, BioCyc correctly predicted that only the five expected rfb genes rfbABCDX are transcribed as a single transcriptional unit. With this analysis, we then focused on using BioCyc to further investigate to what extent unique rfb-gene-cluster patterns could be predicted as being transcribed with other non-rfb genes in this study. By using representative genomes of B. thetaiotaomicron and P. distasonis, we determined that the rfb-gene-clusters have transcription elements in the surrounding sequences that would make these rfb-gene-clusters to be transcribed as either single transcriptional units with and without other genes, depending on the rfb-gene-cluster/strain. This BioCyc analysis also revealed that when single rfb genes are present, they may be transcribed with other genes as depicted in Supplementary Figure S3, supporting the hypothesis that such isolated genes or rfb-gene-clusters may assume various other roles in the phylum which remain yet to be defined.
Rfb-gene-cluster conservation in P. distasonis encompasses 85 y of history
Using Parabacteroides and Alistipes as genus models for Bacteroidota,Citation1,Citation2 we then quantified the rfb sequence conservation (% similarity among strains),Citation3 since conservation indicates functional/evolutionary advantages.Citation40,Citation41 Analysis showed that, irrespective of orientation, the doublet rfbCB sequences, present in 80% of P. distasonis, and the rfbACD triplets have homologies ranging from 72%–99.8% (77.5 ± 5.15%), contrasting the much lower operon similarities across Alistipes (range 27.5% to 83.7%; 50 ± 16.4%, p < .001; Supplementary Figure S4). We have considered rfbFGC as a triplet operon, even with a 900 bp non-rfb gene present in this operon.
Considering that the P. distasonis strains examined in this study span 85 years and various geographic locations, from ATCC8503 (USA, c.1933) to 82G9 (South Korea, c.2018), the high rfb-gene-cluster sequence similarity (p < .001 ) indicates well-conserved genomic features across time and space. However, this conservation cannot explain the observed rfbFGC->rfbA variability. Instead, it implies that an independent genomic mechanism might be responsible for generating modern rfbFGC->rfbA variants over time, which were not present in the founding strain ATCC8503, 85 years ago.
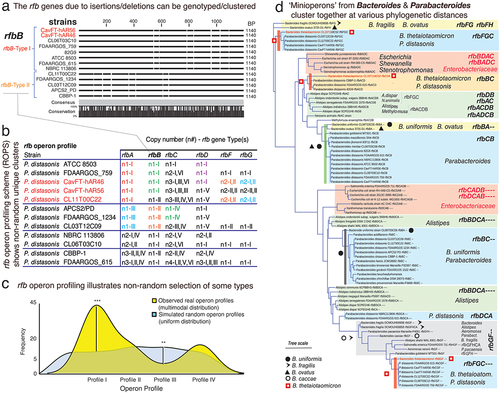
Figure 4. ‘Rfb-operon profiling’ indicates non-random selection and rfb-gene-cluster similarity between Parabacteroides and Bacteroides, namely B. thetaiotaomicron. a) gaps and insertions in rfb gene sequences designate different rfb-types using protocols described for rfbA-typing.Citation3 supplementary figure S5 illustrates the rfb-typing of rfbC/D/F/G. b) exa mple of global rfb operon profiling system (GOPS) for P. distasonis. b) density plots between random and real rfb operon profiles in P. distasonis. Observed types are statistically different from a random (uniform) distribution (**, *** for p<.05 and p<.01, respectively). We generated random and real operon profiles based on the rfb(A,B,C,D,E,F, and G) gene types. For example, P. distasonis ATCC 8503 has rfbA, rfbB, rfbC, and rfbD genes of type I, with the real operon profile represented as 111,100 (indicating types I for rfbA, rfbB, rfbC, and rfbD, and absence of rfbF and rfbG). Each gene type has varying subtypes (e.g., rfbA has 4 types, rfbB has 2 types, rfbC has 6 types, etc. ()). We then created random profiles by permuting combinations of these types, resulting in profiles such as 111,100, 211100, 311100, 411100, and so on. Using these numeric profile we create a density plot using STATA software. d) phylogenetics across Bacteroidota and enterobacteriaceae based on rfb mini/operon sequences. Remarkably, several Bacteroides species, but namely B. thetaiotaomicron CLT5T119C52, cluster together with several Parabacteroides, especially P. distasonis, irrespective of rfb-gene-cluster considered (red squares; further details in Supplementary Figures S6).
Although the operons in Alistipes are more similarly organized (BDCA) than Parabacteroides, the sequences are more variable, depending on operon orientation (). Of interest, duplicated rfb-gene-clusters in Parabacteroides are virtually identical, which contrasts the sequence dissimilarity seen among duplicated rfb operons in Alistipes (T-Test p = .039, ). The variable homologies across Alistipes indicate that they may i) produce a more variable array of LPS/O-antigens, with at least two different types of O-antigens if operons are duplicated, and/or ii) have higher virulence in vivo. Given the recognition of Alistipes as an emerging genus in human diseases,Citation1 we developed PCR primers for rfbBDCA/ACDB operons to facilitate their future study in Alistipes (Supplementary Table S1).
Genome ‘rfb operon profiling’ shows rfb-gene-cluster occurrence is not random
To further characterize the rfb genes and the genome-wide operon patterns in Bacteroidota, we propose a global ‘rfb operon profiling’ system (GOPS). Using our rfbA-typing methodologyCitation3 and P. distasonis strains as a model, we showed that it is possible to discern the strains examined based on distinct genotypes for each rfb gene (rfbB-types n = 2, rfbC-types n = 6, rfbD-types n = 4, rfbF-types n = 2, rfbG-types n = 2, & Supplementary Figure S5). By applying the previous scheme to generate a combined alphabetic-numeric profile of the entire rfb operon, accounting for both copy number and rfb genotype, we generated summary profiles for testing (). Of note, this system may be broadly applied to type other operon genes. Remarkably, the GOPS profiles identified were determined to be reproducible, favoring profiles that are statistically different from random profiles, supporting the assumption that rfb-gene-cluster arrangements have been selected over time ().
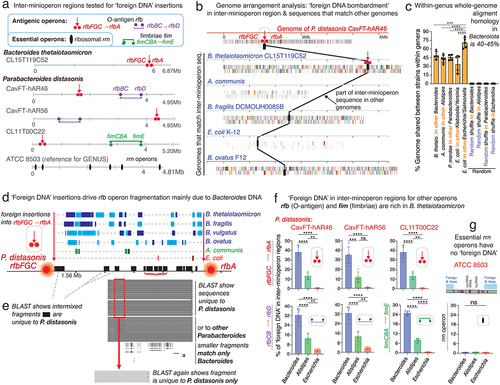
Figure 5. Fragmentation of nonessential operons is driven by insertions of ‘foreign DNA’, mainly Bacteroides. a) Schematics of O-antigen (rfb), fimbriae (fim) and ribosomal (rrn) gene operons tested for DNA insertions. b) example of P. distasonis inter-rfb-gene-cluster DNA fragment found in other genomes. c) % of DNA from one species common to the genome of 4–5 other species within the genus. d) Schematics showing source of ‘foreign DNA’ insertions into the rfbFGC->rfbA space separating the rfb genes in P. distasonis. e) ‘pure’ P. distasonis DNA fragments intermixed within inter-rfb-gene-cluster ‘foreign DNA’ insertions. f) genus sources and % of ‘foreign DNA’ fragmenting the rfb and fim rfb-gene-clusters in P. distasonis, as in . Bacteroides (B. thetaiotaomicron, B. fragilis, B. ovatus, B. vulgatus) are the main source of ‘foreign DNA bombardment’ (p<.0001 vs. Alistipes and Escherichia). Additional information is available in supplementary table S3. g) ribosomal operons (rrn, deemed essential) are not fragmented in P. distasonis, despite presence of ‘foreign DNA’ in vicinity. *, **, ***, for p<.01, p<.001, p<.0001, respectively. Supplementary table S4 shows rrn operons are not fragmented in other genera, i.e., Prevotella, Bacteroides, Alistipes, and Porphyromonas.
Rfb-gene-clusters from Bacteroides thetaiotaomicron and Parabacteroides cluster together
Considering the challenges of examining all Bacteroidota rfb-gene-clusters with current operon mapping tools,Citation38,Citation42–44 and the high prevalence of incomplete draft genomes, we next used the NCBI-BLAST database to further investigate if the observed rfb-gene-cluster patterns were i) nonrandom, and ii) either widespread across various phyla in the NCBI database or exclusive to particular genera or species. Using P. distasonis rfb-gene-cluster sequences, we found that the rfb doublets and triplets are limited to P. distasonis strains exhibiting ≥99% coverage. Low-matching hits for rfb operons/rfb-gene-clusters present with lower (≤24%) sequence coverage were more similar to Bacteroides species (B. thetaiotaomicron, B. caccae, B. cellulosilyticus) and more distant (≤3% coverage) from Proteobacteria, suggesting that the evolution of O-antigen in Parabacteroides has been closer to Bacteroides than to Proteobacteria (Supplementary Table S2). Phylogenetic analysis of mini/operons from Enterobacteriaceae and Bacteroidota with the best BLAST/NCBI sequences (lowest E-scores) further illustrates the well-conserved nature of Bacteroides spp. rfb-gene-clusters across genera, and the little similarity to Proteobacteria ().
For generating the phylogenetic tree of the rfb operons, instead of relying on multiple sequence alignment of the nucleotide sequence for operon to infer genetic distance or clustering, we adopted the maximum likelihood evolutionary approach using nucleotide sequences of the operon to elucidate the evolutionary relationships among them. Of interest, a specific strain of B. thetaiotaomicron (CLT5T119C52/USA/c.2018) clustered in three distinct clades (rfbFGC, BC, and FGD—) with P. distasonis, including CavFT strains. This finding indicates that Parabacteroides/Bacteroides clusters are likely to have a common ancestor or high affinity for DNA exchange and horizontal ‘operon’ transfer (). These Parabacteroides/Bacteroides rfb-gene-cluster clades also harbored B. uniformis or B. ovatus, but not Alistipes or Enterobacteriaceae.
Operon fragmentation through ‘foreign DNA’ insertions from Bacteroides
Given the known high-frequency of horizontal gene transfer (HGT) between Bacteroides and gram-negative microbes in the gut,Citation45 the presence of rfb-gene-clusters in Bacteroidota could indicate that sharing certain rfb patterns could reflect adaptation/selection benefits. However, such HGT exchange theory could not explain the observed genus-specificity and low frequency of rfb-gene-clusters in other taxa. Therefore, we hypothesized that the cumulative insertion of ‘foreign DNA’ between rfb genes could account for the fragmentation of operons and the variable rfbFGC->rfbA separation distances observed in . We further hypothesized that the genetic exchange in Bacteroidota could be a specialized event, confined to niches with compatible species, rather than occurring randomly in the gut. Supported by clades in (containing Bacteroides matching Parabacteroides of CavFT origin), this exchange could explain the low representation of rfb-gene-clusters in NCBI. Testing these hypotheses, we first performed whole-genome rearrangement analysis to quantify the percentage of genome matching sequences and their fragment sizes. We then examined the sequences located within fragmented rfbFGC->rfbA rfb-gene-clusters and select surface antigenic fimbriae inter-rfb-gene-cluster regions as depicted in .
Genome rearrangement analyses showed that 30–45% of the P. distasonis genome sequence, in various fragment sizes, match that of B. thetaiotaomicron, confirming the two strains belong to different clades at genome scales. However, such finding also emphasizes the potential for disruptive random ‘foreign DNA’ insertions into operons, since genome alignment showed that the largest shared DNA fragment was also present in other Bacteroides (). Of mechanistic interest, we found that the DNA present in the inter-rfb-gene-cluster rfbFGC->rfbA sequences represent an overlapping mixture of DNA that matched primarily Bacteroides (27–45%; B. thetaiotaomicron, B. fragilis, P. ovatus), with limited similarity to Alistipes or E. coli, and no evidence of similarity to random sequences (shuffled genomes; ). Notably for P. distasonis, the entire inter-rfb-gene-cluster sequence represents the combination of ‘foreign’ Bacteroides DNA intermixed with DNA sequences verified by BLAST as pure P. distasonis (). This suggests that, over time, such inter-rfb-gene-cluster sequences have either become specific for P. distasonis, or they represent insertions of ‘foreign DNA’ from other P. distasonis. Examination of inter-rfb-gene-cluster regions in other strains and in the antigenic fimbriae (fim) gene-clusters in P. distasonis strain CL11T00C22 (strain with most complete set of fim genes,Citation46 ) confirmed the same pattern of Bacteroides predominance.
No fragmentation in ribosomal operons
Lastly, to determine if operon fragmentation also affected essential genes, we examined ribosomal rrn operons (~5000bp containing highly conserved 16S, 23S, and 5S rRNA/tRNA genes). We i) assessed rrn operon integrity, and ii) quantified the amount of Bacteroides (B. thetaiotaomicron) DNA found in or near the rrn operons of diverse genomes; as an analytical control, we also quantified the amount of B. thetaiotaomicron DNA found in randomly generated 5000bp-loci. Analysis revealed that none of the P. distasonis rrn operons studied (encompassing 21 rRNAs and interspersed tRNA genes) were fragmented or contained B. thetaiotaomicron (Fisher’s exact p = .0081, vs. randomly selected loci; 0/7 vs. 12/20; ), despite being exposed to the same ‘foreign DNA insertion pressure’, which was inferred by comparing the average distance of the nearest B. thetaiotaomicron DNA insertion to the rrn operon (vs. distances to randomly generated locus coordinates; T-test p = .79). Interestingly, multiple large spans (50,000+bp) of bacterial genome were found to be free of B. thetaiotaomicron DNA which could be considered as a future strategy to identify essential operons.
‘Foreign DNA’ insertions in Bacteroidota impact metagenomic statistics and interpretation
Ranking analysis of all DNA fragments from 15 genomes that aligned to P. distasonis CL11T00C22, shown as dot plots in , illustrates that Bacteroides are the most likely sources of recent genomic exchange with Parabacteroides based on the number and size of homologous DNA fragments. Especially intriguing is that B. thetaiotaomicron strain CLT5T119C52, again, has the most distinctive sharing pattern (largest number/longest fragments) with Parabacteroides, suggesting that DNA exchange is an active, still ongoing interspecies process. Similar insertions of B. thetaiotaomicron DNA were also observed in other inter-rfb-gene-cluster regions in other genera (B. fragilis, Barnesiella viscericola, P. intermedia, and A. shahii). Analysis indicates that not all species are equally likely to receive B. thetaiotaomicron, further supporting that inter-species exchange is not random across the phylum, but rather, driven by yet unknown pairing factors among species that share environments where other Bacteroides may be DNA donors (B. fragilis, B. ovatus; Supplementary Figure S7 & Supplementary Table S5).
Figure 6. Impact of ‘foreign DNA’ insertions in Bacteroidota on metagenomics, antigenic operons and bacteria-bacteria inflammatory interactions in a ‘Bacteroidota and Enterobacteriaceae model of gut microlesions’. a) plot of DNA fragments that align between parabacteroides CL11T00C22 and Bacteroides, Alistipes, Escherichia genomes (n=16) ordered by length of each aligning fragment. Notice genus-genus differences. B. thetaiotaomicron CL15T119C52 has unique pattern of abundant fragments (maximum fragment sizes and average % of genome shared, inset bar plots). b) BlastX (protein) and metagenomic (nucleotide) taxonomic analyses of 250bp-fragmented P. distasonis genome. Notice ‘extra species’ assigned by metagenomics (inflation), reflecting ‘foreign DNA’ insertions/exchange across Bacteroidota, and not real presence of species (inset bar plot). The n of ‘extra species’ varied with fragment length (Pearson corr. 0.84, p<.05). The performance of BLAST and BLASTX depends on bacterial genome (supplementary figure S9). c) metagenomic community simulation with Bacteroides, Parabacteroides, Alistipes, and Escherichia (1:1:1:1 genomes). Krona plot (relative abundances within hierarchies of metagenomic classificationsCitation47 illustrates E. coli sequences are poorly assigned to E. coli leading to relative ratio overestimation of Bacteroidota abundance (1:17:12:18; see krona plots for individual genomes (‘alone’) in supplementary figure S10). Bacteroides is commonly listed as ‘extra species’ (bar plot; complete list in supplementary figure S8). d) KEGG pathway and total gene counts in Enterobacteriaceae and Bacteroidota, highlighting the significant differences for Bacteroidota (details in supplementary table S7). e) Stereomicroscopic 3D-apperance of an intestinal cavernous fistulous tract (CavFT) micropathology in a Crohn’s disease patient. Reproduced for illustration from Rodriguez-Palacios, A. et al.Citation23 Diagram illustrates co-existence of Enterobacteriaceae (red) and Bacteroidota (blue) in microlesions, and a peritonitis experiment in which mice received P. distasonis/Bacteroidota or E. coli/Enterobacteriaceae. f) average pro-inflammatory cytokine secretion by bacteria derived from in the Raw murine macrophage stimulation assays, indicating that Bacteroidota whole-cell heat killed extract trigger the release of pro-inflammatory TNF-a cytokines to a lower extent compared to Enterobacteriaceae. g) hypothetical model of gut microlesions with colonization of commensal/pathobionts modulating inflammation. Peritonitis model showed mice with enterobacteriaceae had fatal peritonitis, but not if receiving Bacteroidota B. thetaiotaomicron, B. fragilis, or P. distasonis. *p<.01; ****p<.00001.
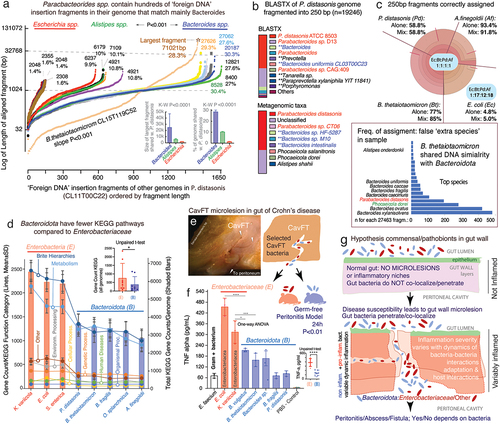
With a large number of ‘foreign DNA’ insertions in the genome that could disrupt operons (n = 1450–1650, x-axis ), there is also potential for impacting metagenomic results. In examining the taxonomic assignment of inter-rfb-gene-cluster sequences using BLAST, we first illustrated the potential for metagenomic overestimation (‘inflation’: calling of ‘extra species’ in a sample when they are not there). BLAST suggests this could be important for strains avid to share DNA, but not for non-avid strains. While numerous inter-rfb-gene-cluster sequences matched to several B. thetaiotaomicron strains in NCBI with 100% coverage and >99% identity (81% of top hits), in a few cases (<4%) DNA matched other Bacteroides with lower similarity (<85%), such as B. longzhouii or B. faecis, and B. fragilis (21.4% top hits, Supplementary Table S6). Together, as illustrated in , analyses indicate DNA insertion similarity is more likely with Bacteroides, being restricted to Bacteroidota. To expand these BLAST-derived inferences, we used the BV-BRC metagenomics workflow for taxonomic classification of DNA sequences ‘in bulk’ to inspect entire genomes. By fragmenting the genome of selected strains into equal nonoverlapping 250 bp simulated ‘reads’, we determined, in-silico, that the species inferred from inter-rfb-gene-cluster sequence queries in NCBI were also reproducible by metagenomics.
Since metagenomics uses sequences to deduce i) taxonomic composition using BLAST (nucleotide database), or ii) taxonomic composition and pathways/functions using BLASTX (protein database), ‘foreign DNA’ insertions in Bacteroidota affect the accuracy of these tools. The impact of ‘foreign DNA’ insertions can be visualized for both ‘individual absolute metagenomic’ analyses (fragmented genomes analyzed individually, ), and on ‘relative metagenomic analyses’ for a simulated community with four genomes (A. finegoldii, B. thetaiotaomicron, P. distasonis, and E. coli, 1:1:1:1, 1 x and 20 x, ). Although metagenomics aligns fragments/reads into contigs (algorithm assumption) and then finds their best match in a database with genes representing selected reference strains (BV-BRC n = 24868; 16840 species; 30573 taxon units), analysis shows the presence of DNA from numerous Bacteroidota, especially the species shown by NCBI-BLAST in inter-rfb-gene-cluster regions. Top species for B. thetaiotaomicron included B. xylannisolvens, B. ovatus, P. dorei and P. distasonis, further confirming inter-species affinity for selective genetic sharing. Diagnostically relevant, and as anticipated, results also illustrate that metagenomic workflows could overestimate diversity by suggesting the presence of ‘extra species’ not actually present in a sample. Of note, the calling of ‘extra species’ by metagenomics depends largely on the fragment length used; however, while the number of fragments called ‘extra species’ is reduced as fragments get longer (from 100 bp to 16000 bp), the relative presence of ‘extra species’ increases with fragment length (Pearson’s p < .05), confirming overestimation and suggesting the need of algorithm revision in current workflows.
Assessing communities in confined micro-niches will remain challenging using current metagenomics, since underestimation of present species (‘deflation’: species underestimated in abundance or deemed absent from sample) could also occur in cases where pathobionts/commensals coexist. For example, our relative community analysis showed that if a species is largely under-assigned by metagenomics (E. coli, ~4%, ), there will be further overestimation of Bacteroidota ratios (12-to-18-fold magnification vs. E. coli, and Supplementary Figure S8).
Rfb genes from P. distasonis not found in metagenome databases suggests niche-specific strains
The Bacteroides genus is predominantly anaerobic and is primarily associated with the gut environment, making it challenging to find outside this context. However, when we conducted a JGI-IMG BLAST using metagenome data from other environmental sources, with the rfbA-D genes of P. distasonis ATCC8503 as the reference genome, we identified significant hits in marine water and cattle rumen metagenome data in case of rfbA and rfbB (79%–14% of identify, with 29% query coverage. In contrast, during the NCBI-BLAST against the WGS database, we primarily observed outputs related to the human and gut microbiome (Supplementary Tables S8–S9).
Reduced KEGG pathways, TNF-alpha induction, and peritonitis by (CavFT) Bacteroidota
Since a widespread process of DNA insertions throughout the genome could affect the functionality of various operons and genetic pathways, we next tested if such a phenomenon of operon disruption could be visualized by observing a lower number of functional pathways using a pathways database. By using the Kyoto Encyclopedia of Genes and Genomes (KEGG), a large-scale molecular dataset generated by genome sequencing, high-throughput experiments, and manual curation to infer enzymatic pathways across bacterial genomes,Citation48 we confirmed that Bacteroidota have significantly less pathways than Enterobacteriaceae (T-test, p < .05). Although the database may be biased toward Enterobacteriaceae due to more published evidence, demonstrates that selected Bacteroidota have significantly fewer functional pathways responsible for ‘metabolism’ and ‘environmental information processing’, while there was no difference for pathways responsible for ‘organismal systems,’ ‘human diseases,’ ‘genetic information processing,’ and ‘cellular processes’, supporting that the findings are well-controlled for basic pathways functions.
Of interest, when testing representative CavFT bacterial isolates derived from the gut wall of patients with CD in our laboratory (), we determined that the overall inflammatory potential of bacteria on murine macrophages was significantly reduced compared to Enterobacteriaceae. shows that TNF-alpha production by macrophages cultured in vitro (RAW264.7 cells) exposed to heat-treated bacterial extracts was about half the immune-proinflammatory potential observed for E. coli and Klebsiella variicola. Controlling for the apoptotic effect that bacterial extracts could have on macrophages at different extract dilutions (measured using cell viability MTT assay), as LPS represents the primary constituent derived from gram-negative bacteria responsible for triggering the release of TNF-alpha by macrophages,Citation35,Citation49 TNF-alpha data shows that Bacteroidota isolates from CavFT have non-inflammatory antigenic phenotypes compared to Enterobacteriaceae (e.g., O-antigen rfb operons) or Enterococcus faecium (gram-positive bacteria). However, we acknowledge that this cell stimulation assay was conducted using whole-cell heat-killed extracts of bacteria, and we recognize that LPS is the component responsible for triggering TNF-alpha release from macrophage cells,Citation35,Citation49 but, it is not the sole factor contributing to TNF-alpha production; other factors may also play a role in TNF-alpha release (). To further validate in vivo the sub-inflammatory potential of CavFT Bacteroidota, we injected suspensions of live bacteria into the peritoneal cavity of germ-free Swiss Webster mice to quantify their inflammatory potential. Our peritonitis model, based on 270,000 real-time telemetry data points, revealed that the mice receiving E. coli or K. variicola became febrile, then hypothermic, lethargic, and moribund within 24 h post injection, while mice receiving CavFT Bacteroidota only became transiently hypothermic following the injection, overall being clinically normal or telemetrically less active until the end of study 24 h post injection.
Based on evidence of antigenic operon fragmentation by Bacteroides and the reduced proinflammatory potential of Bacteroidota in vitro and in vivo, depicts a hypothesis where Bacteroidota invade, interact, and adapt to gut wall micro-niches where other enteric bacteria (e.g., Enterobacteriaceae and Bacteroidota) may be present and dynamically fluctuate to explain the cyclical remission-flare dynamics of gut wall inflammation and complications in chronic bowel diseases like CD.
Discussion
To explore the causes of pro-inflammatory surface antigen variability affecting gut pathobiont dualism within Bacteroidota, this study initially assessed the integrity of the rfb operon utilizing a selected set of complete genomes. Validation of findings was achieved by extending the analysis to other genomes and annotated sequence repositories, mainly using NCBI/BLAST. Of note, Bacteroidota have their rfb operons either intact (Odoribacter, Porphyromonas gingivalis), duplicated (Alistipes) or, mainly, completely or partially fragmented into ‘rfb-gene-clusters’, which can be classified into at least six (n = 6) categories using a broadly-applicable operon cataloging system (). Overall, rfb-gene-clusters are highly conserved, non-random, rare features within genera and across NCBI databases, and form distinctive patterns sporadically shared among distant species, indicating a common mechanism for operon fragmentation within the phylum. The mechanism of transcription and the role of the transcribed genes in the biology of Bacteroidota remains to be explored via RNA sequence transcriptomic intervention experiments and mutagenesis, which are outside of the scope of the present report.
The objective of this study was to identify potential genomic structural signatures that could be used to classify bacteria and guide mechanistic studies that could be impacting surface antigens favoring Bacteroidota switches from commensalism to pathobiont dualism. By characterizing and cataloging rfb operon fragmentation patterns, we determined that the insertion of ‘foreign DNA’ from other bacteria, mainly Bacteroides, could explain operon fragmentation as observed in P. distasonis (supra-numerary rfb-gene-cluster fragmentation), which was used together with Alistipes spp. as model bacteria for hypothesis testing. Of note, by using operon transcription and mapping analysis, we observed that when two or more rfb genes were present together as rfb-gene-clusters, they were consistently transcribed as a single transcriptional unit. This finding suggests that the presence of multiple rfb genes (e.g., duplicated singlets/rfb-gene-clusters or operons) in the same genome may have a functional impact on LPS formation and could play a crucial role in the pathogenesis of bacterial strains.
Intriguingly, the presence of Bacteroides ‘foreign DNA’ insertions occurred within the antigenic operon genes (rfb, fim), but not in essential ribosomal operons (rrn). In B. thetaiotaomicron, studies have shown significant changes in gene usage during the initial days of experimental colonization, with high expression of diverse polysaccharide utilization loci like operons (BT1871–BT1877), due to mutations detected in the BT1871 gene which encodes for α-galactosidase, but no spontaneous mutations in the rrn operon located downstream of the operon.Citation50 Our data suggest that DNA insertions/operon damage could favor the selection of better adaptive bacteria if the insertions promote bacterial survival. Indeed, previously observed patterns of operon disruption in Bacteroides spp. have been linked to functional themes related to niche-habitat survival,Citation51,Citation52 suggesting that a similar phenomenon may occur across rfb operons in other genera in the phylum Bacteroidota. Notably, in Staphylococcus aureus, studies revealed that mutations in operons confer adaptive benefits for pathogen adaptation within the host.Citation53 In contrast a loss-of-function study in E. coli analyzing the conservation of the lac operon, showed that lac-negative E. coli strains face minimal survival chances in the intestinal environment, demonstrating that the benefits of an operon alteration may have drastic effects depending on the environment, e.g. the diet and microbiota composition in the gut.Citation54
Furthermore, prior literature supports that, in addition to reductions/loss of the O-antigen,Citation55,Citation56 rfb gene mutations influence bacterial survivalCitation57 and bacteriophage infection.Citation58 While more research is needed to elucidate how rfb gene dosage and structure influence LPS and/or other cellular KEGG ontology maps and functions in Bacteroidota, it is important to highlight that likely most Bacteroidota are under-studied genomes with unverified annotations (e.g. hypothetical genes) and unknown protein functions. Our analyses demonstrated the lack of O-antigen production by P. distasonis under different growth conditions and the lower TNF-alpha production induced by CavFT Bacteroidota isolated from CD patients in our laboratory,Citation14,Citation15 or the lack of induction of peritonitis, contrast numerous reports of severe peritonitis due to this and other species within the phylum (B. thetaiotaomicron or B. fragilis) which is commonly seen in immunocompromised individuals.Citation59–62 Therein, further in vivo validation is required to investigate the potential pathobiont role of Bacteroidota species in IBD.
Of evolutionary interest, several Bacteroides, including the bacterium B. thetaiotaomicron CL15T119C52 (human/feces/2018) were remarkably noted to cluster with P. distasonis CavFT strains (CD patients/2019) based on conserved rfb-gene-clusters (). This suggests there is preferential DNA exchange among certain strains (namely Bacteroides as shown throughout the study, ). Together, our in-silico analyses, manual annotation, and experimental observations serve as a proof-of-principle for the (primarily) Bacteroides DNA insertion mechanism of operon fragmentation and its potential impact on metagenomics. To further validate the impact of gene insertion on microbiome analysis outcomes, future studies could be conducted with mono-strain and multi-strain microbial colonization of germ-free mice to determine with more precision how such a Bacteroides DNA insertion mechanism of operon fragmentation impair our ability to predict bacterial community compositions accurately using metagenomics. Our genomic analysis showed that the essential rrn ribosomal operons are not affected by such Bacteroides DNA insertion indicating that 16S rRNA microbiome analysis could be superior to metagenomics at predicting Bacteroidota community composition. Our data indicates that composition predictions based on 16S rRNA, deemed as suboptimal in the past compared to metagenomics, could be actually accurate and not suboptimal as described in studies comparing the performance of both methods, including a recent study on chicken microbiota that stated that 16S rRNA gene sequencing captures only a fraction of the microbiota community when compared to shotgun sequencing.Citation63 Based on our analysis, the use of 16S rRNA analysis would be less confounded because bacteria containing foreign DNA from other species or genera would not affect the integrity of the rrn (16S rRNA) operons.
Our findings provide novel insights and opportunities for diagnostics and therapeutic developments, especially considering that the most remarkable findings, such as the distancing of the rfbFGC->rfbA rfb-gene-clusters, involve DNA from bacteria that we have isolated from CavFT lesions as understudied chronic inflammatory microenvironments in CD.Citation25 The relevance of these findings needs to be assessed in future studies. Our study, for the first time examines and reports the genomic features of CavFTs in context with other members of the phylum, also isolated from CavFTs, which could evolve and adapt into lineages that may survive on/inside micro-niches in the inflamed gut wall.Citation24,Citation25
In conclusion, herein, we present a system for profiling the antigenic operon integrity in Bacteroidota, primarily focusing on the O-antigen rfb operon. We offer novel insights into operon fragmentation and explore the potential impact that such disruption in integrity could have on gut commensalism and intestinal inflammation. This is particularly relevant at micro-niche levels, which may be significant for bacteria derived from the purulent debris contents in CavFT in patients with CD. Our findings highlight that operon fragmentation provides novel mechanistic insights for commensal adaptation and metagenomic applications. An improved understanding of Bacteroidota rfb operons can provide valuable insights into bacterial genetics and their role in human health, as well as help refine experimental strategies for studying host–pathogen interactions. Future studies on these interactions or disease causality and operon integrity would benefit from combining bacterial isolation with genomic and transcriptomic sequencing to assess KEGG-pathways functionality.
With respect to our study limitations and future needs, herein, we propose a cataloging system for operon integrity and diversity applicable to multiple species based on the fragmentation patterns observed in Bacteroidota. This system could have significant implications for understanding the variability of pro-inflammatory surface antigens in gut commensals, especially considering they could have functional advantages on the ecology of microbial translocation and microbe gut inflammatory microniches. The presence of Bacteroidota and cohabitation with species from other phyla, such as Pseudomonadota, affect immune cells and other species in such microscopic lesions in patients prone to CD could create complex scenarios where inflammation or silent growth occur and varies or oscillates over time. This study further supports that the potential combination of bacteria with varied operon fragmentation patterns in confined (CavFT) inflammatory niches, likely to be associated with flares or remission as we earlier proposed.Citation3 Future functional studies are essential to elucidate the specific role that fragmentation and diversification play in influencing intestinal health both in vitro and in vivo.
We have discovered that Bacteroidota coexist with E. coli/Klebsiella within CavFT microlesions. This cohabitation has been shown to induce immune cell cytotoxicity in SAMP1/YitFc mice, which are susceptible to CD-like ileitis, through multifactorial pathogenic mechanisms aggravated by exposure to succinate.Citation25 This finding is particularly significant because recent studies have shown that succinate levels are elevated in patients with IBD.Citation64,Citation65 Interestingly, we determined that succinate is a primary byproduct metabolite produced by Bacteroidota species isolated from such gut wall cavitations. The compromised integrity of the O-antigen operon in Bacteroidota, which could make these species less pro-inflammatory yet capable of producing succinate, in conjunction with E. coli metabolites, could represent a novel multifactorial mechanism underlying disease complications in CD.Citation25 The present study highlights how such characteristics could be applied to a wide range of species, where fragmented operons and diversifications could act as a patterned mechanism for disease or commensalism mediation in intestinal homeostasis.
To overcome the inherent limitations of this study, particularly regarding the extrapolation of results from genomic and metagenomic data to functional impacts in vivo, there will be need for additional experimental validation of the proposed mechanisms in model organisms or human microbiome samples. It is of utmost importance to clarify that the pathobiont or commensalism role of Bacteroidota, might not be merely determined by its rfb operon. As demonstrated recently in our laboratory,Citation25 it might be locally variable and systematically determined by homeostasis of gut ecosystem alterations in the host or even whole immune/physiological system defects of the host. Some rfb operons bearing DNA insertions, which might increase the ability of a Bacteroidota species to produce TNF-alpha, would not significantly change the fate/role of a Bacteroidota species as a pathobiont or commensalism because even in its “worst” proinflammatory situation, compared with some other highly pathogenic bacteria, Bacteroidota species as commensals and promoters of TNF-alpha might not be detrimental on their own. However, in some microenvironment, for example, in a highly inflamed gut mucosal tissues with disrupted mucosal integrality, any commensal bacterial species (because of exposure and stimulation of bacterial LPS or other cell componentsCitation3 might induce further production of TNF-alpha or other inflammatory cytokines, or cause immune cell cytotoxicity as we have shown. In that scenario, such commensals promoting further disease would be with no doubt pathogenic, invading microorganisms and promoters of chronic inflammation.
Materials and methods
Genome databases, data collection, and rfb gene nomenclature
Genome-wide genetic analyses for the rfb operon were conducted on publicly available datasets and on reference strains sequenced in our laboratory that we isolated from intramural cavernous lesions in the damaged bowel of Crohn’s disease patients as previously reported.Citation5,Citation25 Only complete reference genomes of selected strains of human-derived Enterobacteriaceae (e.g., E. coli K12, as reference for the rfb operon), selected strains for representative genera of the Bacteroidota phylum, all available reference strains for all species within the Alistipes and Parabacteroides, and all available strains for the Parabacteroides distasonis species were used in this study. National Center for Biotechnology Information (NCBI) GenBank and the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) were accessed for bacterial genomic data. In this study, we conducted a comprehensive search for the gene names, employing different synonyms of the rfb genes gathered from BioCyc,Citation39 in the NCBI and BV-BRC databases within each individual genome. We used the rfb nomenclature because rfb is the official name of the gene that is also known by the different synonyms according to BioCyc.Citation39 For example, with rfbA we used glucose-1-phosphate thymidylyltransferase 1 along with rmlA and dTDP-glucose pyrophosphorylase. Following that strategy, we precisely mapped the locations of the rfb genes in the genome that were utilized in our research. To broaden the searchability for homologue genes that might have not been automatically annotated by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP),Citation5 we employed BLAST using rfb genes of the bacteria E. coli K-12 substr. MG1655, S. enterica FDAARGOS_711, A. finegoldii DSM 17,242, B. thetaiotaomicron CL15T119C52 and P. distasonis CavFT-hAR46 as input sequence with default settings to explore homologs of the rfb genes within the genomes used in this study. Data collected from each genome includes accession number, genome length, rfb gene copy number, nucleotide sequence, location, and direction within the genome. All results of the rfb gene query were manually verified using NCBI annotated graphical portals. Additional data was collected to find the closest homolog of conserved rfb-gene-clusters in P. distasonis; We used the previously obtained FASTA sequences of P. distasonis rfb-gene-clusters as input queries in the nucleotide Basic Local Alignment Search Tool (BLAST) using default settings under the nr/nt database, and BLAST hit result data and rfb gene sequence data for matching organisms from homologue queries were subsequently collected.
The rfb genes search in metagenome databases
To identify the rfb gene in the gut microbiome and other environments, we conducted NCBI Blastn searches using whole-genome shotgun contigs (wgs) for the rfbA, rfbB, rfbC, and rfbD genes of P. distasonis ATCC8503. The organisms included in the analysis were sourced from the human microbiome (Taxid 646,099), gut microbiome (Taxid 749,906), human oral microbiome (Taxid 410,658), aquaculture metagenome (Taxid 2,714,431), aquaculture system metagenome (Taxid 27,144,341), anaerobic metagenome (Taxid 1,263,854), and alkali sediment metagenome (Taxid 1,680,915). Additionally, we utilized metagenome data in IMG-JGI and performed BLAST searches for the rfbA, rfbB, rfbC, and rfbD genes of P. distasonis ATCC8503 using host-associated metagenome data in IMG-JGI to identify significant hits in metagenome data associated with both the environment and host.
Visualization of rfb gene dosage and location
The rfb gene and rfb-gene-cluster/operon copy numbers for each genome were used to generate heatmaps demonstrating relative rfb gene dosages. Heatmaps for rfb gene dosages in P. distasonis strains were created using ClustVisCitation66 and a summary heatmap of rfb gene dosages for all genomes was generated with the web tool Morpheus (https://software.broadinstitute.org/morpheus).
Homology analyses
Sequences of recurring doublets; rfbCB(++) and rfbBC(–), and of recurring triplets; rfbACD(+++), rfbDCA(–), rfbFGC(+++), and rfbCGF(–), were collected and aligned with both their respective doublets/triplets in Parabacteroides genomes as well as their respective reverse complements. In Alistipes spp., sequences of its rfbACDB(++++)/BDCA(----) operons were aligned to determine homologies at the species level. Of note, no consistent patterns of rfb-gene-clusters were observed in the selected Bacteroides or Prevotella spp., thus no analyses could be performed to determine their respective homologies. Sequence homology was performed using the Sequence Identity and Similarity (SIAS) tool (http://imed.med.ucm.es/Tools/sias.html).
Global rfb operon profiling system (GOPS) design
DNA sequences for each rfb gene in complete P. distasonis strain genomes were collected and aligned respectively (e.g., rfbB gene alignment, rfbC gene alignment, etc.) in CLC Genomic Workbench (commercially available). Following our previously developed rfbA-typing protocol,Citation3 rfb-gene-types were designated for each rfb gene alignment. The aggregate results of the copy number(s) and rfb-type(s) for each genome were used to construct an example rfb operon profiling system, utilizing the nomenclature system previously proposed for rfbA-type reporting in Bacteroidota.Citation3
Intergene and rfb loci statistics
The linear distance between each rfb gene was determined by calculating the number of base pairs in between consecutive genes in each genome. Rfb loci were counted in each genome, where a locus was determined to be a discrete location of either a single rfb gene or a cluster of contiguous rfb genes (operons/rfb-gene-clusters). Statistical analyses were performed to determine if rfb intergene linear distances and number of rfb gene loci differed significantly between the genera and phyla examined. The range and variance of rfb intergene distances was also determined at the genus and species level for Parabacteroides and Alistipes. Prior to statistical analysis, rfb intergene linear distance data was log transformed. Transformed rfb intergene linear distances and rfb gene loci data were analyzed using Brown-Forsythe ANOVA and Welch’s ANOVA to determine if statistically significant differences existed between the respective mean values of these data for Enterobacteriaceae, Parabacteroides, Bacteroides, Prevotella, and Alistipes.
Construction of phylogenetic trees
The phylogenetic tree of whole genomes was made by Bacterial and Viral Bioinformatics Resource Center (BV-BRC), under the default setting for codon tree (which uses 100 amino acids) and nucleotide sequences from BV-BRC’s global Protein Families to build an alignment and then generate a tree based on the differences within those selected sequences. For the phylogeny of rfb gene clusters (operons and rfb-gene-clusters), the nucleotide FASTA sequences encoding the rfb operons and rfb-gene-clusters were downloaded from NCBI database. The multiple sequence alignments of all nucleotide sequences were performed using the clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) which was used for the construction of phylogenetic trees with the maximum likelihood methods for evolutionary analysis by using Webserver IQ-Tree (http://iqtree.cibiv.univie.ac.at/) under default parameters of ultrafast bootstrap. The phylogenetic trees with branches were built with iTOL (https://itol.embl.de/).
Primer design for amplification of the alistipes spp. rfbBDCA/ACDB operon
Primer design was conducted by identifying left and right flanking regions of the Alistipes spp. rfbBDCA/ACDB operon alignment of which were whole (i.e., no gaps or deletions) throughout all sequences. Then, from the corresponding regions of the rfbBDCA/ACDB operon sequence alignment consensus sequence, left and right flanks of approximately 20 base pair sequences were selected and entered into the Basic Local Alignment Search Tool (BLAST) to confirm accuracy in identifying Alistipes spp. utilized in this study.
Random sequences
Random generation of genomes and the random shuffling of complete genomes were conducted using Sequence Manipulation Suite software (https://www.bioinformatics.org/sms2/shuffle_dna.html) with parameters that matched the %GC content of relevant and selected species, including 43% GC content to mimic Bacteroides genomes (B. thetaiotaomicron CL15T119C52: 43.07%, B. fragilis DCMOUH0085B: 43.61%, B. ovatus F-12: 41.98%, B. vulgatus NCTC10583: 42.03%, and B. dorei MGYG-HGUT-02478: 42.04% for an average of 42.55%).
Calculation of the gene combination of rfb-gene-clusters
For the bacterial genomes used in this study, the number of potential rfb-gene-cluster combinations for each bacterium was determined by calculating the sum of the permutations of these rfb-gene-cluster gene combinations.
LPS extraction and glycoprotein staining
P. distasonis and E. coli were cultured in five different conditions (BHI+5% Yeast Extract, 50% BHI Broth, BHI Broth + 5% Yeast Extract with normal person heat-killed feces, BHI + 5% Yeast Extract with 4% bile and 50% BHI Broth with CD patient heat-killed feces) and centrifuged at 2000 g for 10 minutes. The supernatant was decanted and the masses of the pellets were obtained. The LPS was extracted by LPS extraction kit (Sigma Aldrich Catalogue Number: MAK339) using the manufacturer’s instructions. In short, the cell pellets were resuspended in lysis buffer at a ratio of 100 μL of buffer for every 10 mg of cell pellet. The bacterial cells were lysed using a MP Fast-Prep 24 Homogenizer, using 4 rounds of shaking at 4 m/s for a duration of 20 seconds. The lysed pellets were then centrifuged at 10,000 g for 5 minutes to sediment the cell debris. The supernatant containing LPS was collected in a separate tube, to which proteinase K was added for a final concentration of 0.01 mg proteinase K/ml. The solution was heated to 60°C and held for 60 minutes. Following this, the solution was centrifuged at 10,000 g for 5 minutes, and the supernatant containing the free LPS was collected. The LPS extracts were loaded into a NuPAGE 4–12% Bis-Tris Gel (NP0335BOX) along with commercially available LPS (Sigma) derived from E. coli, as well as a pre-stained protein ladder. The gel bands were fixed by incubating the gel in 50% Methanol for 30 minutes at room temperature. The gel was stained using a commercially available Pierce™ Glycoprotein Staining Kit according to the manufacturer’s instructions. Briefly, the gel was then immersed in a 3% acetic acid solution and incubated for 10 minutes at room temperature. The solution was removed and replaced with fresh acetic acid for another 10 minutes. The gel was then submerged in the oxidizing solution and incubated for 15 minutes with agitation using an orbital shaker. The gel was washed three times with 3% acetic acid for minutes. The gel was then transferred to the glycoprotein staining solution and incubated for 15 minutes on an orbital shaker.
TNF-α stimulation assay in RAW 264.7 cells
The RAW 264.7 cells were flushed and cultured in DMEM (Dulbecco’s Modified Eagle’s Medium (DMEM)) and supplemented with 10% FCS, NEAA, glutamax, penicillin–streptomycin. Cells were plated for experiments after 6 d. Cells were plated at 4 × 104 cells per well of a 96-well plate, and all cell lines were seeded 16 hours prior to challenging. The bacteria were cultured, and pellets were resuspended in PBS. Each pellet was heat killed by placing in a heating block for 30 min at 95°C. To normalize the concentrations of each bacterial suspension, the OD600 value was taken. The different dilutions (1:1, 1:5, 1:25 and 1:125 dilution) of the heat-killing bacterial extract were added to the RAW 264.7 cells, then the medium was collected after 18 h for testing by TNF enzyme-linked immunosorbent assay.
Peritonitis model
To quantify the impact of bacteria on the ability to trigger inflammation in vivo, we conducted studies with mice using a peritonitis model. Using fresh anaerobic bacterial preparations (10^8 CFU/mL), each animal received an intraperitoneal injection of selected bacteria and underwent continuous monitoring for 24 h prior to being euthanized. Real-time mobility in the cage measured with subcutaneous RFID tags, response to stimuli, body temperature, mortality, and bacterial viability in the peritoneal fluid were measured as main outcomes.
Statistics
Data analysis was conducted using parametric and non-parametric statistics using parametric or non-parametric methods (e.g., student T-tests, ANOVAs) using the software STATA (v17), R, and GraphPad, which was used primarily to make bar plots or boxplot illustrations where significance is represented as asterisks based on the level of significance which was held at p < .05(*), <.001(**) and < .0001(***). Stata functions were used to assess multimodality (rfb-gene-cluster types observed vs. simulated random) as previously reported in our laboratory.Citation67 Together, data analysis represents over 636 analytical submissions made to the various bioinformatic resources described in this study.
Assessing operon fragmentation
Using the Bacterial and Viral Bioinformatics Resource Center (BV-BRC; Bacterial and Viral Bioinformatics Resource Center) as the data source and genome alignment tool, genomic homologies were first assessed between P. distasonis and representative members of Bacteroides, Alistipes, and Escherichia, respectively, to test the hypothesis that genomic insertions were not from random sources but rather derived primarily from Bacteroides. We then identified unique rfb order series that were present across seemingly unrelated genomes (chronologically, geographically), including the series ‘rfbFGC ➔ rfbA’, wherein a triplet (rfbFGC) is followed by a nearby separated singlet gene (rfbA). Focused on Bacteroides and P. distasonis as recipient genomes, including CavFT strains, we examined inter-rfb-gene-cluster DNA sequences (e.g., between rfbFGC and rfbA) to conduct genome-wide arrangement analysis between such regions in P. distasonis and the genomes of B. thetaiotaomicron, other Bacteroidota, and E. coli to quantify their genus-dependent potential as donors of ‘foreign DNA’. Intra-phylum genome homologies of Enterobacteriaceae were also measured as a referent. We assessed the genetic homologies of three inter-rfb-gene-cluster segments (rfbFGC ➔ rfbA, rfbCB ➔rfbD, fimCBA ➔fimE) present either completely or uniquely across three P. distasonis strains (CavFT-hAR46, CavFT-hAR56, and CL11T00C22). Whole-genome homologies were then compared to inter-rfb-gene-cluster genetic homologies. Next, selectivity of operon fragmentation was assessed in the P. distasonis ATCC 8503 reference genome, for which 7 16S rRNA loci were annotated in BV-BRC. These 7 loci were compared with 20 randomly generated loci of similar size to compare the amount of B. thetaiotaomicron DNA in or near each locus.
Genome fragmentation alignment analysis
We quantified and plotted the distribution of DNA fragments detected by genome alignment ranked by fragment number (x-axis) vs. the size of each fragment (y-axis) across 16 genomes (4 Escherichia spp., 5 Alistipes spp., and 7 Bacteroides spp.) individually aligned to P. distasonis in BV-BRC.
Assessment of fragmentation in other antigenic (fimbriae, fim) operons
Recently, researchers investigating the antigenic cell surface structure of P. distasonis identified fimbriae (fim) operon genes in several strains,Citation46 though there was again ample heterogeneity in the type and quantity of these genes amongst the strains investigated, suggesting operon fragmentation could also occur for other functions. For this analysis we used the P. distasonis CL11T00C22 strain genome as it contains the most complete set of identified fim operon genes.
Metagenomics of Bacteroidota
Whole-genome sequences of select Bacteroidota (and Escherichia coli K-12 as referent) downloaded from NCBI were split into 250 bp sequences using FASTA-splitting software (https://www.bioinformatics.org/sms2/split_fasta.html) to approximate the range of in vivo DNA samples which are commonly used for shotgun sequencing.Citation68 The sequences for each respective genome were then used as inputs for taxonomic classification in BV-BRC as well as for assessment of matching protein sequences using BLASTX.
We validated our hypothesis using principles of genomic and metagenomics data analysis using well-referenced and established methods that are readily accessible over the internet to the scientific community.Citation69–71 We used the Bacteria and Virus Bioinformatics Resources Center (BV-BRC) web server as it integrates multiple analysis steps into single workflows,Citation71 by combining the resources from the former Pathosystems Resource Integration Center (PATRIC), the Virus Pathogen Database and Analysis Resource (ViPR) and the Influenza Research Database (IRD).Citation69,Citation70 Analysis tools include pipelines for read assembly, open reading frame prediction, and annotation with BLAST, and GO pathways classifiers. The BV-BRC web server is an easy-to-use web-based interface for processing, annotation, and visualization of genomic and functional metagenomics sequencing data, designed to facilitate the analysis of data by non-bioinformaticians.Citation71 Metagenomics results derived from Kraken2, are visualized via Krona as a dynamic online feature for hierarchical data and prediction confidence.Citation47 The BV-BRC online setting provides scientists a fast open-source strategy to analyze raw sequences and generate complex comparative analysis by selecting reference genomes or genomes uploaded by the user to an academic account. Genome arrangement analysis was conducted using the tool ‘Comparative Analysis tools’ available in beta version. Sequence and genome coordinate data can be examined as summary tables or visually through colored interactive arrangement plots. BV-BRC facilitates effortless inspection of gene function, clustering, and distribution. The webserver is available at https://www.bv-brc.org/.
Contributors
NCB and ARP conceptualized the analysis, analyzed the data and wrote the manuscript. VS contributed with data analysis and manuscript writing. VS and KR assisted with LPS experiments. BG assisted with peritonitis model experiments. BM and AB assisted with cytokine quantification in cultures of mouse ATCC raw.264 macrophages. ARP leads the program for identification of pathogenic bacteria in severe surgical Inflammatory Bowel Disease/Crohn’s Disease. All authors approved the final version of the manuscript.
Supplemental Material
Download MS Word (9.9 MB)Acknowledgments
This project was supported by 1) NIH grant R21 DK118373 to ARP, entitled “Identification of pathogenic bacteria in Crohn’s disease”, 2) a Crohn’s & Colitis Foundation Student Research Fellowship Award #932611 to NB/ARP, entitled “Molecular Immunology of Parabacteroides distasonis in Crohn’s Disease and Role of Strain Diversity in Macrophage Activation”, 3) a Crohn’s & Colitis Foundation Litwin Pioneers Award to ARP, and 4) an earlier Career Development Award from the Crohn’s and Colitis Foundation to ARP.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2024.2350150.
References
- Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The genus alistipes: gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol. 2020;11:906. doi:10.3389/fimmu.2020.00906.
- Ezeji J, Sarikonda D, Hopperton A, Erkkila H, Cohen D, Martinez S, Cominelli F, Kuwahara T, Dichosa A, Good C. et al., Parabacteroides distasonis: intriguing aerotolerant gut anaerobe with emerging antimicrobial resistance and pathogenic and probiotic roles in human health. Gut Microbes. 2021;13(1):1922241. doi:10.1080/19490976.2021.1922241.
- Bank NC, Singh V, Rodriguez-Palacios A. Classification of parabacteroides distasonis and other Bacteroidetes using O- antigen virulence gene: RfbA-typing and hypothesis for pathogenic vs. probiotic strain differentiation. Gut Microbes. 2022;14(1):1997293. doi:10.1080/19490976.2021.1997293.
- Nayfach S, Pollard KS. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015;16(1):51. doi:10.1186/s13059-015-0611-7.
- Yang F, Kumar A, Davenport KW, Kelliher JM, Ezeji JC, Good CE, Jacobs MR, Conger M, West G, Fiocchi C. et al., Complete genome sequence of a Parabacteroides distasonis Strain (CavFT hAR46) isolated from a gut wall-cavitating microlesion in a patient with severe Crohn’s disease. Microbiol Resour Announc. 2019;8(36):36. doi:10.1128/MRA.00585-19.
- Di Lorenzo F, Pither MD, Martufi M, Scarinci I, Guzmán-Caldentey J, Łakomiec E, Jachymek W, Bruijns SCM, Santamaría SM, Frick J-S. et al., Pairing bacteroides vulgatus LPS structure with its immunomodulatory effects on human cellular models. ACS Cent Sci. 2020;6(9):1602–26. doi:10.1021/acscentsci.0c00791.
- Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hämäläinen AM. et al., Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165(4):842–853. doi:10.1016/j.cell.2016.04.007.
- Lindberg AA, Weintraub A, Zähringer U, Rietschel ET. Structure-activity relationships in lipopolysaccharides of bacteroides fragilis. Rev Infect Dis. 1990;12 Suppl 2(Supplement_2):SS133–S141. doi:10.1093/clinids/12.supplement_2.s133.
- Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38(5):996–1047. doi:10.1111/1574-6976.12075.
- Eggerth AH, Gagnon BH. The bacteroides of human feces. J Bacteriol. 1933;25(4):389–413. doi:10.1128/jb.25.4.389-413.1933.
- Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7(4):265–276. doi:10.1016/j.chom.2010.03.004.
- Awadel-Kariem FM, Patel P, Kapoor J, Brazier JS, Goldstein EJ. First report of parabacteroides goldsteinii bacteraemia in a patient with complicated intra-abdominal infection. Anaerobe. 2010;16(3):223–225. doi:10.1016/j.anaerobe.2010.01.001.
- Henthorne JC, Thompson L, Beaver DC. Gram-negative bacilli of the genus bacteroides. J Bacteriol. 1936;31(3):255–274. doi:10.1128/jb.31.3.255-274.1936.
- Sun H, Guo Y, Wang H, Yin A, Hu J, Yuan T, Zhou S, Xu W, Wei P, Yin S. et al., Gut commensal parabacteroides distasonis alleviates inflammatory arthritis. Gut. 2023;72(9):1664–1677. doi:10.1136/gutjnl-2022-327756. © Author(s) (or their employer(s)) 2023. No commercial re-use. See rights and permissions. Published by BMJ. 2023.
- Lai H-C, Lin T-L, Chen T-W, Kuo Y-L, Chang C-J, Wu T-R, Shu C-C, Tsai Y-H, Swift S, Lu C-C. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory parabacteroides goldsteinii lipopolysaccharide. Gut. 2022;71(2):309. doi:10.1136/gutjnl-2020-322599.
- Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol. 2012;30(1):759–795. doi:10.1146/annurev-immunol-020711-074937.
- Kverka M, Zakostelska Z, Klimesova K, Sokol D, Hudcovic T, Hrncir T, Rossmann P, Mrazek J, Kopecny J, Verdu EF. et al., Oral administration of parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol. 2011;163(2):250–259. doi:10.1111/j.1365-2249.2010.04286.x.
- Kelly D, Conway S, Aminov R. Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 2005;26(6):326–333. doi:10.1016/j.it.2005.04.008.
- Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-γ and RelA. Nat Immunol. 2004;5(1):104–112. doi:10.1038/ni1018.
- Di Lorenzo F, De Castro C, Silipo A, Molinaro A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol Rev. 2019;43(3):257–272. Accessed 2023 Jan 10. doi:10.1093/femsre/fuz002.
- d’Hennezel E, Abubucker S, Murphy Leon O, Cullen Thomas W, Lozupone C. Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. mSystems. 2017;2(6):e00046–e00017. Accessed 2023 Jan 13. doi:10.1128/mSystems.00046-17.
- Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8(1):1784. doi:10.1038/s41467-017-01973-8.
- Rodriguez-Palacios A., Kodani T., Kaydo, L et al. Diagram illustrates co-existence of Enterobacteriaceae (red) and Bacteroidota (blue) in microlesions, and a peritonitis experiment in which mice received P. distasonis/Bacteroidota or E. coli/Enterobacteriaceae. Stereomicroscopic 3D-pattern profiling of murine and human intestinal inflammation reveals unique structural phenotypes. Nat Commun 6, 7577 (2015). doi:10.1038/ncomms8577.
- Singh V, West G, Fiocchi C, Cominelli F, Good CE, Jacobs MR, Rodriguez-Palacios A. Genomes of bacteroides ovatus, B. cellulosilyticus, B. uniformis, phocaeicola vulgatus, and P. dorei isolated from gut cavernous fistulous tract micropathologies in Crohn’s disease. Microbiol Resour Announc. 2024:e0115223. doi:10.1128/mra.01152-23.
- Singh V, West G, Fiocchi C, Good CE, Katz J, Jacobs MR, Dichosa AEK, Flask C, Wesolowski M, McColl C. et al., Clonal Parabacteroides from Gut Microfistulous Tracts as Transmissible Cytotoxic Succinate-Commensal Model of Crohn’s Disease Complications. bioRxiv. 2024. doi:10.1101/2024.01.09.574896.
- Reeves PR, Hobbs M, Valvano MA, Skurnik M, Whitfield C, Coplin D, Kido N, Klena J, Maskell D, Raetz CR. et al., Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 1996;4(12):495–503. doi:10.1016/s0966-842x(97)82912-5.
- Peterson DA, Planer JD, Guruge JL, Xue L, Downey-Virgin W, Goodman AL, Seedorf H, Gordon JI. Characterizing the interactions between a naturally primed immunoglobulin a and its conserved bacteroides thetaiotaomicron species-specific epitope in gnotobiotic mice. J Biol Chem. 2015;290(20):12630–12649. doi:10.1074/jbc.M114.633800.
- Roelofs KG, Coyne MJ, Gentyala RR, Chatzidaki-Livanis M, Comstock LE, Huffnagle GB. Bacteroidales secreted antimicrobial proteins target surface molecules necessary for gut colonization and mediate competition in vivo. mBio. 2016;7(4):10–128. doi:10.1128/mBio.01055-16.
- Jacobson AN, Choudhury BP, Fischbach MA, Relman DA. The biosynthesis of lipooligosaccharide from bacteroides thetaiotaomicron. mBio. 2018;9(2):10–128. doi:10.1128/mBio.02289-17.
- Li ZZ, Riegert AS, Goneau MF, Cunningham AM, Vinogradov E, Li J, Schoenhofen IC, Thoden JB, Holden HM, Gilbert M. Characterization of the dTDP-Fuc3N and dTDP-Qui3N biosynthetic pathways in campylobacter jejuni 81116. Glycobiology. 2017;27(4):358–369. doi:10.1093/glycob/cww136.
- Koh GY, Kane A, Lee K, Xu Q, Wu X, Roper J, Mason JB, Crott JW. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int J Cancer. 2018;143(7):1797–1805. doi:10.1002/ijc.31559.
- Jacobson AN, Choudhury BP, Fischbach MA, Relman DA. The biosynthesis of lipooligosaccharide from bacteroides thetaiotaomicron. MBio. 2018;9(2):e02289–02217. doi:10.1128/mBio.02289-17.
- D’Souza JM, Samuel GN, Reeves PR. Evolutionary origins and sequence of the Escherichia coli O4 O-antigen gene cluster. FEMS Microbiol Lett. 2005;244(1):27–32. Accessed 2021 Jun 23. doi:10.1016/j.femsle.2005.01.012.
- Bäckhed F, Normark S, Schweda EKH, Oscarson S, Richter-Dahlfors A. Structural requirements for TLR4-mediated LPS signalling: a biological role for LPS modifications. Microbes Infect. 2003;5(12):1057–1063. doi:10.1016/S1286-4579(03)00207-7.
- Di Lorenzo F, Pither MD, Martufi M, Scarinci I, Guzmán-Caldentey J, Łakomiec E, Jachymek W, Bruijns SCM, Santamaría SM, Frick JS. et al., Pairing bacteroides vulgatus LPS structure with its immunomodulatory effects on human cellular models. ACS Cent Sci. 2020;6(9):1602–1616. doi:10.1021/acscentsci.0c00791.
- Yoshida N, Emoto T, Yamashita T, Watanabe H, Hayashi T, Tabata T, Hoshi N, Hatano N, Ozawa G, Sasaki N. et al., Bacteroides vulgatus and bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation. 2018;138(22):2486–2498. doi:10.1161/CIRCULATIONAHA.118.033714.
- Solovyev VV, Salamov A. Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW. editor. Metagenomics and its applications in agriculture, biomedicine and environmental studies. NY, USA: Nova Science Publishers; 2011. pp. 61–78.
- Taboada B, Estrada K, Ciria R, Merino E, Hancock J. Operon-mapper: a web server for precise operon identification in bacterial and archaeal genomes. Bioinformatics. 2018;34(23):4118–4120. Accessed 2023 Apr 18. doi:10.1093/bioinformatics/bty496.
- Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, Keseler IM, Krummenacker M, Midford PE, Ong Q. et al., The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2019;20(4):1085–1093. doi:10.1093/bib/bbx085.
- Bertani B, Ruiz N, Slauch JM. Function and biogenesis of Lipopolysaccharides. EcoSal Plus. 2018;8(1):10–128. doi:10.1128/ecosalplus.ESP-0001-2018.
- Rocha EPC. The organization of the bacterial genome. Annu Rev Genet. 2008;42(1):211–233. doi:10.1146/annurev.genet.42.110807.091653.
- Okuda S, Yoshizawa AC. ODB: a database for operon organizations, 2011 update. Nucleic Acids Res. 2011;39(Database):D552–D555. doi:10.1093/nar/gkq1090. From NLM.
- Taboada B, Ciria R, Martinez-Guerrero CE, Merino E. ProOpDB: Prokaryotic Operon DataBase. Nucleic Acids Res. 2012;40(D1):D627–D631. doi:10.1093/nar/gkr1020.
- Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, Mao F, Lai W, Xu Y. DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2013;42(D1):D654–D659. Accessed 2023 Jun 18. doi:10.1093/nar/gkt1048.
- Groussin M, Poyet M, Sistiaga A, Kearney SM, Moniz K, Noel M, Hooker J, Gibbons SM, Segurel L, Froment A. et al., Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell. 2021;184(8):2053–2067.e2018. doi:10.1016/j.cell.2021.02.052.
- Chamarande J, Cunat L, Alauzet C, Cailliez-Grimal C. In silico study of cell surface structures of parabacteroides distasonis involved in its maintenance within the gut microbiota. Int J Mol Sci. 2022;23(16):9411. doi:10.3390/ijms23169411.
- Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a web browser. BMC Bioinf. 2011;12(1):385. doi:10.1186/1471-2105-12-385.
- Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30(1):42–46. doi:10.1093/nar/30.1.42.
- Cui W, Morrison DC, Silverstein R, Moore RN. Differential tumor necrosis factor alpha expression and release from peritoneal mouse macrophages in vitro in response to proliferating gram-positive versus gram-negative bacteria. Infect Immun. 2000;68(8):4422–4429. doi:10.1128/IAI.68.8.4422-4429.2000.
- Kennedy MS, Zhang M, DeLeon O, Bissell J, Trigodet F, Lolans K, Temelkova S, Carroll KT, Fiebig A, Deutschbauer A. et al., Dynamic genetic adaptation of bacteroides thetaiotaomicron during murine gut colonization. Cell Rep. 2023;42(8):113009. doi:10.1016/j.celrep.2023.113009.
- Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10(5):323–335. doi:10.1038/nrmicro2746.
- Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex Glycan Catabolism by the human gut microbiota: the Bacteroidetes Sus-like Paradigm. J Biol Chem. 2009;284(37):24673–24677. Accessed 2023 Mar 13. doi:10.1074/jbc.R109.022848.
- Giulieri SG, Guérillot R, Duchene S, Hachani A, Daniel D, Seemann T, Davis JS, Tong SYC, Young BC, Wilson DJ. et al., Niche-specific genome degradation and convergent evolution shaping. Elife. 2022;11:11. doi:10.7554/eLife.77195.
- Pinto C, Melo-Miranda R, Gordo I, Sousa A. The selective advantage of the lac operon for Escherichia coli is conditional on diet and microbiota composition. Front Microbiol. 2021;12:709259. doi:10.3389/fmicb.2021.709259.
- Klena JD, Schnaitman CA. Function of the rfb gene cluster and the rfe gene in the synthesis of O antigen by shigella dysenteriae 1. Mol Microbiol. 1993;9(2):393–402. doi:10.1111/j.1365-2958.1993.tb01700.x.
- Liu D, Reeves PR. Escherichia coli K12 regains its O antigen. Microbiol. 1994;140(Pt 1):49–57. doi:10.1099/13500872-140-1-49. From NLM.
- Boels IC, Beerthuyzen MM, Kosters MH, Van Kaauwen MP, Kleerebezem M, De Vos WM. Identification and functional characterization of the Lactococcus lactis rfb operon, required for dTDP-rhamnose biosynthesis. J Bacteriol. 2004;186(5):1239–1248. doi:10.1128/JB.186.5.1239-1248.2004.
- Liang J, Li X, Zha T, Chen Y, Hao H, Liu C, Duan R, Xiao Y, Su M, Wang X. et al., DTDP-rhamnosyl transferase RfbF, is a newfound receptor-related regulatory protein for phage phiYe-F10 specific for Yersinia enterocolitica serotype O: 3. Sci Rep. 2016;6(1):22905. doi:10.1038/srep22905.
- Chao CM, Liu WL, Lai CC. Peritoneal dialysis peritonitis caused by bacteroides thetaiotaomicron. Perit Dial Int. 2013;33(6):711–712. doi:10.3747/pdi.2012.00229.
- Faur D, García-Méndez I, Martín-Alemany N, Vallès-Prats M. Mono-bacterial peritonitis caused by bacteroides thetaiotaomicron in a patient on peritoneal dialysis. Nefrología (English Edition). 2012;32(5):694. doi:10.3265/Nefrologia.pre2012.Jun.11568.
- Chao CT, Lee SY, Yang WS, Chen HW, Fang CC, Yen CJ, Chiang CK, Hung KY, Huang JW. Peritoneal dialysis peritonitis by anaerobic pathogens: a retrospective case series. BMC Nephrol. 2013;14(1):111. doi:10.1186/1471-2369-14-111. From NLM.
- Montravers P, Gauzit R, Muller C, Marmuse JP, Fichelle A, Desmonts JM. Emergence of antibiotic-resistant bacteria in cases of peritonitis after intraabdominal surgery affects the efficacy of empirical antimicrobial therapy. Clin Infect Dis. 1996;23(3):486–494. doi:10.1093/clinids/23.3.486. From NLM.
- Durazzi F, Sala C, Castellani G, Manfreda G, Remondini D, De Cesare A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci Rep. 2021;11(1):3030. doi:10.1038/s41598-021-82726-y.
- Fernández-Veledo S, Vendrell J. Gut microbiota-derived succinate: friend or foe in human metabolic diseases? Rev Endocr Metab Disord. 2019;20(4):439–447. doi:10.1007/s11154-019-09513-z.
- Connors J, Dawe N, Van Limbergen J. The role of succinate in the Regulation of Intestinal Inflammation. Nutrients. 2018;11(1):25. doi:10.3390/nu11010025.
- Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal component analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–W570. Accessed 2021 Nov 11. doi:10.1093/nar/gkv468.
- Basson AR, Cominelli F, Rodriguez-Palacios A. ‘Statistical irreproducibility’ does not improve with larger sample size: how to quantify and address disease data multimodality in human and animal research. J Pers Med. 2021;11(3):234. doi:10.3390/jpm11030234.
- Alneberg J, Karlsson CMG, Divne A-M, Bergin C, Homa F, Lindh MV, Hugerth LW, Ettema TJG, Bertilsson S, Andersson AF. et al., Genomes from uncultivated prokaryotes: a comparison of metagenome-assembled and single-amplified genomes. Microbiome. 2018;6(1):173. doi:10.1186/s40168-018-0550-0.
- Parrello B, Butler R, Chlenski P, Pusch GD, Overbeek R, Kalendar R. Supervised extraction of near-complete genomes from metagenomic samples: a new service in PATRIC. PLOS ONE. 2021;16(4):e0250092. doi:10.1371/journal.pone.0250092.
- Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM. et al., The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 2020;48(D1):D606–D612. doi:10.1093/nar/gkz943.
- Olson RD, Assaf R, Brettin T, Conrad N, Cucinell C, Davis JJ, Dempsey DM, Dickerman A, Dietrich EM, Kenyon RW. et al., Introducing the bacterial and viral bioinformatics resource center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023;51(D1):D678–D689. doi:10.1093/nar/gkac1003.