
Abstract
Mice treated with anthrax lethal toxin (LT) exhibit hemorrhage caused by unknown mechanisms. Moreover, LT treatment in mice induced liver damage. In this study, we hypothesized that a suppressed coagulation function may be associated with liver damage, because the liver is the major producing source of coagulation factors. The hepatic expression of coagulant factors and the survival rates were analyzed after cultured cells or mice were exposed to LT. In agreement with our hypothesis, LT induces cytotoxicity against hepatic cells in vitro. In addition, suppressed expression of coagulation factor VIII (FVIII) in the liver is associated with a prolonged plasma clotting time in LT-treated mice, suggesting a suppressive role of LT in coagulation. Accordingly, we further hypothesized that a loss-of-function approach involving treatments of an anticoagulant should exacerbate LT-induced abnormalities, whereas a gain-of-function approach involving injections of recombinant FVIII to complement the coagulation deficiency should ameliorate the pathogenesis. As expected, a sublethal dose of LT caused mortality in the mice that were non-lethally pretreated with an anticoagulant (warfarin). By contrast, treatments of recombinant FVIII reduced the mortality from a lethal dose of LT in mice. Our results indicated that LT-induced deficiency of FVIII is involved in LT-mediated pathogenesis. Using recombinant FVIII to correct the coagulant defect may enable developing a new strategy to treat anthrax.
Introduction
Anthrax refers to diseases caused by infection from the Gram-positive bacterium Bacillus anthracis. Comprising 2 protein components, protective antigen (PA) and lethal factor (LF), anthrax lethal toxin (LT) is one of the major virulence factors of B. anthracis.Citation1-3 Injection of LT is sufficient to induced mortality in rodents such as mice and rats.Citation4,5 A specific protease of mitogen-activated protein kinase kinase (MAPKK, also known as MEK), the LT component LF cleaves MEK, resulting in the inhibition of MAPK signaling pathways and, thus, inhibiting vital functions of target cells.Citation1-3 Although LT treatments cannot address the full complexity of B. anthracis infections, reductionist approaches that involve using LT to treat cells or animals enable clarifying how LT exerts pathogenic roles. LT causes multiorgan dysfunction, the major targets of which seem to be the heart and smooth muscles.Citation6 However, the reason both clinical and animal studies have revealed that hemorrhagic disorder is one of the major manifestations of LT-treated or B. anthracis-infected hosts remains unclear.Citation7-13 Recent studies have shown that LT might contribute to hemorrhage by inducing thrombocytopenia and megakaryocytic suppression.Citation14-16 The mechanism underlying coagulation suppression, however, remains undetermined.
Blood-derived hemostatic machinery consists of platelets and coagulation factors.Citation17 Our previous study demonstrated an inhibitory role of LT on platelet activation.Citation14 In the present study, we further found that LT challenges suppressed and prolonged the plasma clotting of mice. In this experiment, platelet-poor plasma was affected, suggesting that LT suppresses not only platelets but also coagulant factors of the plasma. The parenchymal cells of the liver synthesized most coagulant and anticoagulant factors.Citation18 Various types of liver damage induced hemostatic abnormalities and hemorrhage.Citation19 Previous studies have revealed that anthrax induces liver damage; invasions of bacterium B. anthracis have been observed frequently in the liver of infected patientsCitation20-22 and animals.Citation15,23,24 These results prompted us to investigate whether coagulant factor defects are associated with LT-induced liver damage. Herein, we determined that LT challenges induce toxicity toward hepatic cells in vitro and in vivo. In addition, such suppression is associated with a prolonged plasma clotting time and the suppression of coagulant factor VIII (FVIII) in mice. Furthermore, administrations of recombinant FVIII (rFVIII) reduced the mortality of mice challenged with a lethal dose of LT. This paper discusses the potential use of rFVIII for treating LT-induced coagulant deficiency.
Results
Lethal toxin-induced liver damage
We examined LT-induced liver damage in mice, noting a difference between lethal and sublethal LT treatments in C57Bl/6J mice (LT, lethal dose 4.4 mg/kg, sublethal dose 3 mg/kg; LF:PA = 1:5 [w/w]; ). Hematoxylin and eosin stained liver sections revealed that lethal LT treatments, but not PA treatments, induced a leakage of red blood cells into the interstitial space of the liver (), indicating that blood vessels ruptured in the liver. To investigate the levels of extravascular plasma, the Evans blue dye that remained in the mouse liver was recorded after systemic perfusion. The extravascular plasma in the liver significantly increased after LT treatments, but not after PA treatments (; **P < 0.01 and, *P < 0.05). To evaluate the extent of liver damage, we measured the levels of hepatic enzyme aspartate aminotransferase and alanine aminotransferase (AST/ALT) in mouse sera. In agreement with previous reports,Citation15,25 both ASL and ALT levels were substantially elevated (; PA vs. LT in 96-h groups) at the time point proximal to the occurrence of mortality (; day 4).
Figure 1. LT-induced liver damage. Mortality of mice treated with PA (4.4 mg/kg) and LT (4.4 mg/kg, lethal dose; or 3 mg/kg, sublethal dose) (A) (n = 8) was plotted as Kaplan–Meier curves (PA vs. LT, **P < 0.01). Analyses data were obtained from animals with the same type of treatments as indicated in Panel A (B–E). Hematoxylin and eosin staining of liver sections from PA (B) and lethal dose LT (C) treated mice (96-h treatment). Arrows indicate the blood cell leaked into liver sinusoid (C). Liver retained Evans blue levels, which indicates the plasma leakage (D) (96-h treatment). Serum aspartate aminotransferase (ASL) and alanine aminotransferase (ALT) levels of mice treated with PA and LT for 0, 72, and 96 h, respectively; *P < 0.05, compared with respective PA groups (E). n = 6 (D, E).
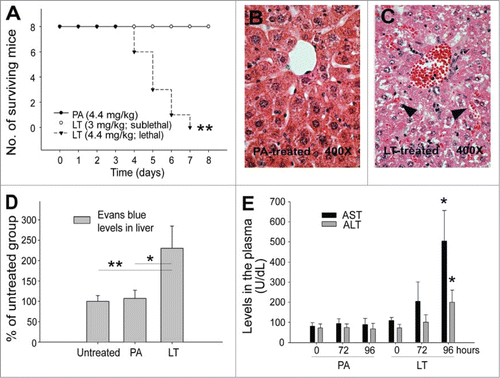
LT treatments prolong plasma clotting
The D-dimer is a fibrinolytic degradation product of fibrinogen (coagulation factor I).Citation26 The reaction of thrombin (coagulation factor II) with its major inhibitor antithrombin III causes formation of thrombin–antithrombin (TAT).Citation27 Therefore, D-dimer and TAT are both proteolytic products of coagulation factors after the activation of coagulation and subsequent fibrinolytic cascades.Citation26,28 Consequently, elicitation of circulating D-dimer and TAT suggests an induction of coagulation activation or disseminated intravascular coagulation (DIC).Citation26,28 To investigate role of LT on the induction of DIC, which was reported in B. anthracis-challenged animals,Citation29-31 we analyzed the plasma levels of D-dimer and TAT complex (). We noted that unlike in lipopolysaccharide (LPS)-challenged control groups, LT treatments did not elicit considerable plasma D-dimer or TAT (), indicating a lack of coagulation activation and coagulopathy. The data of plasma recalcification clotting time further indicated that LT treatments suppressed the clotting of plasma (, LT vs. other groups). Such suppressive effects were observed only after LT treatments in vivo (); hence, LT might not suppress coagulant factors directly through the protein cleavage by the LF protease. These results implied that LT treatments elicit coagulation suppression rather than coagulation activation or DIC in mice, a phenomenon that is in agreement with a recent report that B. anthracis cell-wall components, but not LT, induces DIC.Citation30 The coagulation initiation cascade can be divided into intrinsic (activated inside the blood vessel) and extrinsic (activated outside the blood vessel, e.g., in tissues) pathways.Citation32 To determine whether an intrinsic or extrinsic coagulation pathway is involved in LT-mediated suppression, analyses of the activated partial thromboplastin time (APTT; normal reaction requires the presence of intrinsic coagulation factors VIII, IX, X, XI, and XII) and prothrombin time (PT; requires extrinsic coagulation factor VII) were conducted.Citation32 Our data indicated that LT mediates differential suppressive effects (APTT vs. PT) via different time courses. The intrinsic pathway was suppressed significantly earlier, 72 h after LT treatments, whereas the extrinsic pathway was suppressed much later, 96 h after LT treatments, a time point corresponding to the occurrence of mortality (, ; day 4 and 96-h groups). Because the LT treatments indirectly suppressed plasma clotting, and the intrinsic pathway was more sensitive to LT than was the extrinsic pathway, we hypothesized that LT might suppress the liver production of at least one key factor in the intrinsic pathway.
Figure 2. D-dimer, TAT and clotting time analysis. D-dimer analysis of plasma from untreated mice or mice treated with PA (4.4 mg/kg), LT (4.4 mg/kg), and LPS (5 ng/kg) for 0, 6, 24 and 72 h, respectively (A). TAT analysis of plasma from untreated, PA, LT, and LPS treated mice (72 h) (B). Recalcification plasma clotting time analysis of plasma samples collected from untreated, PA, LT, and LPS treated mice (72 h) (C), or normal mice plasma treated with or without PA, LT or anti-PA Ig preincubated LT (LT + anti-PA) (2 h in vitro) (D). Activated partial thromboplastin time (APTT) and prothrombin time (PT) analysis of plasma from untreated, PA, and LT treated mice (E). *P < 0.05, **P < 0.01, compared to respective PA groups. n = 6 (A–E).
LT-mediated suppression of the liver
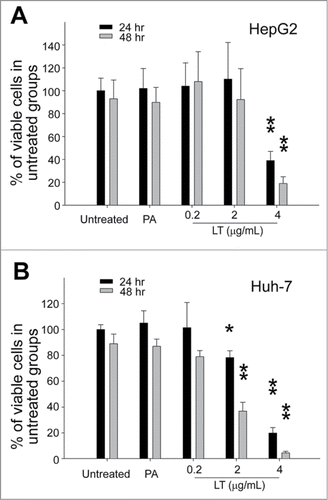
A cell culture model and a mouse model were further used to investigate whether LT treatments could disturb hepatic cell function. Human hepatoma HepG2 and Huh-7 cell lines were used to determine the cytotoxicity of LT against hepatic cells in vitro. Both hepatic cell lines died rapidly when treated with a high dose (; 4 μg/mL), and Huh-7 cells were more sensitive to LT treatments at lower doses (; 0.2 μg/mL, 2 μg/mL).
Figure 3. Cytotoxicity analysis. Relative cellular surviving rates of human hepatoma HepG2 (A) or Huh-7 (B) cells treated with or without PA (4 μg/mL), or LT (0.2, 2, and 4 μg/mL). *P < 0.05, **P < 0.01, compared to respective PA groups. n = 6 (A, B).
LT is a pan-MEK/MAPK axis inhibitor.Citation2,3 However, during LT treatments, inhibition of each MAPK pathway (p42/44 MAPK, p38 MAPK, and c-Jun N-terminal kinase [JNK]) may not contribute equally to the suppression of cellular function in various types of cells. For example, among 3 MAPK pathways, only p38 MAPK is involved in LT-induced macrophage apoptosis,Citation33 whereas both p42/44 and p38 MAPKs are associated with LT-mediated suppression on platelets.Citation14 Consequently, we applied a pharmacological approach to investigate which MAPK pathway is involved in the suppression of hepatic cells. The selective inhibitors U0126 (MEK1/2 inhibitor; MEK1/2 are upstream regulators of p42/44 MAPK), SB202190 (p38 inhibitor) and SP600125 (JNK inhibitor), all of which have been compared with LT in inhibiting MAPKs,Citation34,35 were employed. The cell survival and caspase-3 activities of HepG2 cells were examined. Analysis results revealed that only treatments of U0126, but not SB202190 and SP600125, significantly induced cell death (Fig. S1A, B), suggesting that MEK1/2 and the down-stream p42/44 MAPK pathways are involved. Similarly, Western blotting analysis revealed that MEK1 and the pp42/44 MAPK were both suppressed after LT treatment (Fig. S1C, D; quantified data). Because the inhibitor SB202190 did not induce cell death (Fig. S1A, B), the inhibition of pp38 MAPK (Fig. S1C; LT) may not pertain to cell survival.
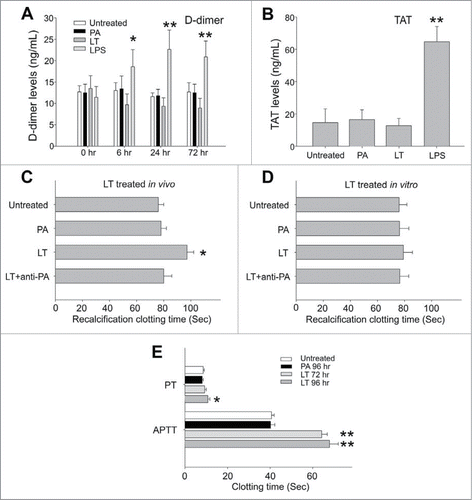
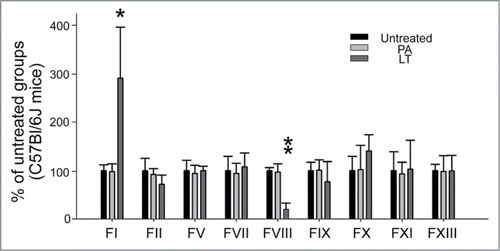
To investigate whether LT caused the abnormal expression of coagulation factors in the liver, coagulation factors factor I (FI; fibrinogen), FII (thrombin), FV, FVII, FVIII, FIX, FX, FXI, and FXIII were analyzed in a mouse model (). Among these, only the mRNA expressions of FI and FVIII were significantly disturbed by LT treatments; FI was upregulated and FVIII was downregulated (). FI is the most abundant downstream coagulation factor in circulation.Citation36 A plasma FI level of 1 g/L is sufficient for adequate hemostasis.Citation37,38 We found that FI was expressed in an excessive level in the circulation of normal mice (C57Bl/6J, 1.8 ± 0.3 g/L; n = 6), suggesting that the upstream and less abundant factors are more critical for controlling clotting efficiency. Notably, only the suppressed coagulant FVIII played a critical role in the intrinsic coagulation pathway (; FVIII groups). This result was consistent with the prolonged APTT, used for analyzing intrinsic coagulation pathways, that was observed after LT treatments (; 72-h groups). To analyze whether LT caused the abnormal expression of coagulation factors in cultured hepatic cells, a HepG2 cell line was employed. Compared with that of Huh-7 cells, the behavior of HepG2 cells in response to sub-cytotoxic LT doses may be more relevant to a pathway-specific inhibition of the toxin because of the lower sensitivity, but not because of the cell death response. Therefore, an RT-PCR analysis of sub-cytotoxic (2 μg/mL) LT-treated HepG2 cells was investigated. The data showed that the mRNA expression of FI was upregulated and the FII was downregulated after LT treatments (Supplementary Fig. 2A, B); responses were partly similar to those in the in vivo liver analysis (; FI was upregulated and FII tended to be downregulated). Because the mRNA expression levels were low, we could not obtain RT-PCR data for other coagulation factors (including FVIII) in the HepG2 cell line.
Figure 4. mRNA expression of coagulant factors in mice liver. The RT-PCR analysis for coagulant-factor-mRNA was measured according to gel intensities. The mRNA levels of coagulant factors FI, FII, FV, FVII, FVIII, FIX, FX, FXI, and FXIII were detected using RT-PCR and liver samples from mice treated with or without PA or LT for 72 h. Data were first normalized with internal control actin; untreated groups were subsequently adjusted to 100% during each time course. *P < 0.05, **P < 0.01, compared to respective PA-treated groups (n = 6).
LT-induced FVIII deficiency
Factor VIII deficiency could result in coagulant defects and a prolonged APTT.Citation39 Commercial and laboratory prepared coagulation-factor-deficient plasma has been used to analyze and ensure a single factor deficiency of tested plasma.Citation40-42 For example, when commercial-derived FVIII-deficient plasma is mixed with patient plasma samples from a normal donor and a patient with hemophilia A (FVIII deficient), because the normal plasma complements functional FVIII, the APTT of the mixed plasma differs significantly between the normal donor and the patient. By contrast, when FVIII-deficient plasma is mixed with plasma samples from a normal donor and a patient with hemophilia B (FIX deficient), because both normal and hemophilia B plasma contain functional FVIII, and FVIII-deficient plasma contains functional FIX, the clotting time of these 2 mixed plasma does not differ significantly. Accordingly, analysis using FVIII-deficient plasma was further employed to verify whether LT suppresses FVIII at the protein level. Theoretically, the prolonged APTT of FVIII-deficient plasma can be corrected through premixing with normal plasma.Citation41 Our data showed that the plasma from PA-treated mice, but not that from LT-treated mice, significantly corrected the prolonged APTT of FVIII-deficient plasma (; PA vs. LT, FVIII-deficient plasma groups), indicating that a FVIII deficiency occurred after the LT treatments. This result was further verified using enzyme-linked immunosorbent assay (ELISA) kits. The data revealed that the LT-treatments significantly suppressed the expression levels of FVIII, but not FVII, in the plasma of LT-treated mice (; *P < 0.05), a result that corresponds with the RT-PCR () and FVIII-deficient plasma () analyses results.
Figure 5. Analysis of FVIII deficiency. The FVIII defects were analyzed using FVIII-deficient plasma (A) and an FVIII ELISA kit (B), respectively. After treatment with PA or LT for 72 h, APTT analysis was performed using various ratios of combined FVIII-deficient plasma and mouse sample plasma (A). The mice plasma + normal plasma groups (mice plasma/normal plasma = 1/5 [v/v]) served as a basal level, in which the deficiency of coagulation factor in mice plasma was complemented by normal plasma and thus exhibited the fastest clotting response (n = 6). For ELISA analyses, plasma levels of factor VIII (FVIII) and factor VII (FVII) were measured (B) (n = 4). Respective untreated groups were normalized to 100%. *P < 0.05, **P < 0.01, compared to respective PA-treated groups.
![Figure 5. Analysis of FVIII deficiency. The FVIII defects were analyzed using FVIII-deficient plasma (A) and an FVIII ELISA kit (B), respectively. After treatment with PA or LT for 72 h, APTT analysis was performed using various ratios of combined FVIII-deficient plasma and mouse sample plasma (A). The mice plasma + normal plasma groups (mice plasma/normal plasma = 1/5 [v/v]) served as a basal level, in which the deficiency of coagulation factor in mice plasma was complemented by normal plasma and thus exhibited the fastest clotting response (n = 6). For ELISA analyses, plasma levels of factor VIII (FVIII) and factor VII (FVII) were measured (B) (n = 4). Respective untreated groups were normalized to 100%. *P < 0.05, **P < 0.01, compared to respective PA-treated groups.](/cms/asset/d8ca5a5d-37cd-4dd3-a877-ab8d20908578/kvir_a_1031454_f0005_b.gif)
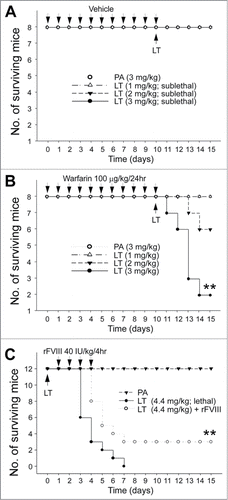
Improving or reducing the survival rate of LT-treated mice through coagulation intervention
The aforementioned results suggested that coagulation suppression is part of LT-mediated pathophysiology, prompting us to hypothesize that anticoagulant treatments may exacerbate and recombinant FVIII (rFVIII) treatments may ameliorate LT-mediated pathogenesis. Warfarin, an inhibitor of vitamin K dependent coagulation factors, is an anticoagulant normally used in preventing thrombosis.Citation43 Pretreatments of sublethal doses of warfarin were used to investigate whether hypocoagulant stress could influence the survival rate of LT-treated mice. The analysis results revealed that when compared with the mice in the PA control groups, only warfarinized mice exhibited sublethal LT-induced mortality dose dependently (, no mortality; , 1, 2, and 3 mg/kg of LT induced no mortality, 2/8 mortality, and 6/8 mortality, respectively). Recombinant FVIII was also used to investigate whether the correction of FVIII deficiency can increase the survival rate. Our results suggested that the rFVIII treatments considerably improved the survival rate of mice treated with a lethal dose of LT (; **P < 0.01, LT vs. LT + rFVIII groups).
Figure 6. Mortality analysis. Sublethal doses PA (3 mg/kg) and LT (1, 2, and 3 mg/kg) were used to challenge mice with (warfarin) or without (vehicle) 10-d pretreatment of sublethal warfarin (100 mg/kg/day) (A, B) (n = 8), plotted as Kaplan–Meier curves (PA vs. LT, **P < 0.01). No mortality occurred when sublethal doses of LT (1, 2, and 3 mg/kg) were used to challenge mice without warfarin preconditioning (A). Recombinant FVIII (rFVIII; 40 IU/kg/4hr) was used to rescue lethal-dose LT (4.4 mg/kg) challenged mice (C) (n = 12). **P < 0.01, LT vs. LT + rFVIII.
Discussion
Hemorrhage is a common manifestation of anthrax. In clinical studies, hemorrhage occurs in the draining lymph nodes, bowel segments, and meninges.Citation11,44,45 In animal models, hemorrhage was observed in mediastinal, mesenteric, and tracheobronchial lymph nodes, as well as in the meninges, lungs, and small intestinal serosas of B. anthracis-infected animals.Citation11 Because LT treatments induce hemorrhage,Citation14,46 these results might indicate that anthrax LT is involved in the hemorrhagic pathogenesis. Previously, we determined that LT suppresses platelet functionsCitation14; however, the influence of LT treatments on coagulant factors remained unclear. In the present study, we found that the expression of FVIII in mouse livers was suppressed using LT treatments at a disease stage before to the occurrence of mortality. This is the first study indicating that an acquired FVIII deficiency is induced by a pathogen. Anthrax LT blocks MAPK-C/EBP–dependent pathways (Fig. S1C).Citation1-3 Because the FVIII acute phase response requires the participation of C/EBP,Citation47 the inhibition of FVIII expression by LT treatments is reasonable.
Coagulation inhibition is the phenomenon of bacteria spreading in the host.Citation48 An example of the primitive association between the host coagulation system and the pathogens occurs in the horseshoe crab.Citation49 Endotoxin can induce a clotting response that presumably inhibits the spreading of bacteria, providing an initial defense against invasion.Citation48,49 In higher mammals, the entrapment of bacteria in a fibrin deposit might diminish the magnitude of bacterial dissemination; the host might therefore benefit from the encapsulation of the microorganism using fibrin and platelet clots.Citation48,50,51 This is probably why various pathogenic bacteria have evolved molecular mechanisms to interfere with the coagulation and fibrinolysis systems.Citation48 Accordingly, the anticoagulant property of LT is theoretically more relevant to the beneficial LT-mediated effects during real B. anthracis infections in which LT is used to untangle the constraints of blood clots surrounding the invading bacteria and facilitates the spreading of bacteria in the host. Consequently, correction for coagulant defects might be used passively for preventing hemorrhage and actively for enhancing immunity. This hypothesis remains unconfirmed; however, the findings from one of our previous studies might support this hypothesis.Citation52 LT treatments can greatly enhance the mortality of mice with sublethal Escherichia coli.Citation52 The suppression of macrophage is likely involved, and constructing a reduced coagulation system remains feasible.Citation52
We suggest a novel strategy for coagulation intervention to treat anthrax. Approved by the US Food and Drug Administration, rFVIII is a drug used for genetic and acquired FVIII deficiencies.Citation53,54 The drug rFVIII may enable enhancing coagulation in patients with anthrax who exhibit severe hemorrhage and coagulopathy, as described in clinical settings.Citation9-12,31 To develop an effective treatment against anthrax, combining other anti-anthrax agents with a coagulation intervention strategy may be a feasible approach.
Materials and Methods
Bacterial strain, toxin, mice, and recombinant factor VIII
The Bacillus anthracis toxin protective antigen (PA) and lethal factor (LF) were purified from B. anthracis (ATCC 14186) culture supernatants as described in previous studies.Citation4,5,14,16,52,55,56 For cell and animal studies, high-performance liquid chromatography (HPLC)-purified LF and PA were reconstituted as LT in a ratio of 1:5, which resembles the ratio of PA and LF in the culture supernatants of B. anthracis. The LPS contamination was monitored using a Limulus Amoebocyte Lysate QCL-1000 kit (Lonza, Walkersville, MD, USA).Citation4,57 Batches of purified LT, in which LPS contamination was <1 endotoxin unit (EU)/mg LT, were used, because treatments of LPS at this level cannot elicit significant inflammatory cytokine IL-1β and TNF-α secretion in vitro and in vivo (data not shown). An anti-PA antibody that neutralizes the LT-mediated cytotoxicity was purified from rabbit sera that were immunized with PA (100 µg per cycle at 3-wk intervals for 6 cycles). C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and injected intravenously with sublethal or lethal doses of LT according to different experimental settings (sublethal dose: 3 mg/kg LT, LF:PA = 1:5 [w/w]; lethal dose: 4.4 mg/kg LT; the mice completely succumbed at approximately 168 h post treatment). All animal experimental procedures described in this report were performed according to the approved guidelines of the Animal Care and Use Committee of Tzu-Chi University (approval ID: 97060 and 98104). Anthrax LT treatments in mice did not induce obvious discomfort except a reduction in activity during initial treatments. However, activity reduction was a general phenomenon, appearing in all mice, including those that survived the LT-treatments. This suggested that reduced activity cannot serve as a predictor of mortality. We measured the body weight 69 h after LT treatments; no significant body weight decrease in the LT-challenged mice was observed, suggesting that these mice maintained a normal food and water uptake during the treatments. After the experiments, surviving mice were euthanized using CO2 according to a National Institutes of Health (NIH) guideline (http://oacu.od.nih.gov/ARAC/documents/Rodent_Euthanasia_Adult.pdf). The mice were continually examined every 8–12 h for as many as 15 d during the mortality experiments. The surviving mice were monitored every day for 2 mo. Chemicals and proteins such as lipopolysaccharide (LPS, E. coli) and bovine serum albumin were purchased from Sigma–Aldrich (St. Louis, MO, USA), and recombinant factor VIII (rFVIII) was purchased from Baxter (Deerfield, IL, USA).
Histopathology
Tissue samples were prepared from mice exhibiting symptoms of paralysis approximately 90–100 h post-lethal LT treatments. Samples were fixed by immersion in 10% neutral buffered formalin and then dehydrated and embedded in paraffin. Tissues were sectioned in 5–6 μm slices and stained with hematoxylin and eosin for light microscopy.
Plasma leakage analysis
To measure the plasma leakage, each of the mice received a single intravenous injection of Evans blue dye followed by lethal LT treatments.Citation58,59 After 72 h post-LT treatment (4.4 mg/kg), the mice were euthanized and the tissues were removed and minced before being incubated with formamide-water (1:1) for 48 h at 37°C. The concentration of dye was determined according to a standard curve of Evans blue in formamide using a spectrophotometer (Hitachi, Japan).
Clotting time analysis
The plasma recalcification clotting time, activated partial thromboplastin time (APTT) and prothrombin time (PT) of mice treated with lethal LT were measured. The plasma recalcification clotting time was measured according to previously described methods.Citation60 APTT and PT analyses were performed to differentiate the intrinsic and extrinsic coagulation pathways using a coagulometer (ACL -Futura Plus, Instrumentation Laboratory, Milan, Italy), following the manufacturer's instructions.Citation4,14 APTT was used to determine the intrinsic pathway (also known as the contact activation pathway) of coagulation; to activate this pathway, phospholipid and an activator (e.g., silica) were added to the plasma samples.Citation61 PT was used to determine the extrinsic pathway (also known as the tissue factor pathway) of coagulation; to activate this pathway, tissue factor was added.Citation62 The time was optically measured until a thrombus (clot) formed. For in vitro analysis, the plasma samples were subjected to LT treatments (2 µg/mL) for 2 h at 37°C. To analyze recalcification clotting time, mouse plasma was collected 72 h post-LT treatment (4.4 mg/kg). To evaluate APTT and PT, mouse plasma was collected 0, 72 and 96 h post-LT treatment (4.4 mg/kg).
Aspartate aminotransferase and alanine transaminase analyses
To analyze the liver function, whole blood samples (50–100 μL) of mice were collected from the retro-orbital venous plexus and mixed with anticoagulant citrate dextrose solution (38 mM citric acid, 75 mM sodium citrate, 100 mM dextrose) in Eppendorf tubes.Citation14,63,64 The levels of aspartate aminotransferase (AST) and alanine transaminase (ALT) in the plasma of mice were then measured using a clinical biochemistry analysis system (COBAS INTEGRA® 800, Roche)Citation63,65 at various time intervals (0, 72, 96 h after LT treatments).
Cell viability analysis
The analysis was performed as previously described.Citation52 Human hepatoma cell lines HepG2 (ATCC HB8055) and Huh-7 were maintained in a Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. After the cells were changed to a serum-free medium, the cells (2 × 104/well) were treated with various concentrations (0.2, 2, or 4 μg/mL) of LT (LF:PA = 1:5) in a 96-well cell culture dish. The level of viable cells was analyzed using the WST-1 kit (Roche Diagnostics, Taipei, Taiwan) 3 h after the treatments, according to manufacturer's instructions.Citation52 Tetrazolium salt WST-1 reduced to formazan by cellular dehydrogenases in viable cells generates yellow formazan, which is measurable at 450 nm and correlated to the number of cells. A standard curve plotted using WST-1 values to indicate seriously diluted cells (0–5 × 105) was obtained first; the relative number of viable cells was then calculated accordingly.
RT-PCR analysis
LT-treated HepG2 cells (2 μg/mL, 48 h) or mouse livers from LT-treated mice (4.4 mg/kg, 72 h; a lethal dose) were collected and stored in RNAlater (Ambion–Life Technologies, Carlsbad, CA, USA) at −80°C before the total RNA was extracted using an RNAqueous-4PCR kit (Ambion–Life Technologies). Two micrograms of total RNA were reverse transcribed into cDNA using SuperScript II (Invitrogen–Life Technologies) and amplified using an Advantage-GC 2 PCR kit (BD Biosciences, San Jose, CA, USA). The PCR products were then analyzed through electrophoresis using 2% agarose gels. Table S1 lists the primer sequences used in this study.
Western blotting analysis
Western blotting analysis was performed as previously described.Citation14 Antibodies against MAPK pathway kinases (MEK1, p42/44, phospho-p42/44 [pp42/44], and p38) were purchased from Cell Signaling Technology (Danvers, MA, USA), and anti-pp38 immunoglobulin was purchased from Promega (Madison, WI, USA). The gel intensities were measured using ImageJ (Version 1.32; NIH). The vehicle was dimethyl sulfoxide, PBS, or both, or, in different experiments, a solvent control. The doses of reagents used were as follows: U0126, 40 µmol/L; SB202190, 20 µmol/L; and SP600125, 50 µmol/L.
Factor VIII, factor VII, factor-deficient plasma, and ELISA analyses
Blood samples were obtained from the experimental mice via the retro-orbital plexus route. The samples were collected in polypropylene Eppendorf tubes containing a 2/10 volume of 3.2% (wt/vol) trisodium citrate. Plasma was prepared by centrifuging blood (2000 g) for 10 min at 4°C that was subsequently immediately snap-frozen in liquid nitrogen and stored at −80°C before analysis. The FVIII-deficient plasma was purchased from Sigma–Aldrich (St. Louis, MO, USA). Mouse plasma FVIII and FVII levels were measured using a USCN Life Science FVIII ELISA kit (Houston, TX, USA) and an Antibodies-Online FVII ELISA kit (Atlanta, GA, USA), respectively. Both analyses were performed according to the instructions from the suppliers. An enzygnost thrombin-antithrombin complex (TAT) kit (Dade Behring, Newark, DE, USA) and a D-dimer kit (Imuclone; American Diagnostica, Stamford, CT, USA) were used to detect plasma TAT and D-dimer levels. LPS was used as a positive control to induce D-dimer (5 ng/kg). For D-dimer analysis, plasma was collected from the mice 0, 6, 24, and 72 h after PA, LT (4.4 mg/kg), and LPS treatments. For TAT analysis, plasma was collected from the mice 72 h after PA, LT (4.4 mg/kg), and LPS treatments.
Influence of warfarin and rFVIII treatments in LT-induced mortality
C57BL/6J mice were treated intravenously with a sublethal dose (90 mg/kg/day) of the anticoagulant drug warfarin (3-[acetonylbenzyl]-4-hydroxycumarin) for 10 consecutive days. This course of treatment resulted in no mortalities and led to the development of a hypocoagulant state with a prolonged plasma clotting time. Sublethal doses of LT (1, 2, 3 mg/kg) were used 10 d after the first warfarin treatments, and the mortality was recorded. In the rFVIII rescue experiment, a lethal dose of LT (4.4 mg/kg) was injected into the C57Bl/6J mice. The rFVIII was then administered 24-h later at a dose of 40 international unit (IU)/kg/4 h for 3 consecutive days (a total of 18 injections). Again, the mortality of the mice was recorded.
Statistics
To calculate the means, standard deviations, and statistics for the quantifiable data, Microsoft Office Excel 2003, SigmaPlot 10, and SPSS 17 were used. The statistical significance of the data was determined using a one-way analysis of variance (ANOVA) followed by a post hoc Bonferroni-corrected t test. Univariate Kaplan–Meier analysis was applied to compare the difference in survival rates between groups with various treatments. To determine statistical significance, P values were calculated and log-rank tests were performed. A probability of type 1 error α = 0.05 was determined to be the threshold of statistical significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
DSS designed experiments and analyzed data; PCL, JHK, YLS, HHH, CRL, CCL, YPW and KCC performed part of experiments and analyzed data; and HHC did original experimental design, data analyses and manuscript drafting.
1031454_Supplemental_files.zip
Download Zip (437.1 KB)Acknowledgments
We are grateful to Professor Wang MH and his team at the Experimental Animal Center of Tzu-Chi University for animal care.
Funding
This work was supported by the grants of National Science Council (grants 96–2311-B-320–005-MY3 and 99–2311-B-320–003-MY3) and Tzu-Chi University (grants 610400130, TCIRP 95002; TCIRP 98001; TCIRP 101001).
Supplemental Materia
Supplemental data for this article can be accessed on the publisher's website.
References
- Dixon TC, Meselson M, Guillemin J, Hanna PC. Anthrax. N Engl J Med 1999; 341:815-26; PMID:10477781; http://dx.doi.org/10.1056/NEJM199909093411107
- Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol 2003; 19:45-70; PMID:14570563; http://dx.doi.org/10.1146/annurev.cellbio.19.111301.140655
- Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol 2004; 7:19-24; PMID:15036135; http://dx.doi.org/10.1016/j.mib.2003.12.001
- Kau JH, Shih YL, Lien TS, Lee CC, Huang HH, Lin HC, Sun DS, Chang HH. Activated protein C ameliorates Bacillus anthracis lethal toxin-induced lethal pathogenesis in rats. J Biomed Sci 2012; 19:98; PMID:23170801; http://dx.doi.org/10.1186/1423-0127-19-98
- Kau JH, Sun DS, Huang HH, Wong MS, Lin HC, Chang HH. Role of visible light-activated photocatalyst on the reduction of anthrax spore-induced mortality in mice. PloS One 2009; 4:e4167; PMID:19132100; http://dx.doi.org/10.1371/journal.pone.0004167
- Liu S, Zhang Y, Moayeri M, Liu J, Crown D, Fattah RJ, Wein AN, Yu ZX, Finkel T, Leppla SH. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013; 501:63-8; PMID:23995686; http://dx.doi.org/10.1038/nature12510
- Fritz DL, Jaax NK, Lawrence WB, Davis KJ, Pitt ML, Ezzell JW, Friedlander AM. Pathology of experimental inhalation anthrax in the rhesus monkey. Lab Invest 1995; 73:691-702; PMID:7474943
- Vasconcelos D, Barnewall R, Babin M, Hunt R, Estep J, Nielsen C, Carnes R, Carney J. Pathology of inhalation anthrax in cynomolgus monkeys (Macaca fascicularis). Lab Invest 2003; 83:1201-9; PMID:12920249; http://dx.doi.org/10.1097/01.LAB.0000080599.43791.01
- Abramova FA, Grinberg LM, Yampolskaya OV, Walker DH. Pathology of inhalational anthrax in 42 cases from the Sverdlovsk outbreak of 1979. Proc Natl Acad Sci U S A 1993; 90:2291-4; PMID:8460135; http://dx.doi.org/10.1073/pnas.90.6.2291
- Barakat LA, Quentzel HL, Jernigan JA, Kirschke DL, Griffith K, Spear SM, Kelley K, Barden D, Mayo D, Stephens DS, Popovic T, et al. Fatal inhalational anthrax in a 94-year-old Connecticut woman. Jama 2002; 287:863-8; PMID:11851578; http://dx.doi.org/10.1001/jama.287.7.863
- Shafazand S, Doyle R, Ruoss S, Weinacker A, Raffin TA. Inhalational anthrax: epidemiology, diagnosis, and management. Chest 1999; 116:1369-76; PMID:10559102; http://dx.doi.org/10.1378/chest.116.5.1369
- Shieh WJ, Guarner J, Paddock C, Greer P, Tatti K, Fischer M, Layton M, Philips M, Bresnitz E, Quinn CP, et al. The critical role of pathology in the investigation of bioterrorism-related cutaneous anthrax. Am J Pathol 2003; 163:1901-10; PMID:14578189; http://dx.doi.org/10.1016/S0002-9440(10)63548-1
- Suffin SC, Carnes WH, Kaufmann AF. Inhalation anthrax in a home craftsman. Hum Pathol 1978; 9:594-7; PMID:101438; http://dx.doi.org/10.1016/S0046-8177(78)80140-3
- Kau JH, Sun DS, Tsai WJ, Shyu HF, Huang HH, Lin HC, Chang HH. Antiplatelet activities of anthrax lethal toxin are associated with suppressed p42/44 and p38 mitogen-activated protein kinase pathways in the platelets. J Infect Dis 2005; 192:1465-74; PMID:16170766; http://dx.doi.org/10.1086/491477
- Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest 2003; 112:670-82; PMID:12952916; http://dx.doi.org/10.1172/JCI17991
- Chen PK, Chang HH, Lin GL, Wang TP, Lai YL, Lin TK, Hsieh MC, Kau JH, Huang HH, Hsu HL, et al. Suppressive effects of anthrax lethal toxin on megakaryopoiesis. PloS One 2013; 8:e59512; PMID:23555687; http://dx.doi.org/10.1371/journal.pone.0059512
- Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 2013; 93:327-58; PMID:23303912; http://dx.doi.org/10.1152/physrev.00016.2011
- DeSancho MT, Pastores SM. The liver and coagulation. Textbook of Hepatology: From Basic Science to Clinical Practice. In: Faber KN, Donaldson PT, Rodes J, Benhamou JP, Blei A, Reichen J, et al., eds. New York: Blackwell Publishers; 2007:255-63.
- Kelly DA, Tuddenham EG. Haemostatic problems in liver disease. Gut 1986; 27:339-49; PMID:3084341; http://dx.doi.org/10.1136/gut.27.3.339
- Jackson PJ, Hugh-Jones ME, Adair DM, Green G, Hill KK, Kuske CR, Grinberg LM, Abramova FA, Keim P. PCR analysis of tissue samples from the 1979 Sverdlovsk anthrax victims: the presence of multiple Bacillus anthracis strains in different victims. Proc Natl Acad Sci U S A 1998; 95:1224-9; PMID:9448313; http://dx.doi.org/10.1073/pnas.95.3.1224
- Guarner J, Jernigan JA, Shieh WJ, Tatti K, Flannagan LM, Stephens DS, Popovic T, Ashford DA, Perkins BA, Zaki SR, et al. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am J Pathol 2003; 163:701-9; PMID:12875989; http://dx.doi.org/10.1016/S0002-9440(10)63697-8
- Grinberg LM, Abramova FA, Yampolskaya OV, Walker DH, Smith JH. Quantitative pathology of inhalational anthrax I: quantitative microscopic findings. Mod Pathol 2001; 14:482-95; PMID:11353060; http://dx.doi.org/10.1038/modpathol.3880337
- Prince AS. The host response to anthrax lethal toxin: unexpected observations. J Clin Invest 2003; 112:656-8; PMID:12952914; http://dx.doi.org/10.1172/JCI200319581
- Altunkaynak BZ, Ozbek E. Histopathological effects of anthrax lethal factor on rat liver. J Immunotoxicol 2015; 12:33-9; PMID:24344743
- Sweeney DA, Cui X, Solomon SB, Vitberg DA, Migone TS, Scher D, Danner RL, Natanson C, Subramanian GM, Eichacker PQ. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J Infect Dis 2010; 202:1885-96; PMID:21067373; http://dx.doi.org/10.1086/657408
- Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998; 59:65-73; PMID:9723580; http://dx.doi.org/10.1002/(SICI)1096-8652(199809)59:1%3c65::AID-AJH13%3e3.0.CO;2-0
- Hoek JA, Sturk A, ten Cate JW, Lamping RJ, Berends F, Borm JJ. Laboratory and clinical evaluation of an assay of thrombin-antithrombin III complexes in plasma. Clin Chem 1988; 34:2058-62; PMID:3168216
- Lippi G, Cervellin G, Franchini M, Favaloro EJ. Biochemical markers for the diagnosis of venous thromboembolism: the past, present and future. J Thromb Thrombolysis 2010; 30:459-71; PMID:20213258; http://dx.doi.org/10.1007/s11239-010-0460-x
- Stearns-Kurosawa DJ, Lupu F, Taylor FB, Jr., Kinasewitz G, Kurosawa S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am J Pathol 2006; 169:433-44; PMID:16877346; http://dx.doi.org/10.2353/ajpath.2006.051330
- Qiu P, Li Y, Shiloach J, Cui X, Sun J, Trinh L, Kubler-Kielb J, Vinogradov E, Mani H, Al-Hamad M, et al. Bacillus anthracis cell wall peptidoglycan but not lethal or edema toxins produces changes consistent with disseminated intravascular coagulation in a rat model. J Infect Dis 2013; 208:978-89; PMID:23737601; http://dx.doi.org/10.1093/infdis/jit247
- Coggeshall KM, Lupu F, Ballard J, Metcalf JP, James JA, Farris D, Kurosawa S. The sepsis model: an emerging hypothesis for the lethality of inhalation anthrax. J Cell Mol Med 2013; 17:914-20; PMID:23742651; http://dx.doi.org/10.1111/jcmm.12075
- Raber MN. Coagulation Tests. Clinical Methods: The History, Physical, and Laboratory Examinations. In: Walker HK, Hall WD, Hurst JW, eds. Boston: Butterworths; 1990:739-42.
- Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002; 297:2048-51; PMID:12202685; http://dx.doi.org/10.1126/science.1073163
- Chow EM, Batty S, Mogridge J. Anthrax lethal toxin promotes dephosphorylation of TTP and formation of processing bodies. Cell Microbiol 2010; 12:557-68; PMID:19995385; http://dx.doi.org/10.1111/j.1462-5822.2009.01418.x
- Batty S, Chow EM, Kassam A, Der SD, Mogridge J. Inhibition of mitogen-activated protein kinase signalling by Bacillus anthracis lethal toxin causes destabilization of interleukin-8 mRNA. Cell Microbiol 2006; 8:130-8; PMID:16367872; http://dx.doi.org/10.1111/j.1462-5822.2005.00606.x
- Ariens RA. Fibrin(ogen) and thrombotic disease. J Thromb Haemost 2013; 11(Suppl 1):294-305; PMID:23809133; http://dx.doi.org/10.1111/jth.12229
- Ehmann WC, al-Mondhiry H. Congenital afibrinogenemia and splenic rupture. AmJ Med 1994; 96:92-4; PMID:8304369; http://dx.doi.org/10.1016/0002-9343(94)90123-6
- Shima M, Tanaka I, Sawamoto Y, Kanehiro H, Matsuo N, Nishimura A, Giddings JC, Yoshioka A. Successful treatment of two brothers with congenital afibrinogenemia for splenic rupture using heat- and solvent detergent-treated fibrinogen concentrates. J Pediatr Hematol Oncol 1997; 19:462-5; PMID:9329471; http://dx.doi.org/10.1097/00043426-199709000-00011
- Webert KE. Acquired hemophilia A. Semin Thromb Hemost 2012; 38:735-41; PMID:22941793; http://dx.doi.org/10.1055/s-0032-1327775
- Mackie I, Cooper P, Lawrie A, Kitchen S, Gray E, Laffan M; British Committee for Standards in Haematology. Guidelines on the laboratory aspects of assays used in haemostasis and thrombosis. IntJ Lab Hematol 2013; 35:1-13; PMID:22978493; http://dx.doi.org/10.1111/ijlh.12004
- Verbruggen B, Giles A, Samis J, Verbeek K, Mensink E, Novakova I. The type of factor VIII deficient plasma used influences the performance of the Nijmegen modification of the Bethesda assay for factor VIII inhibitors. Thromb Haemost 2001; 86:1435-9; PMID:11776311
- Hornsey VS, Waterston YG, Prowse CV. Artificial factor VIII deficient plasma: preparation using monoclonal antibodies and its use in one stage coagulation assays. J Clin Pathol 1988; 41:562-7; PMID:3133400; http://dx.doi.org/10.1136/jcp.41.5.562
- O'Reilly RA. Drugs used in disorders of coagulation. Basic and Clinical Pharmacology. In: Katzung BG, ed. San Francisco: Prentice Hall International; 1998.
- Oncu S, Oncu S, Sakarya S. Anthrax–an overview. Med Sci Monit 2003; 9:RA276-83; PMID:14586293
- Meyer MA. Neurologic complications of anthrax: a review of the literature. Arch Neurol 2003; 60:483-8; PMID:12707059; http://dx.doi.org/10.1001/archneur.60.4.483
- Kuo SR, Willingham MC, Bour SH, Andreas EA, Park SK, Jackson C, Duesbery NS, Leppla SH, Tang WJ, Frankel AE. Anthrax toxin-induced shock in rats is associated with pulmonary edema and hemorrhage. Microb Pathog 2008; 44:467-72; PMID:18222626; http://dx.doi.org/10.1016/j.micpath.2007.12.001
- Begbie M, Notley C, Tinlin S, Sawyer L, Lillicrap D. The Factor VIII acute phase response requires the participation of NFkappaB and C/EBP. Thromb Haemost 2000; 84:216-22; PMID:10959692
- Sun H. The interaction between pathogens and the host coagulation system. Physiology (Bethesda) 2006; 21:281-8; PMID:16868317; http://dx.doi.org/10.1152/physiol.00059.2005
- Muta T, Iwanaga S. The role of hemolymph coagulation in innate immunity. Curr Opin Immunol 1996; 8:41-7; PMID:8729445; http://dx.doi.org/10.1016/S0952-7915(96)80103-8
- Tapper H, Herwald H. Modulation of hemostatic mechanisms in bacterial infectious diseases. Blood 2000; 96:2329-37; PMID:11001879
- Degen JL, Bugge TH, Goguen JD. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 2007; 5(Suppl 1):24-31; PMID:17635705; http://dx.doi.org/10.1111/j.1538-7836.2007.02519.x
- Kau JH, Sun DS, Huang HS, Lien TS, Huang HH, Lin HC, Chang HH. Sublethal doses of anthrax lethal toxin on the suppression of macrophage phagocytosis. PloS one 2010; 5:e14289; PMID:21170330; http://dx.doi.org/10.1371/journal.pone.0014289
- Pipe SW. The promise and challenges of bioengineered recombinant clotting factors. J Thromb Haemost 2005; 3:1692-701; PMID:16102035; http://dx.doi.org/10.1111/j.1538-7836.2005.01367.x
- Kaufman RJ, Powell JS. Molecular approaches for improved clotting factors for hemophilia. Blood 2013; 122:3568-74; PMID:24065241; http://dx.doi.org/10.1182/blood-2013-07-498261
- Chang HH, Wang TP, Chen PK, Lin YY, Liao CH, Lin TK, Chiang YW, Lin WB, Chiang CY, Kau JH, et al. Erythropoiesis suppression is associated with anthrax lethal toxin-mediated pathogenic progression. PloS One 2013; 8:e71718; PMID:23977125; http://dx.doi.org/10.1371/journal.pone.0071718
- Chang HH, Chiang YW, Lin TK, Lin GL, Lin YY, Kau JH, Huang HH, Hsu HL, Wang JH, Sun DS. Erythrocytic mobilization enhanced by the granulocyte colony-stimulating factor is associated with reduced anthrax-lethal-toxin-induced mortality in mice. PloS One 2014; 9:e111149; PMID:25384016; http://dx.doi.org/10.1371/journal.pone.0111149
- Chang HH, Shyu HF, Wang YM, Sun DS, Shyu RH, Tang SS, Huang YS. Facilitation of cell adhesion by immobilized dengue viral nonstructural protein 1 (NS1): arginine-glycine-aspartic acid structural mimicry within the dengue viral NS1 antigen. J Infect Dis 2002; 186:743-51; PMID:12198607; http://dx.doi.org/10.1086/342600
- Inoue H, Aizawa H, Matsumoto K, Shigyo M, Takata S, Hara M, Hara N. Effect of beta 2-agonists on histamine-induced airway microvascular leakage in ozone-exposed guinea pigs. Am J Respir Crit Care Med 1997; 156:723-7; PMID:9309985; http://dx.doi.org/10.1164/ajrccm.156.3.9606019
- Sun DS, King CC, Huang HS, Shih YL, Lee CC, Tsai WJ, Yu CC, Chang HH. Antiplatelet autoantibodies elicited by dengue virus non-structural protein 1 cause thrombocytopenia and mortality in mice. J Thromb Haemost 2007; 5:2291-9; PMID:17958746; http://dx.doi.org/10.1111/j.1538-7836.2007.02754.x
- Andre P, Hartwell D, Hrachovinova I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci U S A 2000; 97:13835-40; PMID:11095738; http://dx.doi.org/10.1073/pnas.250475997
- van den Besselaar AM, Neuteboom J, Meeuwisse-Braun J, Bertina RM. Preparation of lyophilized partial thromboplastin time reagent composed of synthetic phospholipids: usefulness for monitoring heparin therapy. Clin Chem 1997; 43:1215-22; PMID:9216459
- Tripodi A, Arbini A, Chantarangkul V, Mannucci PM. Recombinant tissue factor as substitute for conventional thromboplastin in the prothrombin time test. Thromb Haemost 1992; 67:42-5; PMID:1615481
- Chang WK, Sun DS, Chan H, Huang PT, Wu WS, Lin CH, Tseng YH, Cheng YH, Tseng CC, Chang HH. Visible light-responsive core-shell structured In2O3@CaIn2O4 photocatalyst with superior bactericidal properties and biocompatibility. Nanomedicine 2012; 8:609-17; PMID:22033083; http://dx.doi.org/10.1016/j.nano.2011.09.016
- Huang HS, Sun DS, Lien TS, Chang HH. Dendritic cells modulate platelet activity in IVIg-mediated amelioration of ITP in mice. Blood 2010; 116:5002-9; PMID:20699442; http://dx.doi.org/10.1182/blood-2010-03-275123
- Chen YL, Chen YS, Chan H, Tseng YH, Yang SR, Tsai HY, Liu HY, Sun DS, Chang HH. The use of nanoscale visible light-responsive photocatalyst TiO2-Pt for the elimination of soil-borne pathogens. PloS One 2012; 7:e31212; PMID:22384003; http://dx.doi.org/10.1371/journal.pone.0031212