
ABSTRACT
Mechanisms that favor Hepatitis C virus (HCV) persistence over clearance are unclear, but involve defective innate immunity. Chronic infection is characterized by hepatic iron overload, hyperferraemia and hyperferittinaemia. Hepcidin modulates iron egress via ferroportin and its storage in ferritin. Chronic HCV patients have decreased hepcidin, while HCV replication is modified by HAMP silencing. We aimed to investigate interactions between HCV and hepcidin, during acute and chronic disease, and putative alterations in cellular iron homeostasis that enhance HCV propagation and promote viral persistence. Thus, we used HCV JFH-1-infected co-cultures of Huh7.5 hepatoma and THP-1 macrophage cells, HCV patients’ sera and Huh7 hepcidin-expressing cells transfected with HCV replicons. Hepcidin levels were elevated in acutely infected patients, but correlated with viral load in chronic patients. HAMP expression was up-regulated early in HCV infection in vitro, with corresponding changes in ferritin and FPN. Hepcidin overexpression enhanced both viral translation and replication. In HCV-infected co-cultures, we observed increased hepcidin, reduced hepatoma ferritin and a concurrent rise in macrophaghic ferritin over time. Altered iron levels complemented amplified replication in hepatoma cells and one replication round in macrophages. Iron-loading of macrophages led to enhancement of hepatic HCV replication through reversed ferritin “flow.” Viral transmissibility from infected macrophages to naïve hepatoma cells was induced by iron. We propose that HCV control over iron occurs both by intracellular iron sequestration, through hepcidin, and intercellular iron mobilisation via ferritin, as means toward enhanced replication. Persistence could be achieved through HCV-induced changes in macrophagic iron that enhances viral replication in these cells.
Introduction
HCV is a positive-stranded RNA virus of the Flaviviridae family. Increased intracellular iron may be linked to enhanced HCV IRES-dependent translation, since cellular factors implicated in promoting HCV translation bind specifically to HCV RNA in an iron-dependent manner.Citation1,2 In contrast, the effect of iron on HCV replication is obscure, with opposing data from in vitro studies that describe either promotion or inhibition of HCV replication in the presence of exogenous iron.Citation3-5 In clinical studies, about 30–40% of chronic HCV patients possess elevated serum iron, ferritin and transferrin saturation.Citation6 A correlation between increased cellular and systemic iron levels and HCV patients with poor prognosis and severe deterioration in liver metabolic processes has been previously reported.Citation6 However, it remains unclear whether there is any correlation between HCV replication and increased intrahepatic or serum iron in chronic HCV patients.
Hepcidin is a 25 aa cysteine-rich bioactive peptide found in human serum and urine.Citation7 It is mainly synthesized by hepatocytes and to a lesser extent by monocytes, macrophages, adipocytes and brain cells. Hepcidin represents the key peptide hormone that modulates iron homeostasis by binding to the only known cell surface non-haeme iron exporter FPN and causing its internalisation and degradation, thus inhibiting iron efflux.Citation8 FPN is expressed abundantly in duodenal enterocytes and macrophages. Therefore, hepcidin inhibits iron absorption by the duodenum and macrophage-dependent iron recycling. It is moderately expressed in hepatocytes, where the extent of its involvement in iron mobilisation from hepatic stores is unknown.Citation9 Despite references to FPN-independent mechanisms for hepatocyte iron export, there is at least enough evidence to implicate FPN in hepatic iron homeostasis, under certain conditions such as dietary iron deficiency.Citation10 Interestingly, functional genomic screening for cellular cofactors of HCV replication underscored the absence of hepcidin as deleterious for successful viral replication.Citation11 Thus, the link between altered iron state and HCV infection could be hepcidin, due to its role as a potent regulator in iron homeostasis. However, in most cases, hepcidin was found suppressed during chronic HCV infection,Citation12,13 possibly because of ROS-mediated reduction in HAMP gene expression.Citation14 On the other hand, HCV-driven inflammation may counteract ROS-induced hepcidin repression, since elevated IL-6 stimulates HAMP gene transcription.Citation15 This second level of dichotomy merely stresses our incomplete understanding of the nature of the interactions between HCV and iron homeostasis at various disease stages. Therefore, the present study aims to delineate putative interactions between the virus, hepcidin and other cellular components of systemic iron homeostasis that may play a significant role in HCV life cycle.
Results
HAMP gene expression is up-regulated in early HCV infection in vitro
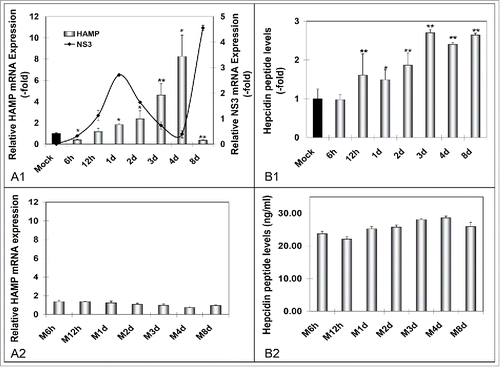
Perplexed by the literary conflict concerning hepcidin levels in HCV infection,Citation11-13,Citation16 we examined HAMP gene expression in vitro, using the JFH-1 infectious system. Thus, we infected Huh7.5 hepatoma cells that were harvested at early (6 h - 4 d) and established (8 d) intervals p.i. Subsequently, qRT-PCR and ELISA tests were performed for RNA and secreted peptide levels. HAMP mRNA increased progressively from 12 h to 4 d p.i., with peak levels 8-fold higher than the baseline of mock infected cells, then plummeted to approximately 40% of control on the eighth day (). NS3 mRNA was measured in order to monitor infection progress. Furthermore, hepcidin secreted peptide levels were increased maximally at 3 d - 4 d p.i. However, they did not mirror the extraordinary changes observed in mRNA, instead they peaked 2-3-fold compared to control and remained stable until 8 d p.i. (). ELISA measurements at 12 d - 16 d p.i. revealed that the great mRNA decline at 8 d p.i. was belatedly translated to decreased protein (data not shown). Hepcidin peptide expression did not change significantly in mock-infected cells with time ().
Figure 1. HCV-mediated regulation of hepcidin gene expression in vitro. A. qRT-PCR analysis of HAMP mRNA levels in (A1) HCV JFH-1- and (A2) mock-infected Huh7.5 cells. B. ELISA measurements of secreted hepcidin peptide in supernatants isolated from (B1) HCV-JFH-1- and (B2) mock-infected Huh7.5 cell cultures. Fold- changes in the infected cultures are presented as ratios of the mock-infected control (black bar). HCV NS3 mRNA expression (continuous line) was used to monitor HCV infection.
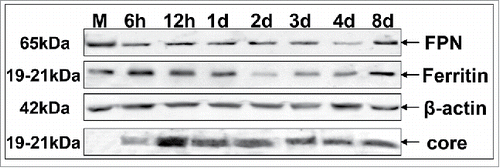
Consequently, we investigated FPN and ferritin expression during HCV infection. Western blotting in whole cell extracts from HCV- and mock-infected Huh7.5 cells showed that FPN levels were decreasing with time () while an increase at 8 d p.i. suggests that FPN regulation may be hepcidin-independent during established infection. Moreover, we observed an early increase in ferritin expression that would slowly decline and reach a minimum at 2 d p.i., when NS3 expression peaked, then a gradual rise toward 8 d p.i. (). This distinct pattern may be partially explained by increased hepcidin and reduced FPN, more so after the first 2 d p.i.. Core expression was used to monitor viral infection ().
Figure 2. HCV modulates the expression of major iron homeostasis proteins. Representative Western blot analysis of expressed FPN, ferritin and HCV core in whole cell extracts from HCV JFH-1-infected hepatoma cells and respective molecular weights. β-actin was used as a loading control. (M: mock-infected control).
HAMP overexpression enhances HCV translation and replication in hepatoma cells
We hypothesized that the iron dependency of HCV IRES Citation1,2 might be fulfilled through an HCV-orchestrated manipulation of iron homeostasis. To test this hypothesis, we engineered the H21 hepcidin-expressing hepatoma cell line. Compared to V24 control cells, this cell line demonstrated a 12-fold increase in HAMP mRNA expression (), which resulted to a 4-fold induction in secreted peptide (). Secreted hepcidin of the correct molecular weight, as compared against purified recombinant peptide,Citation17 was immunoprecipitated successfully with a monoclonal hepcidin-specific antibody ().
Figure 3. Hepcidin overexpression modulates HCV propagation. A. Validation of the hepcidin-overexpressing hepatoma cell line H21 by measurement of HAMP mRNA levels with qRT-PCR (A1), secreted hepcidin peptide levels with ELISA (A2), as compared to the “empty vector” cells (V24, black bar). A3. Immunoprecipitation of hepcidin peptide secreted by H21 cells with a monoclonal anti-hepcidin antibody. B. Electroporation of H21 cells with defective (B1) or wt (B2) HCV replicon RNA and RLA determination at various time points p.t. demonstrates viral translation and replication efficiency under conditions of overexpressed hepcidin. (M: marker).
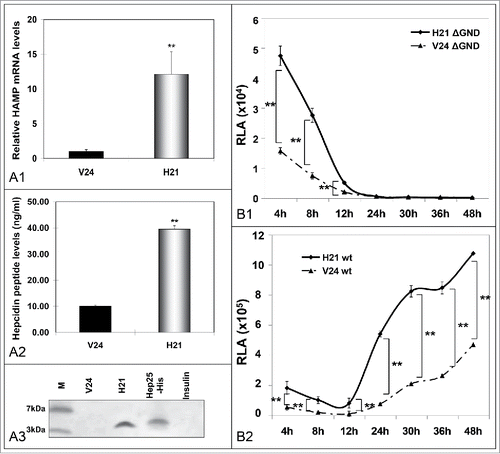
H21 hepcidin-overexpressing and V24 control hepatoma cells were electrotransfected with in vitro transcribed RNA from the plasmid coding for the replication-defective HCV replicon ΔGND, which expresses firefly luciferase under the instruction of HCV IRES. Cells were harvested between 4 h - 48 h, when HCV translation was expected to have finished, and subjected to luciferase activity assays. indicates that luciferase activity was significantly enhanced in H21 cells, at early times between 4 h - 12 h p.t.. Subsequently, we used a replication-competent HCV replicon (wt) in order to investigate the effect of hepcidin overexpression on HCV replication. shows significantly elevated luciferase activity in H21 extracts 2 d p.t., when HCV replication peaked according to infected hepatoma NS3 levels (see ). Similar data were produced with a different hepcidin-expressing Huh7 clone (data not shown).
Are macrophages involved in a hepcidin-mediated advancement of HCV propagation?
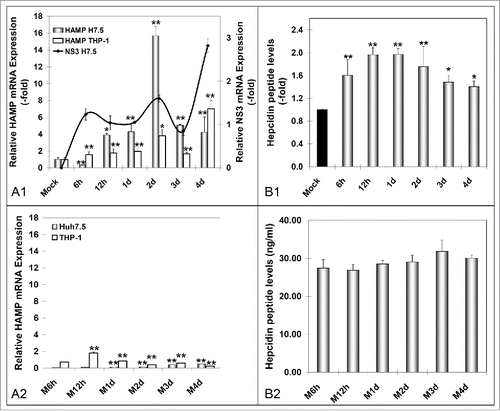
Iron storage and recycling are largely mediated by macrophages.Citation18 To examine whether they act as “elicitors” of hepcidin production and promote HCV propagation upon infection, we co-cultured HCV JFH-1-infected Huh7.5 cells with differentiated THP-1 macrophages, for 6 h - 4 d. Both cell lines were subjected to HAMP mRNA qRT-PCR, while supernatants pooled from upper and lower chamber were used to measure secreted hepcidin peptide by ELISA. shows that when compared to the hepatoma monoculture system (), Huh7.5 cells expressed approximately twice as much HAMP mRNA in the presence of macrophages, with a faster ‘turnover’ in HAMP gene transcription that shifted the RNA peak from 4 d in the Huh7.5 monoculture to 2 d p.i. in the co-cultured Huh7.5 cells. Notably, NS3 levels indicated swifter HCV replication cycles in the presence of macrophages () than in infected hepatoma cells alone. Finally, there was a gradual significant induction of macrophagic HAMP gene expression that would double with every new peak in NS3 mRNA levels. Notably, in mock-infected cells, we observed slight increases in HAMP gene transcription, which we attributed to continued cell growth (). HAMP mRNA was not fully translated into protein, with a modest 1.5- to 2-fold elevation in hepcidin secreted peptide (). Still, they mirrored the RNA shift toward earlier times of infection. There was no overall change in secreted hepcidin peptide ().
Figure 4. Hepcidin expression in HCV-infected hepatoma – macrophage co-cultures. A. Determination of HAMP mRNA from both cell lines following (A1) HCV-JFH-1 infection or (A2) mock infection. B. Measurement of secreted peptide levels from pooled supernatants in (B1) HCV-JFH-1- or (B2) mock-infected co-cultures at the given time points. NS3 mRNA expression in hepatoma cells was used to monitor HCV infection.
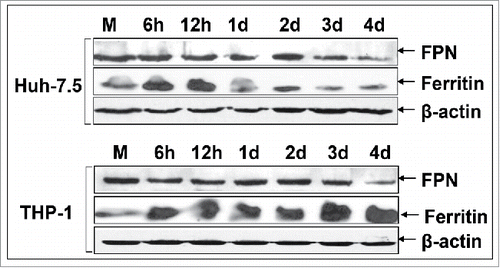
Next, we assessed FPN and ferritin protein expression by Western blotting. FPN expression clearly declined during late infection in both cell lines (), pointing toward functional hepcidin secreted by the co-culture system. Conversely, ferritin followed an inversely proportional pattern of expression between hepatoma cells and macrophages. Increased during the early 12 h p.i. in both cell lines, it dropped thereafter in Huh7.5 cells, while it kept accumulating dramatically in THP-1 cells (). Biochemical measurements carried out on an Abbott biochemical analyzer verified the observed ferritin pattern at 12 h - 24 h p.i. for Huh7.5 cells (88.6 and 21.0 μg/dl, respectively), as well as 6 h - 12 h for THP-1 cells (32.7 and 129.5 μg/dl, respectively). Overall, there is a fundamental difference in hepatoma ferritin expression between the monoculture and co-culture system, in that, in the former, ferritin recovered after the peak of HCV replication (2 d p.i.) had been reached.
Figure 5. Attenuation of ferritin expression in HCV JFH-1-infected hepatoma cells in the presence of macrophages. Representative immunoblotting of whole cell extracts from infected co-cultures with antibodies against FPN, ferritin and β-actin (loading control). (M: mock-infected control).
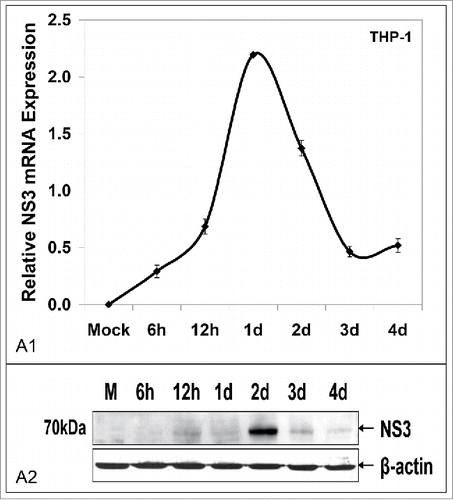
Finally, we investigated whether the elevated iron content in THP-1 macrophages could not only support infection, as reported elsewhere,Citation19 but also viral replication. qRT-PCR analysis of macrophage mRNA revealed the presence of NS3 mRNA and its sharp increase at 1 d p.i. (). Immunoblotting showed successful translation into NS3 peptide 24 h later (). The above unexpected data suggested that iron manipulation via HCV-mediated induction of HAMP gene expression resulted in multicycle intrahepatic replication and a single round of intramacrophagic replication.
Figure 6. Macrophages co-cultured with HCV JFH-1-infected hepatoma cells support low levels of viral replication. HCV NS3 mRNA levels measured by qRT-PCR (A1) and HCV NS3 protein expression determined by Western blotting (A2) in co-cultured THP-1 macrophages. β-actin was used as a loading control. (M: mock-infected control).
Iron-loading of THP-1 macrophages results in enhanced HCV replication in Huh7.5 cells. A role for ferritin?
Given that in our co-cultures there was a phenomenal increase in THP-1 iron stores (see ), we wondered whether these ferritin-loaded immune cells would act as ferritin-mediated iron donorsCitation20 and procure any HCV replication alterations in newly infected Huh7.5 cells. However, differentiated THP-1 macrophages have a limited life span in culture, which prohibited their extended use for a second 4-day infection cycle. Therefore, freshly differentiated cells, plated in hanging inserts, were loaded with inorganic iron. Iron-loading was examined by Western blotting of ferritin expression (data not shown). The inserts were subjected to extensive rinsing with phosphate buffer saline, so that residual FAC would be removed, before being placed on top of freshly JFH-1-infected Huh7.5 cells. Upon cell harvest, we measured HCV NS3 expression in hepatoma cells. demonstrates an extraordinary, statistically significant up-regulation in NS3 levels in the presence of FAC-loaded macrophages, starting at the first day p.i. This resulted in increased NS3 translation, 24 h earlier than achieved in hepatoma cells co-cultured without FAC-loaded macrophages (). These profound changes in HCV replication were accompanied by a ferritin “flow” from THP-1 to Huh7.5 cells very early in infection, between 6 h-1 d (), that was supported by intracellular iron measurements (data not shown). Additionally, increased ferritin, assisted in prolonging and enhancing viral replication within macrophages themselves ().
Figure 7. Iron loading of THP-1 macrophages enhances HCV replication in infected cells and promotes HCV infection in naïve cells. A. HCV NS3 mRNA (A1) and protein (A2) levels measured in infected hepatoma cells co-cultured with macrophages. B. Ferritin immunoblots in whole cell extracts from (A). C. HCV NS3 mRNA levels from THP-1 cells isolated from co-cultures of (A). D. HCV NS3 mRNA levels from naïve Huh7.5 cells co-cultured with infected THP-1 macrophages from (A).
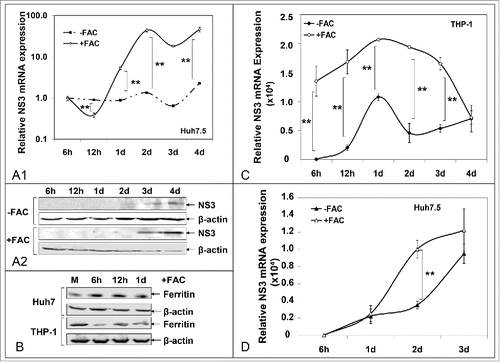
Interestingly, THP-1 cells co-cultured for 2 d with JFH-1-infected Huh7.5 cells were able to infect a naïve Huh7.5 population in a 3-day indirect co-culture experiment. The THP-1 cells had been previously rinsed thoroughly with cold phosphate buffer saline, so that any viral particles found on the cell membrane would be effectively removed. Iron-loaded macrophages promoted more efficient HCV infection of the hepatoma cells that resulted in higher NS3 expression at least for the first 2 d p.i. ().
Finally, given that HCV infection spurs liver inflammationCitation21 and IL-6 is a positive regulator of HAMP gene expressionCitation15,22 we monitored IL-6 levels by qRT-PCR in co-cultured THP-1 and infected Huh7.5 cells, in the presence and absence of iron loading. IL-6 mRNA was originally noted in hepatoma cells at 12 h p.i., but was then decreased to background. Subsequently, it peaked at 1 d p.i. in macrophages, but was abolished later (Suppl. ). Notably, iron loading resulted in a statistically significant difference in top IL-6 levels in both cell populations, possibly linking iron toxicity to inflammation; however these changes levelled out later in the time course (Suppl. ).
Differential hepcidin regulation in HCV patients. Dependence on viral infection parameters
In our hands, HAMP gene expression has been shown consistently increased during early HCV infection with the laboratory strain JFH-1, then dropped as infection progressed. Given the conflicting results reported in literature, which refer mostly to chronic HCV infection,Citation12,13 we suspected that hepcidin increase may be more relevant to acute infection than chronic infection. Therefore, we determined hepcidin serum levels in 7 acutely infected (A) and 20 chronic HCV patients by ELISA. Half of the chronic patients had high (H) and the other half low (L) viral load. Sera from 17 healthy donors (N) were used as control. The demographic data of the patients are recorded in . shows that when compared to healthy donors with an average hepcidin serum concentration of 27.3 ng/ml, both acutely infected and chronic patients with high viral load possess statistically significant elevated hepcidin levels of 81.0 ng/ml and 39.2 ng/ml, respectively. Conversely, chronic HCV patients with low viral load had reduced hepcidin levels (11.5 ng/ml), as reported elsewhere.Citation12,13 A Spearman's correlation test conducted on our data, showed a strong positive relationship between viral load and hepcidin serum levels in chronic HCV patients (rs = 0.786, p < 0.005)
Figure 8. Differential regulation of secreted hepcidin peptide in vivo. ELISA measurements of hepcidin levels in sera of acute and chronic HCV patients.
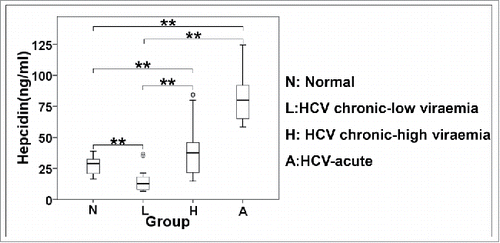
Table 1. Clinical and biochemical characteristics of enrolled subjects.
Discussion
In this study, we report novel cellular interactions between HCV and the host iron homeostasis and demonstrate the increase of hepcidin expression during early HCV infection in vitro. We have previously showed that HCV core induces HAMP gene expression in hepatoma cellsCitation23 and this may also apply to early HCV infection. Conversely, upon established infection, hepcidin expression dropped in our in vitro model and in patient sera,Citation12,13 in step with studies from chronic HCV patients with low intrahepatic HAMP mRNACitation24 and decreased serum hepcidin,Citation12,13 and earlier in vitro RNA data.Citation25,26 Secreted protein expression was also increased throughout infection, albeit not as dramatically as mRNA. The latter may be due to post-transcriptional regulation of HAMP gene expression, including mRNA destabilisation Citation27 and secretion modulation.Citation28
FPN and ferritin are crucial for iron homeostasis. In strong support of hepcidin’s role as an autocrine FPN regulator in hepatocytes,Citation29 FPN was reduced during early infection. However, it recovered at 8 d p.i., despite constant hepcidin levels, but in correlation with increased ferritin, during that time. FPN and ferritin are translationally regulated by iron in liver, macrophages and intestine through binding of IRPs to an IRE element in their 5′-UTR.Citation30, Citation31 IRP2 is up-regulated by HCV,Citation25 while FPN internalisation and degradation may also be hepcidin-independent.Citation32 Therefore, we can not exclude the possibility that the observed FPN levels in our experiments may be due to a combination of regulatory mechanisms employed by HCV. Ferritin, on the other hand, was elevated early, dropped at 2 d p.i., when HCV replication peaked, and started increasing again in accordance with hepcidin levels. This expression pattern could imply HCV-mediated manipulation of intracellular iron content through hepcidin modulation, so that the virus secures higher propagation levels. We tested this hypothesis by using a non-replicating sub-genomic replicon in hepcidin-overexpressing hepatoma cells and observed significantly enhanced HCV translation. Thus, inasmuch as hepcidin regulates intracellular iron, either through FPN or an alternative unknown hepatocyte-specific mechanism, our data agree with previous studies that have demonstrated increased HCV IRES function in iron-loaded cells. Citation1,2,Citation33 Others were unable to discern an association between iron and HCV translation,Citation34 but this discrepancy may be due to varied experimental conditions and cellular systems used. Furthermore, hepcidin overexpression resulted in significantly elevated HCV replication levels at 24 h - 48 h p.t. This interval coincides with the substantial decrease in ferritin following infection and could signify a viral need for iron. Although past research showed that hepcidin suppressed HCV replication, acting as an antiviral agent,Citation26 our data remain in agreement with researchers who suggested a positive regulatory role for hepcidin on HCV replication.Citation3,11
Macrophages induce hepcidin transcription in Huh7 cells,Citation22 while macrophagic hepcidin may regulate iron uptake and release in an autocrine manner.Citation35 Furthermore, the role of macrophages in HCV infection is obscure, with altered cytokine and chemokine profile upon infection. Citation36 We investigated putative interactions between macrophages, HCV and iron by utilizing an indirect co-culture system of THP-1 macrophages with Huh7.5 hepatoma cells. Such in vitro systems have been extensively used to unravel cellular communication through soluble factorsCitation37, including hepcidin,Citation22,38 as well as other aspects of liver/macrophage biology.Citation39 Evidently, hepatoma hepcidin expression was increased faster in the co-culture than the monoculture system. Furthermore, macrophagic HAMP mRNA was induced gradually throughout the time course, closely linked to NS3 expression. The early increase in hepcidin expression was accompanied by a faster HCV replication pace in hepatoma cells and evidence for low levels of replication in macrophages. In line with these results and our previous observations about the existence of HCV negative strands in PBMCs isolated from HCV patients,Citation40 several studies offer evidence for intramacrophagic HCV.Citation19, Citation41, Citation42, Citation43
Surprisingly, we observed cell-specific changes in ferritin expression in the co-culture system that could be perceived as a ferritin “flow” from hepatoma cells to macrophages. Previous studies have recorded elevated ferritin protein expression in THP-1 macrophages upon HCV infection.Citation19 Thus, it is possible that HCV-mediated hepcidin modulation, fine-tuned by the crosstalk between the cell populations, may affect iron content in the co-cultured cells in such a way that intramacrophagic HCV replication and the establishment of a viral reservoir are promoted. Despite the fact that the very existence of long-term occult extra-hepatic HCV reservoirs is strongly debated,Citation44 they would explain several clinical manifestations of the disease, like HCV persistence and recurrence following treatment or liver transplantation.Citation40,45, Citation46-49
Due to their role in iron homeostasis, reticuloendothelial macrophages, including Kupffer cells, may be full of iron at any given time.Citation18 Iron loading of co-cultured macrophages resulted in enhanced HCV replication in both cell populations. Importantly, a reversed ferritin “flow” was recorded very early in infection from macrophages toward hepatoma cells. Such mobilisation of iron intercellularly, could boost HCV replication in liver cells, while iron-loading itself, promoted HCV propagation in macrophages. The release/secretion and uptake of ferritin from both hepatocytes and macrophages, which may be controlled by extracellular stimuli, has been proposed before,Citation10,50, Citation51 as has the notion that ferritin acts both as an iron carrier and iron storage protein.Citation52,53 Combined with the HCV-mediated increase in hepcidin, these data indicate that the virus may use both hepcidin-dependent and independent pathways to manipulate iron homeostasis and set up a macrophagic reservoir that would permit viral transmission, at least in vitro. Attesting to that notion was the ability of macrophages taken from HCV-infected co-cultures to “de novo” infect naïve hepatoma cells. Notably, iron-loaded macrophages conferred higher infectivity, as compared to control cells. Earlier experiments demonstrated that viral particles shed from infected hepatoma cells could infect naïve hepatoma populations in a cell-free manner. Citation54 However, we provide data for the first time to the best of our knowledge, that HCV uses macrophages as an intermediate host in an in vitro cell culture system. (see for a schematic representation of our proposed model).
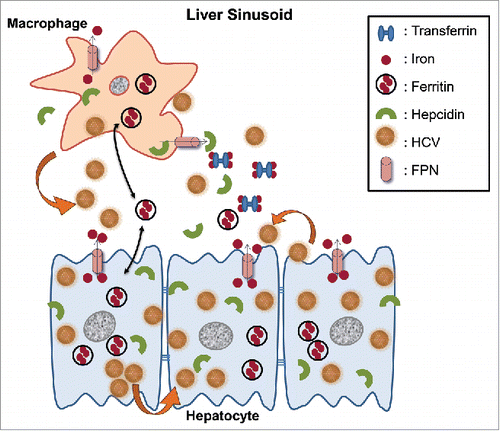
Figure 9. Schematic representation of putative interactions between HCV and the iron homeostasis network in hepatoma-macrophage co-cultures. We used an indirect hepatoma-macrophage co-culture system to simulate the co-existence of Kupffer cells and hepatocytes within the liver sinusoid. Upon viral egress, HCV particles infect a neighboring hepatocyte in a cell-free manner through specific plasma membrane receptors or in a direct cell-to-cell route via gap junction proteins (depicted by thick arrows). Iron is stored intracellularly in ferritin, leaves the cell through iron exporter ferroportin and circulates in blood bound to transferrin, or secreted ferritin. Hepcidin is produced by both hepatocytes and macrophages and regulates iron homeostasis via degradation of ferroportin, thus keeping iron within the cell. In our model, HCV mediated hepcidin up-regulation in hepatoma cells and accompanying macrophages, resulting in reduced ferroportin, elevated ferritin and iron levels intracellularly very early in infection. Later, a ferritin “flow” was observed from hepatoma cells toward macrophages (double pointed arrows). Hepcidin overexpression enhanced HCV translation and replication, and accompanied low levels of viral replication within macrophages. The ferritin “flow” could be reversed following macrophagic iron loading, which promoted enhanced viral replication in both cellular populations, as well as infection of naïve hepatoma cells through particle shedding (showed by a thick arrow).
It has been shown that HCV blocks the establishment of a cytokine “storm” during acute infection, with only a minimal elevation in IL-6 levels at 17 d p.i.Citation55 We recorded a significant increase of hepcidin in sera of acutely infected HCV patients. Despite the fact that alanine aminotransferase levels in those patients were indicative of pronounced liver inflammation, a prominent role for IL-6 in enhanced hepcidin expression during acute infection is debatable. This is in agreement with our in vitro data, where enhanced IL-6 expression in the presence or absence of iron was abolished earlier than the observed changes in hepcidin expression. Surprisingly, increased serum hepcidin in chronic HCV patients was dependent on high viral load. Few studies have mentioned such an association, with no clear reference to viral load levels in chronic HCV patients and contradictory data concerning the relationship between HCV viraemia and hepcidin expression.Citation12,24,Citation56 On the other hand, our observation hints at a putative differential regulation of hepcidin by the virus depending on its replication capacity and, in that respect, is in accordance with the hepcidin levels observed in our in vitro model.
In conclusion, we propose that HCV conditionally up-regulates HAMP gene expression both in vitro and in vivo, depending on viral parameters such as infection acuteness and increased viral load. In turn, induced hepcidin expression was correlated to enhanced HCV translation and replication in vitro. The ability of the virus to control iron levels in order to achieve more efficient replication both by intracellular iron sequestration, through hepcidin, and intercellular iron mobilisation, via ferritin, portrays a novel role for iron homeostasis that could provide a mechanistical insight for viral persistence. Alterations in the iron network may promote the utilization of macrophages as viral reservoirs that can infect hepatoma cells through viral particle shedding and assist further with viral propagation.
Materials and Methods
Materials
Human sera were obtained from chronically HCV infected patients with high and low viral loads who had not received previous treatment, patients with acute HCV infection and healthy blood donors, following their consent and approval by the Ethical and Scientific Committee of Hippokration Hospital of Athens.
The antibodies used were: β-actin (MAB1501, Millipore), Ferroportin 1 (MTP11, Alpha Diagnostics), ferritin (A0133, Dako) and HCV-NS3 (1B6, Adipogen). The polyclonal HCV-1a core antibody used in this study has been described elsewhere.Citation57 Huh7.5 and Huh7-Lunet cells were supplied by Dr R. Bartenschlager (University of Heidelberg, Germany).
Cells lines and co-cultures
The hepatoma cell lines used were maintained in Dulbecco’s modified Eagle medium (high glucose for Huh7.5/ Huh7-Lunet and low glucose for Huh7 cells) supplemented with 2 mM glutamine, 10% (v/v) heat-inactivated foetal calf serum and 100 U/ml penicillin/streptomycin and non-essential aminoacids. THP-1 monocytes were fed with RPMI 1640, supplemented as above, and differentiated to macrophages with 0.16 μM phorbol 12-myristate 13-acetate for 24 h.
A Huh7 stable cell line expressing hepcidin (H21) was generated by electrotransfecting 3 μg of linearized pCDNA3.1 plasmid that contained the prohepsidin coding sequence.Citation17 The transfected cells were grown in the presence of medium with 500 μg/ml G418 and individual clones were obtained by limited dilution. An empty vector cell line (V24) was also constructed, using just pCDNA3.1.
For the indirect co-culture experiments, Huh7.5 hepatoma cells, seeded in the appropriate multiwell plates, were overlaid with hanging inserts (Greiner) that did not allow cell-cell contact. THP-1 macrophages were seeded on the top surface of the inserts at hepatoma:macrophage ratio of 5:1, which resembles normal livers with standard resident macrophage populations.Citation58 Iron loading of THP-1 macrophages was achieved by incubation with 100 μM FAC for 24 h prior to them being placed above Huh7.5 cells. Trypan blue exclusion assay was performed to ascertain that iron-mediated cytotoxicity resulted in the death of less than 1% of cultured cells at all times.
Viruses and RNA transfections
Viral infections of permissive Huh7.5 cells with the JFH-1 infectious clone were carried out essentially as described in ref.Citation59 at multiplicity of infection = 1. The protocols used for in vitro transcription of the pFK-I341PI-Luc/NS3-3′/JFH1 (wt) and the corresponding non - replicating construct (ΔGND),Citation60 as well as transfection of the RNA transcripts into the H21 and V24 cell lines, are described in ref.Citation61 Briefly, 100,000 cells/well were seeded in 48-well plates 24 h prior to the experiment. Cells were electrotransfected with 10 μg RNA and equal amounts of carrier t-RNA. Following transfection, cells were harvested at the appropriate time points and subjected to luciferase and β-galactosidase activity determinations with commercially available kits (Promega). Luciferase activity was normalized to β-galactosidase activity in order to yield RLA. Each transfection was carried out at least 3 times in triplicate, unless otherwise stated.
mRNA and protein expression analyses
Total cellular RNA and protein were isolated simultaneously using the dual NucleoSpin® RNA/protein kit (Macherey - Nagel), according to manufacturer’s instructions. Then, the RNA fraction was subjected to qRT-PCR, essentially as described in ref.Citation23 The gene specific primers used for HAMP, 18S rRNA, IL-6 and NS3-2A have been published elsewhere.Citation23,59 Results were analyzed with the internal standard-curve method and normalized to 18S rRNA to provide the relative mRNA expression. In all PCR experiments, unless otherwise stated, the relative mRNA expression of mock (mean±SD) was set as 100% or fold-change and all other values were depicted as a ratio of this.
The protein fraction was subjected to protein determination with the Protein Quantification Assay (Macherey - Nagel). 40 μg of cell lysates were used in Western blotting, as published before.Citation23,59 Gel photographs presented in each figure panel are representative results from samples that were derived from the same experiment and processed in parallel. All experiments were performed at least in triplicate.
Secreted hepcidin levels were quantitated ELISA, as stated in refs.Citation23,59 The percentage change in protein expression of the control cells was set as 100% and all other values were a percentage of the control, except when absolute peptide concentration values were measured in ng/ml. The assays were repeated at least 3 times in quadruplicates.
Statistical analysis
Unless otherwise shown, statistical analysis was performed between mock and treated cells using One-Way ANOVA and Student’s t-test. A Spearman’s correlation test was carried out to investigate a putative correlation between viral load and hepcidin levels in chronic HCV patients. In all cases p-value ≤ 0.05 were considered as statistically significant (*, p-value ≤ 0.05; **, p-value ≤ 0.005).
Abbreviations
| ANOVA | = | analysis of variance |
| HCV | = | Hepatitis C virus |
| HAMP | = | hepcidin antimicrobial peptide |
| ELISA | = | enzyme-linked immunosorbent assay |
| FPN | = | ferroportin |
| FAC | = | ferric ammonium citrate |
| IL-6 | = | interleukin-6 |
| IRES | = | internal ribosomal entry site |
| IRPs | = | iron regulatory proteins |
| p.i. | = | post-infection |
| p.t. | = | post-transfection |
| RLA | = | relative luciferase activity |
| qRT-PCR | = | quantitative reverse transcription - polymerase chain reaction |
| ROS | = | reactive oxygen species |
| wt | = | wild type |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KVIR_S_1175700.zip
Download Zip (27.4 KB)Acknowledgments
We thank Prof. R. Bartenschlanger for providing the replicon constructs and Prof T. Wakita for providing the pJFH1 construct
Funding
This project was supported by grants from the GSRT No. 09SYN-12-68, the “InfeNeuTra project (MIS 450598) GSRT’s KRIPIS” co-financed by the European Union and Greek national funds through the National Strategic Reference Framework (NSRF) and the Postdoc Fellowships of Excellence – Siemens (2014/15) of the State Scholarships Foundation/IKY.
References
- Cho H, Lee HC, Jang SK, Kim YK. Iron increases translation initiation directed by internal ribosome entry site of hepatitis C virus. Virus Genes 2008; 37:154-60; PMID:18566883; http://dx.doi.org/10.1007/s11262-008-0250-0
- Theurl I, Zoller H, Obrist P, Datz C, Bachmann F, Elliott RM, Weiss G. Iron regulates hepatitis C virus translation via stimulation of expression of translation initiation factor 3. J Infect Dis 2004; 190:819-25; PMID:15272411; http://dx.doi.org/10.1086/422261
- Bartolomei G, Cevik RE, Marcello A. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J Gen Virol 2011; 92:2072-81; PMID:21593278; http://dx.doi.org/10.1099/vir.0.032706-0
- Fillebeen C, Pantopoulos K. Iron inhibits replication of infectious hepatitis C virus in permissive Huh7.5.1 cells. J Hepatol 2010; 53:995-9; PMID:20813419; http://dx.doi.org/10.1016/j.jhep.2010.04.044
- Kakizaki S, Takagi H, Horiguchi N, Toyoda M, Takayama H, Nagamine T, Mori M, Kato N. Iron enhances hepatitis C virus replication in cultured human hepatocytes. Liver 2000; 20:125-8; PMID:10847480; http://dx.doi.org/10.1034/j.1600-0676.2000.020002125.x
- Fontana RJ, Israel J, LeClair P, Banner BF, Tortorelli K, Grace N, Levine RA, Fiarman G, Thiim M, Tavill AS, et al. Iron reduction before and during interferon therapy of chronic hepatitis C: results of a multicenter, randomized, controlled trial. Hepatology 2000; 31:730-6; PMID:10706565; http://dx.doi.org/10.1002/hep.510310325
- Steinbicker AU, Muckenthaler MU. Out of balance–systemic iron homeostasis in iron-related disorders. Nutrients 2013; 5:3034-61; PMID:23917168; http://dx.doi.org/10.3390/nu5083034
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306:2090-3; PMID:15514116; http://dx.doi.org/10.1126/science.1104742
- Drakesmith H, Nemeth E, Ganz T. Ironing out Ferroportin. Cell Metab 2015; 22:777-87; PMID:26437604; http://dx.doi.org/10.1016/j.cmet.2015.09.006
- Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 2012; 56:961-71; PMID:22473803; http://dx.doi.org/10.1002/hep.25746
- Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009; 5:298-307; PMID:19286138; http://dx.doi.org/10.1016/j.chom.2009.02.001
- Girelli D, Pasino M, Goodnough JB, Nemeth E, Guido M, Castagna A, Busti F, Campostrini N, Martinelli N, Vantini I, et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J Hepatol 2009; 51:845-52; PMID:19729219; http://dx.doi.org/10.1016/j.jhep.2009.06.027
- Tsochatzis E, Papatheodoridis GV, Koliaraki V, Hadziyannis E, Kafiri G, Manesis EK, Mamalaki A, Archimandritis AJ. Serum hepcidin levels are related to the severity of liver histological lesions in chronic hepatitis C. J Viral Hepat 2010; 17:800-6; PMID:20002304; http://dx.doi.org/10.1111/j.1365-2893.2009.01244.x
- Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008; 48:1420-9; PMID:18671304; http://dx.doi.org/10.1002/hep.22486
- Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101:2461-3; PMID:12433676; http://dx.doi.org/10.1182/blood-2002-10-3235
- Miyachi H, Kobayashi Y, Relja B, Fujita N, Iwasa M, Gabazza EC, Takei Y. Effect of suppressor of cytokine signaling on hepcidin production in hepatitis C virus replicon cells. Hepatol Res 2011; 41:364-74; PMID:21348906; http://dx.doi.org/10.1111/j.1872-034X.2011.00777.x
- Koliaraki V, Marinou M, Samiotaki M, Panayotou G, Pantopoulos K, Mamalaki A. Iron regulatory and bactericidal properties of human recombinant hepcidin expressed in Pichia pastoris. Biochimie 2008; 90:726-35; PMID:18295606; http://dx.doi.org/10.1016/j.biochi.2008.01.012
- Gammella E, Buratti P, Cairo G, Recalcati S. Macrophages: central regulators of iron balance. Metallomics 2014; 6:1336-45; PMID:24905850; http://dx.doi.org/10.1039/C4MT00104D
- Ponzetto A, Gennero L, Cutufia M, Beltramo T, Enrietto M, Pescarmona P, Pugliese A. Effect of HCV infection on THP-1 monocytoid cells. Cell Biochem Funct 2005; 23:347-52; PMID:15515125; http://dx.doi.org/10.1002/cbf.1158
- Alvarez-Hernandez X, Felstein MV, Brock JH. The relationship between iron release, ferritin synthesis and intracellular iron distribution in mouse peritoneal macrophages. Evidence for a reduced level of metabolically available iron in elicited macrophages. Biochim Biophys Acta 1986; 886:214-22; PMID:3697381; http://dx.doi.org/10.1016/0167-4889(86)90139-4
- Zampino R, Marrone A, Restivo L, Guerrera B, Sellitto A, Rinaldi L, Romano C, Adinolfi LE. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J Hepatol 2013; 5:528-40; PMID:24179612; http://dx.doi.org/10.4254/wjh.v5.i10.528
- Matak P, Chaston TB, Chung B, Srai SK, McKie AT, Sharp PA. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica 2009; 94:773-80; PMID:19454498; http://dx.doi.org/10.3324/haematol.2008.003400
- Foka P, Dimitriadis A, Kyratzopoulou E, Giannimaras DA, Sarno S, Simos G, Georgopoulou U, Mamalaki A. A complex signaling network involving protein kinase CK2 is required for hepatitis C virus core protein-mediated modulation of the iron-regulatory hepcidin gene expression. Cell Mol Life Sci 2014; 71:4243-58; PMID:24718935; http://dx.doi.org/10.1007/s00018-014-1621-4
- Sikorska K, Romanowski T, Stalke P, Izycka Swieszewska E, Bielawski KP. Association of hepcidin mRNA expression with hepatocyte iron accumulation and effects of antiviral therapy in chronic hepatitis C infection. Hepat Mon 2014; 14:e21184; PMID:25598789; http://dx.doi.org/10.5812/hepatmon.21184
- Fillebeen C, Pantopoulos K. Hepatitis C virus infection causes iron deficiency in Huh7.5.1 cells. PLoS One 2013; 8:e83307; PMID:24349485; http://dx.doi.org/10.1371/journal.pone.0083307
- Liu H, Trinh TL, Dong H, Keith R, Nelson D, Liu C. Iron regulator hepcidin exhibits antiviral activity against hepatitis C virus. PLoS One 2012; 7:e46631; PMID:23110054; http://dx.doi.org/10.1371/journal.pone.0046631
- Volke M, Gale DP, Maegdefrau U, Schley G, Klanke B, Bosserhoff AK, Maxwell PH, Eckardt KU, Warnecke C. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS One 2009; 4:e7875; PMID:19924283; http://dx.doi.org/10.1371/journal.pone.0007875
- Chachami G, Lyberopoulou A, Kalousi A, Paraskeva E, Pantopoulos K, Simos G. Oxygen-dependent secretion of a bioactive hepcidin-GFP chimera. Biochem Biophys Res Commun 2013; 435:540-5; PMID:23665013; http://dx.doi.org/10.1016/j.bbrc.2013.04.085
- Ramey G, Deschemin JC, Durel B, Canonne-Hergaux F, Nicolas G, Vaulont S. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 2010; 95:501-4; PMID:19773263; http://dx.doi.org/10.3324/haematol.2009.014399
- Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000; 275:19906-12; PMID:10747949; http://dx.doi.org/10.1074/jbc.M000713200
- Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol 2003; 39:710-5; PMID:14568251; http://dx.doi.org/10.1016/S0168-8278(03)00408-2
- Ward DM, Kaplan J. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta 2012; 1823:1426-33; PMID:22440327; http://dx.doi.org/10.1016/j.bbamcr.2012.03.004
- Wang Q, Liu Y, An D, Diao H, Xu W, He X, Sun R, Wei L, Li L. Regulation of hepatitis C virus translation initiation by iron: role of eIF3 and La protein. Virus Res 2012; 167:302-9; PMID:22634302; http://dx.doi.org/10.1016/j.virusres.2012.05.014
- Fillebeen C, Rivas-Estilla AM, Bisaillon M, Ponka P, Muckenthaler M, Hentze MW, Koromilas AE, Pantopoulos K. Iron inactivates the RNA polymerase NS5B and suppresses subgenomic replication of hepatitis C Virus. J Biol Chem 2005; 280:9049-57; PMID:15637067; http://dx.doi.org/10.1074/jbc.M412687200
- Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, Zoller H, Bellmann-Weiler R, Niederegger H, Talasz H, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008; 111:2392-9; PMID:18073346; http://dx.doi.org/10.1182/blood-2007-05-090019
- Kochlios E, Foka P, Mavromara P. Modulation of monocyte/macrophage-derived cytokine and chemokine expression profile by persistent Hepatitis C virus (HCV) infection leads to chronic inflammation. J Mol Biochem 2012; 1:40-53
- Miki Y, Ono K, Hata S, Suzuki T, Kumamoto H, Sasano H. The advantages of co-culture over mono cell culture in simulating in vivo environment. J Steroid Biochem Mol Biol 2012; 131:68-75; PMID:22265957; http://dx.doi.org/10.1016/j.jsbmb.2011.12.004
- Andriopoulos B, Pantopoulos K. Hepcidin generated by hepatoma cells inhibits iron export from co-cultured THP1 monocytes. J Hepatol 2006; 44:1125-31. Epub 2005 Dec 5; PMID:16460831; http://dx.doi.org/10.1016/j.jhep.2005.10.025
- Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 2013; 87:1315-530; PMID:23974980; http://dx.doi.org/10.1007/s00204-013-1078-5
- Koskinas J, Tibbs C, Saleh MG, Pereira LM, McFarlane IG, Williams R. Effects of ribavirin on intrahepatic and extrahepatic expression of hepatitis C virus in interferon nonresponsive patients. J Med Virol 1995; 45:29-34; PMID:7714490; http://dx.doi.org/10.1002/jmv.1890450106
- Pawelczyk A, Kubisa N, Jablonska J, Bukowska-Osko I, Caraballo Cortes K, Fic M, Laskus T, Radkowski M. Detection of hepatitis C virus (HCV) negative strand RNA and NS3 protein in peripheral blood mononuclear cells (PBMC): CD3+, CD14+ and CD19+. Virol J 2013; 10:346; PMID:24279719; http://dx.doi.org/10.1186/1743-422X-10-346
- Revie D, Salahuddin SZ. Role of macrophages and monocytes in hepatitis C virus infections. World J Gastroenterol 2014; 20:2777-84; PMID:24659871; http://dx.doi.org/10.3748/wjg.v20.i11.2777
- Radkowski M, Bednarska A, Horban A, Stanczak J, Wilkinson J, Adair DM, Nowicki M, Rakela J, Laskus T. Infection of primary human macrophages with hepatitis C virus in vitro: induction of tumour necrosis factor-alpha and interleukin 8. J Gen Virol 2004; 85:47-59; PMID:14718619; http://dx.doi.org/10.1099/vir.0.19491-0
- Bare P. Hepatitis C virus and peripheral blood mononuclear cell reservoirs. World J Hepatol 2009; 1:67-71; PMID:21160967; http://dx.doi.org/10.4254/wjh.v1.i1.67
- Laskus T, Radkowski M, Wilkinson J, Vargas H, Rakela J. The origin of hepatitis C virus reinfecting transplanted livers: serum-derived versus peripheral blood mononuclear cell-derived virus. J Infect Dis 2002; 185:417-21; PMID:11865392; http://dx.doi.org/10.1086/338635
- Lee WM, Polson JE, Carney DS, Sahin B, Gale M, Jr. Reemergence of hepatitis C virus after 8.5 years in a patient with hypogammaglobulinemia: evidence for an occult viral reservoir. J Infect Dis 2005; 192:1088-92; PMID:16107964; http://dx.doi.org/10.1086/432917
- Lin A, Thadareddy A, Goldstein MJ, Lake-Bakaar G. Immune suppression leading to hepatitis C virus re-emergence after sustained virological response. J Med Virol 2008; 80:1720-2; PMID:18712814; http://dx.doi.org/10.1002/jmv.21257
- Parodi C, Garcia G, Monzani MC, Culasso A, Aloisi N, Corti M, Campos R, de EdBMM, Bare P. Hepatitis C virus long-term persistence in peripheral blood mononuclear cells in patients with haemophilia. Detection of occult genotype 1. J Viral Hepat 2015; 22:607-16; PMID:25431170; http://dx.doi.org/10.1111/jvh.12363
- Sawada K, Masaki N, Hayashi S, Zeniya M, Ishikawa T, Takahashi H, Ohnishi K, Fukunaga K, Hara N, Yamamoto T, et al. Immunomodulatory effects of selective leucocytapheresis as a new adjunct to interferon-alpha2b plus ribavirin combination therapy: a prospective study in patients with high plasma HCV viraemia. J Viral Hepat 2005; 12:274-82; PMID:15850468; http://dx.doi.org/10.1111/j.1365-2893.2005.00577.x
- Tran TN, Eubanks SK, Schaffer KJ, Zhou CY, Linder MC. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997; 90:4979-86; PMID:9389717
- Zhang H, Potter BJ. The effect of ethanol metabolism on ferritin uptake by freshly isolated rat hepatocytes: is acetaldehyde responsible for this alteration? Alcohol Clin Exp Res 1992; 16:301-7; PMID:1590551; http://dx.doi.org/10.1111/j.1530-0277.1992.tb01381.x
- Meyron-Holtz EG, Moshe-Belizowski S, Cohen LA. A possible role for secreted ferritin in tissue iron distribution. J Neural Transm 2011; 118:337-47; PMID:21298454; http://dx.doi.org/10.1007/s00702-011-0582-0
- Sibille JC, Kondo H, Aisen P. Interactions between isolated hepatocytes and Kupffer cells in iron metabolism: a possible role for ferritin as an iron carrier protein. Hepatology 1988; 8:296-301; PMID:3356411; http://dx.doi.org/10.1002/hep.1840080218
- Liu Z, He JJ. Cell-cell contact-mediated hepatitis C virus (HCV) transfer, productive infection, and replication and their requirement for HCV receptors. J Virol 2013; 87:8545-58; PMID:23720720; http://dx.doi.org/10.1128/JVI.01062-13
- Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol 2009; 83:3719-33; PMID:19176632; http://dx.doi.org/10.1128/JVI.01844-08
- Fujita N, Sugimoto R, Motonishi S, Tomosugi N, Tanaka H, Takeo M, Iwasa M, Kobayashi Y, Hayashi H, Kaito M, et al. Patients with chronic hepatitis C achieving a sustained virological response to peginterferon and ribavirin therapy recover from impaired hepcidin secretion. J Hepatol 2008; 49:702-10; PMID:18620776; http://dx.doi.org/10.1016/j.jhep.2008.05.014
- Tsitoura P, Georgopoulou U, Petres S, Varaklioti A, Karafoulidou A, Vagena D, Politis C, Mavromara P. Evidence for cellular uptake of recombinant hepatitis C virus non-enveloped capsid-like particles. FEBS Lett 2007; 581:4049-57; PMID:17678898; http://dx.doi.org/10.1016/j.febslet.2007.07.032
- Kuiper J, Brouwer A, Knook DL, Van Berkel, TJC. Kupffer and sinusoidal endothelial cells: In The Liver: Biology and Pathobiology. 1994; 3rd edn, 791-818, New York: Raven Press.
- Foka P, Karamichali E, Dalagiorgou G, Serti E, Doumba PP, Pissas G, Kakkanas A, Kazazi D, Kochlios E, Gaitanou M, et al. Hepatitis C virus modulates lipid regulatory factor Angiopoietin-like 3 gene expression by repressing HNF-1alpha activity. J Hepatol 2014; 60:30-8; PMID:23978712; http://dx.doi.org/10.1016/j.jhep.2013.08.016
- Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol 2008; 82:7034-46; PMID:18480457; http://dx.doi.org/10.1128/JVI.00118-08
- Lohmann, V. HCV replicons: overview and basic protocols. In: Tang H ed. Hepatitis C: Methods and Protocols 510 2nd ed 2009; Humana Press 2009: 145-62