
ABSTRACT
We previously identified a series of inositol polyphosphate kinases (IPKs), Arg1, Ipk1, Kcs1 and Asp1, in the opportunistic fungal pathogen Cryptococcus neoformans. Using gene deletion analysis, we characterized Arg1, Ipk1 and Kcs1 and showed that they act sequentially to convert IP3 to PP-IP5 (IP7), a key metabolite promoting stress tolerance, metabolic adaptation and fungal dissemination to the brain. We have now directly characterized the enzymatic activity of Arg1, demonstrating that it is a dual specificity (IP3/IP4) kinase producing IP5. We showed previously that IP5 is further phosphorylated by Ipk1 to produce IP6, which is a substrate for the synthesis of PP-IP5 by Kcs1. Phenotypic comparison of the arg1Δ and kcs1Δ deletion mutants (both PP-IP5-deficient) reveals that arg1Δ has the most deleterious phenotype: while PP-IP5 is essential for metabolic and stress adaptation in both mutant strains, PP-IP5 is dispensable for virulence-associated functions such as capsule production, cell wall organization, and normal N-linked mannosylation of the virulence factor, phospholipase B1, as these phenotypes were defective only in arg1Δ. The more deleterious arg1Δ phenotype correlated with a higher rate of arg1Δ phagocytosis by human peripheral blood monocytes and rapid arg1Δ clearance from lung in a mouse model. This observation is in contrast to kcs1Δ, which we previously reported establishes a chronic, confined lung infection. In summary, we show that Arg1 is the most crucial IPK for cryptococcal virulence, conveying PP-IP5–dependent and novel PP-IP5–independent functions.
Introduction
Cryptococcus neoformans is a major opportunistic fungal pathogen in immunocompromised individuals, especially those with HIV/AIDS (reviewed inCitation1,2,3). Although the primary site of infection is the lung, immunosuppressed patients typically present with meningoencephalitis, which is fatal without treatmentCitation1,2,3. Even with appropriate treatment, (amphotericin B and 5 flucytosine, followed by long-term fluconazole), unacceptably high mortalities are reportedCitation1,2,3, highlighting the need to develop more effective antifungal therapies. Current drugs target the ergosterol biosynthetic pathway/cell membrane (azoles, amphotericin B), RNA (5 flucytosine) or the cell wall biosynthetic enzyme, glucan 3 synthase (echinocandins). Amphotericin B exhibits significant haematological and nephrological toxicityCitation4, while echinocandins are ineffective against the Cryptococcus complexCitation5,6. Furthermore, fluconazole-resistant C. neoformans has been reported in Sub-Saharan Africa and South East AsiaCitation7,8. A detailed understanding of the biology of C. neoformans and other pathogenic fungi is essential for the identification of new drug targets.
The ability of C. neoformans to adapt to the host environment is mediated by several key signalling pathways (reviewed inCitation9). These pathways also fine-tune secretory mechanisms to facilitate export of virulence factors and cell wall remodelling enzymesCitation10,11,12. Secreted virulence factors include the cell-wall associated enzyme, laccase (produces melanin)Citation13, the cell wall-associated and secreted invasin, phospholipase B1 (Plb1)Citation14, and polysaccharide capsule building blocksCitation15,16 which attach to the cell wallCitation17,18. Secretion is therefore essential for regulating fungal virulence and surface architecture, the latter of which impacts recognition by the innate immune systemCitation19.
Using a combination of gene deletion analysis, inositol polyphosphate (IP) and phenotype profiling and enzyme activity assays, we identified and characterized the phospholipase C1/inositol polyphosphate kinase (Plc1/IPK) pathway, and showed that it is essential for the production of several virulence traits and for regulating cell wall integrity. Specifically, we showed that Plc1, the homologue of the PLC1δ isoform in mammalian cells, hydrolyzes the membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2) to generate inositol 1,4,5-trisphosphate (IP3)Citation20. Several IPK enzymes, i.e. Arg1, Ipk1, Kcs1 and Asp1, then act sequentially to generate a series of IPs and inositol pyrophosphates (PP-IPs).
IPs and PP-IPs are present in all eukaryotes and play critical roles in cellular function (reviewed inCitation21,22). Several lines of evidence support fungal IPKs as being candidate targets for antifungal drug development. Firstly, the IPK pathway is non-linear and more complex in mammalian cells than it is in fungi, and generates a wider variety of IP species. Secondly, some mammalian enzymes exhibit functional redundancy, e.g. inositol polyphosphate multikinase (IPMK) and IP3K both convert IP3 to IP4. In contrast, each IP conversion step in C. neoformans is catalyzed by a single IPK. Thirdly, IPKs share a low sequence homology with their mammalian counterparts (e.g. human IPMK and C. neoformans Arg1 are only 19% identical). Finally, a homology search reveals that IPKs are also non-redundant in other fungal pathogens, for example Candida albicans, suggesting that IPKs are a possible target for panfungal therapy.
The first IP produced by the Plc1/IPK pathway is IP3, which we showed serves as a substrate for Arg1Citation20. In comparison with WT, IP3 accumulated in the ARG1 deletion mutant (arg1Δ), whereas all IP and PP-IP species downstream of IP3 were depleted, confirming, indirectly, that Arg1 is an IP3 kinaseCitation23. We also identified a homolog of Arg1 in the cryptococcal genome, Arg2, but found that it neither played a role in IP3 conversion nor in virulenceCitation20. Ipk1, Kcs1, and Asp1 were later identified as IP5, IP6 and PP-IP5 kinases, respectively, acting downstream of Arg1Citation23,24.
Phenotypic characterization of the arg1Δ mutant demonstrated that Arg1 is essential for thermotolerance, cell wall integrity and vacuolar homeostasisCitation20. The arg1Δ mutant was also hypovirulent in a Galleria melonella infection modelCitation20. In contrast, the terminal IPK, Asp1, and its product, (PP)2-IP4, were dispensable for cellular function and virulenceCitation23. Using the kcs1Δ mutant, which is deficient in PP-IP5 and (PP)2-IP4, we showed that PP-IP5 is crucial for cell wall integrity, response to stress, metabolic adaptation to the utilization of diverse carbon sources and dissemination of infection in a mouse inhalation modelCitation20,23,24. Although the ipk1Δ deletion mutant is also deficient in PP-IP5 and IP6, attenuation of its phenotypes was not as severe as in the kcs1Δ mutant due to the accumulation of PP-IP4, which we concluded partially compensates for the absence of PP-IP524. Similar to kcs1Δ, the arg1Δ mutant does not produce PP-IP5, suggesting that the absence of PP-IP5 is responsible, at least in part, for the phenotypic defects of arg1Δ.
In this study we demonstrate that recombinant Arg1 is a dual specificity kinase, converting IP3 to IP4 and IP4 to IP5. We also perform extensive phenotypic comparison of the arg1Δ and kcs1Δ mutants, and identify Arg1 functions that are independent of PP-IP5. These functions include capsule production, N-linked mannosylation and secretion of the virulence determinant Plb1 and cell wall organization. We show that via these functions, Arg1 affects recognition of C. neoformans by the host immune system and its ability to establish a lung infection.
Results
Cryptococcal Arg1 restores defective phenotypes in the S. cerevisiae arg82Δ mutant
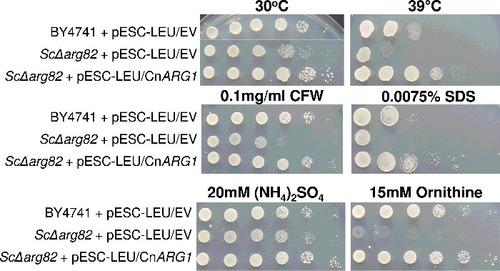
To assess the degree of functional similarity between Arg1 and its orthologue in S. cerevisiae, Arg82, we tested whether ARG1 could restore the growth defects observed in the S. cerevisiae arg82Δ mutant in a complementation assay. Arg82 acts as both an IP3 and an IP4 kinase to produce IP5. Both the arg1Δ mutant and the arg82Δ mutant exhibit a pleiotropic phenotypeCitation25,26. Phenotypes that are shared by arg1Δ and arg82Δ include defective mating and high temperature sensitivityCitation20. The arg82Δ mutant is also auxotrophic for arginine and ornithineCitation25-27. In , we show that the introduction of cryptococcal ARG1 into the S. cerevisiae arg82Δ mutant restored its growth at elevated temperature and on ornithine as a sole nitrogen source. Furthermore, ARG1 restored arg82Δ tolerance to SDS and the cell wall perturbing agent calcofluor white (CFW). These findings support the role of Arg1 as an IP3 and an IP4 kinase, similar to Arg82.
Figure 1. Arg1 from C. neoformans restores defective phenotypes in the S. cerevisiae arg82Δ mutant. ARG1 cDNA was cloned into the pESC-LEU vector, which was then used to transform the S. cerevisiae arg82Δ mutant. Cells containing cloned ARG1 or empty vector were spotted onto plates containing the various media, as indicated, from 106 cells per drop to 10 cells per drop. Plates were incubated at either 30°C or 39°C
Arg1 and Arg82 contribute differentially to stress adaptation of C. neoformans and S. cerevisiae, respectively
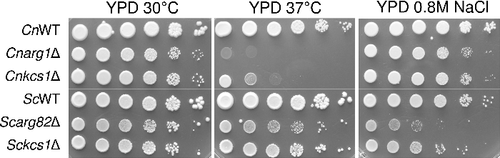
To compare the contribution of Arg1 and Arg82 to stress tolerance in C. neoformans and S. cerevisiae, respectively, the deletion mutants were exposed to elevated temperature and ionic stress. demonstrates that cryptococcal Arg1 and Kcs1 contribute more significantly to temperature tolerance in C. neoformans than Arg82 and Kcs1 do in S. cerevisiae. However, in contrast to Arg82 and Kcs1 in S. cerevisiae, cryptococcal Arg1 and Kcs1 contribute very little to adaptation to ionic stress in C. neoformans.
Figure 2. Arg1 and Arg82 differentially contribute to stress adaptation of C. neoformans and S. cerevisiae. WT and the indicated mutant strains were spotted onto plates containing the various media, as indicated, from 106 cells per drop to 10 cells per drop, and incubated for 2–4 days
Recombinant Arg1 has IP3 and IP4 kinase activities
In our previous study, we used HPLC-based IP profiling of WT and arg1Δ to demonstrate indirectly that Arg1 is an IP3 kinase: the arg1Δ mutant accumulated IP3, while IP4-6, PP-IP5 and (PP)2-IP4 were absentCitation23. To obtain direct evidence that, similar to Arg82, Arg1 has both IP3 and IP4 kinase activities, we produced recombinant His-tagged Arg1 in E. coli (). We found that overnight induction of His-Arg1 expression at 22°C resulted in the highest yield of soluble, full-length fusion protein. The enzyme was then purified in two steps using cobalt affinity and anion exchange chromatography.
Figure 3. Purified Arg1 specifically phosphorylates IP3 to produce IP4 and IP5. (A) Recombinant Arg1 (49 kDa, including the His6 tag) was purified by TALON cobalt affinity chromatography followed by ion exchange chromatography (IEC). (B) Purified Arg1 (1–10 ng/µl reaction, as indicated) was incubated with ATP and IP3 and the reaction products analyzed by polyacrylamide gel electrophoresis and Toluidine Blue staining. Bands representing different IP species and ATP are indicated. (C) Kinetics of Arg1 activity. For these assays, purified Arg1 (20 ng) was incubated with ATP (50–500 µM) and 200 µM of IP3 in 10 µL reactions at room temperature for 0–30 min. The consumption of ATP was monitored using bioluminescence. The assay was performed twice, each time in triplicate. V: reaction velocity
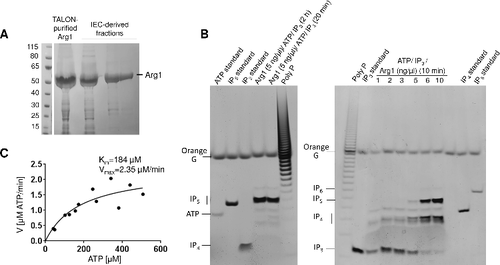
To establish IP5 as the end-product of Arg1 catalysis, purified enzyme (final concentration 5 ng/µl) was incubated with IP3 and ATP for 20 min and 2 hours. Phosphorylation products were analyzed by polyacrylamide gel electrophoresis followed by Toluidine Blue staining. The results in , left panel demonstrate the complete conversion of IP3 to IP5 by Arg1 in 20 minutes. A faint band migrating at a rate similar to that of IP6 was observed in both Arg1 reactions. This band is most likely PP-IP4, which is also a product of purified IPMKCitation28. To confirm IP4 as the intermediate product in Arg1-mediated IP5 production, purified Arg1 at different concentrations (1–10 ng/µl) was incubated with IP3 and ATP for 10 minutes. The results in , right panel demonstrate direct correlation between the amount of Arg1 and the amount of IP4 and IP5 produced, establishing Arg1 as a dual specificity (IP3 and IP4) kinase.
To determine the kinetic properties of Arg1, we used a bioluminescent assay to quantify Arg1-mediated ATP consumption (). The Km of Arg1 for ATP was estimated to be 184 µM and the Vmax 2.35 µM/min.
Sensitivity of cryptococcal arg1Δ and kcs1Δ to nutrient and cell wall stress highlights the importance of PP-IP5 for stress adaptation
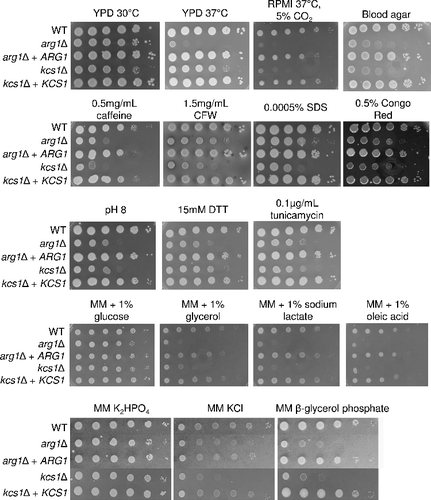
We previously described the defective phenotype of the arg1Δ mutantCitation20. To distinguish PP-IP5-dependent and -independent defects, we performed a comprehensive phenotypic comparison of the arg1Δ and kcs1Δ mutants using drop dilution assays (). The results demonstrate that both mutants are hypersusceptible to multiple stress conditions including alkaline pH, cell wall perturbing agents and ER stress (tunicamycin and DTT). Growth of both mutants was also slower on blood agar, on carbon sources other than glucose, in an environment deficient in free phosphate and at high temperature (37°C). At 37°C, arg1Δ growth was more severely compromised than kcs1Δ growth, while under cell culture conditions (37°C/5% CO2, pH 7.4), growth of kcs1Δ and arg1Δ was equally compromised. This was likely due to the basic pH, to which both mutants are equally sensitive (). Taken together, these results confirm the importance of PP-IP5 in stress adaptation.
Figure 4. Comparative phenotypic testing of arg1Δ and kcs1Δ. WT, mutant and reconstituted strains were spotted onto plates containing the various media as indicated, from 106 cells per drop to 10 cells per drop, and incubated for 2–4 days
arg1Δ and kcs1Δ mutants differ in cell wall organisation, vacuolar morphology and capsule production
Cell wall organization: The hypersusceptibility of the arg1Δ and kcs1Δ mutants to cell wall perturbing agents prompted us to visualize cell wall ultrastructure using transmission electron microscopy (TEM). In comparison to WT and the kcs1Δ mutant, arg1Δ cells were often enlarged. The cell walls of the enlarged cells were disorganized and multi-layered (). In WT, the cell wall thickness ranged between 100 and 150 nm, while in the enlarged arg1Δ cells it often reached 300–500 nm. Unexpectedly, given that the hypersusceptibility of the kcs1Δ mutant to cell wall perturbing agents is greater than or equal to that of the arg1Δ mutant, the cell walls of this mutant were similar to those of the WT in thickness and morphology ().
Figure 5. Transmission electron microscopy (TEM) demonstrating that arg1Δ cell walls are thicker than, and structurally different to, those of WT and kcs1Δ, and that arg1Δ cells have larger vacuoles. N: nucleus; CW: cell wall; V: vacuole
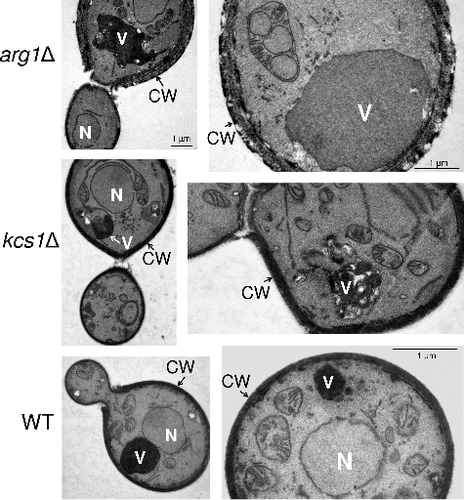
Vacuolar morphology: Using differential interference contrast and fluorescence microscopy we previously demonstrated that a significant proportion of arg1Δ cells were larger than those of WT, and that arg1Δ cells had enlarged vacuoles, which were likely to have formed by fusion of smaller vacuolesCitation20. We therefore used the lipophilic dye, FM 4–64, to label the endocytic pathway in WT, arg1Δ and kcs1Δ as described inCitation20. We found that in contrast to arg1Δ, kcs1Δ cells rarely produced enlarged vacuoles and their cell size was similar to that of WT (). Images obtained by TEM () similarly show enlarged vacuoles in arg1Δ cells, as reported previouslyCitation20.
Figure 6. arg1Δ cells, but not kcs1Δ cells, are enlarged and produce bigger vacuoles. Cells were allowed to endocytose the lipophilic dye, FM 4–64, for 40 min to label vacuoles (indicated by arrows in panels on the right). Differential interference contrast images are shown on the left

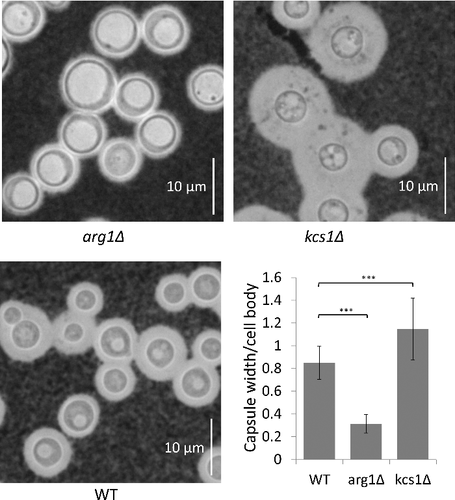
Capsule production: Capsules were induced following growth in minimal medium, and visualized under the light microscope following negative staining with India Ink (). Statistical analysis of capsule size showed that arg1Δ capsules are smaller than those of WT and kcs1Δ, and that kcs1Δ capsules are larger than those of WT and arg1Δ. We also note that vacuoles are significantly larger in arg1Δ than in WT and kcs1Δ, supporting the FM 4–64 data in .
Figure 7. arg1Δ and kcs1Δ capsule size is reduced and increased, respectively, relative to WT. Cells were grown in minimal medium (broth) overnight. Capsules were visualized with India Ink stain under a light microscope at 100X magnification. The graph represents the mean ratio of capsule width to cell body diameter in each strain ± standard deviation (n = 51 cells). ###, P<0.001 using a Tukey-Kramer multiple comparison test
Phospholipase B1 secretion is blocked in arg1Δ
To further investigate the differences in cell wall architecture between arg1Δ and kcs1Δ, we focused on the secretory pathway, which is essential for the export of mannoproteins involved in cell wall integrity. One of these mannoproteins is the virulence factor Plb1Citation29,30. Not only is Plb1 a fungal ‘invasin’ with a role in cell wall integrityCitation14, it is also a marker of the classical secretion pathway in C. neoformansCitation31,32. Plb1 acquires a glycosylphosphatidylinositol (GPI) anchor in the ER/Golgi to enable Plb1 attachment to the cell surfaceCitation14,33. However, Plb1 can also become detached from the cell surface and released to the external milieuCitation14,34.
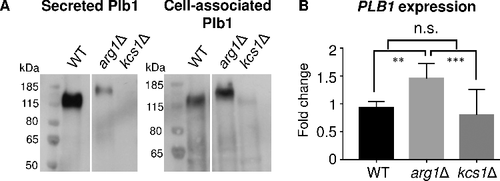
The levels of secreted and cell-associated Plb1 in WT, arg1Δ and kcs1Δ were assessed by Western blotting with the anti-Plb1 antibody. demonstrates that the majority of Plb1 in WT samples is released extracellularly. Conversely, the arg1Δ mutant secreted very little Plb1 but produced more cell-associated Plb1 than WT, indicative of a Plb1 secretion block. In contrast, no Plb1 was detected in the secretions and very little Plb1 was produced intracellularly in kcs1Δ, despite the PLB1 mRNA level being similar to that of the WT ().
Figure 8. Blocked secretion of Plb1 in arg1Δ coincides with an increase in Plb1 molecular weight as assessed by Western blotting. (A) Plb1 in secretions and cell lysates was resolved by SDS-PAGE and detected using anti-Plb1 antibody. (B) PLB1 gene expression was quantified by qPCR. The results are the average of three biological replicates ± SD. ##, P < 0.01; ###, P < 0.001; n.s, not significant
Plb1 is hyper N-linked mannosylated in arg1Δ but not in kcs1Δ
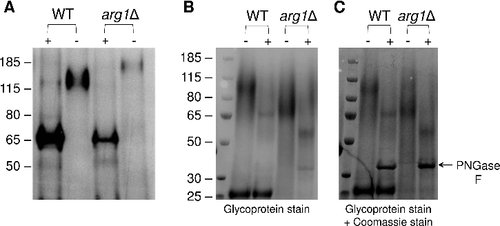
In , we observed that Plb1 produced by arg1Δ has a higher apparent molecular weight on SDS-PAGE (170 kDa) than WT (120 kDa). We showed previously that approximately 30% of the molecular weight of Plb1 is attributable to N-linked mannosylationCitation35, and that preventing the acquisition of N-linked mannosylation by site-directed mutagenesis prevents Plb1 transport to the cell surface in a heterologous expression systemCitation30. To determine whether the increase in Plb1 size in the arg1Δ mutant () is due to abnormal processing of N-linked mannose sugars in the Golgi, we treated the secretions collected from WT and arg1Δ with N-glycosidase F (PNGase F) and assessed protein size by anti-Plb1 Western blotting (). Secretions were chosen over cell-associated fractions as TRIzol-extracted cellular proteins are less amenable to enzymatic treatment due to the need to solubilize them in a strong detergent. Although arg1Δ secretes less Plb1 than WT, sufficient Plb1 protein was present in arg1Δ secretions to observe a shift in molecular weight following PNGase F treatment. As shown in , PNGase F treatment reduced the size of Plb1 in both WT and arg1Δ to approximately 65 kDa, consistent with the molecular weight of Plb1 as deduced from its amino acid sequence. These results confirm that Plb1 is hyper N-mannosylated in the arg1Δ mutant.
Figure 9. N-linked mannosylation of Plb1 and secretome composition are altered in arg1Δ. Cryptococcal secretions, each containing 40 µg of protein, were collected from WT and arg1Δ, treated with PNGase F (+), resolved by SDS-PAGE and subjected to either (A) anti-Plb1 Western blotting, (B) glycoprotein staining or (C) glycoprotein staining followed by Commassie staining. In (C), the arrow indicates the position of the PNGase F enzyme
To determine whether hyper N-linked mannosylation in the arg1Δ mutant extends to all secreted proteins, PNGase F treated and untreated secretions were resolved by SDS-PAGE. The gels were then stained consecutively with a glycoprotein-specific stain () and Coomassie Blue (). Differences were observed in the glycoprotein profile of the secretomes from untreated WT and arg1Δ (). PNGase F-treated WT and arg1Δ secretomes had a fainter stain than the corresponding untreated sample, indicating that most of the proteins in each secretome are N-linked mannosylated. However, in contrast to Plb1 in , Coomassie staining unexpectedly demonstrated differences in the profiles of PNGase F-treated WT and arg1Δ secretions, indicating that the composition of the arg1Δ secretome is altered; this prevented assessment of whether N-linked mannosylation, in general, is also affected. Interestingly, we observed a highly abundant protein with a MW of 25 kDa in WT, but not arg1Δ. This protein appeared to be glycosylated as it stained strongly with the glycoprotein stain, but its MW was unchanged following PNGase F treatment, consistent with the presence of O-linked sugars.
Arg1 is essential for lung colonization and dissemination in a murine inhalation model
To assess the impact of ARG1 deletion on cryptococcal virulence, we compared the survival of mice infected with WT, arg1Δ and arg1Δ + ARG1 using the inhalation model, which mimics the natural route of infection in humans (). All arg1Δ-infected mice were healthy and had lost no weight by 60-days post-infection. In comparison, all WT and arg1Δ + ARG1-infected mice succumbed to infection by day 21. The median survival time of the WT and arg1Δ + ARG1-infected mice was 15 and 16.5 days, respectively. The difference in survival of arg1Δ-infected mice versus WT- and arg1Δ + ARG1-infected mice was statistically significant ().
Figure 10. Arg1 is essential for lung colonization and dissemination in a murine inhalation model of cryptococcosis. (A) arg1Δ is avirulent compared to WT and arg1Δ + ARG1. Mice (n = 10 per strain) were inoculated intranasally with 5× 105 cells/20 µL and monitored daily. Difference in survival between WT/arg1Δ + ARG1-infected mice and arg1Δ-infected mice is statistically significant (####P < 0.0001). (B) and (C) arg1Δ infection is rapidly cleared from mouse lung between 3 and 7 days post-infection. Mice (n = 3 per strain) were inoculated intranasally with 5× 105 cells/20 µL and sacrificed on the days indicated. Lung and brain were harvested for quantitative culture (CFUs) (B) or histopathological analysis for lung only by Periodic acid–Schiff (PAS) staining (C). In (B), the difference in lung CFUs for WT and arg1Δ-infected mice at day 3 is statistically significant (#P < 0.05). In (C), lung histopathology is shown at 3 days post-infection (PAS stain, 40x magnification). In WT-infected lung tissue, arrows indicate cryptococcomas where capsular material is represented by white halos surrounding the cell bodies (stained magenta). In arg1Δ-infected tissue, arrows indicate single acapsular cryptococcal cells
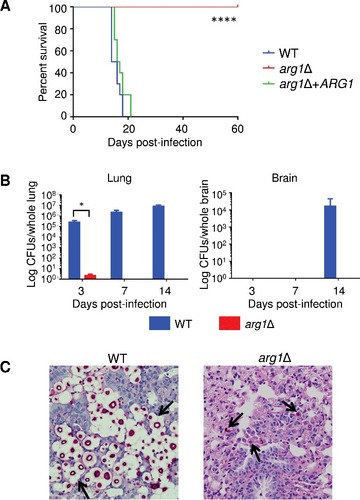
Fungal burdens in the lung and brain of WT and arg1Δ–infected mice were also assessed. In contrast to kcs1Δ, which was avirulent, but established a chronic, localized, asymptomatic lung infection that persisted for 50-days post-inoculationCitation23, arg1Δ infection was cleared from mouse lung between 3 and 7-days post-inoculation (). Consistent with the absence of established lung infection, no arg1Δ CFUs were detected in the brain by day 14, which is the time when WT brain burdens were the highest (). Brain tissue harvested from arg1Δ-infected mice upon termination of the study at day 60 revealed no evidence of cryptococcal dissemination to the CNS (data not shown). Histological analysis showed that by 3-days post-inoculation, WT cryptococci had colonized the lung parenchyma (). In contrast, only a few small acapsular cryptococci are observed in the lung parenchyma of arg1Δ-infected lungs at this time point, with lung pathology appearing otherwise healthy.
We also assessed the survival and organ burdens of mice infected with WT and arg1Δ in a model of disseminated cryptococcosis where mice were infected via the retro-orbital plexus as previously describedCitation36. In this model, WT-infected mice all succumbed to infection by day 10 as previously reportedCitation36. However, following arg1Δ infection, mice remained asymptomatic over a 40-day infection period and there was no evidence of infection in blood, spleen, lungs or brain (data not shown).
Phagocytosis of arg1Δ is greater than that of kcs1Δ
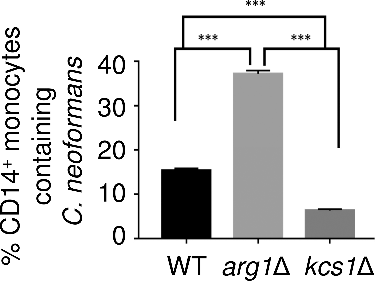
Studies in mouse infection models have demonstrated that monocytes are the first line of defence against C. neoformans, either containing or clearing the infectionCitation37,38. Given that the arg1Δ mutant has an altered surface (, and ), we compared the phagocytosis of WT, arg1Δ and kcs1Δ by human peripheral blood monocytes (PBMCs) using flow cytometry. demonstrates that the extent of phagocytosis of arg1Δ was 7 and 2.5 times greater than that of kcs1Δ and WT, respectively.
Figure 11. arg1Δ is more readily phagocytosed by human PBMCs. Results are expressed as the mean % of CD14+ monocytes containing FITC-labelled C. neoformans ± SD (n = 3). Statistical significance is indicated as follows: ###P < 0.001
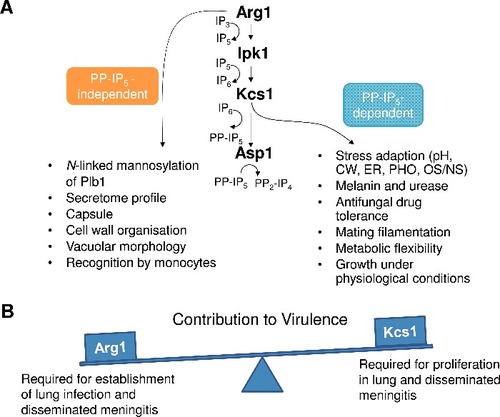
The PP-IP5-dependent functions of Arg1 involved in stress adaptation, and its novel PP-IP5-independent functions relating to cell wall homeostasis, cell surface architecture and host recognition, are summarized in . The combination of these functions contributes to the essential role of Arg1 in establishment of lung infection.
Figure 12. Summary of PP-IP5-dependent and –independent functions of Arg1 (A), and the contribution of Arg1 and Kcs1 to virulence (B). pH – alkaline pH stress; CW – cell wall stress induced by calcofluor white, Congo red, SDS, caffeine; ER – endoplasmic reticulum stress caused by tunicamycin and DTT; PHO – phosphate deprivation; OS/NS – oxidative and nitrosative stress, respectively
Discussion
Arg1 is a dual-specificity IP3/IP4 kinase
Complementation studies demonstrated functional similarities between Arg1 and the IP3–4 kinase in S. cerevisiae (Arg82), as expression of Arg1 in the arg82Δ mutant restored Arg82-dependent phenotypes. We also showed directly that recombinant Arg1 is a dual specificity kinase that rapidly converts IP3 to IP5 (via IP4) in the presence of ATP. Characterization of the enzymatic properties of Arg1 determined a Km of 184 µM for ATP as compared to 74 µM and 17 µM for chicken GgIP3K-A and human IPMK, respectivelyCitation39. These values are all significantly lower than the estimated intracellular concentration of ATP (∼1 mM), suggesting that these IP3 kinases are continuously occupied by ATP. In contrast to IP3 kinases, the Km of Kcs1 enzymes for ATP is in the millimolar rangeCitation40,41. The lower affinity of Kcs1 for ATP is therefore consistent with the biosynthesis of PP-IP5, but not IP5, being linked to the availability of cellular ATP and hence PP-IP5 acting as a sensor of cellular energy statusCitation21.
Similarity of the arg1Δ and kcs1Δ stress sensitivity phenotypes confirms the importance of PP-IP5 for stress adaptation
Using the kcs1Δ mutant, we showed that PP-IP5 is essential for stress tolerance, metabolic adaptation and dissemination of infection in a mouse inhalation modelCitation23. Our previous studies also showed that the arg1Δ mutant, which is deficient in all IPs downstream of IP3, was defective in many cellular functions and virulence-related phenotypesCitation20. However, direct comprehensive comparison of arg1Δ and kcs1Δ phenotypes to assess the PP-IP5-dependent and -independent contribution of Arg1 to cellular function had not been undertaken. We therefore performed extensive phenotypic analysis of arg1Δ and kcs1Δ and demonstrated that the phenotypes of both mutants overlap significantly. This included hypersusceptibility to cell wall perturbing and other stress-inducing agents. Furthermore, the ability of arg1Δ and kcs1Δ to metabolize alternative carbon sources was diminished. This was supported by our published RNAseq dataCitation23. This data demonstrated that the expression of genes encoding mitochondrial enzymes essential for the utilization of alternative carbon source is down-regulated in arg1Δ and kcs1Δ, while the expression of glycolytic enzymes is increased. Given that PP-IP5 is absent in arg1Δ and kcs1Δ, the results demonstrate the importance of PP-IP5 for adaptation to diverse stress conditions, potentially via pyrophosphorylation of key regulatory proteins. In support of this, the presence of inositol pyrophosphates leads to a decrease in the glycolytic/mitochondrial metabolic ratio in S. cerevisiae. This effect is attributable, at least in part, to pyrophosphorylation of the major glycolytic transcription factor Gcr1: pyrophosphorylated Gcr1 binds Gcr2 with lower affinity, leading to reduced transcription of genes encoding glycolytic enzymesCitation42.
demonstrates that PP-IP5 is required for growth under physiological conditions (RPMI media, pH7.4 5%CO2/37°C). Despite this, the kcs1Δ mutant persisted in the lungs of infected miceCitation23, while arg1Δ was cleared (). This result suggests that PP-IP5-independent factors contribute to arg1Δ clearance from lung in vivo.
Arg1 regulates capsule production, N-linked mannosylation, secretion and cell wall morphology independently of PP-IP5
We identified several phenotypes that were specifically defective in arg1Δ, but not kcs1Δ, and which must therefore result from factors other than the absence of PP-IP5. These phenotypes include diminished capsule production, hyper N-linked mannosylation of Plb1, that was associated with failure of its release from the cell, an altered general secretion profile, and cell walls with abnormal morphology. Arg1Δ was also more rapidly phagocytozed by monocytes than kcs1Δ. Given that cell wall integrity is dependent on secretory functions, such as correct mannosylation and secretion, an alteration in these functions in arg1Δ potentially contributes to altered cell wall morphology and to smaller capsules, since capsule is associated with the cell wallCitation17,18. This in turn could affect recognition by the immune system. arg1Δ-specific phenotypes coincided with avirulence of the arg1Δ mutant in a mouse model and rapid clearance of arg1Δ from the lungs (). This is in contrast to kcs1Δ, which had normal cell wall morphology and established a subclinical infection that was confined to the lungCitation23. It is possible that the arg1Δ mutant is rapidly cleared from the lung because it is more rapidly phagocytozed by cells of the innate immune system. Reduced capsule size in the arg1Δ mutant is likely to expose more surface β-glucan and mannoprotein to the binding of opsonising antibody, thereby increasing phagocyte recognition and phagocytosis, as has previously been reported for acapsular mutantsCitation43.
Interestingly, less apparent alteration in cell wall organization was detected in kcs1Δ by TEM analysis as compared to arg1Δ. This was despite kcs1Δ displaying a more severe growth defect in the presence of cell wall perturbing reagents. Compensatory changes, such as alteration of cell wall architecture, may therefore be essential for maintaining the viability of arg1Δ. As the absence of Kcs1 is less detrimental, resulting in loss of only PP-IP5-dependent functions, the compensatory changes are less apparent, or not required, in kcs1Δ. Compensatory changes in the cell wall have been reported in other fungi where cell wall stress caused by mutations or antifungal drugs, triggered increased deposition of chitin in the cell wallCitation44,45,46.
In contrast to arg1Δ, Plb1 protein was barely detectable in kcs1Δ despite the PLB1 mRNA level being similar to that of the WT (). Why Plb1 protein production is reduced in kcs1Δ, but not in arg1Δ is unknown but is most likely due to a defect in translation of the PLB1 mRNA. In our previous publicationCitation23, we demonstrated that exposure of mannoproteins on the surface of the kcs1Δ mutant, as detected by concanavalin A binding, was reduced, and that this coincided with the reduced expression of a subset of mannoprotein-encoding genes. RNAseq analysis also shows that expression of these mannoproteins, but not Plb1, is reduced in the arg1Δ mutant (GEO accession GSE78824). We propose that expression of other mannoproteins, including Plb1, could be upregulated in the arg1Δ mutant (but not in the kcs1Δ mutant) in an attempt to compensate for a defective cell wall, with Plb1 being more highly associated with the cell periphery.
We propose that excess IP3 and/or the absence of IP4/IP5 are likely to contribute to the PP-IP5-independent phenotypes observed in arg1Δ. An excess of IP3 might be detrimental by competing with phosphoinositides and IPs for binding to the pleckstrin homology (PH) domain, and/or other specialized protein domains, thus interfering with the regulation of their functionsCitation47,48. Alternatively, an excess of IP3 could potentially inhibit Plc1 activity via feedback inhibition, since IP3 is a product of Plc1-mediated PIP2 hydrolysisCitation20. In support of this, the PLC1 deletion mutant (plc1Δ) and the arg1Δ mutant have a similar phenotype, including cell walls with abnormal morphology, blocked release of Plb1 from the cell periphery and rapid pulmonary clearance in a mouse modelCitation34, which are all PP-IP5 independent. The absence of IP4 could affect gene expression since IP4 is required for the incorporation of the histone deacetylase HDAC3 into a repressive complex, and for its enzymatic activity in mammalian cellsCitation49,50. Both IP4 and IP5 have been reported to bind to PH domainsCitation49,50,51,52. Hence the absence of IP4/IP5 in the arg1Δ mutant could affect the function of proteins containing PH domains.
In summary, the combination of PP-IP5 -dependent and -independent functions render Arg1 the most important IPK for the virulence of C. neoformans in animal models. Its PP-IP5-independent roles in regulating cryptococcal surface topology likely affect recognition by the host immune system, resulting in clearance in mouse models. The key role of Arg1 in virulence, coupled with its low sequence similarity and non-redundant function as compared to mammalian cells, make Arg1 a candidate target for antifungal drug development.
Material and methods
Fungal strains and media
Cryptococcal strains used in this study were wild-type (WT) C. neoformans var. grubii strain H99 (serotype A, MATα) and the isogenic strains, arg1Δ, kcs1Δ and reconstituted arg1Δ (arg1Δ + ARG1), which were created as previously described inCitation20,23. Strains were routinely grown on YPD (1% yeast extract, 2% peptone and 2% dextrose) or Sabouraud (SAB) agar (1% peptone, 4% glucose, 1.5–2% agar). The S. cerevisiae deletion mutants, arg82Δ (#4003531) and kcs1Δ (#4003956), which were derived from the parental strain BY4741, were obtained from the ATCC.
Expression, purification and enzymatic characterisation of recombinant Arg1
Arg1 cDNA was amplified using primers ARG1-BgIII -s (tacgagatctGACCTGCCCCTCACCCTCG) and ARG-EcoRI-a (CAAGGAATTCTCAAACACAACCCCGTTCAACC) and cloned into the pRSET expression vector to allow production of a 6 histidine-tagged Arg1 fusion protein in Rosetta™ (DE3) E. coli strain. Cultures were incubated overnight at 37°C, diluted 1:100 and incubated at 37°C until the OD600 reached 0.6–0.8. 1 mM IPTG was then added to induce expression of His-tagged Arg1 and the cultures were incubated overnight at ambient temperature. The cells were washed with water, resuspended in lysis buffer (500 mM NaCl; 25 mM Tris–HCl pH 8.0), sonicated and centrifuged to remove debris.
His-Arg1 fusion protein in the supernatant then underwent two rounds of purification. The first round involved using a His GraviTrap TALON affinity column (GE Healthcare, Cat. No. 29-0005-94). The column loaded with His-Arg1 was washed with 10 mL of wash buffer I (600 mM NaCl; 0.1% (v/v) Trition X-100; 25 mM Tris–HCl pH 8.0), and then with 10 mL of wash buffer II (100 mM NaCl; 0.1% (v/v) Trition X-100; 25 mM Tris–HCl pH 7.4). His-Arg1 was eluted from the column with the elution buffer (100 mM NaCl, 200 mM imidazole, 25 mM Tris pH 7.5). His-Arg1 protein was then diluted 3.5-fold in a buffer containing 20 mM Tris pH 7.4, 1 mM DTT and 1 mM MgSO4 and concentrated using an Amicon concentrator (10 kDa cutoff). The second round of purification involved using an anion exchange column; His-Arg1 was eluted from this column using a NaCl gradient (buffer A: 30 mM NaCl, 20 mM Tris pH 7.4, 1 mM MgSO4, 1 mM DTT; buffer B: similar to A, but with NaCl 1000 mM). The purified His-Arg protein was then diluted in a storage buffer (100 mM NaCl; 1 mM MgSO4; 1 mM DTT; 0.05% (v/v) CHAPS; 20 mM Tris–HCl pH 7.4, 40% glycerol) at 500 µg/mL, snap-frozen and stored in aliquots at -80°C.
Routinely, Arg1 activity assay (10 µL) contained 5 µL 2X reaction buffer (40 mM Hepes pH 6.8; 200 mM NaCl; 12 mM MgCl; 2 mM DTT), 200 µM IP3, 500 µM ATP and 20 ng purified Arg1. The reactions were incubated at ambient temperature for up to 30 min, and ATP consumption was measured using a bioluminescent ATP assay kit (Kinase-Glo® Max Luminescent Kinase Assay Kit, Promega). To determine the kinetic properties of Arg1, the reactions were performed using starting ATP concentrations ranging between 50–500 µM ATP, and the ATP concentration was assessed after 0, 10, 20 and 30 min. The assay was performed twice, each in triplicate. Km and Vmax were determined using the Michaelis-Menten model.
To establish that Arg1 is an IP3–4 kinase using PAGE, 40 µl reactions were set up as described above, using different final concentrations of Arg1 (1 to 10 ng/µl). The reactions were terminated after 10 min, 20 min or 2 hours, as indicated in , by the addition of EDTA (10 mM). Visualization of IPs (Arg1 substrates/products) using PAGE and Toluidine Blue staining was performed as described inCitation53.
Yeast complementation assay
ARG1 cDNA from C. neoformans was amplified using the forward primer ccaactagtATGGACCTGCCCCTCACCCT and reverse primer cgcttaattaaTCAAACACAACCCCGTTCAAC, and cloned into the pESC-LEU expression vector. The vectors were used to transform strain BY4741 (S. cerevisiae) and the arg1Δ mutant using a Frozen-EZ Yeast Transformation II Kit (Zymo Research). Complemented and control strains were tested by growing on standard SD medium without leucine at high temperature (39°C) or in the presence of 0.1 mg/mL Calcofluor white (CFW) or 0.0075% SDS. To test growth on different nitrogen sources, SD was prepared without amino acids and ammonium sulfate, and supplemented with ornithine or ammonium sulfate as the sole nitrogen source. Stress tolerance of the S. cerevisiae and C. neoformans mutant and WT strains was compared on YPD plates supplemented with 0.8M NaCl or incubated at 37°C.
Virulence studies
This study was approved and performed in accordance with the recommendations and guidelines of the Western Sydney Local Health District Animal Ethics Committee, Department of Animal Care, Westmead Hospital. The virulence of the strains in 7-week-old female BALB/c mice (from the Animal Resource Centre, Floreat Park, Western Australia) was tested as previously describedCitation24.
Survival: 10 mice/group were inoculated intranasally with either the WT, arg1Δ or the arg1Δ + ARG1 strain (5× 105 cells/20 µL PBS), observed daily for signs of infection over a 60-day period, and sacrificed at predetermined clinical endpoints.
Organ burden: 3 mice/group were inoculated intranasally with either the WT or the arg1Δ strain, and lung and brain were collected at 3, 7 and 14 days post infection to assess fungal load and histopathology.
Histopathology: Lungs were removed and fixed in 4% formalin. Sections (5 µm) were cut with a microtome and stained with Periodic Acid–Schiff (PAS) stain to identify infecting C. neoformans cells. Images were obtained as various magnifications using an Olympus BX43 light microscope fitted with an Olympus DP21 digital camera.
Phagocytosis assay
Fungal cells were grown overnight in YPD broth, washed with water, and heat-killed by incubating at 56°C for 30 min to prevent replication. Cells were then labelled with 0.5 mg/mL FITC (Sigma) in PBS for 30 min. Excess stain was removed by washing 4 times with PBS and the final cell pellet was resuspended in PBS. FITC-labeled fungal cells were opsonized for 30 min by incubation with 10% human serum in PBS (106 cells/100 µL), pelleted, and then resuspended in RPMI 1640–10% FBS medium.
Peripheral blood mononuclear cells (PBMCs) were isolated from ∼25 mL human blood using Ficoll-Paque PLUS (GE Healthcare) following the manufacturer's instructions. The PBMC layer was extracted and resuspended in RPMI/10% FBS. PBMCs (4× 106) were mixed with cryptococci (4× 106) in 1:1 ratio in 100 µL of RPMI 1640–10% FBS medium, and co-incubation was allowed for 2 h at 37°C/5% CO2.
The co-culture was washed once with PBS, and treated with 50 µg Fc blocking agent (mouse IgG Life Technologies, Cat. No. 02–6502; prepared working solution of 100 µg/mL in 0.5% BSA PBS with 0.05% sodium azide). The monocytes in the PBMC population were labelled with anti-CD14 BV421-conjugated antibodies (BioLegend, Cat. No. 301830). The extent of phagocytosis in 50 µL of the co-culture was determined by flow cytometry (BD FACSCanto™ II).
Comparing cell-associated and secreted Plb1 protein by Western blotting
Each strain was cultured overnight in YNB + 2% glucose. The cultures were adjusted to equal cell numbers and grown for a further 18 hours. Equal cell numbers were pelleted by centrifugation and the supernatants (protein secretions) were collected and dialyzed using SnakeSkin® Dialysis Tubing with a 10K molecular weight cut-off (Thermo Fisher Scientific). The dialyzed supernatants were snap-frozen in liquid nitrogen before being lyophilized within a Christ Alpha 2–4 freeze dryer (Martin Christ, Osterode, Germany). The dried pellets were then resuspended in 125 mM imidazole buffer (pH 4.0) and precipitated with 90% trichloroacetic acid (TCA).
Total protein and RNA from the cell pellets were extracted with TRIzol® (Thermo Fisher Scientific, Cat. No. 15596026) and used to assess the level of cell-associated Plb1 protein and PLB1 mRNA, respectively. Briefly, the fungal cell pellets were resuspended in TRizol® and homogenized in the presence of glass beads (426–600 µm, Sigma) using a bead beater (4 cycles, 30 sec beating, 1 min rest). After homogenization, RNA and protein were prepared according to the manufacturer's instructions.
Proteins in the secreted and cell-associated fractions were dissolved in NuPAGE® sample loading buffer and reducing agent (Life Technologies). They were then resolved by SDS-PAGE using NuPAGE® 4–12% bis-Tris gels (Life Technologies) at 170V for 55 min, and transferred to a PVDF membrane (Immobilon-P 0.45 µm transfer membrane, Millipore). To detect Plb1 protein the blots were incubated with an anti-Plb1 peptide specific antibody: 1 µg/mL in Tris-buffered saline with 0.05% Tween-20 (TBST) and 2% BSA. Production of anti-CnPlb1 antibody was described inCitation54. The membranes were washed four times with TBST and then incubated with donkey-anti-goat secondary antibody (Santa Cruz Biotechnology), diluted 1:5000 in TBST with 2% BSA. Chemiluminescence was detected by exposing the membrane to Amersham Hyperfilm™ ECL (GE Healthcare) and developing the film using an X-ray developer.
PLB1 gene expression
Total RNA extracted as described above was treated with DNase and used for oligo-dT-primed cDNA synthesis. The primer pairs for amplifying PLB1 (target gene) and ACT1 (reference gene) cDNA were prepared at a final concentration of 2 µM each. Primer sequences were for a) PLB1: TCATTAGCGCACCGAGTTTG (forward) and AATGGCAAGTGGTGGTAGCC (reverse) and b) ACT1: ATGGTATTGCCGACCGTATG (forward) and CTCTTCGCGATCCACATCTG (reverse). PLB1 gene expression levels in each sample, relative to ACT1, were then determined using SYBR Green qRT-PCR (Corbett Rotor-Gene 6000). Relative quantification of PLB1 expression was determined using the ΔΔCt methodCitation55.
Enzymatic removal of N-linked glycans
Secretions containing 40 µg protein were resuspended in 25 µL 20 mM sodium phosphate buffer (pH 7.5) and treated with 2U PNGase F (New England Biolabs P0704S). A PNGase F negative control was prepared by substituting PNGase F with 2 µL 20 mM sodium phosphate buffer. Samples were incubated at 37°C for 18 hours, after which they were dried using an Eppendorf Concentrator. Samples were then subjected to SDS-PAGE and then either stained for glycoproteins according to the manufacturer's instructions (Pierce Glycoprotein Staining Kit, catalogue number 24562) or subjected to Western blotting as per the Plb1 Western blotting protocol above.
Microscopy
Sample preparation for Transmission Electron Microscopy was carried out as described inCitation34. Briefly, overnight YPD cultures of C. neoformans were prepared for TEM using 2% glutaraldehyde pre-fixation and 2% KMnO4 post-fixation. Following resuspension in 1:1 acetone:epoxy resin, cell pellets were embedded in 100% epoxy resin. 60–80 nm sections were cut using the Leica Ultracut UCT ultramicrotome (Leica Microsystems, Bannockburn, IL), stained with uranyl acetate-lead citrate and viewed with a JEM 1200EX transmission electron microscope (JEOL, USA).
Vacuole labeling with FM 4–64 was carried out as described inCitation20. Briefly, cells were grown in YPD overnight, pelleted and resuspended in fresh medium (1 ml, OD600 ∼0.5). After addition of FM 4–64 (10 µM final concentration), the cells were stained for 5 min, pelleted, resuspended in fresh YPD medium and incubated for an additional 40 min at 30°C with shaking. The tubes were stored on ice until visualization.
Statistical analysis
Statistical analysis was carried out using SPSS Statistics software (version 24). Specifically, the Kaplan Meier Log-ranked Mantel-Cox test and the Mann-Whitney U test were used to assess survival and organ burden differences, respectively, in the mice studies. One-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison was used to assess PLB1 gene expression levels and phagocytosis.
Disclosure of potential conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Virginia James from the histopathology facility at The Westmead Institute for Medical Research for technical assistance.
Funding
This work was supported by a National Health and Medical Research Council of Australia (NHMRC) project grant (APP1058779_Djordjevic). CL is supported by an Australian Postgraduate Award. SL is supported by a Westmead Institute for Medical Research (WIMR) Career Development award. AS is supported by the Medical Research Council (MRC) core support to the MRC/UCL Laboratory for Molecular Cell Biology University Unit (MC_UU_1201814). JPM is supported by a Senior Research Fellowship from the NHMRC. TCS is supported by the Sydney Medical School Foundation.
Related Research Data
References
- Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A, Boulware DR. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;873–881. doi:https://doi.org/10.1016/S1473-3099(17)30243-8.
- Williamson PR. The relentless march of cryptococcal meningitis. Lancet Infect Dis. 2017;790–791. doi:https://doi.org/10.16/S1473-3099(17)30245-1.
- Williamson PR, Jarvis JN, Panackal AA, Fisher MC, Molloy SF, Loyse A, Harrison TS. Cryptococcal meningitis: epidemiology, immunology, diagnosis and therapy. Nat Rev Neurol. 2017;13:13–24. doi:https://doi.org/10.38/nrneurol.2016.167.
- Falci DR, da Rosa FB, Hematological toxicities associated with amphotericin B formulations. Leuk Lymphoma. 2015;56:2889–94. doi:https://doi.org/10.9/10428194.2015.1010080.
- Abruzzo GK, Flattery AM, Gill CJ, Kong L, Smith JG, Pikounis VB, Bouffard AF, Dropinski JF, Rosen H, et al. Evaluation of the echinocandin antifungal MK-0991 (L-743,872): efficacies in mouse models of disseminated aspergillosis, candidiasis, and cryptococcosis. Antimicrob Agents Chemother. 1997;41:2333–8.
- Feldmesser M, Kress Y, Mednick A, Casadevall A. The effect of the echinocandin analogue caspofungin on cell wall glucan synthesis by Cryptococcus neoformans. J Infect Dis 2000;182:1791–5. doi:https://doi.org/10.1086/317614.
- Smith KD, Achan B, Hullsiek KH, McDonald TR, Okagaki LH, Alhadab AA, Akampurira A, Rhein JR, Meya DB, Boulware DR, et al. Increased Antifungal Drug Resistance in Clinical Isolates of Cryptococcus neoformans in Uganda. Antimicrob Agents Chemotherr. 2015;59:7197–204. doi:https://doi.org/10.1128/AAC.01299-15.
- Pan W, Khayhan K, Hagen F, Wahyuningsih R, Chakrabarti A, Chowdhary A, Ikeda R, Taj-Aldeen SJ, Khan Z, Imran D, et al. Resistance of Asian Cryptococcus neoformans serotype A is confined to few microsatellite genotypes. PloS one. 2012;7:e32868. doi:https://doi.org/10.1371/journal.pone.0032868.
- Kozubowski L, Lee SC, Heitman J. Signalling pathways in the pathogenesis of Cryptococcus. Cell Microbiol. 2009;11:370–80. doi:https://doi.org/10.1111/j.1462-5822.2008.01273.x.
- McCotter SW, Horianopoulos LC, Kronstad JW. Regulation of the fungal secretome. Current genetics. 2016;62:533–45. doi:https://doi.org/10.1007/s00294-016-0578-2.
- Rodrigues ML, Djordjevic JT. Unravelling secretion in Cryptococcus neoformans: more than one way to skin a cat. Mycopathologia. 2012;173:407–18. doi:https://doi.org/10.1007/s11046-011-9468-9.
- Geddes JM, Croll D, Caza M, Stoynov N, Foster LJ, Kronstad JW. Secretome profiling of Cryptococcus neoformans reveals regulation of a subset of virulence-associated proteins and potential biomarkers by protein kinase A. BMC Microbiol. 2015;15:206. doi:https://doi.org/10.1186/s12866-015-0532-3.
- Waterman SR, Hacham M, Panepinto J, Hu G, Shin S, Williamson PR. Cell wall targeting of laccase of Cryptococcus neoformans during infection of mice. Infect Immun. 2007;75:714–22. doi:https://doi.org/10.1128/IAI.01351-06.
- Siafakas AR, Sorrell TC, Wright LC, Wilson C, Larsen M, Boadle R, Williamson PR, Djordjevic JT. Cell wall-linked cryptococcal phospholipase B1 is a source of secreted enzyme and a determinant of cell wall integrity. J Biol Chem. 2007;282:37508–14. doi:https://doi.org/10.1074/jbc.M707913200.
- Yoneda A, Doering TL. A eukaryotic capsular polysaccharide is synthesized intracellularly and secreted via exocytosis. Mol Biol Cell. 2006;17:5131–40. doi:https://doi.org/10.1091/mbc.E06-08-0701.
- Rodrigues ML, Nakayasu ES, Oliveira DL, Nimrichter L, Nosanchuk JD, Almeida IC, Casadevall A. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryotic cell. 2008;7:58–67. doi:https://doi.org/10.1128/EC.00370-07.
- Reese AJ, Doering TL. Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Eukaryot Cell. 2003;50:1401–9. doi:https://doi.org/10.1046/j.1365-2958.2003.03780.x.
- Reese AJ, Yoneda A, Breger JA, Beauvais A, Liu H, Griffith CL, Bose I, Kim MJ, Skau C, Yang S, et al. Loss of cell wall alpha(1-3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol Microbiol. 2007;63:1385–98. doi:https://doi.org/10.1111/j.1365-2958.2006.05551.x.
- Levitz SM. Innate recognition of fungal cell walls. PLoS Pathog. 2010;6:e1000758. doi:https://doi.org/10.1371/journal.ppat.1000758.
- Lev S, Desmarini D, Li C, Chayakulkeeree M, Traven A, Sorrell TC, Djordjevic JT. Phospholipase C of Cryptococcus neoformans regulates homeostasis and virulence by providing inositol trisphosphate as a substrate for Arg1 kinase. Infect Immun. 2013;81:1245–55. doi:https://doi.org/10.1128/IAI.01421-12.
- Wilson MS, Livermore TM, Saiardi A. Inositol pyrophosphates: between signalling and metabolism. Biochem J. 2013;452:369–79. doi:https://doi.org/10.1042/BJ20130118.
- Shears SB. Inositol pyrophosphates: why so many phosphates? Adv Biol Regul. 2015;57:203–16. doi:https://doi.org/10.1016/j.jbior.2014.09.015.
- Lev S, Li C, Desmarini D, Saiardi A, Fewings NL, Schibeci SD, Sharma R, Sorrell TC, Djordjevic JT. Fungal Inositol Pyrophosphate IP7 Is Crucial for Metabolic Adaptation to the Host Environment and Pathogenicity. mBio. 2015;6:e00531–15. doi:https://doi.org/10.1128/mBio.00531-15.
- Li C, Lev S, Saiardi A, Desmarini D, Sorrell TC, Djordjevic JT. Identification of a major IP5 kinase in Cryptococcus neoformans confirms that PP-IP5/IP7, not IP6, is essential for virulence. Sci Rep. 2016;6:23927. doi:https://doi.org/10.1038/srep23927.
- Dubois E, Messenguy F. Pleiotropic function of ArgRIIIp (Arg82p), one of the regulators of arginine metabolism in Saccharomyces cerevisiae. Role in expression of cell-type-specific genes. Mol Gen Genet: MGG. 1994;243:315–24.
- Dubois E, Scherens B, Vierendeels F, Ho MM, Messenguy F, Shears SB. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J Biol Chem. 2002;277:23755–63. doi:https://doi.org/10.1074/jbc.M202206200.
- Seeds AM, Bastidas RJ, York JD. Molecular definition of a novel inositol polyphosphate metabolic pathway initiated by inositol 1,4,5-trisphosphate 3-kinase activity in Saccharomyces cerevisiae. J Biol Chem. 2005;280:27654–61. doi:https://doi.org/10.1074/jbc.M505089200.
- Saiardi A, Nagata E, Luo HR, Sawa A, Luo X, Snowman AM, Snyder SH. Mammalian inositol polyphosphate multikinase synthesizes inositol 1,4,5-trisphosphate and an inositol pyrophosphate. Proc Natl Acad Sci U S A. 2001;98:2306–11. doi:https://doi.org/10.1073/pnas.041614598.
- Cox GM, McDade HC, Chen SC, Tucker SC, Gottfredsson M, Wright LC, Sorrell TC, Leidich SD, Casadevall A, Ghannoum MA, et al. Extracellular phospholipase activity is a virulence factor for Cryptococcus neoformans. Mol Microbiol. 2001;39:166–75. doi:https://doi.org/10.1046/j.1365-2958.2001.02236.x.
- Turner KM, Wright LC, Sorrell TC, Djordjevic JT. N-linked glycosylation sites affect secretion of cryptococcal phospholipase B1, irrespective of glycosylphosphatidylinositol anchoring. Biochim Biophys Acta. 2006;1760:1569–79. doi:https://doi.org/10.1016/j.bbagen.2006.07.002.
- Chayakulkeeree M, Johnston SA, Oei JB, Lev S, Williamson PR, Wilson CF, Zuo X, Leal AL, Vainstein MH, Meyer W, et al. SEC14 is a specific requirement for secretion of phospholipase B1 and pathogenicity of Cryptococcus neoformans. Mol Microbiol. 2011;80:1088–101. doi:https://doi.org/10.1111/j.1365-2958.2011.07632.x.
- Lev S, Crossett B, Cha SY, Desmarini D, Li C, Chayakulkeeree M, Wilson CF, Williamson PR, Sorrell TC, Djordjevic JT. Identification of Aph1, a phosphate-regulated, secreted, and vacuolar acid phosphatase in Cryptococcus neoformans. mBio. 2014;5:e01649–14. doi:https://doi.org/10.1128/mBio.01649-14.
- Djordjevic JT, Del Poeta M, Sorrell TC, Turner KM, Wright LC. Secretion of cryptococcal phospholipase B1 (PLB1) is regulated by a glycosylphosphatidylinositol (GPI) anchor. Biochem J. 2005;389:803–12. doi:https://doi.org/10.1042/BJ20050063.
- Chayakulkeeree M, Sorrell TC, Siafakas AR, Wilson CF, Pantarat N, Gerik KJ, Boadle R, Djordjevic JT. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol Microbiol. 2008;69:809–26.
- Chen SC, Wright LC, Golding JC, Sorrell TC. Purification and characterization of secretory phospholipase B, lysophospholipase and lysophospholipase/transacylase from a virulent strain of the pathogenic fungus Cryptococcus neoformans. Biochem J. 2000;347:431–9. doi:https://doi.org/10.1042/bj3470431 https://doi.org/10.1042/0264-6021:3470431.
- Lev S, Kaufman-Francis K, Desmarini D, Juillard PG, Li C, Stifter SA, Feng CG, Sorrell TC, Grau GE, Bahn YS, et al. Pho4 Is Essential for Dissemination of Cryptococcus neoformans to the Host Brain by Promoting Phosphate Uptake and Growth at Alkaline pH. mSphere. 2017;2. doi:https://doi.org/10.1128/mSphere.00381-16.
- Osterholzer JJ, Milam JE, Chen GH, Toews GB, Huffnagle GB, Olszewski MA. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect Immun. 2009;77:3749–58. doi:https://doi.org/10.1128/IAI.00454-09.
- Feldmesser M, Kress Y, Novikoff P, Casadevall A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun. 2000;68:4225–37. doi:https://doi.org/10.1128/IAI.68.7.4225-4237.2000.
- Mayr GW, Windhorst S, Hillemeier K. Antiproliferative plant and synthetic polyphenolics are specific inhibitors of vertebrate inositol-1,4,5-trisphosphate 3-kinases and inositol polyphosphate multikinase. J Biol Chem. 2005;280:13229–40. doi:https://doi.org/10.1074/jbc.M500545200.
- Voglmaier SM, Bembenek ME, Kaplin AI, Dorman G, Olszewski JD, Prestwich GD, Snyder SH. Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc Natl Acad Sci U S A. 1996;93:4305–10. doi:https://doi.org/10.1073/pnas.93.9.4305.
- Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9:1323–6. doi:https://doi.org/10.1016/S0960-9822(00)80055-X.
- Szijgyarto Z, Garedew A, Azevedo C, Saiardi A. Influence of inositol pyrophosphates on cellular energy dynamics. Science. 2011;334:802–5. doi:https://doi.org/10.1126/science.1211908.
- Cross CE, Bancroft GJ. Ingestion of acapsular Cryptococcus neoformans occurs via mannose and beta-glucan receptors, resulting in cytokine production and increased phagocytosis of the encapsulated form. Infect Immun. 1995;63:2604–11.
- Popolo L, Gilardelli D, Bonfante P, Vai M. Increase in chitin as an essential response to defects in assembly of cell wall polymers in the ggp1delta mutant of Saccharomyces cerevisiae. J Bacteriol. 1997;179:463–9. doi:https://doi.org/10.1128/jb.179.2.463-469.1997.
- Terashima H, Yabuki N, Arisawa M, Hamada K, Kitada K. Up-regulation of genes encoding glycosylphosphatidylinositol (GPI)-attached proteins in response to cell wall damage caused by disruption of FKS1 in Saccharomyces cerevisiae. Mol Gen Genet: MGG. 2000;264:64–74. doi:https://doi.org/10.1007/s004380000285.
- Lagorce A, Le Berre-Anton V, Aguilar-Uscanga B, Martin-Yken H, Dagkessamanskaia A, Francois J. Involvement of GFA1, which encodes glutamine-fructose-6-phosphate amidotransferase, in the activation of the chitin synthesis pathway in response to cell-wall defects in Saccharomyces cerevisiae. Eur J Biochem / FEBS. 2002;269:1697–707. doi:https://doi.org/10.1046/j.1432-1327.2002.02814.x.
- Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci U S A. 1995;92:10472–6. doi:https://doi.org/10.1073/pnas.92.23.10472.
- Takeuchi H, Kanematsu T, Misumi Y, Sakane F, Konishi H, Kikkawa U, Watanabe Y, Katan M, Hirata M. Distinct specificity in the binding of inositol phosphates by pleckstrin homology domains of pleckstrin, RAC-protein kinase, diacylglycerol kinase and a new 130 kDa protein. Biochim Biophys Acta. 1997;1359:275–85. doi:https://doi.org/10.1016/S0167-4889(97)00109-2.
- Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JW. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. doi:https://doi.org/10.1016/j.molcel.2013.05.020.
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–40.
- Fukuda M, Kojima T, Kabayama H, Mikoshiba K. Mutation of the pleckstrin homology domain of Bruton's tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5-tetrakisphosphate binding capacity. J Biol Chem. 1996;271:30303–6. doi:https://doi.org/10.1074/jbc.271.48.30303.
- Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, Peter Downes C, Safrany ST, Alessi DR, van Aalten DM. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004;23:3918–28. doi:https://doi.org/10.1038/sj.emboj.7600379.
- Losito O, Szijgyarto Z, Resnick AC, Saiardi A. Inositol pyrophosphates and their unique metabolic complexity: analysis by gel electrophoresis. PloS one. 2009;4:e5580. doi:https://doi.org/10.1371/journal.pone.0005580.
- Siafakas AR, Wright LC, Sorrell TC, Djordjevic JT. Lipid rafts in Cryptococcus neoformans concentrate the virulence determinants phospholipase B1 and Cu/Zn superoxide dismutase. Eukaryot Cell. 2006;5:488–98. doi:https://doi.org/10.1128/EC.5.3.488-498.2006.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi:https://doi.org/10.1006/meth.2001.1262.