
ABSTRACT
Staphylococcus epidermidis accounts for the majority of cases of neonatal sepsis. Moreover, it has been demonstrated to be associated with neonatal morbidities, such as bronchopulmonary dysplasia (BPD), white matter injury (WMI), necrotizing enterocolitis (NEC) and retinopathy of prematurity (ROP), which affect short-term and long-term neonatal outcome. Imbalanced inflammation has been considered to be a major underlying mechanism of each entity. Conventionally regarded as a harmless commensal on human skin, S. epidermidis has received less attention than its more virulent relative Staphylococcus aureus. Particularities of neonatal innate immunity and nosocomial environmental factors, however, may contribute to the emergence of S. epidermidis as a significant nosocomial pathogen. Neonatal host response to S. epidermidis sepsis has not been fully elucidated. Evidence is emerging regarding the implication of S. epidermidis sepsis in the pathogenesis of neonatal inflammatory diseases. This review focuses on the interplay among S. epidermidis, neonatal innate immunity and inflammation-driven organ injury.
Introduction
Steady progresses in the fields of perinatology and neonatology have improved neonatal survival, especially in very immature preterm infants [Citation1,Citation2]. However, the burden of neonatal morbidities, such as bronchopulmonary dysplasia (BPD), white matter injury (WMI), necrotizing enterocolitis (NEC) and retinopathy of prematurity (ROP), remains substantial [Citation1,Citation2]. Besides these inflammatory disorders, neonatal sepsis is ranked as one of the major morbidities in preterm neonates [Citation1,Citation2]. Staphylococcus epidermidis, the leading species of coagulase-negative staphylococci (CONS), has emerged as the predominant pathogen of sepsis in preterm infants [Citation3]. Due to its ubiquitous colonization on human skin, S. epidermidis has been conventionally considered as a harmless commensal [Citation4]. However, mortality due to S. epidermidis sepsis, defined as one or more positive blood cultures with corresponding clinical signs, ranges from 1.9% to 4.8% in the general neonatal population, and may be as high as 9.4% in very low birth weight (VLBW) infants [Citation5-9]. Moreover, a growing body of evidence implicates a strong association between S. epidermidis sepsis and inflammation-related neonatal morbidities, such as BPD, WMI, NEC and ROP [Citation10-16]. Although not fully delineated, in each entity, the pathogenesis is considered to be a multiple-hit process with inflammation being a principal downstream mechanism [Citation17-20]. Prenatal and postnatal injurious events, such as chorioamnionitis, resuscitation, oxygen toxicity, mechanical ventilation and neonatal infection, have been demonstrated to contribute to dysregulated and sustained inflammation that may severely impair organ development, especially in very immature preterm infants [Citation17-20]. Maturation-dependent characteristics and genetic propensity may underlie the individual vulnerability for extrinsic insults [Citation17-20].
Neonatal sepsis is a major risk factor of adverse neonatal inflammation, and considerably contributes to neonatal short-term and long-term morbidity by inducing, exacerbating or perpetuating an imbalanced host inflammatory response [Citation17-21]. S. epidermidis is likely to be implicated in sepsis-induced neonatal inflammation, and thus might play an important role in inflammatory-driven organ injury. As compared to other pathogens, such as Staphylococcus aureus and Escherichia coli, S. epidermidis may be underestimated in terms of its role in neonatal morbidity. This review summarizes data from current clinical and experimental studies to better elucidate the pathogenic mechanisms of S. epidermidis, host response to S. epidermidis, and the role of S. epidermidis sepsis-induced inflammation in the pathogenesis of neonatal inflammatory disorders. A thorough understanding in this area may help to optimize current anti-infectious strategies and to improve neonatal outcomes, especially in very immature preterm infants.
Epidemiology and population structure of S. epidermidis in neonates
The colonization of S. epidermidis in humans starts immediately after birth [Citation22]. Despite wide interpersonal variations and differences in terms of body sites and age within the same host, S. epidermidis remains to be the most frequently isolated species of skin microbiome [Citation23]. Recent data indicate that S. epidermidis may belong to the abundant bacterial genera in airway and gut microbiomes, especially of hospitalized preterm and term neonates [Citation24-26]. Multilocus sequence typing (MLST), a nucleotide sequence based technology, is an advanced tool of investigating bacterial evolution and epidemiological pattern [Citation8,Citation25,Citation27-30]. Current data demonstrate that S. epidermidis strains isolated from hospitalized neonates are not widely diverse among various regions, but are represented by a limited number of clones [Citation8,Citation25,Citation28,Citation30]. The hands of health-care personnel are the major sources of S. epidermidis transmission among neonates [Citation8,Citation25,Citation28,Citation30].
Incidence of S. epidermidis sepsis and neonatal inflammatory diseases
The emergence of S. epidermidis as the most common pathogen of neonatal sepsis is closely associated with paradigm shifts in perinatal and neonatal medicine. While the universal prenatal screening and treatment protocol for Group B Streptococcus (GBS) have contributed to a significant reduction in early-onset sepsis (EOS), the incidence of nosocomial late-onset sepsis (LOS) has significantly increased [Citation3, Citation31]. LOS is most frequently defined as sepsis occurring after 72 h of age, and has been well recognized to be associated with prematurity, invasive interventions like intravascular catheterization, failure in early enteral feeding, prolonged antibiotic treatment and hospitalization, as well as underlying respiratory and cardiovascular diseases [Citation3]. The omnipresence of S. epidermidis on human skin and host susceptibility may allow S. epidermidis to easily invade into the bloodstream through indwelling catheters [Citation3,Citation4]. Despite intensive efforts to reduce nosocomial infections, an average of one third of VLBW infants would still have at least one episode of LOS [Citation9, Citation32-43]. The incidence of LOS increases up to 50% in very immature preterm infants with a gestational age (GA) ≤ 24 weeks, compared to 24–30% and 8–19% in neonates with GA 25–26 weeks and 27–28 weeks, respectively [Citation2]. Over the last 5–10 years, the proportion of LOS caused by S. epidermidis in most countries remained rather unchanged at around 50% () [Citation9, Citation32-43].
Table 1. The proportion of culture-proven late-onset sepsis due to coagulase-negative Staphylococci, predominantly Staphylococcus epidermidis, among different geographical areas.
Accumulating data demonstrate that S. epidermidis sepsis confers an increased risk of adverse short-term and long-term neonatal outcome, especially in very immature preterm infants. Although prematurity, genetic predisposition, nutritional deficits and hemodynamic instability may also contribute to BPD, WMI, NEC and ROP [Citation17-20], S. epidermidis sepsis has been demonstrated to be an independent risk factor of all these neonatal morbidities [Citation10-16]. Neonates with S. epidermidis sepsis were demonstrated to have a significantly higher risk of BPD compared to neonates without sepsis. The relative risk (RR) for BPD varied from 2.6 (95% CI: 1.5-4.6) to 9.40 (95% CI: 3.83-23.08) among different studies [Citation12,Citation16]. In a recent meta-analysis, the odds ratios (ORs) for neurodevelopmental impairment (NDI) and cerebral palsy were found to be 1.31 (95% CI: 1.09-1.57) and 1.7 (95% CI: 1.02-2.87), respectively, in neonates with S. epidermidis sepsis compared to non-septic controls [Citation10]. Additionally, white and grey matter abnormality was demonstrated in neonates with S. epidermidis sepsis by means of magnetic resonance imaging (MRI) [Citation14]. It has been shown that the incidence of NEC and severe ROP ≥ stage 3 in extremely preterm infants with S. epidermidis sepsis was 21.9% and 37.1%, respectively, nearly 2 times higher than non-sepsis controls [Citation13]. Apart from these, there might be an enhanced risk of growth retardation in neonates with S. epidermidis sepsis compared to uninfected neonates (95% CI of OR: 1.1-1.7) [Citation15]. Notably, the sepsis-associated risk of BPD, NDI and growth retardation does not seem to differ between S. epidermidis and other pathogens, namely coagulase-positive staphylococci, gram-negative bacteria and fungi [Citation11,Citation12,Citation14,Citation15]. This suggests that a common pathway of inflammation initially triggered by diverse pathogens may contribute to neonatal long-term adverse outcomes. In general, these results challenge the conventional conception of S. epidermidisis being a non-virulent microorganism, and underscored a significant role of S. epidermdis sepsis in the pathogenesis of neonatal inflammatory diseases.
Putative virulent factors of S. epidermidis involved in neonatal sepsis
Abundant research has demonstrated that most of the pathogenic determinants of S. epidermidis have their original roles in a commensal life, including outcompeting pathogens for physiological niches in the host, inhibition of other harmful microorganisms and priming of innate immune responses [Citation44-46]. Factors associated with nosocomial environmental, such as skin-breaching interventions, and individual susceptibility may contribute to the role switch of S. epidermidis from a commensal to a nosocomial pathogen [Citation36, Citation47]. According to investigations carried out in the last two decades, S. epidermids sequence type 2 (ST2) appears to be the most widespread nosocomial clone in neonatal ward, and may account for the majority of cases of neonatal sepsis [Citation8, Citation25,Citation28,Citation30]. ST2 is characterized by biofilm forming capacity, antibiotic resistance and a highly flexible genetic background [Citation27-30]. This sends an alarming signal that pathogenicity of S. epidermidis may evolve with the progress in medical technology and changes in patient demographics, and indicates the necessity of reevaluating the role of S. epidermidis in nosocomial infections. In the following, putative virulent factors implicated in S. epidermidis sepsis will be reviewed, with a special focus on their roles in bacteria-host interaction. For a more comprehensive understanding of S. epidermidis pathogenic factors, readers are referred to a review elsewhere (see [Citationref. 48).
Biofilms
Bacterial biofilm is a highly organized society of bacteria embedded in self-produced extracellular polymeric matrix [Citation49]. Its development follows a three-stage process comprising attachment, accumulation and detachment [Citation49]. The attachment of bacteria on a biotic surface is mainly mediated via microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) [Citation50]. Cell surface hydrophobicity is another important mediating factor in bacterial interaction with abiotic surfaces [Citation51]. Subsequently, both bacterial proliferation and the secretion of various extracellular polymers contribute to the formation of small aggregates, which then mature into biofilm with a three-dimensional structure and enhanced resistance against antibiotics and immune cells [Citation49,Citation52,Citation53]. S. epidermidis has been shown to form biofilms on medical devices within 24 h of invasion [Citation51]. As maturation proceeds, bacteria may disperse from the biofilm and colonize new infection sites [Citation52]. Although chronic and indolent by nature, biofilm formation should not be excluded from the pathogenic mechanisms of neonatal sepsis, which is otherwise considered to be an acute disease. Bacteremia and the subsequent inflammation are considered to partly originate from the dispersal of bacteria from biofilms on indwelling medical devices [Citation48,Citation54]. Moreover, the persistence of biofilms may provide a base for a continuous seeding of bacteria into the bloodstream [Citation48,Citation54]. This may largely explain why persistent S. epidermidis sepsis is not rare among very immature preterm neonates during hospitalization [Citation16,Citation55].
Extracellular polymers and surface components of S. epidermidis
Polysaccharide intercellular adhesin (PIA), also named poly-N-acetylglucosamine in terms of its basic structure, is the best characterized S. epidermidis extracellular polymer and a major constituent of S. epidermidis biofilms [Citation56]. The icaADBC operon regulates the synthesis of PIA, and a small insertion sequence (IS) element IS256 may switch biofilm phenotype through reversible transposition into the icaADBC locus [Citation27,Citation30]. S. epidermidis strains positive for ica and IS256 are preferentially found in nosocomial epidemic clones like ST2, emphasizing the significance of PIA and genetic flexibility in S. epidermidis pathogenicity [Citation27-30]. PIA has been demonstrated to increase the amount of C5a in human serum, reduce the sensitivity of S. epidermidis to human antimicrobial peptides (AMPs) such as LL-37 and β-defensin 3, inhibit the phagocytosis of human neutrophils and macrophages, and enhance bacterial survival in phagosomes [Citation57-59]. Compared to PIA-negative S. epidermidis strains, PIA-positive strains seem to induce lower levels of pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, IL-12p40 and IL-12p70, but a higher level of IL-8, a potent neutrophil recruiting factor [Citation57-59]. In contrast to PIA, poly-γ-DL-glutamic acid (PGA) is produced by strains both of clinical and commensal origins, and may have minimal impact on S. epidermidis biofilm formation [Citation60]. Nonetheless, S. epidermidis strains incapable of producing PGA were more likely to be phagocytosed by human neutrophils and killed by LL-37 and β-defensin 3 than PGA-positive strains [Citation60]. Recently, a novel lipopeptide (LP01) produced by S. epidermdis has been identified to be a Toll-like receptor 2 (TLR2) agonist, and may trigger the expression of β-defensins [Citation61]. In addition, S. epidermidis surface components lipoteichoic acid (LTA) and peptidoglycan (PGN) may increase the production of TNF-α, IL-1β, IL-6 and nitric oxide (NO) in murine macrophages [Citation62]. The overall pro-inflammatory effect of S. epidermidis, however, was demonstrated to be weaker than that provoked by E. coli [Citation62]. These results underscore that the interaction between S. epidermidis and the host immunity originally relies on immune evasion, rather than aggressive invasion [Citation54].
Toxins, exoenzymes and other virulent factors
For pathogenic microorganisms, toxins are considered to be major virulence factors. In contrast to the vast repertoire of toxins produced by other virulent pathogens, such as S. aureus and E. coli, toxin production in S. epidermidis is predominantly restricted to phenol-soluble modulins (PSMs) [Citation63]. PSMs comprise a group of amphipathic, α-helical peptides which are widely produced by staphylococcal species, and can be categorized into shorter α-type peptides (20-25 amino acids) and longer β-type peptides (43-45 amino acids) [Citation63]. PSMs of S. epidermidis include PSMα, PSMδ, PSMϵ, δ-toxin and PSM-mec, which all belong to α-type peptides, as well as PSMβ1 and PSMβ2 [Citation63]. PSMs may contribute to S. epidermidis pathogenicity in several ways. First, PSMs have surfactant-like properties, which may shape biofilm into a mature three-dimensional structure and facilitate bacterial dispersal [Citation52]. Second, some α-type PSMs of S. epidermidis, especially PSMδ, are potent cytolytic agents of human neutrophils at micromolar concentrations [Citation64]. However, the composition of α-type PSMs in relation to non-cytolytic β-type PSMs in S. epidermidis was found to be lower than in S. aureus [Citation64]. Additionally, the production profile of PSMs appears to be highly strain-dependent, and some clinical invasive strains may have an overall enhanced production of PSMs [Citation64,Citation65]. Third, some PSMs produced by S. epidermidis, such as PSMδ, PSMϵ, PSM-mec and PSMβ1, are powerful pro-inflammatory agents which may stimulate IL-8 release even at nanomolar concentrations [Citation64,Citation65]. Fourth, certain PSM of S. epidermidis was demonstrated to be enteropathic, and may contribute to the development of NEC in preterm infants [Citation66]. The production of most PSMs is regulated by agr quorum-sensing system, a core-genome regulator which also influences the expression of MSCRAMMs and exoenzymes [Citation67]. In contrast, PSM-mec is encoded by psm-mec gene locus, located within the mobile genetic element staphylococcal chromosome cassette mec (SCCmec), which mediates methicillin resistance in S. epidermidis [Citation27,Citation65]. The emergence of psm-mec by combining two pathogenic elements in one genetic exchange event is a good example of S. epidermidis genetic flexibility [Citation65]. In a mouse model of neonatal sepsis, PSM-mec significantly increased the expression of IL-1β, TNF-α and the mouse IL-8 homologue CXCL1, leading to increased mortality [Citation68]. This recent study by Qin et al [Citation68]. was the very first to suggest an important role of PSM-mec in the pathogenesis of S. epidermidis sepsis.
Other virulent factors of S. epidermidis, such as exoenzymes, have been demonstrated to be mainly involved in S. epidermidis immune evasion. Protease SepA may contribute to the resistance of S. epidermidis against human AMPs and neutrophils [Citation64]. Endopeptidase Esp was found to degrade host fibronectin and complement factors [Citation69]. Furthermore, S. epidermidis can produce lipases GehC and GehD to inactivate host-derived fatty acids [Citation70]. As an alternative strategy to circumvent host immunity, S. epidermidis may employ Aps AMP sensor/resistance regulator to repel AMPs and enhance its survival after being phagocytosed [Citation64].
Taken together, the generally passive nature of S. epidermidis pathogenicity is consistent with its commensal origin. However, the nosocomial environment and individual state of host immunity may drive S. epidermidis to transform into a nosocomial pathogen by adjusting its expression of pathogenic components. S. epidermidis-induced neonatal inflammation may reflect the net effect of a consortium of intricately orchestrated virulent factors interacting with the host immune system.
Neonatal innate immune response to S. epidermidis
As S. epidermidis sepsis is acute by nature, neonatal host immune response to S. epidermidis is considered to be mainly mediated by innate immunity [Citation71,Citation72]. Accumulating data have demonstrated that neonatal innate immunity may be partially deficient in quantity and/or quality, with a distinctive regulation pattern of inflammatory response [Citation72,Citation73]. Until now, neonatal host response to S. epidermidis sepsis has not been fully elucidated.
Phagocytosis and intracellular killing of S. epidermidis
Compared to term neonates and adults, preterm neonates seem to be deficient in serum levels of complement and AMPs, as well as in the proportion of phagocytic neutrophils and monocytes [Citation74-76]. However, both phagocytosis and intracellular killing of S. epidermidis were demonstrated to be similar between preterm and term neonatal monocytes [Citation76]. Upon stimulation with S. epidermidis, neutrophils of preterm infants may exert a lower oxidative burst activity than those of term neonates [Citation77]. This indicates that oxygen radical-independent pathways might contribute to the uncompromised capacity of preterm neonatal neutrophils to kill S. epidermidis intracellularly. Future studies are warranted to confirm this assumption. Notably, neutrophil oxidative burst responses to S. epidermidis seem to vary among strains [Citation77]. Sepsis-associated strains tend to induce an enhanced oxidative burst activity compared to non-sepsis strains [Citation77]. In order to accurately interpret bacteria-host interactions, strain phenotype of S. epidermidis should be characterized to identify clinical strains which are more virulent than others. On account of the often unspecific information on strain characteristics in current studies, the phagocytosis and intracellular killing capacity of S. epidermidis by neonatal phagocytes remain to be elucidated.
S. epidermidis-driven cytokine and chemokine responses
Data concerning S. epidermidis-induced inflammatory mediators in neonates are predominantly based on in vitro experiments (). S. epidermidis was demonstrated to induce the release of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, IL-8 and IL-12 [Citation78-83]. This induction of pro-inflammatory cytokines appears to be dependent on time, bacterial inoculum and strain phenotype. The levels of TNF-α, IL-1β, IL-6 and IL-8 may show a rapid rise by several folds within the first 4 h upon S. epidermidis exposure [Citation79]. Intracytoplasmic IL-6 production was demonstrated to be nearly 4 times higher at a multiplicity of infection value of 10:1 (colony forming unit: white blood cell) than at 1:1 [Citation81]. Furthermore, S. epidermidis strains positive for icaADBC seem to induce a higher level of IL-8 while inhibiting the release of IL-6 as compared to icaADBC negative strains [Citation81,Citation83]. This may be associated with the pathogenicity of clinically invasive S. epidermidis clones.
Table 2. In vitro and in vivo studies reporting neonatal host inflammatory responses to Staphylococcus epidermidis.
Discussions are ongoing regarding the pro-inflammatory capacity of S. epidermidis compared to other pathogens. It has been demonstrated that levels of TNF-α, IL-1β, IL-6 and IL-8 induced by S. epidermidis at 2 h and 4 h upon stimulation were several times lower than those induced by E. coli and GBS [Citation79]. In contrast, another study showed that the release of IL-6, IL-8 and IL-12 at 18 h of infection did not differ among S. epidermidis, E. coli and GBS [Citation82]. Discrepant results also exist concerning S. epidermidis-induced pro-inflammatory response in relation to neonatal maturity. Some studies reported a GA-dependent increase of S. epidermidis-induced TNF-α, IL-1β, IL-6 and IL-8 [Citation78,Citation81], while others demonstrating similar concentrations of IL-6, IL-8 and IL-12 in preterm and term neonates [Citation80,Citation82]. Anti-inflammatory IL-10 was consistently shown to be produced in a GA-independent manner [Citation80-82]. The net effect of counterbalancing pro- and anti-inflammatory cytokines was mostly unexplored in in vitro studies of S. epidermidis, with only one study demonstrating a higher production of pro-inflammatory factors than anti-inflammatory factors [Citation80]. It should be noted that perinatal factors such as in utero exposure to infection/inflammation, hyperoxia/hypoxia, and temperature instability are likely to affect the signaling of immune cells [Citation84]. These aspects may, to some extent, explain the inconsistency among studies. Furthermore, most in vitro studies used heat-killed S. epidermidis, which has been demonstrated to mount a different pattern of cytokine production compared to live bacteria [Citation85]. Other confounding factors may include TLR2 gene polymorphism [Citation84], S. epidermidis strain phenotypes [Citation83], and experimental methods.
Performance of in vitro studies using cell lines may not accurately reflect the bacteria-host interaction. By comparison, in vivo animal studies have the advantage to explore the release pattern of inflammatory cytokines and the impact of inflammatory response on host. Currently, there is a dearth of studies providing such information [Citation86,Citation87]. Kronforst et al. and Bi et al. are the first to establish a mouse model of neonatal S. epidermidis sepsis [Citation86,Citation87]. At 2–6 h post infection, S. epidermidis nosocomial strain 1457 was demonstrated to induce a rapid production of cytokines, such as TNF-α, IL-1β, IL-6, IL-12p70 and IL-10, as well as chemoattractant protein-1 (MCP-1), granulocyte-colony stimulating factor (G-CSF), CCL2 and CXCL1 [Citation86,Citation87]. Moreover, the production profile of cytokines was found to increase with bacterial inoculum, and to shift towards pro-inflammation as IL-10 was down-regulated compared to pro-inflammatory cytokines [Citation87]. As a result, newborn mice suffered significant weight loss at 24 h and 48 h post infection [Citation87], followed by white and grey matter injury observed at the 14th day after birth [Citation86].
Signaling pathways involved in the host immune response to S. epidermidis sepsis
The host immune response against S. epidermidis is found to be largely mediated by TLR2 [Citation88]. The pathogenic factors PIA, LTA, PGN, lipopeptides and PSMs of S. epidermidis are well-recognized ligands for TLR2 [Citation61,Citation89-91]. Recently, formyl peptide receptor 2 (FPR2) and nucleotide oligomerization domain (NOD)-like receptor NOD2 have been shown to be additional receptors for PSMs and PGN, respectively [Citation92-94]. Activation of TLR2, FPR2 and NOD2 uniformly culminates in nuclear factor-κB (NF-κB) transcription and the synthesis of inflammatory mediators [Citation84,Citation95,Citation96]. The expression of TLR2 and downstream signaling molecules appears to be similar between neonates of different GA and adults [Citation97-98]. The same pattern of expression has been demonstrated for NOD2 [Citation99]. Moreover, TLR2 expression may be up-regulated in neonates with sepsis, supporting the hypothesis of dysregulated neonatal inflammatory response in favor of hyper-inflammation [Citation100,Citation101]. As for FPR2, we found no data concerning its expression in neonates. A study using a mouse model of neonatal sepsis indicated that at high bacterial concentrations, FPR2 and NOD2 may be more involved in the recognition of S. epidermidis than TLR2 [Citation86].
In general, in the scenario of established sepsis caused by clinically invasive S. epidermidis strains, neonatal immune response to S. epidermidis appears to skew towards hyper-inflammation, potentially leading to tissue damages and long-term morbidity. However, more studies are warranted to verify this pattern of S. epidermidis-induced inflammatory response in neonates.
S. epidermidis-induced inflammation and neonatal diseases
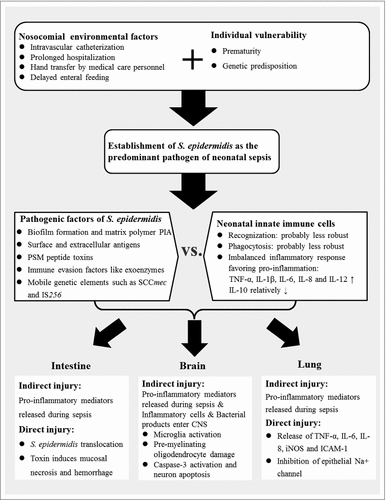
The role of S. epidermidis sepsis in neonatal inflammatory diseases, such as BPD, WMI, NEC and ROP, has just begun to be unraveled. So far, data concerning the mechanisms of S. epidermidis sepsis-driven ophthalmologic injury are lacking. In regard of other inflammatory disorders, exposure to pro-inflammatory factors such as TNF-α, IL-1β, IL-6 and IL-8, in combination with a deficiency of anti-inflammatory factor IL-10, has been demonstrated to cause lung tissue damages as well as the breakdown of intestinal epithelial integrity and blood-brain barrier [Citation17,Citation18,Citation102,Citation103]. This may confer indirect evidence of inflammation-induced organ injuries associated with S. epidermidis sepsis (). Moreover, there is emerging evidence of S. epidermidis directly interacting with neonatal tissues, which will be elaborated in this part of the review.
Bronchopulmonary dysplasia
BPD is characterized by dysregulated alveolarization and impaired angiogenesis, and has become the most common complication of preterm births [Citation17]. The incidence of BPD is estimated to be 50% in extremely preterm neonates [Citation2]. So far, direct evidence linking S. epidermidis infection and lung injury is scarce. One in vitro study has shown that S. epidermidis may stimulate higher levels of IL-8 and intercellular adhesion molecule-1 (ICAM-1) than S. aureus in vascular endothelial cells and small airway epithelial cells, whereas IL-10 was undetected [Citation104]. As IL-8 and ICAM-1 are strong chemoattractants, the authors speculated that S. epidermidis sepsis might contribute to the pathogenesis of BPD by generating a persistent recruitment of inflammatory cells into lung tissues [Citation104]. In another in vitro study using bronchial epithelial cells, S. epidermidis was demonstrated to increase the production of IL-6, IL-8, TNF-α and inducible nitric oxide synthase (iNOS) in a time-dependent manner [Citation105]. Furthermore, activities of epithelial Na+ channel (ENaC) and cystic fibrosis transmembrane conductance regulator (CFTR), both being critical in the regulation of lung liquid homeostasis, were enhanced upon stimulation of S. epidermidis [Citation105]. The role of S. epidermidis infection in the pathogenesis of BPD remains controversial. Some suggested that S. epidermidis might colonize in the lung and persistently produce a low-grade inflammation to cause BPD [Citation104]. Another assumption is based on the finding that infection-induced inflammatory mediators, such as neutrophils and cytokines, may not be detected several days after the onset of infection [Citation106]. Therefore, a pro-inflammatory response initiated by S. epidermidis sepsis may act as a contributing hit. Subsequently, sustained activation of the lung inflammatory cascade in the context of prematurity and dysregulated neonatal immunity may contribute to the pathogenesis of BPD.
White matter injury
The most common form of preterm brain injury is white matter injury (WMI), characterized by focal and diffuse abnormalities in cerebral white matter with loss of pre-myelinating oligodendrocytes, inhibition of neuronal precursor cell proliferation, and excessive activation of microglia [Citation18,Citation107,Citation108]. Neonatal brain injury following bacteremia/sepsis may be due to the penetration of bacteria into central nervous system (CNS), as in the case of meningitis caused by E. coli and GBS [Citation109]. In contrast, S. epidermidis is less virulent and invasive, and barely causes meningitis [Citation4]. However, S. epidermidis-induced systemic cytokines, inflammatory cells, and bacterial products, such as toxins and surface components, may enter the CNS through a disrupted blood-brain barrier [Citation18,Citation86]. The study conducted by Bi et al. is the first to provide evidence of a causal relationship between S. epidermidis sepsis and neonatal brain injury [Citation86]. In this model, culture results from the brain as well as the cerebrospinal fluid (CSF) were persistently negative of S. epidermidis [Citation86]. However, the white blood cell count in CSF and CCL2 in the brain increased upon S. epidermidis sepsis at 2 h [Citation86]. At 6 h post infection, the expression of caspase-3, which may activate microglia and trigger brain apoptosis, was enhanced in brain tissues [Citation86]. Of note, 14 days postnatally, decreased volumes of white and grey matter as well as disruption of myelination at the cortical region were detected [Citation86]. In light of the above, S. epidermidis may directly trigger an imbalanced systemic pro-inflammatory reaction, and may also indirectly stimulate resident cerebral immune cells to release cerebral inflammatory mediators, leading to the damage of pre-myelinating oligodendrocytes and neurons [Citation18,Citation86]. These effects may act in concert to result in white and grey matter abnormalities later in life [Citation18,Citation86].
Necrotizing enterocolitis
NEC is the most common gastrointestinal emergency among premature infants [Citation19,Citation110]. Characterized by intestinal inflammation that can progress to necrosis and perforation, NEC often confers life-long sequelae in survivors [Citation19,Citation110]. Although bacteria, including S. epidermidis, have been clearly demonstrated to be involved in the pathogenesis of NEC, there is not a single species that has been found to be the determinant pathogen [Citation19]. Instead, microbial dysbiosis and disrupted gut intestinal barrier are speculated to be major underlying mechanisms [Citation111]. The expression of TLR2 on intestinal epithelia seems to be up-regulated during NEC [Citation19], which may further enhance the susceptibility of the immature gut to pathogens. Besides indirect evidence linking S. epidermidis sepsis and gut injury, there is also direct evidence indicating that S. epidermidis was capable to translocate into the gut cavity through injured intestine [Citation112]. Results from very recent studies corroborated this finding and demonstrated that the causative agent of LOS, including S. epidermidis, may correspond to the abundant bacterial genera in gut microbiome at the diagnosis of LOS [Citation24,Citation25]. The gut-colonizing S. epidermidis seems to be characterized by the carriage of pathogenic factors, such as icaA, IS256, SCCmec and toxins [Citation25,Citation66]. Toxins of S. epidermidis were found to induce mucosal necrosis and hemorrhage in the bowel [Citation66]. Taken together, S. epidermidis may be implicated in the pathogenesis of NEC, probably not as a causative pathogen but as a conspirator. The contribution of S. epidermidis sepsis to NEC remains to be elucidated.
Retinopathy of prematurity
As a retinal vascular disorder most commonly seen in preterm neonates, ROP is the major cause of visual impairment or blindness in children [Citation113]. The development of ROP comprises an initial phase of inhibited retinal vessel growth followed by a second phase of abnormal vasoproliferation [Citation114]. Although prematurity and oxygen exposure are central in the etiology of ROP, neonatal inflammation and infection may have a deleterious impact on retinal angiogenesis, aggravating the risk of ROP [Citation114,Citation115]. Pro-inflammatory cytokines, such as TNF-α and IL-6, as well as vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF-1) are associated with dysregulated vascularization [Citation20,Citation114]. S. epidermidis is one of the prevalent pathogens of endophthalmitis in adults, indicating its potential to cause ocular inflammatory injury [Citation116]. However, direct evidence linking S. epidermidis to ROP has not been found yet. Based on current knowledge, S. epidermidis is not likely to play a key role in the development of ROP following S. epidermidis sepsis, which predominantly occurs in very immature preterm infants who are also at an enhanced risk of oxygen exposure [Citation2]. Conversely, S. epidermidis sepsis-driven inflammation may, through indirect mechanisms, exacerbate the dysregulated process of angiogenesis leading to ROP.
Future perspectives
Despite being the most frequently isolated pathogen of neonatal sepsis, the significance of S. epidermidis in neonatal diseases is recognized to a lesser extent compared to other virulent pathogens, such as S. aureus and E. coli. However, due to the enhanced awareness that S. epidermidis may considerably contribute to neonatal short-term and long-term morbidity via inflammation-induced organ injury, epidemiological and experimental studies are warranted in the future.
How can current knowledge be implicated in the clinical management of neonatal sepsis? Close surveillance of the distribution of epidemic clones and characterization of pathogenic elements of S. epidermidis strains may improve infection control. Moreover, the prevention of preterm births as well as the reduction of invasive procedures and strict hand hygiene will still constitute the cornerstone of preventive measures against S. epidermidis sepsis [Citation3]. For patients with established sepsis, our armamentarium of anti-sepsis strategies is still highly restricted to antibiotics. Given the potential role of sepsis-induced inflammation in the pathogenesis of neonatal morbidity, new strategies of immunomodulation aiming at restoring the balance of neonatal inflammatory response might add to current sepsis treatment protocols [Citation21].
Figure 1. Putative mechanisms of Staphylococcus epidermidis-induced neonatal inflammatory response and consecutive organ injuries. PIA: polysaccharide intercellular adhesin; PSMs: phenol-soluble modulins; SCCmec: staphylococcal chromosome cassette mec; IS: insertion sequence; TNF-α: tumor necrosis factor-α; IL: interleukin; CNS: central nervous system; iNOS: inducible nitric oxide synthase; ICAM-1: intercellular adhesion molecule-1.
Disclosure of potential conflicts of interests
No potential conflicts of interests were declared.
Additional information
Funding
References
- Liu L, Oza S, Hogan D, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet. 2016;388:3027–35. doi:10.1016/S0140-6736(16)31593-8
- Stoll BJ, Hansen NI, Bell EF, et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA. 2015;314::1039–51. doi:10.1001/jama.2015.10244
- Dong Y, Speer CP. Late-onset neonatal sepsis: recent developments. Arch Dis Child Fetal Neonatal Ed. 2015;100:F257–63. doi:10.1136/archdischild-2014-306213
- Dong Y, Speer CP. The role of Staphylococcus epidermidis in neonatal sepsis: guarding angel or pathogenic devil? Int J Med Microbiol. 2014;304:513–20. doi:10.1016/j.ijmm.2014.04.013
- Isaacs D; Australasian Study Group For Neonatal Infections. A ten year, multicentre study of coagulase negative staphylococcal infections in Australasian neonatal units. Arch Dis Child Fetal Neonatal Ed. 2003;88:F89–93. doi:10.1136/fn.88.2.F89
- Al-Taiar A, Hammoud MS, Cuiqing L, et al. Neonatal infections in China, Malaysia, Hong Kong and Thailand. Arch Dis Child Fetal Neonatal Ed. 2013;98:F249–55. doi:10.1136/archdischild-2012-301767
- Hornik CP, Fort P, Clark RH, et al. Early and late onset sepsis in very-low-birth-weight infants from a large group of neonatal intensive care units. Early Hum Dev. 2012;88 (Suppl 2):S69–74. doi:10.1016/S0378-3782(12)70019-1
- Hira V, Sluijter M, Estevão S, et al. Clinical and molecular epidemiologic characteristics of coagulase-negative staphylococcal bloodstream infections in intensive care neonates. Pediatr Infect Dis J. 2007;26:607–12. doi:10.1097/INF.0b013e318060cc03
- Lee SM, Chang M, Kim KS. Blood culture proven early onset sepsis and late onset sepsis in very-low-birth-weight Infants in Korea. J Korean Med Sci. 2015;30(Suppl 1):S67–74. doi:10.3346/jkms.2015.30.S1.S67
- Alshaikh B, Yusuf K, Sauve R. Neurodevelopmental outcomes of very low birth weight infants with neonatal sepsis: systematic review and meta-analysis. J Perinatol. 2013;33:558–64. doi:10.1038/jp.2012.167
- Schlapbach LJ, Aebischer M, Adams , et al. Impact of sepsis on neurodevelopmental outcome in a Swiss National Cohort of extremely premature infants. Pediatrics. 2011;128:e348–57. doi:10.1542/peds.2010-3338
- Liljedahl M, Bodin L, Schollin J. Coagulase-negative staphylococcal sepsis as a predictor of bronchopulmonary dysplasia. Acta Paediatr. 2004;93:211–5. doi:10.1111/j.1651-2227.2004.tb00708.x
- Alshaikh B, Yee W, Lodha A, et al. Coagulase-negative staphylococcus sepsis in preterm infants and long-term neurodevelopmental outcome. J Perinatol. 2014;34:125–9. doi:10.1038/jp.2013.155
- Shah DK, Doyle LW, Anderson PJ, et al. Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J Pediatr. 2008;153:170–5. 175.e1. doi:10.1016/j.jpeds.2008.02.033
- Stoll BJ. Hansen NI, Adams-Chapman I, Fanaroff AA, Hintz SR, Vohr B, Higgins RD; National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292:2357–65. doi:10.1001/jama.292.19.2357
- Anderson-Berry A, Brinton B, Lyden E, et al. Risk factors associated with development of persistent coagulase-negative staphylococci bacteremia in the neonate and associated short-term and discharge morbidities. Neonatology. 2011;99:23–31. doi:10.1159/000292567
- Speer CP. Chorioamnionitis, postnatal factors and proinflammatory response in the pathogenetic sequence of bronchopulmonary dysplasia. Neonatology. 2009;95:353–61. doi:10.1159/000209301
- Strunk T, Inder T, Wang X, et al. Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect Dis. 2014;14:751–62. doi:10.1016/S1473-3099(14)70710-8
- Nanthakumar N, Meng D, Goldstein AM, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One. 2011;6:e17776. doi:10.1371/journal.pone.0017776
- Hartnett ME. Role of cytokines and treatment algorithms in retinopathy of prematurity. Curr Opin Ophthalmol. 2017;28:282–88. doi:10.1097/ICU.0000000000000360
- Wynn JL, Neu J, Moldawer LL, et al. Potential of immunomodulatory agents for prevention and treatment of neonatal sepsis. J Perinatol. 2009;29:79–88. doi:10.1038/jp.2008.132
- Capone KA, Dowd SE, Stamatas GN, et al. Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131:2026–32. doi:10.1038/jid.2011.168
- Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–92. doi:10.1126/science.1171700
- Stewart CJ, Embleton ND, Marrs ECL, et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome. 2017;5:75. doi:10.1186/s40168-017-0295-1
- Soeorg H, Huik K, Parm Ü, et al. Molecular epidemiology of Staphylococcus epidermidis in neonatal intensive care units. APMIS. 2017;125:63–73. doi:10.1111/apm.12637
- Lohmann P, Luna RA, Hollister EB, et al. The airway microbiome of intubated premature infants: characteristics and changes that predict the development of bronchopulmonary dysplasia. Pediatr Res. 2014;76:294–301. doi:10.1038/pr.2014.85
- Kozitskaya S, Olson ME, Fey PD, et al. Clonal analysis of Staphylococcus epidermidis isolates carrying or lacking biofilm-mediating genes by multilocus sequence typing. J Clin Microbiol. 2005;43:4751–57. doi:10.1128/JCM.43.9.4751-4757.2005
- Klingenberg C, Rønnestad A, Anderson AS, et al. Persistent strains of coagulase-negative staphylococci in a neonatal intensive care unit: virulence factors and invasiveness. Clin Microbiol Infect. 2007;13:1100–11. doi:10.1111/j.1469-0691.2007.01818.x
- Li M, Wang X, Gao Q, et al. Molecular characterization of Staphylococcus epidermidis strains isolated from a teaching hospital in Shanghai, China. J Med Microbiol. 2009;58:456–61. doi:10.1099/jmm.0.007567-0
- Schoenfelder SM, Lange C, Eckart M, et al. Success through diversity – how Staphylococcus epidermidis establishes as a nosocomial pathogen. Int J Med Microbiol. 2010;300:380–6. doi:10.1016/j.ijmm.2010.04.011
- Shah BA, Padbury JF. Neonatal sepsis: an old problem with new insights. Virulence. 2014;5:170–8. doi:10.4161/viru.26906
- Claessens LC, Zonnenberg IA, van den Dungen FA, et al. Cerebral ultrasound abnormalities in preterm infants caused by late-onset sepsis. PLoS One. 2017;12:e0173227. doi:10.1371/journal.pone.0173227
- The Canadian Neonatal Network. The Canadian Neonatal Network Annual Report 2015. http://www.canadian neonatal network.org/portal.
- Mitha A, Foix-L'Hélias L, Arnaud C, et al. Neonatal infection and 5-year neurodevelopmental outcome of very preterm infants. Pediatrics. 2013;132:e372–80. doi:10.1542/peds.2012-3979
- Gowda H, Norton R, White A, et al. Late-onset neonatal sepsis – A 10-year review from north Queensland, Australia. Pediatr Infect Dis J. 2017; 36:883–8. doi:10.1097/INF.0000000000001568
- Greenberg RG, Kandefer S, Do BT, et al. Late-onset sepsis in extremely premature infants: 2000–2011. Pediatr Infect Dis J. 2017;36:774–9. doi:10.1097/INF.0000000000001570
- Davis JW, Odd D, Jary S, et al. The impact of a sepsis quality improvement project on neurodisability rates in very low birth weight infants. Arch Dis Child Fetal Neonatal Ed. 2016; pii: fetalneonatal-2015–309804. doi:10.1136/archdischild-2015-309804
- Tröger B, Härtel C, Buer J, et al. Clinical relevance of pathogens detected by multiplex PCR in blood of very-low-birth weight infants with suspected sepsis – multicentre study of the German Neonatal Network. PLoS One. 2016;11:e0159821. doi:10.1371/journal.pone.0159821
- Wójkowska-Mach J, Gulczyńska E, Nowiczewski M, et al. Late-onset bloodstream infections of Very-Low-Birth-Weight infants: data from the Polish Neonatology Surveillance Network in 2009–2011. BMC Infect Dis. 2014;14:339. doi:10.1186/1471-2334-14-339
- Labi AK, Obeng-Nkrumah N, Bjerrum S, et al. Neonatal bloodstream infections in a Ghanaian Tertiary Hospital: Are the current antibiotic recommendations adequate? BMC Infect Dis. 2016;16:598. doi:10.1186/s12879-016-1913-4
- Lu Q, Zhou M, Tu Y, et al. Pathogen and antimicrobial resistance profiles of culture-proven neonatal sepsis in Southwest China, 1990–2014. J Paediatr Child Health. 2016;52:939–43. doi:10.1111/jpc.13278
- Ozkan H, Cetinkaya M, Koksal N, et al. Culture-proven neonatal sepsis in preterm infants in a neonatal intensive care unit over a 7 year period: coagulase-negative Staphylococcus as the predominant pathogen. Pediatr Int. 2014;56:60–6. doi:10.1111/ped.12218
- Boo NY, Cheah IG. Factors associated with inter-institutional variations in sepsis rates of very-low-birth-weight infants in 34 Malaysian neonatal intensive care units. Singapore Med J. 2016;57:144–52. doi:10.11622/smedj.2016056
- Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–82. doi:10.1038/nm.2062
- Iwase T, Uehara Y, Shinji H, et al. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465:346–9. doi:10.1038/nature09074
- Cogen AL, Yamasaki K, Sanchez KM, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol. 2010;130:192–200. doi:10.1038/jid.2009.243
- Duckney P, Wong HK, Serrano J, et al. The role of the skin barrier in modulating the effects of common skin microbial species on the inflammation, differentiation and proliferation status of epidermal keratinocytes. BMC Res Notes. 2013;6:474. doi:10.1186/1756-0500-6-474
- Otto M. Molecular basis of Staphylococcus epidermidis infections. Semin Immunopathol. 2012;34:201–14. doi:10.1007/s00281-011-0296-2
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–22. doi:10.1126/science.284.5418.1318
- Bowden MG, Chen W, Singvall J, et al. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology. 2005;151:1453–64. doi:10.1099/mic.0.27534-0
- Patel JD, Ebert M, Ward R, et al. S. epidermidis biofilm formation: effects of biomaterial surface chemistry and serum proteins. J Biomed Mater Res A. 2007;80:742–51. doi:10.1002/jbm.a.31103
- Le KY, Dastgheyb S, Ho TV, Otto M. Molecular determinants of staphylococcal biofilm dispersal and structuring. Front Cell Infect Microbiol. 2014;4:167.
- Yao Y, Sturdevant DE, Otto M. Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J Infect Dis. 2005;191:289–98. doi:10.1086/426945
- Cheung GY, Otto M. Understanding the significance of Staphylococcus epidermidis bacteremia in babies and children. Curr Opin Infect Dis. 2010;23:208–16. doi:10.1097/QCO.0b013e328337fecb
- Linder N, Hernandez A, Amit L, et al. Persistent coagulase-negative staphylococci bacteremia in very-low-birth-weight infants. Eur J Pediatr. 2011;170:989–95. doi:10.1007/s00431-010-1387-0
- Mack D, Fischer W, Krokotsch A, et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–83. doi:10.1128/jb.178.1.175-183.1996
- Satorius AE, Szafranski J, Pyne D, et al. Complement c5a generation by staphylococcal biofilms. Shock. 2013;39(4):336–42. doi:10.1097/SHK.0b013e31828d9324
- Vuong C, Voyich JM, Fischer ER, et al. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004;6:269–75. doi:10.1046/j.1462-5822.2004.00367.x
- Spiliopoulou AI, Kolonitsiou F, Krevvata MI, et al. Bacterial adhesion, intracellular survival and cytokine induction upon stimulation of mononuclear cells with planktonic or biofilm phase Staphylococcus epidermidis. FEMS Microbiol Lett. 2012;330:56–65. doi:10.1111/j.1574-6968.2012.02533.x
- Kocianova S, Vuong C, Yao Y, et al. Key role of poly-gamma-DL-glutamic acid in immune evasion and virulence of Staphylococcus epidermidis. J Clin Invest. 2005;115:688–94. doi:10.1172/JCI200523523
- Li D, Lei H, Li Z, et al. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS One. 2013;8:e58288. doi:10.1371/journal.pone.0058288
- Jones KJ, Perris AD, Vernallis AB, et al. Induction of inflammatory cytokines and nitric oxide in J774.2 cells and murine macrophages by lipoteichoic acid and related cell wall antigens from Staphylococcus epidermidis. J Med Microbiol. 2005;54:315–21. doi:10.1099/jmm.0.45872-0
- Cheung GY, Joo HS, Chatterjee SS, et al. Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38:698–719. doi:10.1111/1574-6976.12057
- Cheung GY, Rigby K, Wang R, et al. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog. 2010;6:e1001133. doi:10.1371/journal.ppat.1001133
- Queck SY, Khan BA, Wang R, et al. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5:e1000533. doi:10.1371/journal.ppat.1000533
- Scheifele DW, Bjornson GL, Dyer RA, et al. Delta-like toxin produced by coagulase-negative staphylococci is associated with neonatal necrotizing enterocolitis. Infect Immun. 1987;55:2268–73.
- Le KY, Otto M. Quorum-sensing regulation in staphylococci-an overview. Front Microbiol. 2015;6:1174.
- Qin L, Da F, Fisher EL, et al. Toxin mediates sepsis caused by methicillin-resistant Staphylococcus epidermidis. PLoS Pathog. 2017;13:e1006153. doi:10.1371/journal.ppat.1006153
- Dubin G, Chmiel D, Mak P, et al. Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol Chem. 2001;382:1575–82. doi:10.1515/BC.2001.192
- Longshaw CM, Farrell AM, Wright JD, et al. Identification of a second lipase gene, gehD, in Staphylococcus epidermidis: comparison of sequence with those of other staphylococcal lipases. Microbiology. 2000;146:1419–27. doi:10.1099/00221287-146-6-1419
- Nguyen TH, Park MD, Otto M. Host response to Staphylococcus epidermidis colonization and infections. Front Cell Infect Microbiol. 2017;7:90. doi:10.3389/fcimb.2017.00090
- Power Coombs MR, Kronforst K, Levy O. Neonatal host defense against Staphylococcal infections. Clin Dev Immunol. 2013;2013:826303. doi:10.1155/2013/826303
- Levy O, Wynn JL. A prime time for trained immunity: innate immune memory in newborns and infants. Neonatology. 2014;105:136–41. doi:10.1159/000356035
- Strunk T, Doherty D, Richmond P, et al. Reduced levels of antimicrobial proteins and peptides in human cord blood plasma. Arch Dis Child Fetal Neonatal Ed. 2009;94:F230–1. doi:10.1136/adc.2008.143438
- Fleer A, Gerards LJ, Aerts P, et al. Opsonic defense to Staphylococcus epidermidis in the premature neonate. J Infect Dis. 1985;152:930–7. doi:10.1093/infdis/152.5.930
- Prosser A, Hibbert J, Strunk T, et al. Phagocytosis of neonatal pathogens by peripheral blood neutrophils and monocytes from newborn preterm and term infants. Pediatr Res. 2013;74:503–10. doi:10.1038/pr.2013.145
- Björkqvist M, Jurstrand M, Bodin L, et al. Defective neutrophil oxidative burst in preterm newborns on exposure to coagulase-negative staphylococci. Pediatr Res. 2004;55:966–71. doi:10.1203/01.pdr.0000127018.44938.89
- Strunk T, Prosser A, Levy O, et al. Responsiveness of human monocytes to the commensal bacterium Staphylococcus epidermidis develops late in gestation. Pediatr Res. 2012;72:10–8. doi:10.1038/pr.2012.48
- Mohamed MA, Cunningham-Rundles S, Dean CR, et al. Levels of pro-inflammatory cytokines produced from cord blood in-vitro are pathogen dependent and increased in comparison to adult controls. Cytokine. 2007;39:171–7. doi:10.1016/j.cyto.2007.07.004
- Tatad AM, Nesin M, Peoples J, et al. Cytokine expression in response to bacterial antigens in preterm and term infant cord blood monocytes. Neonatology. 2008;94:8–15. doi:10.1159/000112541
- Härtel C, Osthues I, Rupp J, et al. Characterisation of the host inflammatory response to Staphylococcus epidermidis in neonatal whole blood. Arch Dis Child Fetal Neonatal Ed. 2008;93:F140–5. doi:10.1136/adc.2007.124685
- Peoples JD, Cheung S, Nesin M, et al. Neonatal cord blood subsets and cytokine response to bacterial antigens. Am J Perinatol. 2009;26:647–57. doi:10.1055/s-0029-1220788
- Haase B, Faust K, Heidemann M, et al. The modulatory effect of lipids and glucose on the neonatal immune response induced by Staphylococcus epidermidis. Inflamm Res. 2011;60:227–32. doi:10.1007/s00011-010-0258-5
- Glaser K, Speer CP. Toll-like receptor signaling in neonatal sepsis and inflammation: a matter of orchestration and conditioning. Expert Rev Clin Immunol. 2013;9:1239–52. doi:10.1586/1744666X.2013.857275
- Strunk T, Richmond P, Prosser A, et al. Method of bacterial killing differentially affects the human innate immune response to Staphylococcus epidermidis. Innate Immun. 2011;17:508–16. doi:10.1177/1753425910379840
- Bi D, Qiao L, Bergelson I, et al. Staphylococcus epidermidis bacteremia induces brain injury in neonatal mice via Toll-like receptor 2-dependent and -independent pathways. J Infect Dis. 2015;212:1480–90. doi:10.1093/infdis/jiv231
- Kronforst KD, Mancuso CJ, Pettengill M, et al. A neonatal model of intravenous Staphylococcus epidermidis infection in mice <24 h old enables characterization of early innate immune responses. PLoS One. 2012;7:e43897. doi:10.1371/journal.pone.0043897
- Strunk T, Power Coombs MR, Currie AJ, et al. TLR2 mediates recognition of live Staphylococcus epidermidis and clearance of bacteremia. PLoS One. 2010;5:e10111. doi:10.1371/journal.pone.0010111
- Stevens NT, Sadovskaya I, Jabbouri S, et al. Staphylococcus epidermidis polysaccharide intercellular adhesin induces IL-8 expression in human astrocytes via a mechanism involving TLR2. Cell Microbiol. 2009;11:421–32. doi:10.1111/j.1462-5822.2008.01264.x
- Schwandner R, Dziarski R, Wesche H, et al. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. 1999;274:17406–9. doi:10.1074/jbc.274.25.17406
- Hajjar AM, O'Mahony DS, Ozinsky A, et al. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–9. doi:10.4049/jimmunol.166.1.15
- Natsuka M, Uehara A, Yang S, et al. A polymer-type water-soluble peptidoglycan exhibited both Toll-like receptor 2- and NOD2-agonistic activities, resulting in synergistic activation of human monocytic cells. Innate Immun. 2008;14:298–308. doi:10.1177/1753425908096518
- Kretschmer D, Rautenberg M, Linke D, et al. Peptide length and folding state govern the capacity of staphylococcal β-type phenol-soluble modulins to activate human formyl-peptide receptors 1 or 2. J Leukoc Biol. 2015;97:689–97. doi:10.1189/jlb.2A0514-275R
- Kretschmer D, Nikola N, Dürr M, et al. The virulence regulator Agr controls the staphylococcal capacity to activate human neutrophils via the formyl peptide receptor 2. J Innate Immun. 2012;4:201–12. doi:10.1159/000332142
- Shaw MH, Reimer T, Kim YG, et al. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol. 2008;20:377–82. doi:10.1016/j.coi.2008.06.001
- Cattaneo F, Parisi M, Ammendola R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int J Mol Sci. 2013;14:7193–230. doi:10.3390/ijms14047193
- Sadeghi K, Berger A, Langgartner M, et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis. 2007;195:296–302. doi:10.1086/509892
- Dasari P, Zola H, Nicholson IC. Expression of Toll-like receptors by neonatal leukocytes. Pediatr Allergy Immunol. 2011;22:221–8. doi:10.1111/j.1399-3038.2010.01091.x
- Granland C, Strunk T, Hibbert J, et al. NOD1 and NOD2 expression and function in very preterm infant mononuclear cells. Acta Paediatr. 2014;103:e212–8. doi:10.1111/apa.12559
- Zhang JP, Yang Y, Levy O, et al. Human neonatal peripheral blood leukocytes demonstrate pathogen-specific coordinate expression of TLR2, TLR4/MD2, and MyD88 during bacterial infection in vivo. Pediatr Res. 2010;68:479–83. doi:10.1203/PDR.0b013e3181f90810
- Viemann D, Dubbel G, Schleifenbaum S, et al. Expression of toll-like receptors in neonatal sepsis. Pediatr Res. 2005;58:654–9. doi:10.1203/01.PDR.0000180544.02537.FD
- Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi:10.4049/jimmunol.171.11.6164
- Emami CN, Chokshi N, Wang J, et al. Role of interleukin-10 in the pathogenesis of necrotizing enterocolitis. Am J Surg. 2012;203:428–35. doi:10.1016/j.amjsurg.2011.08.016
- Ivarsson M, Schollin J, Björkqvist M. Staphylococcus epidermidis and Staphylococcus aureus trigger different interleukin-8 and intercellular adhesion molecule-1 in lung cells: implications for inflammatory complications following neonatal sepsis. Acta Paediatr. 2013;102:1010–6. doi:10.1111/apa.12350
- Hussain R, Oliynyk I, Roomans GM, et al. Modulation of ENaC, CFTR, and iNOS expression in bronchial epithelial cells after stimulation with Staphylococcus epidermidis (94B080) and Staphylococcus aureus (90B083). APMIS. 2013;121:814–26. doi:10.1111/apm.12138
- Balany J, Bhandari V. Understanding the impact of infection, inflammation, and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med (Lausanne). 2015;2:90.
- Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008;93:F153–61. doi:10.1136/adc.2006.108837
- Ferriero DM. The vulnerable newborn brain: imaging patterns of acquired perinatal injury. Neonatology. 2016;109:345–51. doi:10.1159/000444896
- Huang SH, Stins MF, Kim KS. Bacterial penetration across the blood-brain barrier during the development of neonatal meningitis. Microbes Infect. 2000;2:1237–44. doi:10.1016/S1286-4579(00)01277-6
- Shin SH, Kim EK, Yoo H, et al. Surgical necrotizing enterocolitis versus spontaneous intestinal perforation in white matter injury on brain magnetic resonance imaging. Neonatology. 2016;110:148–54. doi:10.1159/000444387
- Sharma R, Tepas JJ 3rd. Microecology, intestinal epithelial barrier and necrotizing enterocolitis. Pediatr Surg Int. 2010;26:11–21. doi:10.1007/s00383-009-2536-2
- Luo CC, Shih HH, Chiu CH, et al. Translocation of coagulase-negative bacterial staphylococci in rats following intestinal ischemia-reperfusion injury. Biol Neonate. 2004;85:151–4. doi:10.1159/000075065
- Hellstrom A, Smith LE, Dammann O. Retinopathy of prematurity. Lancet. 2013;382:1445–57. doi:10.1016/S0140-6736(13)60178-6
- Cayabyab R, Ramanathan R. Retinopathy of prematurity: therapeutic strategies based on pathophysiology. Neonatology. 2016;109:369–76. doi:10.1159/000444901
- Lundgren P, Lundberg L, Hellgren G, et al. Aggressive posterior retinopathy of prematurity is associated with multiple infectious episodes and thrombocytopenia. Neonatology. 2017;111:79–85. doi:10.1159/000448161
- Gentile RC, Shukla S, Shah M, et al. Microbiological spectrum and antibiotic sensitivity in endophthalmitis: a 25-year review. Ophthalmology. 2014;121:1634–42. doi:10.1016/j.ophtha.2014.02.001