
ABSTRACT
Pathogens enhance their survival during infections by manipulating host defenses. Francisella tularensis evades innate immune responses, which we have found to be dependent on an understudied gene ybeX (FTL_0883/FTT_0615c). To understand the function of YbeX, we sought protein interactors in F. tularensis subsp. holarctica live vaccine strain (LVS). An unstudied Francisella protein co-immunoprecipitated with recombinant YbeX, which is a predicted glycosyltransferase with a DXD-motif. There are up to four genomic copies of this gene with identical sequence in strains of F. tularensis pathogenic to humans, despite ongoing genome decay. Disruption mutations were generated by intron insertion into all three copies of this glycosyltransferase domain containing gene in LVS, gdcA1-3. The resulting strains stimulated more cytokines from macrophages in vitro than wild-type LVS and were attenuated in two in vivo infection models. GdcA was released from LVS during culture and was sufficient to block NF-κB activation when expressed in eukaryotic cells. When co-expressed in zebrafish, GdcA and YbeX were synergistically lethal to embryo development. Glycosyltransferases with DXD-motifs are found in a variety of pathogens including NleB, an Escherichia coli type-III secretion system effector that inhibits NF-κB by antagonizing death receptor signaling. To our knowledge, GdcA is the first DXD-motif glycosyltransferase that inhibits NF-κB in immune cells. Together, these findings suggest DXD-motif glycosyltransferases may be a conserved virulence mechanism used by pathogenic bacteria to remodel host defenses.
Introduction
Evasion of host responses is a characteristic shared among many pathogens that successfully colonize and invade their hosts. Examples include IgA protease in Neisseria [Citation1], capsule production in organisms like Streptococcus pneumoniae and Klebsiella pneumoniae [Citation2,Citation3], alteration of phagolysosome maturation by Mycobacterium tuberculosis and Legionella pneumoniae [Citation4,Citation5], and escape from a phagosome into the host cell’s cytosol by Listeria monocytogenes and F. tularensis [Citation6,Citation7]. Delineating these mechanisms of evasion is important to understand pathogenesis, to probe host cell biology, and ultimately to develop antimicrobial interventions, such as vaccines against pneumococcal capsular polysaccharides [Citation8].
Francisella tularensis is a bacterial pathogen known for its ability to cause severe disease in a wide range of hosts, including humans [Citation9]. Because of the critical disease caused in humans and its ease of cultivation and dissemination, F. tularensis was a preferred biowarfare weapon [Citation10]. More recently, these attributes have raised concerns that F. tularensis could be intentionally released as a bioterrorism agent [Citation10]. Because of these threats, considerable effort has gone into studying the pathogenesis of tularemia, the disease caused by F. tularensis.
Important characteristics of F. tularensis include its ability to infect and replicate in a diverse assortment of eukaryotic cells and to weakly activate cells of the mammalian innate immune system compared to other Gram-negative pathogens. Limited activation of innate immunity has been ascribed to passive and active mechanisms [Citation11–Citation14]. Passive mechanisms include LPS that does not trigger inflammation [Citation15]. The pathogen also actively suppresses host cell responses [Citation16–Citation22]. Accumulating evidence, therefore, suggests F. tularensis has multiple modes of evading host defenses.
We found that ybeX contributes to F. tularensis pathogenesis and evasion of innate immunity [Citation23]. Wild-type F. tularensis subsp. holarctica live vaccine strain (LVS) and the virulent F. tularensis subsp. tularensis Schu S4 strain elicit a limited pro-inflammatory innate immune response, but this response increases significantly after disruption or deletion of ybeX in LVS and Schu S4, FTL_0883 and FTT_0615c, respectively [Citation23,Citation24]. Importantly, strains without these genes are attenuated in vivo [Citation23,Citation24]. Further supporting the significance of ybeX, the homolog in F. novicida was identified in two independent in vivo negative selection screens [Citation25,Citation26]. Despite accumulating evidence for its relevance, the mechanism(s) by which YbeX contributes to Francisella pathogenesis are not understood.
FTL_0883 in LVS and FTT_0615c in Schu S4 have homology with ybeX found in many other bacteria. In Escherichia coli, the gene was originally named ybeX because the open reading frame (ORF) was functionally uncharacterized [Citation27,Citation28]. ybeX was subsequently identified as a member of the σ32-regulon associated with the E. coli response to heat stress [Citation29]. The homolog in Salmonella enterica serovar Typhimurium was recovered in a screen for resistance to cobalt and named corC [Citation30]. The authors concluded that corC and related loci contribute to efflux of divalent cations, either by coupling the major cation transport CorA to an energy source or by sensing cations [Citation30]. Francisellae and other bacteria, including Neisseria gonorrhoeae and Legionella pneumophila, have homologs to corC but not to the cation transporter corA. Therefore, YbeX/CorC may have a broader role in microbial physiology than ion transport. corC has also been described in other reports [Citation31,Citation32], but there is either no definitive function ascribed to the gene [Citation32] or there is ambiguity between corC and a related gene, tlyC, which shares two similar protein domains [Citation31]. Because the function of this protein is uncertain, the E. coli ybeX/YbeX nomenclature will be used in this manuscript.
The objective of this research was to gain a better understanding of the Francisella ybeX homologs and their specific contribution to F. tularensis immune evasion [Citation23]. We used a biochemical approach to discover protein-protein interactors as a means to characterize the function of F. tularensis YbeX. In this work, we identify and characterize one protein interacting with YbeX that has several notable genomic and physiologic features, including a contribution to suppressing host cell activation. Moreover, YbeX and the interactor act synergistically. These results have broader implications for bacterial pathogenesis since the F. tularensis PPI has homology with DXD-containing glycosyltransferases found in many bacterial genera, some of which have been defined as virulence factors in other pathogens.
Results
Biochemical identification of a Francisella YbeX interactor
As an approach to understand YbeX’s function and how it contributes to the pathogenesis of F. tularensis, we sought to identify proteins that interact with YbeX. Whole-cell lysates of wild-type (WT) LVS were incubated with beads that were coated with recombinant LVS YbeX or with beads alone. Adherent proteins were characterized by silver staining of SDS-PAGE gels. A distinct protein band at 39 kDa was found only on YbeX-coated beads, which was identified by mass spectrometry as the predicted protein product of FTL_0265 (). Additional bands were identified as YbeX migrating at 32.7 kDa (predicted 31.5 kDa) and higher molecular weight forms (42.7 and 45 kDa) (). The protein encoded by FTL_0265 is annotated to contain a glycosyltransferase sugar-binding region, which we have designated glycosyltransferase domain containing protein A (GdcA). To confirm the protein interaction, recombinant YbeX with an S-tag and GdcA with a V5 tag were produced independently in E. coli, purified by affinity chromatography, and tested by co-immunoprecipitation. S-tag specific antibodies immunoprecipitated YbeX-S, but not GdcA-V5 (). GdcA-V5 was detected in the pellet after the anti-S immunoprecipitation only when YbeX-S was included (). This result was consistent with the YbeX–GdcA interaction observed using whole cell lysates () and suggested the interaction is direct without the requirement for other Francisella proteins.
Figure 1. Biochemical isolation and identification of GdcA by its interaction with YbeX. (a) Whole cell lysates of LVS were passed over a column comprised beads or beads coupled with recombinant YbeX. Bands seen only in the eluates from YbeX-beads were identified by mass spectrometry, corresponding to GdcA and YbeX. (b) Immunoprecipitation (i.p.) of V5-tagged recombinant GdcA using S-tagged recombinant YbeX as bait. (c) GdcA is found in culture supernatants from LVS with a plasmid bearing a gdcA allele tagged with Strep Tag II that is used for genetic complementation in . Top: Western blot analysis using a Strep Tag II-reactive antibody identified a band at approximately 39 kD, the predicted molecular weight of GdcA, but not from wild-type LVS. Bottom: Lanes from a parallel SDS-PAGE gel stained for total protein using Sypro Ruby according to the manufacturer’s directions. Location of molecular weight markers, in kDa, is on the left side. Similar results were seen with supernatants derived from three independent experiments.
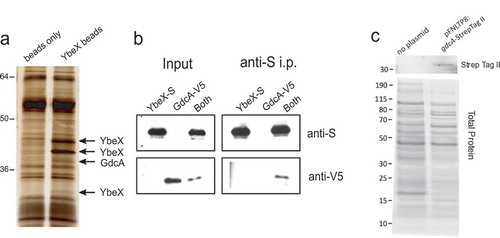
Figure 3. Phenotypes resulting from insertional mutagenesis of gdcA1-3. (a) Cytokine production by human macrophages cultured with media, WT LVS, a deletion mutant of YbeX (∆ybeX), or strains with Targetron insertions in the gdcA paralogs (TT139, TT168). Results are means of TNF levels normalized to the maximal signal within an experiment ± SEM, n = 3 experiments for Targetron strains, n = 2 for ∆ybeX. ** p < 0.01, *** p < 0.001 compared to LVS by one-way ANOVA with Tukey post-test. NS = not significant. (b) Intracellular growth of wild-type LVS and mutant strains TT139 and TT168 in human macrophages. Data are from one experiment with similar results seen in a second iteration. (c) Cytokine production by human macrophages cultured with media (white bar) or LVS strains with no plasmid (black bars), with a plasmid containing gdcA including 354 base pairs of upstream sequence and a Strep Tag II tag (red bars), or with an unrelated construct expressing tdTomato in the same vector [Citation39] (gray bars). Results are means of normalized TNF levels ± SEM, n = 3 experiments. **** p < 0.0001 by one-way ANOVA with Tukey post-test. TNF levels stimulated by complemented Targetron strains were statistically indistinguishable from wild-type LVS. (d) Increased NF-κB activation after culture with TT139. RAW264.7 cells with an NF-κB reporter were cultured overnight with media, wild-type LVS, TT139, or LPS. Wells were imaged using constant exposure times. Similar results were seen in two independent experiments. Scale bar = 100 µm.
![Figure 3. Phenotypes resulting from insertional mutagenesis of gdcA1-3. (a) Cytokine production by human macrophages cultured with media, WT LVS, a deletion mutant of YbeX (∆ybeX), or strains with Targetron insertions in the gdcA paralogs (TT139, TT168). Results are means of TNF levels normalized to the maximal signal within an experiment ± SEM, n = 3 experiments for Targetron strains, n = 2 for ∆ybeX. ** p < 0.01, *** p < 0.001 compared to LVS by one-way ANOVA with Tukey post-test. NS = not significant. (b) Intracellular growth of wild-type LVS and mutant strains TT139 and TT168 in human macrophages. Data are from one experiment with similar results seen in a second iteration. (c) Cytokine production by human macrophages cultured with media (white bar) or LVS strains with no plasmid (black bars), with a plasmid containing gdcA including 354 base pairs of upstream sequence and a Strep Tag II tag (red bars), or with an unrelated construct expressing tdTomato in the same vector [Citation39] (gray bars). Results are means of normalized TNF levels ± SEM, n = 3 experiments. **** p < 0.0001 by one-way ANOVA with Tukey post-test. TNF levels stimulated by complemented Targetron strains were statistically indistinguishable from wild-type LVS. (d) Increased NF-κB activation after culture with TT139. RAW264.7 cells with an NF-κB reporter were cultured overnight with media, wild-type LVS, TT139, or LPS. Wells were imaged using constant exposure times. Similar results were seen in two independent experiments. Scale bar = 100 µm.](/cms/asset/4bd0b653-0517-4e1d-8461-57a930d48eff/kvir_a_1631662_f0003_oc.jpg)
Although the gene encoding GdcA has several notable features described in the next section, little is known about the protein. PSORTb 3.0 reported GdcA’s subcellular location as unknown [Citation33]. An alternative tool, FUEL-mLoc [Citation34], predicted an extracellular location. To resolve this discrepancy, we tested whether GdcA could be found extracellularly in LVS cultures. A construct was created that encoded gdcA bearing a Strep Tag II tag and upstream sequence to capture the native gdcA promoter. Immunoblot analysis performed on cell-free culture supernatants showed a specific band from LVS carrying the gdcA-Strep Tag II construct, but not in LVS (, top). Both lanes contained proteins readily detected by staining for total protein content (, bottom). These results showed GdcA interacted with YbeX, which we have already associated with virulence, and GdcA was found in culture supernatant, raising the possibility it could interact with host cells. Therefore, GdcA was studied further for a potential role in Francisella pathogenesis.
GdcA genomic organization and inactivation
Comparative analysis of 17 Francisella strains revealed two to four copies of genes encoding the putative glycosyltransferase are found in F. tularensis strains that cause disease in humans, but not in the genomes of other Francisella strains found in the environment [Citation35]. The genomic context of the three loci in LVS is shown in . These paralogs are associated with IS elements that may be responsible for gene duplication events [Citation35]. We have designated the three identical copies in LVS as gdcA1, gdcA2, and gdcA3.
Figure 2. Genomic organization and insertional mutagenesis of alleles encoding GdcA. (a) Schematic maps depicting the three gdcA alleles (red) in the LVS genome. The two ORFs comprising the transposase of isftu2 (FTL_1318 and FTL_1319; FTL_1893 and FTL_1894) are represented by a single arrow for simplicity. Intergenic regions are not drawn to scale. (b) Relative locations of Targetron (TT) intron insertions after the 139th (TT139) and 168th (TT168) nucleotides of gdcA1-3. The white segment corresponds to the predicted glycosyltransferase sugar-binding region and the gray segment has high similarity to TcdA/B that includes the DXD motif (pfam04488). The alignment of GdcA (top) and TcdB (bottom) is shown below the diagram with conserved aspartic acids identified with asterisks and a bracket identifying the portion that was deleted in the domain deletion. C. PCR from genomic DNA using locus-specific primers depicting the shift of amplicon size by ~ 1 kb corresponding to intron insertions into each locus.
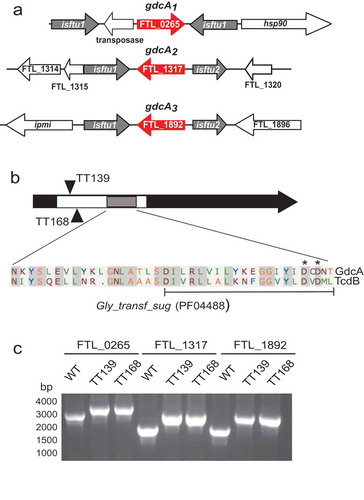
To test the functional role of GdcA, we used Targetron insertional mutagenesis to inactivate all three copies of gdcA in LVS [Citation36]. The Targetron system integrates group II introns into homologous target sites in the bacterial genome [Citation37] and two independent insertion sites were selected (, black triangles). The first insertion placed the intron between the 139 and 140 nucleotides of gdcA1-3, generating LVS strain TT139. An independent strain named TT168 was created using a second insertion site in frame between nucleotides 168 and 169 of the ORF. Both of these targets are in the 5ʹ region of the predicted glycosyltransferase sugar-binding region (white box, ), and the 1 kb insertions were confirmed by PCR ().
Loss of GdcA increases cytokine production by host innate immune cells
We then tested if GdcA contributed to F. tularensis pathogenesis by assessing the interaction of the two mutant strains, TT139 and TT168, with human macrophages. As we have described previously [Citation18,Citation23,Citation38], wild-type LVS stimulates low levels of cytokine production from macrophages in vitro (). In contrast, the ∆ybeX strain of LVS stimulates significantly higher levels of pro-inflammatory cytokines ( and [Citation23]). The LVS strains with disruptions of the gdcA genes showed cytokine phenotypes similar to ∆ybeX; macrophages produced higher levels of TNF when they were co-cultured with TT139 or TT168 (). Therefore, loss of GdcA resulted in strains that were more stimulatory to human macrophages.
Growth within macrophages is another characteristic of wild-type Francisella species that is associated with pathogenicity [Citation14]. We previously found that LVS ∆ybeX replicated poorly in macrophages [Citation23]. gdcA mutants TT139 and TT168, however, grew as well as wild-type LVS within macrophages (). Mutation of gdcA1-3 did not affect intracellular growth of LVS.
Although the genomic organization of the gdcA alleles suggested polar effects would be unlikely (), there was a formal possibility that second site mutations were responsible for the observed phenotypes (). We, therefore, performed genetic complementation to confirm the cytokine stimulation phenotype was attributable to the disruption of the gdcA genes. As seen previously, TT139 and TT168 stimulated more TNF release from macrophages than wild-type LVS (, black bars). Using the construct described in , gdcA in trans reduced the level of TNF stimulated by the gdcA mutant strains (, red bars). Similar results were obtained using non-tagged or hexahistidine-tagged versions of gdcA (data not shown). The same vector plasmid with an irrelevant construct expressing tdTomato, pTCD3 [Citation39], had no demonstrable effect on cytokine stimulation by TT139 and TT168 (, gray bars). Therefore, the expression of gdcA in trans was sufficient to complement the insertion mutations.
TNF is a prototypical, pro-inflammatory gene that is positively regulated by NF-κB [Citation40,Citation41]. LVS downregulates intracellular signaling and cytokine production [Citation42], and we have found that deletion of ybeX leads to increased TNF secretion and NF-κB activation [Citation23]. Since TNF production increased after inactivation of gdcA1-3, we tested whether NF-κB was also affected by the gdcA mutation. For this we used RAW264.7 cells bearing a reporter construct of NF-κB-dependent promoter upstream of a GFP allele [Citation23]. These cells showed low fluorescence when cultured with media, but responded to LPS by producing GFP visualized on fluorescence microscopy (). Wild-type LVS elicited low levels of fluorescence in the cells, similar to our previous observation [Citation23]. In contrast, fluorescence signal stimulated by the TT139 strain was similar to that seen with LPS, consistent with NF-κB activation (). Therefore, both TNF production by macrophages and NF-κB activation by host cells increased after gdcA mutation in LVS.
Loss of GdcA attenuates LVS
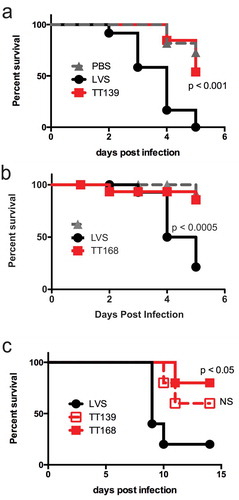
LVS and the insertion mutants were then compared in vivo. We first used the Galleria mellonella model host to assess attenuation of the TT139 and TT168 strains [Citation43]. Injecting wild-type LVS into the hemocoel of the larvae led to nearly 100% mortality in five days (-), similar to previous observations [Citation43]. In contrast, the gdcA insertion mutants survived significantly longer (-). We next tested these strains in a model of pulmonary tularemia in mice. Animals inoculated intratracheally with wild-type LVS succumbed to infection within two weeks, with one outlier that survived the experiment (). Mice survived longer, however, if they were infected with the LVS strains containing insertions in gdcA1-3 (). All of these mice lost weight, indicating that they were infected successfully (data not shown). Together, these results indicated that disruption of gdcA attenuated F. tularensis LVS.
Figure 4. Attenuation of LVS strains with insertions into gdcA1-3. (a) Survival of G. mellonella larvae injected with LVS, TT139, or PBS control. CFU: LVS = 7.2 x 106/larva, TT139 = 7.0 x 106/larva. n = 11 larvae per group. Similar results were seen in three independent experiments. (b) Larva survival injected as above but using TT168. CFU: LVS = 1.7 x 106/larva, TT168 = 2.9 x 106/larva. n = 16 larvae per group. Similar results were seen in two independent experiments. (c) Survival of mice infected intratracheally with LVS, TT139, or TT168. n = 5 mice per group in one iteration. The survival of animals infected with LVS and TT139 or LVS and TT168 were compared using a log-rank test. NS = not significant.
Ectopically expressed GdcA is sufficient to block activation of NFκB
Analysis of the predicted primary structure of GdcA reveals a glycosyltransferase-binding region with a DXD motif (). This region is homologous with other DXD-motif-containing glycosyltransferases, including toxins A and B from Clostridium difficile. An alignment between GdcA and TcdB in the region with the highest amino acid similarity is displayed below the schematic in , which corresponds to the region of TcdA/B’s catalytic domain. Another bacterial glycosyltransferase containing a DXD motif was shown to block signal transduction pathways in eukaryotic cells [Citation44,Citation45]. Since cytokine production increased after disruption of the gdcA alleles (), we hypothesized that GdcA might act by blocking signal transduction that would otherwise lead to synthesis of proinflammatory cytokines. To test this hypothesis, we evaluated the effect of GdcA on NF-κB activation using Jurkat T cells [Citation46]. This approach permitted testing of GdcA in isolation and in a non-macrophage system with different signals that activate NF-κB [Citation47]. Jurkat T cells were transfected with a reporter construct of luciferase cloned behind a promoter with NF-κB binding sites. In addition, Jurkat cells received either a construct expressing GdcA or a negative control, empty vector. After stimulation through the T cell receptor using antibodies targeting the TCR and CD28 (C305/CD28) or bypassing the T cell receptor with phorbol ester and calcium ionophore (PMA/IONO), luciferase was produced by cells transfected with the reporter construct and the negative control vector (). When the GdcA-expressing construct was co-transfected along with the reporter construct, however, there was a dose-dependent reduction in NF-κB activation (). This suggested gdcA was sufficient to disrupt the pathway of NF-κB activation in a mammalian cell.
Figure 5. GdcA inhibits NF-κB activation in Jurkat cells. (a) Cells were transfected with 15 µg of the NF-κB reporter construct and the indicated amounts of gdcA construct. Transfected cells were stimulated through the TCR (C305/CD28) or bypassing the TCR (PMA/IONO). Results are mean ± SEM of percent maximum luminescence with respect to cells receiving vector only. * p < 0.05, ** p < 0.01; n = 4 experiments. (b) Cells were transfected with 15 µg of the NF-κB reporter, 10 µg of plasmids with wild-type gdcA or with alanine substitution or domain deletion mutations. Transfected cells were stimulated as above. Results are mean ± SEM of normalized luminescence as described above. *** p < 0.001; differences between vector, domain deletion, and AXA mutation were not significant. n = 3 experiments. (c) Viability of Jurkat cells after co-transfection of 15 µg of NF-κB reporter, 3 µg of pMAX-GFP, and 10 µg of pCDEF3 constructs. The GFP expressing cells measured by flow cytometry are represented as a percentage with respect to the pCDEF3 vector alone, n = 3 experiments. For all panels, data were analyzed using one-way ANOVA among samples treated with the same stimulus followed by Tukey post-test.
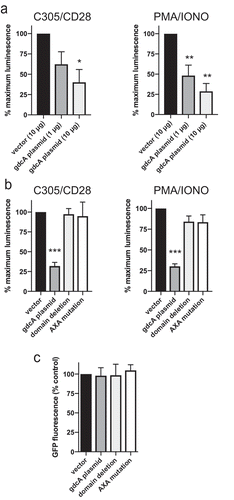
To investigate the inhibition of NF-κB activation further, the putative active site of GdcA was altered with two different mutations. Twenty amino acids of the proposed active site were deleted to create a domain deletion mutant. The deleted residues are identified by the bracket below the GdcA sequence in . In a second construct, alanine residues were substituted for the aspartic acid residues generating the AXA mutant. These residues are identified by the asterisks in . As seen previously, wild-type gdcA significantly reduced NF-κB activation in response to stimulation by either the T cell receptor or bypassing the antigen receptor (). In contrast to the wild-type coding sequence, the domain deletion or the alanine substitution forms of gdcA had no significant effect on NF-κB activation (). As an additional control, cell viability was assessed to determine if death of the Jurkat cells accounted for the decreases of NF-κB activation. Jurkat cells were co-transfected with a construct expressing GFP and either empty vector or one of the constructs expressing gdcA. Flow cytometric analysis of transfected cells showed none of the gdcA constructs affected Jurkat cell survival as assessed by GFP expression (). The mechanism of inhibition, therefore, does not appear to involve death of the Jurkat cells or a non-specific inhibition of transcription or translation. Together, these data suggested an intact coding sequence for GdcA was required to inhibit NF-κB activation.
GdcA and YbeX act synergistically in zebrafish
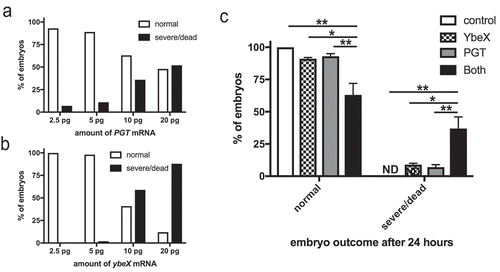
The preceding results demonstrated gdcA was sufficient to exert a biological effect on mammalian cells. Since GdcA was identified by its physical association with YbeX (), we hypothesized that YbeX might enhance the activity of GdcA. To test this hypothesis, we microinjected mRNA encoding GdcA, YbeX, or both into single-cell zebrafish embryos, and allowed them to develop for 24 h. Compared to co-transfection of tissue culture cells, this approach provides more control over protein expression by delivering known amounts of synthetic mRNAs, alone or in combination, and greater confidence that all cells receive the specific mRNA by using direct visualization [Citation48]. Preliminary experiments showed that mRNA encoding GdcA was lethal to the zebrafish embryos in the range of 100–200 pg/embryo (data not shown). Therefore, we tested dilution series with 10 to 100-fold less mRNA. Individually, mRNA encoding GdcA and YbeX had little effect on the embryos at 5 pg doses of mRNA and lower (, ). At 10 pg of mRNA and higher, embryos exhibited more death or severe developmental defects (, ). To test if GdcA and YbeX acted synergistically, therefore, embryos were injected with 1 pg of mRNA encoding either GdcA or YbeX. Alone, this amount of mRNA caused only a small reduction in the number of normal embryos that was not significantly different from control injections (). In contrast, combining the two mRNAs caused a significant reduction in the number of normal embryos with a concomitant increase of severe developmental defects or death (). These results suggest the physical interaction observed with YbeX and GdcA had functional consequences.
Figure 6. GdcA interacts synergistically with YbeX in zebrafish embryos. (a) Titration of mRNA encoding GdcA in embryos and outcomes were scored after 24 h. (b) Titration of ybeX mRNA. (c) Combined effects of RNA encoding GdcA and YbeX where 1 pg of each mRNA were co-injected, and embryos were scored for phenotypes after 24 h. Results are the mean percentage from two independent experiments ± SD, where 25–50 embryos were used per condition per experiment. ** p < 0.01, * p < 0.05 by two-way ANOVA and Tukey post-test. ND = not detected.
Discussion
The mechanisms by which F. tularensis evades host defenses are the subject of ongoing investigation, and this work contributes to our understanding of these mechanisms. We identified GdcA, a proposed glycosyltransferase from F. tularensis that affects host cell stimulation and NF-κB activation in immune cells. Disruption of gdcA1-3 generated strains that were more stimulatory to macrophages and that led to more NF-κB activation. Transfected gdcA was sufficient to disrupt NF-κB activation. GdcA, therefore, appears to be a new mechanism that is necessary and sufficient for LVS to suppress host cell responses.
GdcA has homology with DXD-glycosyltransferases, which is of interest because these enzymes contribute to diseases caused by notable bacterial pathogens. As described above (), GdcA shares homology with the A and B toxins produced by C. difficile. TcdA/B is sufficient to cause disease in vivo through a well-defined mechanism of glucosylating small GTPases that ultimately results in cell death [Citation49]. Cytotoxicity is also associated with other DXD-motif glycosyltransferases, including Lgt1 from Legionella pneumophila and a cytotoxin found in Chlamydia trachomatis [Citation50,Citation51]. Our data indicate that GdcA is distinct from these DXD-glycosyltransferases because viability and morphology are unaffected in cell culture (). Although zebrafish embryos exhibited higher death rates after exposure to GdcA (), this is more likely to be a consequence of the known role of NF-κB in normal embryonic development [Citation52,Citation53].
Another DXD-motif glycosyltransferase from enteropathogenic E. coli (EPEC) has been shown to alter host cell signaling. NleB is an effector protein secreted by the type III secretion system in EPEC, which modifies several death domain proteins in eukaryotic cells to block signal transduction, NF-κB activation, and cell death induced by TNF [Citation44,Citation45]. Our data thus far indicate that GdcA blocks the expression of NF-κB-responsive genes like cytokines, possibly by interfering with signal transduction. GdcA’s activity, however, appears distinct from NleB because NF-κB activation from different receptors and signal transduction pathways are inhibited in different cell types, namely Toll-like receptors in macrophages and the antigen receptor-signaling pathway in T cells. GdcA does not have a membrane translocation domain associated with TcdA/B, and there is no type-3 secretion system in Francisellae to deliver effectors as found in EPEC. GdcA, however, is released into the surrounding media during culture (). Because Francisella escapes from the phagosome during its intracellular life cycle, GdcA release from the bacterium into the host cytosol would give the protein access to the NF-κB signaling apparatus. Taken together, GdcA appears to contribute to LVS pathogenesis differently than TcdA/C and NleB. The mechanism by which GdcA alters the host cell is under investigation.
While the origins of GdcA remain unclear, genomic analysis of F. tularensis strains indicates gdcA has been duplicated and maintained under strong selective pressure [Citation35]. Orthologs of gdcA1-3 are found in Francisella strains that infect humans including F. tularensis subsp. tularensis, where there can be up to four alleles of gdcA homologs. Non-tularensis species also infect humans, notably F. hispaniensis and the F. novicida-like strain 3523 [Citation54,Citation55], each has a single allele with similarity to gdcA. GdcA, therefore, appears to be correlated with Francisella pathogenicity in humans.
In sum, this work identifies a predicted glycosyltransferase that contributes to the pathogenesis of LVS in cellular and animal models of infection. This establishes a new role for this DXD glycosyltransferase and Francisella pathogenesis and underscores the value of comparing related proteins across a spectrum of organisms to delineate mechanisms of pathogenesis.
Materials and methods
Bacterial strains and culture
Routine culture of Francisella was performed by streaking frozen bacterial stocks on chocolate agar that includes GC medium base (BD #228950), hemoglobin (BD #212392), and IsoVitaleX (BD #211876) as previously described [Citation18]. Briefly, bacteria were grown on plates for three days at 37°C with 5% CO2 and used to inoculate overnight cultures. Overnight cultures were grown in trypticase soy broth (BD #211768) supplemented with L-cysteine (Fisher #BP376) (TSB-c) at 37°C and shaken at 250 rpm. Routine cloning was performed using the Escherichia coli DH5α or EC100D strains.
Identification of GdcA
Recombinant YbeX was produced by cloning the FTL_0883 ORF from LVS into pET45b(+), inducing its expression in E. coli ER2566 using IPTG (Thermo Fisher #R0393), and purification by nickel chromatography (GE Healthcare #28401351). This construct incorporated an N-terminal 6His-tag and C-terminal S-tag. After eluting with imidazole, eluate material containing recombinant YbeX was further purified by a second round of affinity purification using magnetic beads (Dynal #112.01D) coated with anti-S antibodies (EMD #71549). This resulted in greater than 95% purity as determined by SDS-PAGE stained with Coomassie blue. This recombinant YbeX preparation was bound to magnetic beads coated with an anti-6His monoclonal antibody (GE Healthcare #27–4710-01) per the protocol of the manufacturer and then blocked with BSA. Whole-cell lysates of LVS were generated by sonication and were passed over magnetic beads with anti-6His or anti-6His coupled with recombinant YbeX. After washing, bound proteins were eluted with 1X Laemmli buffer. Proteins were separated by standard SDS-PAGE followed by silver staining (BioRad #1610449). Bands corresponding to proteins found in the YbeX beads were isolated and identified by LC-MS/MS. Recombinant GDCA was produced by cloning the gdcA ORF into pYes and expression in ER2566, which incorporated an N-terminal His and C-terminal V5-tag. Recombinant GdcA was purified by nickel chromatography followed by affinity chromatography using anti-V5 antibodies (Thermo Fisher #R960-25). For co-immunoprecipitation assays, recombinant, tagged YbeX was bound to anti-S beads (Dynal). Purified recombinant GdcA was added to these beads or beads not treated with recombinant YbeX, followed by washing and elution with 1X Laemmli buffer. Eluates were subjected to SDS-PAGE followed by immunoblotting.
Macrophage culture and in vitro infections
Human macrophages were differentiated from peripheral blood monocytes as described previously [Citation18,Citation56]. Briefly, peripheral blood mononuclear cells (PBMC) were harvested from buffy coats or leukocyte reduction filters followed by density gradient centrifugation over a Ficoll-Paque Plus (GE Healthcare #17–1440-02). Monocytes were purified from 2 × 108 PBMC using Optiprep (Sigma #D1556) and panning in 60 mm tissue culture dishes, followed by culturing for seven days. Macrophages were harvested using 4 mg/ml lidocaine and 5 mM EDTA in PBS and plated into 96-well plates for infection. The optical density of overnight LVS cultures was used to estimate CFU/ml, which were confirmed by plating serial dilutions on chocolate agar plates. Replicate wells of macrophages were infected with a multiplicity of infection of 10 bacteria per cell for cytokine stimulation and 500 for intracellular growth assays as we had done previously [Citation18,Citation56]. CFU analysis showed MOI differences among strains of twofold or less within an experiment. Supernatants from wells were harvested individually for cytokine analysis after 24 h of incubation. Experiments using human cells were approved by the Institutional Review Boards of the University of Pittsburgh School of Medicine and Rhode Island Hospital.
Insertional mutagenesis using group II introns and GdcA cloning
The Targetron system of gene inactivation was used to disrupt all three copies of gdcA, FTL_0265, FTL_1317, and FTL_1892, using published techniques for Francisella [Citation36]. Primers were designed to target two independent locations in gdcA using the proprietary software from Sigma-Aldrich (Supplemental Table 1). These two independent targets the gdcA sequence were selected based on their predicted likelihood of success and their proximity to the 5ʹ end of the ORF. Retargeted PCR products were generated by splice overlap PCR and cloned into pKEK1140 by XhoI/BsrGI restriction digests. The resulting plasmids were electroporated into LVS, followed by serial passage at 30°C. Individual colonies were periodically tested by PCR of genomic DNA for the presence of the 915 bp intron insertion and the wild-type GDCA open reading frame. When the amplicons of these reactions demonstrated intron insertion and no sizes consistent with wild-type template (data not shown), locus-specific PCR reactions were performed to demonstrate insertion into all three genomic sites ().
Complementation of the LVS strains with disruptions in all the gdcA alleles was performed in trans using the plasmid pFNLTP8 [Citation57]. The gdcA open reading frame and 354 nucleotides of upstream sequence were amplified by PCR using a shared 5ʹ primer and one of three 3ʹ primers encoding native GdcA or a Strep Tag II tag (Supplemental Table I). Amplicons were cloned into pBluescript using BamHI and KpnI restriction digests. After sequence confirmation, inserts were subcloned into pFNLTP8 using the same enzymes. Plasmids were introduced into wild-type LVS and gdcA disruption strains by electroporation as we have done previously [Citation56].
NF-κB GFP reporter assay
RAW264.7 macrophage cells stably transfected with an NF-κB-GFP reporter construct were used similarly to our previous work [Citation23,Citation58]. For experiments, the reporter cells were distributed into 24-well Primaria culture dishes (Primaria, BD Biosciences) at 2.5 x 105 cells per well. Wells were treated with media, Escherichia coli lipopolysaccharide (LPS), or strains of LVS. Twenty-two hours post-infection, wells were washed with Hank’s Balanced Salt Solution and GFP fluorescence was visualized on a Zeiss Axiovert 200M microscope. Images were collected by using the Zeiss Axiovision software, converted to tiff files using ImageJ, and the brightness and contrast were adjusted consistently across all images using Microsoft Powerpoint.
Galleria infections
Galleria infection was performed according to previously published protocols [Citation43]. Briefly, Francisella cultures were maintained in TSB supplemented with cysteine. On the day of infection, overnight cultures were diluted in PBS to a density of approximately 1 × 109 CFU/ml. Actual doses were confirmed by plating serial dilutions and counting CFUs 72 h post plating. Galleria mellonella caterpillars in their final larval stage (Grubco) were maintained in wood shavings at RT and protected from sunlight. Larvae were inspected visually to ensure similar sizes were distributed in all treatments. A 50 µl Hamilton syringe was used to inoculate 10 µl aliquots of the inoculum into the hemocoel behind the last left or right proleg. Prior to injection, the area was swabbed with 75% ethanol. After injection, larvae were placed in plastic petri dishes at 37°C and were inspected daily. Larval death was defined as an absence of movement in response to prodding, and numbers of dead larvae were scored daily. PBS injection and no injection control groups were included in all studies.
Mouse infections
Infections of mice were performed as previously described with the approval of the University of Pittsburgh’s Institutional Animal Care and Use Committee. C57BL/6J mice (Jackson Laboratory, female, 6–8 weeks old). The mice were infected intratracheally (i.t.) via oropharyngeal instillation with ~104 CFU of LVS. The health of the mice was monitored using a scoring system once or twice daily, and animals were euthanized after reaching a predetermined score [Citation59].
Jurkat cell transfection and luciferase reporter assays
A eukaryotic expression construct containing the coding sequence of gdcA was generated by PCR amplification to include a 5ʹ Kozak sequence, 2A peptide [Citation60], and mCherry [Citation61]. This construct was cloned behind an EF-1a promoter in pCDEF3 by restriction digest using EcoRI and XbaI [Citation62]. Generation of GdcA mutations was performed using splice-by-overlap PCR of wild-type gdcA [Citation63]. Primers are listed in Supplemental Table 1. After cloning amplicons with pGEM-T, the mutated sequences were mobilized into gdcA/pCDEF3 using restriction digests with HpaI and NdeI.
Jurkat T cells were transfected as described previously using purified plasmids containing NF-κB-luciferase reporter, the constructs in pCDEF3, and a construct constitutively expressing GFP as a control for transfection efficiency and viability [Citation64]. The transfected cells were cultured overnight; then, stimulated with anti-TCR (clone C305, Envigo B.BSI200) and anti-CD28 (clone CD28.2, Tonbo, #70–0289-U100) for 6 h, after which the cells were lysed and luciferase activity was determined with an Orion luminometer (Zylux, Oak Ridge, TN). Normalization was performed to compare across experiments. Background luminescence was first subtracted from raw luminescence values within a treatment, i.e. plasmid, group. Percent maximum of the background-corrected values was then calculated compared to cells receiving the pCDEF3 vector control within a given stimulus. For viability studies, GFP expression was measured using flow cytometry and represented as the percent of cells positive for GFP relative to the cells that received pCDEF3.
Zebrafish embryo studies
Capped mRNAs for injections were synthesized in vitro from linearized pCS2+ plasmid containing open reading frames for GdcA or YbeX using SP6 mMessage mMachine kit (Ambion). The mRNA at 2.5 to 20 pg/nl was microinjected into the embryos at the one-cell stage. For synergy experiments, the two mRNA were mixed at 1 pg/nl concentration and 1 nl was microinjected. After injections, the embryos were incubated in 1xE3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, 0.01% methylene blue) at 28°C overnight, and then assessed for viability and developmental defects at the 24-somite stage. Dead embryos were counted based on lack of movement and the presence of decay. Embryos were scored with a severe development defect if they showed a dorsalized phenotype defined by the presence of dorsal structures without ventral tissues [Citation65]. All other embryos were considered normal.
GdcA release from LVS
LVS with or without the pFNLTP8::gdcA-Strep Tag II complementing construct described above were grown overnight in 15 ml of TSB-c, with densities normalized to an OD600 = 1.4. Bacteria were removed by centrifugation at 4,000 x g for 35 min. The resulting supernatants were passed through a 0.2 µm filter, concentrated using 10 kDa cutoff centrifugal filters (Amicon), and precipitated with acetone. Pellets were resuspended in 150 µl of PBS before separation on SDS-PAGE and immunoblot.
Statistical testing
Statistical testing was performed using GraphPad Prism. For in vitro experiments, groups were compared using one or two-way ANOVA tests and Tukey post-test analysis. Kaplan Meier survival curves and Log-rank tests were used for Galleria and mouse models of infection. A p value of less than 0.05 was considered significant.
Supplemental Material
Download MS Word (15.6 KB)Acknowledgments
The authors thank Dr. Karen Elkins for providing LVS, Dr. Thomas Zahrt for pFNLTP8 used in complementation studies, and Dr. Karl Klose for the Targetron plasmids adapted to Francisella and advice on culturing conditions to disrupt the three gdcA alleles. National Institute of Allergy and Infectious Disease AI060525 “Immunology of Infectious Diseases” provided support for BCR and JH, and AI044821 “Molecular Microbial Persistence and Pathogenesis” for MJB. These studies were also performed with institutional support from the Department of Microbiology and Molecular Genetics at the University of Pittsburgh School of Medicine and the Division of Infectious Diseases at the Alpert School of Medicine at Brown University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Vitovski S, Read RC, Sayers JR. Invasive isolates of Neisseria meningitidis possess enhanced immunoglobulin A1 protease activity compared to colonizing strains. Faseb J. 1999;13:331–337.
- Geno KA, Gilbert GL, Song JY, et al. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev. 2015;28:871–899.
- Li B, Zhao Y, Liu C, et al. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014;9:1071–1081.
- Hmama Z, Pena-Diaz S, Joseph S, et al. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol Rev. 2015;264:220–232.
- Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol. 2010;26:261–283.
- Hamon MA, Ribet D, Stavru F, et al. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 2012;20:360–368.
- Santic M, Al-Khodor S, Abu Kwaik Y. Cell biology and molecular ecology of Francisella tularensis. Cell Microbiol. 2010;12:129–139.
- Bonten MJ, Huijts SM, Bolkenbaas M, et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N Engl J Med. 2015;372:1114–1125.
- Sjostedt A. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci. 2007;1105:1–29.
- Dennis DT, Inglesby TV, Henderson DA, et al. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–2773.
- Bosio CM. The subversion of the immune system by francisella tularensis. Front Microbiol. 2011;2:9.
- Jones BD, Faron M, Rasmussen JA, et al. Uncovering the components of the Francisella tularensis virulence stealth strategy. Front Cell Infect Microbiol. 2014;4:32.
- Jones CL, Napier BA, Sampson TR, et al. Subversion of host recognition and defense systems by Francisella spp. Microbiol Mol Biol Rev. 2012;76:383–404.
- Steiner DJ, Furuya Y, Metzger DW. Host-pathogen interactions and immune evasion strategies in Francisella tularensis pathogenicity. Infect Drug Resist. 2014;7:239–251.
- Ancuta P, Pedron T, Girard R, et al. Inability of the Francisella tularensis lipopolysaccharide to mimic or to antagonize the induction of cell activation by endotoxins. Infect Immun. 1996;64:2041–2046.
- Bauler TJ, Chase JC, Wehrly TD, et al. Virulent francisella tularensis destabilize host mRNA to rapidly suppress inflammation. J Innate Immun. 2014;6:793–805.
- Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547.
- Carlson PE Jr., Carroll JA, D M O, et al. Modulation of virulence factors in Francisella tularensis determines human macrophage responses. Microb Pathog. 2007;42:204–214.
- Chase JC, Celli J, Bosio CM. Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect Immun. 2009;77:180–195.
- Gillette DD, Curry HM, Cremer T, et al. Virulent Type A Francisella tularensis actively suppresses cytokine responses in human monocytes. Front Cell Infect Microbiol. 2014;4:45.
- Ireland R, Wang R, JB A, et al. Francisella tularensis SchuS4 and SchuS4 lipids inhibit IL-12p40 in primary human dendritic cells by inhibition of IRF1 and IRF8. J Iimmunol. 2013;191:1276–1286.
- Telepnev M, Golovliov I, Grundstrom T, et al. Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell Microbiol. 2003;5:41–51.
- Russo BC, Horzempa J, O’Dee DM, et al. A Francisella tularensis locus required for spermine responsiveness is necessary for virulence. Infect Immun. 2011;79:3665–3676.
- Schmitt DM, O’Dee DM, Horzempa J, et al. A francisella tularensis live vaccine strain that improves stimulation of antigen-presenting cells does not enhance vaccine efficacy. PLoS One. 2012;7:e31172.
- Moule MG, Monack DM, Schneider DS. Reciprocal analysis of Francisella novicida infections of a Drosophila melanogaster model reveal host-pathogen conflicts mediated by reactive oxygen and imd-regulated innate immune response. PLoS Pathog. 2010;6:e1001065.
- Weiss DS, Brotcke A, Henry T, et al. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A. 2007;104:6037–6042.
- Rudd KE. Linkage map of Escherichia coli K-12, edition 10: the physical map. Microbiol Mol Biol Rev. 1998;62:985–1019.
- Riley M, Abe T, Arnaud MB, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot–2005. Nucleic Acids Res. 2006;34:1–9.
- Nonaka G, Blankschien M, Herman C, et al. Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev. 2006;20:1776–1789.
- Gibson MM, Bagga DA, Miller CG, et al. Magnesium transport in Salmonella typhimurium: the influence of new mutations conferring Co2+ resistance on the CorA Mg2+ transport system. Mol Microbiol.1991;5:2753–2762.
- Hunnicutt DW, McBride MJ. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldD and gldE. J Bacteriol. 2001;183:4167–4175.
- Pearson MM, Laurence CA, Guinn SE, et al. Biofilm formation by Moraxella catarrhalis in vitro: roles of the UspA1 adhesin and the Hag hemagglutinin. Infect Immun. 2006;74:1588–1596.
- Yu NY, Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615.
- Wan S, Mak MW, Kung SY. FUEL-mLoc: feature-unified prediction and explanation of multi-localization of cellular proteins in multiple organisms. Bioinformatics. 2017;33:749–750.
- Larsson P, Elfsmark D, Svensson K, et al. Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 2009;5:e1000472.
- Rodriguez SA, Yu JJ, Davis G, et al. Targeted inactivation of francisella tularensis genes by group II introns. Appl Environ Microbiol. 2008;74:2619–2626.
- Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 2011;3:a003616.
- Carlson PE Jr., Horzempa J, O’Dee DM, et al. Global transcriptional response to spermine, a component of the intramacrophage environment, reveals regulation of Francisella gene expression through insertion sequence elements. J Bacteriol. 2009;191:6855–6864.
- Horzempa J, Tarwacki DM, Carlson PE Jr., et al. Characterization and application of a glucose-repressible promoter in Francisella tularensis. Appl Environ Microbiol. 2008;74:2161–2170.
- Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–1506.
- Shakhov AN, Collart MA, Vassalli P, et al. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171:35–47.
- Telepnev M, Golovliov I, Sjostedt A. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb Pathog. 2005;38:239–247.
- Aperis G, Fuchs BB, Anderson CA, et al. Galleria mellonella as a model host to study infection by the Francisella tularensis live vaccine strain. Microbes and infection/Institut Pasteur. 2007;9:729–734.
- Li S, Zhang L, Yao Q, et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature. 2013;501:242–246.
- Pearson JS, Giogha C, SY O, et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature. 2013;501:247–251.
- Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–308.
- Cheng J, Montecalvo A, Kane LP. Regulation of NF-kappaB induction by TCR/CD28. Immunol Res. 2011;50:113–117.
- Toyama R, O’Connell ML, Wright CV, et al. Nodal induces ectopic goosecoid and lim1 expression and axis duplication in zebrafish. Development. 1995;121:383–391.
- Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–263.
- Belyi Y, Niggeweg R, Opitz B, et al. 2006. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proceedings of the National Academy of Sciences of the United States of America 103:16953–16958.
- Belland RJ, Scidmore MA, Crane DD, et al. 2001. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proceedings of the National Academy of Sciences of the United States of America 98:13984–13989.
- Correa RG, Tergaonkar V, Ng JK, et al. Characterization of NF-kappa B/I kappa B proteins in zebra fish and their involvement in notochord development. Mol Cell Biol. 2004;24:5257–5268.
- Beg AA, Sha WC, Bronson RT, et al. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170.
- Escudero R, Elia M, Saez-Nieto JA, et al. A possible novel Francisella genomic species isolated from blood and urine of a patient with severe illness. Clin Microbiol Infect. 2010;16:1026–1030.
- Whipp MJ, Davis JM, Lum G, et al. Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J Med Microbiol. 2003;52:839–842.
- Horzempa J, Carlson PE Jr., O’Dee DM, et al. Global transcriptional response to mammalian temperature provides new insight into Francisella tularensis pathogenesis. BMC Microbiol. 2008;8:172.
- Maier TM, Havig A, Casey M, et al. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol. 2004;70:7511–7519.
- Stacey KJ, Young GR, Clark F, et al. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol. 2003;170:3614–3620.
- Russo BC, Brown MJ, Nau GJ. MyD88-dependent signaling prolongs survival and reduces bacterial burden during pulmonary infection with virulent francisella tularensis. Am J Pathol. 2013. DOI:10.1016/j.ajpath.2013.06.013
- Szymczak AL, Workman CJ, Wang Y, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594.
- Shaner NC, Campbell RE, Steinbach PA, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma s. red fluorescent proteinp. Nat Biotechnol. 2004;22:1567–1572.
- de Souza AJ, Oriss TB, K J O, et al. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci U S A. 2005;102:17113–17118.
- Horton RM, Hunt HD, Ho SN, et al. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68.
- Kane LP, Shapiro VS, Stokoe D, et al. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604.
- Mullins MC, Hammerschmidt M, Kane DA, et al. Genes establishing dorsoventral pattern formation in the zebrafish embryo: the ventral specifying genes. Development. 1996;123:81–93.