
ABSTRACT
The virulence behaviors of many Gram-negative bacterial pathogens are governed by quorum-sensing (QS), a hierarchical system of gene regulation that relies on population density by producing and detecting extracellular signaling molecules. Although extensively studied under in vitro conditions, adaptation of QS system to physiologically relevant host environment is not fully understood. In this study, we investigated the influence of lung environment on the regulation of Pseudomonas aeruginosa virulence factors by QS in a mouse model of acute pneumonia. When cultured under laboratory conditions in lysogeny broth, wild-type P. aeruginosa strain PAO1 began to express QS-regulated virulence factors elastase B (LasB) and rhamnolipids (RhlA) during transition from late-exponential into stationary growth phase. In contrast, during acute pneumonia as well as when cultured in mouse bronchial alveolar lavage fluids (BALF), exponential phase PAO1 bacteria at low population density prematurely expressed QS regulatory genes lasI-lasR and rhlI-rhlR and their downstream virulence genes lasB and rhlA. Further analysis indicated that surfactant phospholipids were the primary components within BALF that induced the synthesis of N-(3-oxododecanoyl)-L-homoserine lactone (C12-HSL), which triggered premature expression of LasB and RhlA. Both phenol extraction and phospholipase A2 digestion abolished the ability of mouse BALF to promote LasB and RhlA expression. In contrast, provision of the major surfactant phospholipid dipalmitoylphosphatidylcholine (DPPC) restored the expression of both virulence factors. Collectively, our study demonstrates P. aeruginosa modulates its QS to coordinate the expression of virulence factors during acute pneumonia by recognizing pulmonary surfactant phospholipids.
Introduction
Every day, thousands of microorganisms gain access into respiratory airways and alveolar epithelial surface. Most inhaled microbes are trapped on the mucus layer coating the nasal epithelium and proximal respiratory tract, and are cleared by mucociliary transport [Citation1–Citation5]. Microbes that reach the alveolar spaces are deposited on the pulmonary surfactant, which lines gas-exchange surfaces of the lung epithelium. This contact initiates complex offensive and defensive strategies by both parties. The pulmonary pathogen Pseudomonas aeruginosa expresses large repertoire of virulence factors, among others, cell surface appendages flagellum and type IV pili, secreted exoproteases, phospholipases and rhamnolpids, and type III and type VI secretion system effectors, in order to switch from an unobtrusive soil bacterium to becoming a pathogen for lung colonization and infection [–Citation11]. Diseased lungs infected by P. aeruginosa, including cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), and pneumonias, are associated with altered pulmonary surfactant levels [Citation12–Citation16]. Understanding the complex interactions between the pulmonary epithelium and P. aeruginosa may allow more effective therapeutic strategies against infection in diseased lungs.
Pulmonary surfactant is synthesized and secreted by alveolar type II cells and nonciliated bronchiolar cells lining the terminal bronchioles [Citation17–Citation19]. Pulmonary surfactant is composed of 90% phospholipids and 10% proteins, including surfactant proteins SP-A, SP-B, SP-C, and SP-D. Together with phospholipids, SP-B and SP-C form a lining at the air-liquid interface with critical surface tension-lowering properties that reduce the work of breathing and maintain airspace patency. Traditionally, SP-A and SP-D are thought to be important pulmonary innate immunity proteins that opsonize and facilitate microbial phagocytosis by professional phagocytes [Citation20,Citation21]. Published work by us and others have shown that both SP-A and SP-D also directly kill microbes in a phagocyte-independent manner, by permeabilizing microbial membranes [Citation22–Citation29]. Surfactant phospholipids also modulate host–pathogen interactions within lung [Citation30,Citation31]. Composed of primarily dipalmitoyl phosphatidylcholine (DPPC, 41%) as well as other minor components, including phosphatidylcholine, phosphatidylglycerol, other phospholipids (e.g., sphingomyelin), cholesterol, and neutral lipids, this extracellular lipid reservoir maintain immune quiescence in the lung. However, surfactant phospholipids are continuously exposed to assaults by environmental oxidants, inflammatory agents, and microbial pathogens. These assaults modulate and alter surfactant lipids into bioactive lipid molecules that mediate key roles in inflammation, immunity, and fibrosis [Citation30,Citation31].
Quorum sensing (QS) is a cell-to-cell communication system in Gram-negative bacteria that relies on population density, by producing and detecting the threshold levels of extracellular homoserine lactones, quinolones, and 2-(2-hydroxylphenyl)-thiazole-4-carbaldehyde (IQS) [Citation32–Citation35]. It is well known that the QS network of P. aeruginosa coordinates various activities, including virulence and biofilm formation. The QS network consists of four transcription factors, including LasR, RhlR, PqsR and an unknown transcription factor of the ambBCDE operon. Hierarchically, LasR sits atop of all the regulatory circuits. When its corresponding N-3-oxododecanoyl-homoserine lactone (C12-HSL) encoded by the lasI gene reaches the threshold concentration, LasR activates transcription of RhlR and PqsR, which, in response to their cognate signals C4, N-butanoyl-homoserine lactone (C4-HSL) and 2-heptyl-3-hydroxy-4(1 H)-quinolone (PQS), respectively, activate genes within these regulons that overlap with the LasR regulon. These QS circuits are shut off in the LasR-null mutant. Interestingly, a recent study indicates that under low phosphate conditions, the ambBCDE operon activated by an alternate effector PhoB, synthesizes IQS. In turn, IQS interacts with an unknown transcription factor to activate RhlR and PqsR signaling independent of LasR [Citation34,Citation35]. In this study, we examined the impact of alveolar surfactant on the modulation of QS-regulated virulence factors, including the production of exoproteases and rhamnolipids during acute pneumonia infection.
Materials and methods
Reagents
All chemicals, except where noted, were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Bacterial strains, media and growth conditions
The parental P. aeruginosa wild-type strain PAO1 was provided by Professor Michael Vasil (University of Colorado Health Science Center) as previously published [Citation25–Citation29,Citation36,Citation37]. The isogenic ∆lasI∆rhlI mutant was a gift from Professor Stephen Lory (Harvard Medical School). Bacterial strains were grown in lysogeny broth (LB) for 16 hours at 37°C, resuspended in fresh LB with 20% sterile glycerol and froze in aliquots at −80°C. Before each assay, aliquots of the bacteria were cultured from frozen stocks in appropriate media to desirable growth phase and cell density. The optical density at 600 nm (OD 600 nm) was determined using a spectrophotometer and correlated with numbers of viable bacteria (CFU) after serial dilution plating on agar plates. Whenever appropriate, antibiotics were added to growth media at the following concentrations: carbenicillin (300 µg ml−1), gentamycin (30 µg ml−1), and kanamycin (100 µg ml−1).
Ethics statement
The mouse studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois at Urbana-Champaign (Protocol Numbers: 15171, 18136).
Mouse husbandry and acute pneumonia infection
Six-week-old CD-1 mice (equal ratio of males and females) were purchased from Charles River (Wilmington, MA, USA), and acclimated for 1 week before use. All mice were housed in positively ventilated microisolator cages with automatic recirculating water located in a room with laminar, high efficiency particulate-filtered air. The animals received autoclaved food, water, and bedding. Mice (cohorts of 10) were intranasally infected with 2.5 × 107 CFU (in 50 µl) of wild-type P. aeruginosa PAO1 pre-grown to early exponential phase (OD 600 nm 0.6), mid-exponential phase (OD 600 nm 1.13) and late stationary phase (OD 600 nm 2.73) respectively, as we have previously published [Citation25–Citation29,Citation37]. At indicated time points, mice were euthanized, lungs harvested, homogenized, serially diluted, and plated on Pseudomonas Isolation Agar for determination of bacterial burden, and for isolation of mRNA and assessment of the expression of lasB and rhlA genes by qPCR.
Bronchoalveolar lavage (BAL)
BAL was performed on healthy, 7-week old male and female CD1 mice as we have previously described27,37. The trachea was exposed and intubated with a 1.8-mm outer diameter polyethylene catheter. BAL was performed by instilling sterile 0.9% NaCl in 1 ml aliquots. Two ml of 0.9% NaCl were instilled per mouse. The BAL fluid (BALF) samples were sterilized with 0.22 µm filter and pooled for further experiments. The concentration of each batch of BALF were normalized based on protein concentration as determined by BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).
Exoprotease assays
Exoprotease activities within the supernatants of P. aeruginosa cultured in LB, BALF, or 0.9% NaCl in various experiments were determined as previously described [Citation27,Citation37] by the SensolyteTM Red Protease Assay Kit (AnaSpec Inc; Fremont, CA, USA).
Quantification of rhamnolipids
The concentration of rhamnolipids in P. aeruginosa cultures was measured by the spectrophotometric methylene blue complexation method as described [Citation38], with some modifications. Briefly, filtered culture supernatant of P. aeruginosa were adjusted to pH 2.5 with 1 N HCl, and extracted with five-fold volume of chloroform. The resulting extracts were mixed with equal volume of freshly prepared methylene blue solution (40 µg ml−1). The pH of the mixture was adjusted to 8.5 with 50 mM borax buffer. After vigorous shaking for 4 min, the mixture was left to stand for 15 min. The absorbance of chloroform phase was measured at 638 nm with a spectrophotometer, with chloroform serving as blank. The amount of rhamnolipids was determined by referencing a standard curve constructed with known concentration rhamnolipids (Sigma-Aldrich).
Western blotting
Western blot analyses for LasB were performed using standard protocols as previously described [Citation37]. Briefly, equal concentration of protein samples (from bacterial culture supernatants) were resolved by SDS-PAGE and electro-blotted onto Immobilon P polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). The membranes were then incubated for 60 min at room temperature in blocking solution (3% bovine serum albumin in PBS), followed by a 4-h incubation with polyclonal antibody against P. aeruginosa LasB (a gift from Professor Dennis Ohman, Virginia Commonwealth University). The membranes were hybridized with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody. The immune complexes were visualized using the Amersham ECL Western Blotting Detection System (Danaher Corporation, Washington, D.C., USA) on HyBlot CL autoradiography films (Denville Scientific, InC, Metuchen, NJ, USA). In order to assure reproducibility, western blotting was performed twice with similar results. Protein expression levels were quantified by densitometry normalized to the number of bacteria within each sample by using the ImageJ software.
Quantitative real-time PCR (qPCR) assays
Wild-type PA01 and isogenic ∆lasI∆rhlI mutant cultured to different growth phases (as described above) were inoculated into mouse lungs or under in vitro conditions (BALF, LB, 0.9% NaCl) for a predetermined amount of time. Total RNA was extracted from bacteria pellets or lung homogenates by using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA). cDNA was prepared from the same amount of total RNA as the templates by TaqMan® Real-Time PCR Assays (Thermo Fisher Scientific), with the probes and primers for the lasI, lasR, rhlI, rhlR, lasB and rhlA genes designed by the Applied Biosystems. qPCR wasperformed on an Applied Biosystems GeneAmp PCR System 9700 (Thermo Fisher Scientific). The P. aeruginosa housekeeping gene rpsL was used as endogenous control. All the experiments were independently performed three times in triplicate.
Quantification of C12-HSL
Bacterial cultures were pelleted, and cell-free supernatants were filtered through 0.22 um filters and extracted with two volumes of ethyl acetate containing 0.01% glacial acetic acid. The ethyl acetate phase was collected and dried by using nitrogen gas at room temperature. Dried pellets were dissolved in methanol and solubilized C12-HSL was detected by GC-MS using the Agilent 5975B inert Series GC/MS System at the University of Illinois at Urbana-Champaign Mass Spectrometry Laboratory. Quantification was performed with commercially purchased C12-HSL (Sigma-Aldrich) as a standard.
Phenol:chloroform:isoamyl alcohol extraction of BALF
All proteins and lipids in mouse BALF samples were removed by three successive rounds of extraction with phenol:chloroform:isoamyl alcohol (25:24:1). The aqueous phase was pooled and dried under nitrogen gas at room temperature. The dried components were dissolved in PBS to original volume of BAL for subsequent experiments.
Phospholipase A2 (PLA2) degradation of BALF
BALF was digested with 5 units ml−1 of PLA2 overnight at 37°C. Digested BALF was heated to 100°C for 10 min to inactivate the PLA2. Degradation of phospholipids was confirmed by using a phospholipids assay kit (Sigma-Aldrich, Cat# MAK122) according to manual instruction before and after digestion by PLA2.
Statistical analyses
Quantitative data were expressed as the mean ± standard error (SE). Statistical significance comparisons for samples with equal variances were determined using the parametric Student’s t test for two unpaired samples. For comparing the means of groups of three or more, data were analyzed for statistical significance by the one-way ANOVA followed by Tukey’s tests for comparison between the means. A significant difference was considered to be p < 0.05.
Results
Expression of P. aeruginosa virulence factors in vitro is QS-dependent
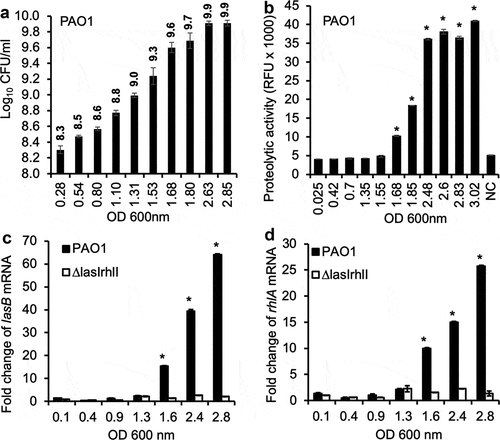
We examined the temporal correlation between viable bacterial count by colony forming units (CFU), bacterial cell density as measured by OD 600 nm, exoprotease activities in culture supernatants, and the expression of two QS-regulated virulence genes lasB and rhlA by quantitative real-time PCR (qPCR) in wild-type P. aeruginosa strain PAO1 versus the QS-deficient mutant ΔlasIΔrhlI. The lasB gene encodes elastase B [Citation39–Citation41] whereas the rhlA gene encodes a protein essential for rhamnolipid biosynthesis [Citation42,Citation43]. At lag, early and mid-exponential phases between OD 600 nm of 0.025 to ~1.3, which corresponded to log10 CFU of 8.3 to 9.0 ()), both the proteolytic activities ()), as well as the expression of lasB and rhlA transcripts were minimal (,d)). At late exponential phase (OD 600 nm of >1.6, log10 CFU of >9.3), both proteolytic activities as well as expression of lasB and rhlA genes began to increase until plateauing at stationary phase (OD 600 nm >2.4, –)). The expression of both lasB and rhlA genes was minimal throughout all growth phases in the ΔlasIΔrhlI mutant, indicating that the expression of LasB and rhamnolipids were regulated by the LasR-LasI and RhlR-RhlI QS circuits.
Figure 1. Expression of QS-regulated P. aeruginosa virulence factors in vitro is growth phase dependent. (a) The CFU of P. aeruginosa strain PAO1 at different OD 600 nm values corresponding to different phases of growth in LB broth. (b) Exoprotease activities of PAO1 cultured in LB broth at different OD 600 nm values. (c–d) Transcription of QS-dependent lasB and rhlA genes in PAO1 and ∆lasI∆rhlI cultured in LB broth. Experiments were performed independently three times in triplicate. Mean ± standard error (SE) from a typical experiment are presented. *p < 0.05 (b–d) when compared against the lowest OD 600 nm by using the one-way ANOVA analysis. *p < 0.05 (b) when compared exoprotease activity in each sample against the sample with the lowest OD 600 nm by using the Tukey’s test. *p < 0.05 (c–d) when compared gene expression in PAO1 against ∆lasI∆rhlI at each OD 600 nm value by using the Tukey’s test. NC: LB alone.
Virulence of P. aeruginosa during acute pneumonia infection is independent of the in vitro growth phase of the inoculum
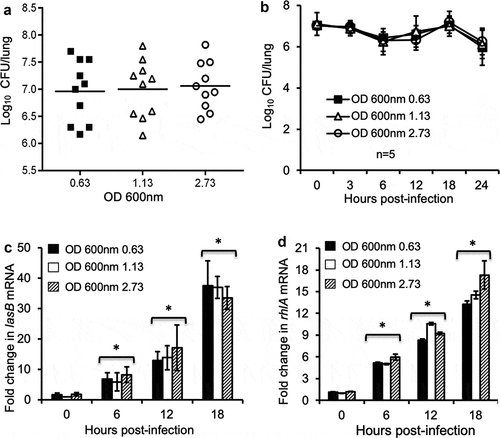
Because the expression of virulence factors including LasB and RhlA began during the transition of late exponential phase into early stationary phase, it was predicted that stationary phase P. aeruginosa bacteria, which harbored transcripts of virulence genes as well as pre-expressed virulence factors, were preconditioned to better initiate and execute infection in the host. By using a mouse model of acute pneumonia, we compared the bacterial burden of lungs infected with equal CFU of P. aeruginosa PAO1 cultured to early exponential phase (OD 600 nm 0.63), mid exponential phase (OD 600 nm 1.13) and late stationary phase (OD 600 nm 2.73). Interestingly at 24-h post-infection, no statistically significant difference was found in the number of PAO1 bacteria recovered from the lungs among the three groups of infected mice ()). Temporal-specific bacterial burden at 0, 3, 6, 12, and 18-h post-infection in mouse lungs also revealed no significant differences between the three groups of mice ()). qPCR analysis of lasB and rhlA in bacteria recovered from BALF of infected lungs showed that these genes were expressed as early as 6 hours, and continued to increase at 18-h post-infection (,d)). However, the expression of both lasB and rhlA genes was not statistically different in all three groups of animals throughout the entire course of infection. Collectively, these results suggest that successful lung infection is independent of the growth phases, and hence, virulence gene expression, of P. aeruginosa inoculum derived from in vitro cultures.
Figure 2. Exponential phase P. aeruginosa bacteria infect mouse lungs as successfully as their stationary phase counterparts. Anesthetized CD1 mice were intranasally inoculated with 2.5 × 10^7 CFU of early exponential phase (OD 0.63 at 600 nm), mid exponential phase (OD 1.13), and stationary phase (OD 2.73) P. aeruginosa PAO1. Mice were sacrificed between 0 to 24 hours post-infection for determination of bacterial burden and virulence gene expression. (a) P. aeruginosa burden in lung homogenates was enumerated 18 hours post-infection. Data are the mean CFU ± SE, n = 10 per group. (b) PAO1 burden in mouse lungs during the course of infection. Data are the mean CFU ± SE, n = 5 per group. (c–d) Changes in the transcription of virulence genes lasB (c) and rhlA (d) in PAO1 bacteria within the homogenates of infected mouse lungs at indicated time points. n = 5 per group. *p < 0.05 when compared the expression of lasB and rhlI at 6, 12 and 18-hour post-infection against Time 0 hour by using the one-way ANOVA analysis.
Mouse BALF induces premature expression of QS-regulated virulence factors in exponential phase P. aeruginosa
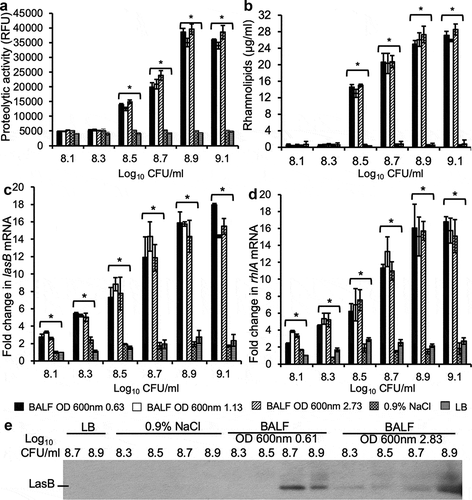
The results in suggest that certain host components within the mouse lungs might induce the exponential phase P. aeruginosa to prematurely express LasB and RhlA, increasing the virulence potential of these bacteria and matching those bacteria from the late stationary phase. To examine this hypothesis, PAO1 bacteria were collected at early exponential (OD 600 nm 0.61), mid exponential (OD 600 nm 1.15), and late stationary phase (OD 600 nm 2.83). Bacteria were washed in 0.9% NaCl and adjusted to the concentration of 2.5 × 10^7 (log10 7.4) CFU ml−1 and cultured in mouse BALF at 37°C while shaking at 120 rpm and collected at cell density of log10 8.1 8.3, 8.5, 8.7, 8.9 and 9.1 CFU ml−1. These concentrations of bacteria were below the necessary threshold needed to induce QS when cultured in LB ()). As controls, the washed stationary phase (OD 600 nm 2.83) PAO1 bacteria (log10 7.4 CFU ml−1) were cultured in LB and collected at cell density from log10 8.1 to 9.1 CFU ml−1, or directly diluted to cell density from log10 8.1 to 9.1 CFU ml−1 in 0.9% NaCl and collected after 3 hours of shaking incubation. Surprisingly, the kinetics of exoprotease activity in cultured supernatants ()), rhamnolipids production ()), as well as the transcripts of lasB and rhlA genes (,d)) in early and mid-exponential phase PAO1 bacteria, were comparable to those from bacteria of late stationary phase. These results indicated that mouse BALF was able to induce a premature expression of exoproteases (including LasB) and rhamnolipids (RhlA) in early and mid-exponential phase PAO1 bacteria, equal to the levels expressed by the late stationary phase bacteria. In contrast, control PAO1 bacteria cultured in LB or 0.9% NaCl did not prematurely express exoproteases or transcription of lasB and rhlA genes (–)). These results were further confirmed by Western blot analysis of LasB produced by early exponential phase PAO1 bacteria as visualized with an anti-LasB antibody ()) and quantified by densitometry normalized to bacterial density (Fig S1).
Figure 3. QS-regulated P. aeruginosa virulence factors are expressed prematurely in mouse BALF. P. aeruginosa strain PAO1 were grown to early exponential phase (OD 600 nm 0.63), mid exponential phase (OD 600 nm 1.13), and stationary phase (OD 600 nm 2.73). The cultured bacteria were washed 3 times at ice-cold sterile 0.9% NaCl with a 4°C microcentrifuge. Then, 2.5 × 107 (log10 7.4) CFU of washed PAO1 were cultured in BALF from healthy CD-1 mice at 37°C while shaking at 120 rpm and collected at different cell density from log10 8.1 to 9.1 CFU. For LB control, 2.5 × 107 washed stationary phase (OD 600 nm 2.73) PAO1 were cultured in LB and collected at cell density from log10 8.1 to 9.1. For 0.9% NaCl control, washed stationary phase (OD 600 nm 2.73) PAO1 diluted to cell density from log10 8.1 to 9.1 were incubated for 3 h respectively and then collected for detection. (a) Exoprotease activities of PAO1 in cultured supernatants. (b) Concentration of rhamnolipids released by PAO1 in cultured supernatants. (c–d) mRNA of lasB and rhlA genes were quantified by qPCR from PAO1 bacteria under indicated culture conditions. Experiments were performed independently three times in triplicate. (a-d) Mean ± SE from a typical experiment are presented. *p < 0.05 when compared BALF samples against both 0.9% NaCl and LB samples by using the one-way ANOVA analysis, as well as when compared each sample against 0.9% NaCl and LB by using the Tukey’s test. (e) Elastase B secreted by PAO1 into supernatant was analyzed by western blot visualized with anti-LasB primary antibody. Western blotting was performed twice with similar results. Expression level was normalized against bacterial numbers and quantified by densitometry and presented in the Fig. S1.
Mouse BALF promotes premature induction of QS regulatory systems in exponential phase P. aeruginosa
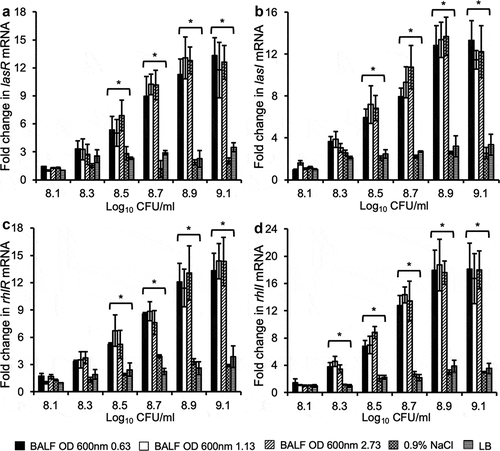
Because the expression of LasB and rhamnolipids are regulated by the LasR/LasI and RhlR/RhlI QS systems, we examined if mouse BALF could induce an early expression of lasR and lasI, as well as rhlR and rhlI genes in bacteria described in . As shown in -), mouse lung BALF induced a premature expression of lasR/lasI and rhlR/rhlI genes in both early and mid-exponential phase PAO1 bacteria, to the levels similar to that of bacteria from late stationary phase. In contrast, the expression of lasR, lasI, rhlR and rhlI genes were not elevated in LB and 0.9% NaCl. These results suggest that mouse BALF induces a premature expression of LasB and RhlA in early and mid-exponential phase PAO1 bacteria by activating both LasR-LasI and RhlR-RhlI QS circuits.
Figure 4. Mouse BALF promotes early expression of QS regulatory genes in P. aeruginosa. PAO1 bacteria were grown to early log (OD 600 nm 0.63), late log (OD 600 nm 1.13), and stationary phase (OD 600 nm 2.73), washed, and subcultured in 0.9% NaCl, LB and mouse BALF as described in . Transcription of various P. aeruginosa QS regulatory genes lasR (a), lasI (b), rhlR (c), and rhlI (d) were examined by qPCR. Experiments were performed independently three times in triplicate. Mean ± SE from a typical experiment are presented. *p < 0.05 when compared BALF samples against the 0.9% NaCl and LB samples by using the one-way ANOVA analysis, as well as when compared each BALF sample against 0.9% NaCl and LB by using the Tukey’s test.
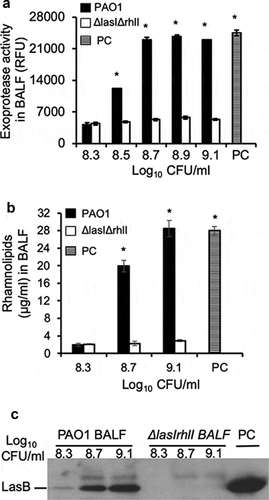
Remodeling of virulence expression in exponential phase P. aeruginosa by mouse BALF is abolished in the ∆lasI∆rhlI bacteria
To further confirm that the premature induction of virulence expression by mouse BALF was indeed dependent on the LasR-LasI and RhlR-RhlI, we compared the expression of exoproteases by early exponential phase wild-type PAO1 versus ∆lasI∆rhlI bacteria (OD 600 nm 0.6, cultured to indicated concentrations as described in ) in mouse BALF. As expected, culture supernatants from late stationary phase PAO1 bacteria grown in LB (OD 600 nm 2.85, PC) exhibited high levels of proteolytic activities ()), rhamnolipid production ()), as well as LasB expression ()). Mouse BALF induced strong proteolytic activities as well as rhamnolipid production in the early exponential phase PAO1 bacteria, which was absent in the ∆lasI∆rhlI bacteria (,b)). In addition, proteolytic activities of PAO1 bacteria were partially dependent on bacterial concentrations as neither proteolytic activities nor rhamnolipid biosynthesis were observed at log10 8.3 CFU. The proteolytic activities and rhamnolipidlevels peaked at PAO1 concentration of ≥ log10 8.7 and 9.1 CFU, respectively. Western blot analysis of LasB (); quantification by densitometry normalized to bacterial density presented in Fig S2) mirrored the proteolytic activities observed in ). Collectively, these results indicate that premature triggering of virulence factor expression by mouse BALF is dependent upon the induction of LasI-LasR and RhlI-RhlR QS circuits. Furthermore, under BALF conditions, a minimum concentration of > log10 8.3 CFU of PAO1 bacteria is needed to achieve threshold levels of signaling molecules required to trigger the QS circuits in exponential phase P. aeruginosa (see ). In contrast, a PAO1 concentration of log10 8.5 CFU is needed to trigger the expression of exoproteases and rhamnolipids in BALF (compare to to ).
Figure 5. Remodeling of virulence expression in exponential phase P. aeruginosa by mouse BALF is dependent on LasR-LasI and RhlR-RhlI QS circuits. P. aeruginosa PAO1 bacteria were grown to early exponential phase (OD 600 nm 0.6), washed, diluted to indicated concentrations, and cultured in mouse BALF as described in . (a) Exoprotease activity of PAO1 and ∆lasI∆rhlI mutant were compared in mouse BALF. (b) The concentration of rhamnolipids released by PAO1 or ∆lasI∆rhlI growing in mouse BALF. Experiments were performed independently three times in triplicate. Mean ± SE from a typical experiment are presented. *p < 0.05 (a–b) when compared PC and PAO1 samples against ∆lasI∆rhlI by using the one-way ANOVA analysis, as well as when compared PC or PAO1 against individual ∆lasI∆rhlI samples by using the Tukey’s test. (c) LasB expressed by PAO1 or ∆lasI∆rhlI in mouse BALF was analyzed by western blot using anti-LasB antibody. PC was positive control from PAO1 grown in LB to late stationary phase (OD 600 nm 2.85). Western blotting was performed twice with similar results. LasB expression level was normalized against bacterial numbers and quantified by densitometry and presented in the Fig. S2.
Remodeling of virulence expression in exponential phase P. aeruginosa by mouse BALF is independent of pulmonary surfactant proteins
We examined which host component within mouse BALF was able to prematurely induce QS-regulated virulence factors. Previously, we and others have shown that pulmonary surfactant proteins A and D are important innate immunity proteins within the alveolar space [Citation20–Citation28]. Exponential phase PAO1 (OD 600 nm 0.8) or late stationary phase (OD 600 nm 2.85) were diluted to log10 7.4 CFU ml−1 and cultured in normal mouse BALF, or in Proteinase K-digested, heat-inactivated BALF, until log10 9.1 CFU/ml. To avoid potential residual protease activity of incompletely inactivated Proteinase-K, which would confound the interpretation of results from protease assays, we examined the production of rhamnolipids. As expected, stationary phase PAO1 bacteria cultured in LB produced high levels of rhamnolipids (Fig S3, PC). Importantly, when compared to normal BALF, Proteinase-K digested BALF induced similar levels of rhamnolipid production in exponential phase PAO1 bacteria (Fig S3). These results suggest that surfactant proteins are not the alveolar surfactant components that cue exponential phase P. aeruginosa to initiate QS-dependent virulence expression.
Pulmonary surfactant phospholipids are responsible for inducing the premature QS-regulated virulence expression
As discussed above, phospholipids are major components of the alveolar surfactant layer. We examined if surfactant phospholipids modulated the premature induction of QS-dependent virulence expression. Exponential phase PAO1 bacteria (OD 600 nm 0.6, log10 7.4 CFU ml−1) were washed and recultured in normal mouse BALF, BALF pre-extracted with phenol:chloroform:isoamyl alcohol, or pre-digested with phospholipase A2 (PLA2), to bacterial densities of log10 8.3, 8.7 and 9.1 CFU ml−1. Both phenol:chloroform:isoamyl alcohol extraction and PLA2 digestion removed surfactant phospholipids. At bacterial densities of log10 CFU of 8.7 and 9.1, BALF induced expression of exoproteases as well as rhamnolipids in the exponential phase bacteria (-)). Importantly, pre-extraction with phenol:chloroform:isoamyl alcohol or pre-digestion with PLA2 inhibited the ability of mouse BALF to induce both exoprotease activities and rhamnolipid production (-)). These data suggest that surfactant phospholipids induce premature expression of QS-dependent exoproteases and rhamnolipids.
Figure 6. Pulmonary surfactant phospholipids are responsible for remodeling of QS-regulated virulence factor expression. P. aeruginosa PAO1 was grown to early exponential phase (OD 600 nm 0.6) and cultured in mouse BALF, BALF pre-extracted with phenol:chloroform:isoamyl alcohol (BALF+phenol) or pre-digested with PLA2 (BALF+PLA2), or BALF with exogenously supplied DPPC (100 µg/ml) after phenol:chloroform:isoamyl alcohol extraction (BALF+phenol+DPPC) or PLA2 digestion followed by heat inactivation (BALF+PLA2+DPPC), as described in . Culture supernatants were collected at different cell density of log10 8.3, 8.7, and 9.1, respectively, for determination of proteolytic activities and rhamnolipid production. (a-b) Exoprotease activities. (c–d) rhamnolipid production. Experiments were performed independently three times in triplicate. Mean ± SE from a typical experiment are presented. *#p < 0.05 when compared proteolytic activities and rhamnolipid production against of BALF+phenol or BALF+PLA2 by using the one-way ANOVA analysis, as well as within individual data set by using the Tukey’s test.
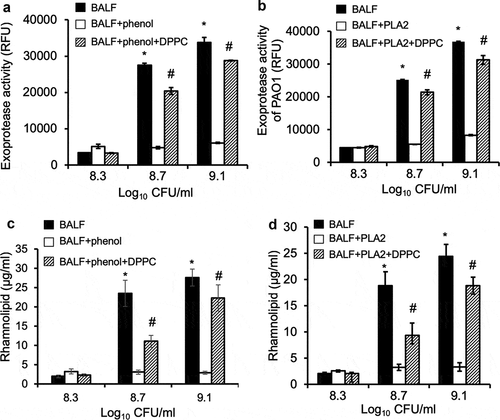
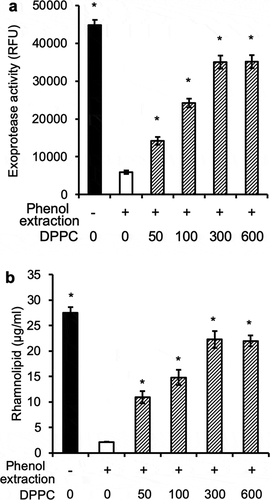
To confirm the aforementioned results, phenol:chloroform:isoamyl alcohol-extracted or PLA2-digested mouse BALF were supplemented with 100 µg ml−1 DPPC, the most abundant component of surfactant phospholipids. Excitingly, provision of DPPC restored the ability of phenol-extracted or PLA2-digested mouse BALF to induce QS-dependent proteolytic activities and rhamnolipids production (-)). Further experiments revealed that DPPC prematurely induced exponential phase PAO1 to express exoproteases and rhamnolipids in a concentration-dependent manner (,b)), which peaked at 300 µg ml−1. Collectively, these results suggest that exponential phase P. aeruginosa bacteria prematurely induce QS-dependent virulence factors to initiate lung infection by detecting surfactant phospholipids, including DPPC.
Figure 7. Dose-dependent restoration of proteolytic activities and rhamnolipid production by dipalmitoylphosphatidylcholine. P. aeruginosa PAO1 bacteria were grown to early exponential phase (OD 600 nm 0.6, log10 7.4 CFU ml−1), washed and cultured as described as Fig 3 to cell density of log10 CFU 9.1 in mouse BALF, BALF pre-extracted with phenol:chloroform:isoamyl alcohol alone, or supplemented with indicated concentrations of DPPC after phenol:chloroform:isoamyl alcohol extraction. (a) Proteolytic activities. (b) rhamnolipid production. *p < 0.05 when compared exoprotease activity and rhamnolipid production against phenol:chloroform:isoamyl alcohol-extracted BALF alone by using the one-way ANOVA analysis, as well as by comparing each sample against phenol:chloroform:isoamyl alcohol-extracted BALF alone by using the Tukey’s test.
Dipalmitoylphosphatidylcholine induces the premature production of N-dodecanoyl-homoserine lactone by P. aeruginosa
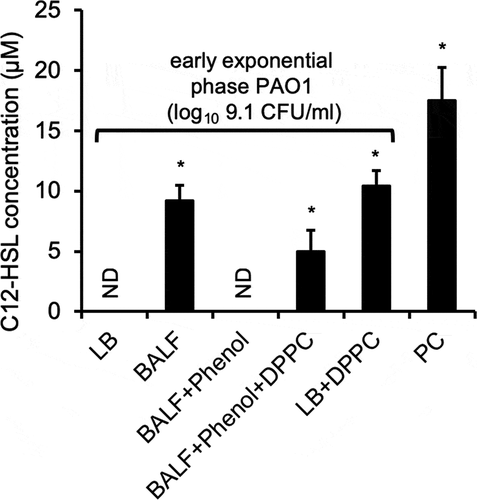
As mentioned above, QS-regulated genes are upregulated when the accumulation of homoserine lactone autoinducers reach threshold concentrations during growth. We examined if provision of DPPC could induce the release of C4-HSL and C12-HSL in log phase low cell density P. aeruginosa. As expected, gas chromatography–mass spectrometry (GC-MS) analysis detected high amount of C12-HSL in the supernatants of late stationary phase PAO1 grown in LB (, PC). In contrast, no C12-HSL was detected in exponential phase PAO1 (log10 9.1 CFU/ml) grown in LB or phenol:chloroform:isoamyl alcohol-extracted BALF (BALF+phenol). Importantly, C12-HSL was detected in the supernatant of exponential phase PAO1 cultured in BALF, phenol:chloroform:isoamyl alcohol-extracted BALF supplemented with DPPC (BALF+phenol+DPPC), and LB+DPPC (; Fig S4A-2 C). However, multiple attempts to detect C4-HSL with GC-MS analysis were unsuccessful (data not shown). Collectively, these results provide strong evidence that P. aeruginosa reprograms its QS in order to adapt to host environments within alveolar space by recognizing surfactant phospholipids.
Figure 8. DPPC alone induces the premature production of homoserine lactone C12-HSL by P. aeruginosa. The levels of C12-HSL as determined by GC-MS produced in supernatant of exponential phase PAO1 (OD 600 nm 0.6, log10 9.1 CFU/ml) grown in LB, BALF, phenol:chloroform:isoamyl alcohol-extracted BALF (BALF+phenol), phenol:chloroform:isoamyl alcohol-extracted BALF supplemented with 100 µg/ml DPPC (BALF+phenol+DPPC), and LB + 100 µg/ml DPPC, as described in Fig 7. PC: Supernatants of stationary phase PAO1 grown in LB (OD 600 nm 2.85, log10 9.9 CFU/ml) was used as positive control. ND: Not detectable. Multiple attempts to detect C4-HSL with GC-MS analysis were unsuccessful (data not shown). *p < 0.05 when compared C12-HSL levels against exponential phase PAO1 grown in LB alone and in BALF+phenol by using the one-way ANOVA analysis, as well as within each data set by using the Tukey’s test.
Discussion
Regulation of virulence gene expression by QS circuits has been studied extensively in vitro. However, the behavior of QS systems and its downstream virulence expression are rarely investigated under physiological host environments. In this study, we examined the expression of P. aeruginosa QS-regulated exoproteases and rhamnolipids in vitro, as well as during acute pneumonia in mice. Contrary to what occurs under in vitro experimental conditions, exponential phase P. aeruginosa at low cell density prematurely express both LasB and RhlA during acute pneumonia, or when grown in the mouse BALF. Premature induction of LasB and RhlA is dependent on the LasI/LasI and RhlI/RhlR circuits. We also showed that surfactant phospholipids are the primary components of mouse BALF triggering the early activation of QS-regulated virulence. Phenol:chloroform:isoamyl alcohol pre-extraction or PLA2 pre-digestion abolish the ability of mouse BALF to promote premature expression of both LasB and RhlA. Conversely, provision of DPPC, a major component of the pulmonary surfactant, restores the expression of both virulence factors. Collectively, our study demonstrates that P. aeruginosa prematurely modulates its QS to coordinate the expression of virulence factors during acute pneumonia by recognizing pulmonary surfactant phospholipids within the distal lung epithelial surface liquid.
Under in vitro experimental conditions, QS allows P. aeruginosa to communicate intercellularly via small, diffusible signaling molecules C12-HSL and C4-HSL, PQS, and IQS [Citation32–Citation35]. Local concentrations of signaling molecules increase at high cell densities, usually at the transition from late exponential to stationary growth phase, enter bacteria, and bind their corresponding transcriptional activators to activate the expression of virulence genes. The precise interplays among these QS systems remain to be defined, but they clearly regulate production of numerous virulence factors, including exoproteases, rhamnolipids, phenazines, exotoxin A, hydrogen cyanide, and biofilm formation [Citation7,Citation32–Citation35]. In contrast, when small numbers of accidental microbes land within alveolar microcompartments, they need to multiply and promptly initiate counter strategies against host innate immune responses, among which include SP-A and SP-D-mediated opsonization and subsequent phagocytosis by residence alveolar macrophages; membrane permeabilization by SPA, SPD and other antimicrobial peptides; and cell wall degradation by lysozymes [Citation20,Citation21,Citation44–Citation48]. For example, we and others have shown that P. aeruginosa elaborates multiple proteases including LasB to degrade SP-A, SP-D, lysozyme, and cytokines to evade host-mediated innate immune responses [Citation27,Citation37,Citation49–Citation54]. To execute these counter measures, our results indicate that P. aeruginosa prematurely activates QS and QS-regulated virulence factors in exponential phase bacteria and at approximately 10-fold lower bacterial density. Premature onset of QS and QS-dependent virulence is expected to confer advantages during acute infection of mouse lungs.
Our results indicate that P. aeruginosa initiates QS circuits early by detecting the presence of surfactant phospholipids. This conclusion is supported by two lines of evidence. Mouse BALF samples pre-extracted with phenol:chloroform:isoamyl alcohol or pre-digested with PLA2 lose their ability to induce the expression of QS-dependent LasB and rhamnolipids, which is restored by the provision of DPPC. Interestingly, DPPC restores only ~80% of the expression of QS-regulated LasB and rhamnolipids in BALF-pre-extracted with phenol:chloroform:isoamyl alcohol or pre-digested with PLA2 ( and ), suggesting that additional surfactant component(s) may play a minor role in inducing QS circuits. However, these components are unlikely to be surfactant proteins as pre-digestion of mouse BALF with Proteinase-K did not destroy its ability to induce rhamnolipid production by log phase P. aeruginosa. More likely, the residual induction of QS may be mediated by other lipids or phospholipids found within the surfactant. For example, sphingomyelin, ceramide, or sphingosine-1-phosphate could be metabolized to sphingosine, which has anti-bacterial activities. A previous study has shown that in response to the modified bovine pulmonary surfactant Survanta (Beractant), P. aeruginosa upregulates SphR-SphA to confer resistance to bacterial killing mediated by sphingosine [Citation55]. Due to reduced activity of acid ceramidase, which generates sphingosine from ceramide, tracheal, and bronchial sphingosine levels are significantly reduced in lung tissues of CF patients [Citation56–Citation59]. Importantly, inhalation of sphingosine, sphingosine analog, or acid ceramidase rescues susceptible mice from P. aeruginosa infection [Citation60]. Currently, it is not clear whether SphR-SphA are regulated by the LasR and RhlR QS circuits.
Although not fully delineated, it is generally accepted that the LasR-LasI sits on top of hierarchical order over RhlR-RhlI, PqsR-PQS, and PhoB-IQS QS circuits [Citation7,Citation31–Citation35,Citation61,Citation62]. Because the expression of LasB and rhamnolipids is not induced by mouse BALF in the ∆lasI∆rhII mutant, it is unlikely that PqsR and IQS signaling pathways, which act downstream of LasR, participate in the initial response to the induction by surfactant phospholipids. In addition, because only C12-HSL but not C4-HSL was detected in the wild-type P. aeruginosa exposed to mouse BALF or DPPC, it is likely that only the LasR-LasI QS circuit exerts the initial regulatory effect on the expression of virulence in response to surfactant phospholipids. However, we could not rule out that C4-HSL was lost during extraction for GC-MS analysis.
Our observation that surfactant phospholipids could prematurely induce QS and the expression of C12-HSL may have broad implications. In recent years, it has been shown that homoserine lactones, especially the P. aeruginosa C12-HSL, has the ability to modulate host immune functions, resulting in either detrimental or beneficial effects. For examples, C12-HSL induces the chemotaxis of neutrophils [Citation63,Citation64], and increases the phagocytic capacity of neutrophils [Citation65] and macrophages [Citation66], which could be due to modulation of (CD)11b/CD18 integrins and CD16 and CD64 immunoglobulin receptors [Citation65], as well as cell volume, morphology, and AQP9 characteristics in phagocytic cells [Citation67]. C12-HSL also disrupts epithelial cell junction and barriers and modulates epithelial cell migration in a dose- and time-dependent manner, most likely by reducing the expression and distribution of ZO-3 and JAM-A through hyperphosphorylation of the aforementioned junction proteins [Citation68,Citation69]. C12-HSL also interacts with the IQ-motif-containing GTPase-activating protein IQGAP1, resulting in the Rac1 and Cdc42-dependent migration of human intestinal epithelial Caco-2 cells [Citation70]. C12-HSL but not C4-HSL was shown to induce apoptosis in bone marrow-derived macrophages, neutrophils, and monocytic cell lines U-937 and P388D1 but not in epithelial cell lines CCL-185 and HEp-2 in concentration- and time-dependent manner [Citation71]. Most recently, C12-HSL has been reported to cause fragmentation of mitochondria, disrupt cristae and inner membrane ultrastructure, alter major characteristics of respiration and energetics, and decrease mitochondrial membrane potential [Citation72]. Another aspect of host response impacted by C12-HSL is the expression and secretion of multiple pro- and anti-inflammatory cytokines and chemokines [Citation73–Citation75]. C12-HSL, but not C4-HSL, was reported to inhibit lymphocyte proliferation and TNFα and IL-12 production by LPS-stimulated macrophages, suggesting that C12-HSL may influence the Th1 - Th2 balance in the infected host [Citation76]. Subsequent in vivo studies suggest that effects of C12-HSL on cytokine production depend on the underlying immune bias of the mouse strain used, with a relative increase of IFN-gamma in Th1-biased C57BL6 mice and a relative increase of IL-4 in Th2-biased BALB/c mice [Citation77], as well as differences in antigen affinity and concentration in these models [Citation78]. However, it should be noted that the aforementioned, primarily in vitro studies, used C12-HSL in concentrations (300–600 μM) mimicking biofilms established in vitro [Citation79]. In contrast, only low μM concentrations of C4-HSL and low nM concentrations of C12-HSL have been detected in the respiratory secretions from bacteria-colonized cystic fibrosis patients [Citation80–Citation82]. Therefore, the results from various experimental studies using high concentrations of C12-HSL need to be interpreted carefully. Nevertheless, in the context of biofilm-mediated infection, it is possible that innate immune cell functions could be modulated by enhanced local concentrations of QS molecules and QS-regulated virulence factors [Citation83,Citation84]. A more extensive discussion on immune modulation by QS molecules can be found in these review articles [Citation85,Citation86]. Our findings, which show that P. aeruginosa is capable of inducing the expression of lasB and rhlA genes as early as 6 hours post-infection (,d)), and that mouse BALF is able to induce the production of μM concentrations of C12-HSL (Figure 8), suggest that bacteria are able to detect the presence of surfactant phospholipids. Additionally, these observations lend credence to a locally enhanced and early QS response that results in expression of virulence factors and modulation of various innate host responses and barrier function.
In summary, the current study demonstrates that P. aeruginosa adapts to host environment in the lung by prematurely upregulating its QS-dependent virulence through the detection of surfactant phospholipids, especially DPPC. Future efforts will focus on identifying of P. aeruginosa receptor(s) responsible for the detection of surfactant phospholipids, as well as delineating whether RhlR-RhlI, PqsR-PQS, and PhoB-IQS are involved in regulation of virulence in response to surfactant phospholipids. Additional effort will determine which minor phospholipids contribute to the residual premature QS induction. Finally, it will be of great interest to identify DPPC analogs that could competitively block the induction of QS and attenuate P. aeruginosa virulence during lung infection.
Author contributions
ZK and GWL conceived and designed and conducted experiments, acquired data, analyzed data, and wrote the manuscript. HTA provided reagents, analyzed data, and reviewed the manuscript. YH and LZ conducted experiments, acquired data, analyzed data, and reviewed the manuscript. RCB and JL analyzed the data, reviewed, and edited the manuscript.
Acknowledgments
We thank Professors Michael Vasil (University of Colorado Health Science Center) for the P. aeruginosa strain PAO1, Stephen Lory (Harvard Medical School) for providing the gift of the ΔlasIΔrhlI mutant, and Dennis Ohman (Virginia Commonwealth University) for the gift of anti-LasB antibodies.
Disclosure statement
The authors have no conflict in financial interests, and are solely responsible for experimental designs and data analysis. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Additional information
Funding
References
- Tilley AE, Walters MS, Shaykhiev R, et al. Cilia dysfunction in lung disease. Annu Rev Physiol. 2015;77:379–406.
- Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol Pathol. 2007;35:116–129.
- Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86:245–278.
- Curran DR, Cohn L. Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol. 2010;42:268–275.
- Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363:2233–2247.
- Huber P, Basso P, Reboud E, et al. Pseudomonas aeruginosa renews its virulence factors. Environ Microbiol Rep. 2016;8:564–571.
- Malhotra S, Hayes D Jr, Wozniak DJ. Cystic fibrosis and pseudomonas aeruginosa: the host-microbe interface. Clin Microbiol Rev. 2019 May 29;32(3):pii: e00138-18. .
- Sana TG, Berni B, Bleves S. The T6SSs of Pseudomonas aeruginosa strain PAO1 and their effectors: beyond bacterial-cell targeting. Front Cell Infect Microbiol. 2016;6:61.
- Pena RT, Blasco L, Ambroa A, et al. Relationship between quorum sensing and secretion systems. Front Microbiol. 2019;10:1100. eCollection 2019. Review. .
- Crousilles A, Maunders E, Bartlett S, et al. Which microbial factors really are important in Pseudomonas aeruginosa infections? Future Microbiol. 2015;10:1825–1836.
- Döring G, Parameswaran IG, Murphy TF. Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol Rev. 2011;35:124–146.
- Baughman RP, Sternberg RI, Hull W, et al. Decreased surfactant protein A in patients with bacterial pneumonia. Am Rev Respir Dis. 1993;147:653–657.
- LeVine AM, Lotze A, Stanley S, et al. Surfactant content in children with inflammatory lung disease. Crit Care Med. 1996;24:1062–1067.
- Griese M, Birrer P, Demirsoy A. Pulmonary surfactant in cystic fibrosis. Eur Respir J. 1997;10:1983–1988.
- Postle AD, Mander A, Reid KB, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol. 1999;20:90–98.
- Noah TL, Murphy PC, Alink JJ, et al. Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. Am J Respir Crit Care Med. 2003;168:685–691.
- Parra E, Pérez-Gil J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem Phys Lipids. 2015;185:153–175.
- Han S, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc. 2015;12:765–774.
- Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol. 2015;10:371–393.
- Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol. 2001;63:521–554.
- Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68.
- Wu H, Kuzmenko A, Wan S, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–1602.
- McCormack FX, Gibbons R, Ward SR, et al. Macrophage-independent fungicidal action of the pulmonary collectins. J Biol Chem. 2003;278:36250–36256.
- Schaeffer LM, McCormack FX, Wu H, et al. Bordetella pertussis lipopolysaccharide resists the bactericidal effects of pulmonary surfactant protein A. J Immunol. 2004;173:1959–1965.
- Zhang S, Chen Y, Potvin E, et al. Comparative signature-tagged mutagenesis identifies Pseudomonas aeruginosa factors conferring resistance to pulmonary collectin SP-A. PLoS Pathog. 2005;1:259–268.
- Zhang S, McCormack FX, Levesque RC, et al. The flagellum of Pseudomonas aeruginosa is required for resistance to clearance by surfactant protein A. PLoS ONE. 2007;2:e564.
- Kuang Z, Hao Y, Hwang S, et al. The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol Microbiol. 2011;79:1220–1235.
- Tan RM, Kuang Z, Hao Y, et al. Type IV pilus of Pseudomonas aeruginosa confers resistance to antimicrobial activities of the pulmonary surfactant protein-A. J Innate Immun. 2014;6:227–239.
- Tan RM, Kuang Z, Hao Y, et al. Type IV pilus glycosylation mediates resistance of Pseudomonas aeruginosa to opsonic activities of the pulmonary surfactant protein A. Infect Immun. 3015;83:1339–1346.
- Fessler MB, Summer RS. Surfactant lipids at the host-environment interface. metabolic sensors, suppressors, and effectors of inflammatory lung disease. Am J Respir Cell Mol Biol. 2016;54:624–635.
- Ghidoni R, Caretti A, Signorelli P. Role of sphingolipids in the pathobiology of lung inflammationRole of sphingolipids in the pathobiology of lung inflammation. Mediators Inflamm. 2015;2015:487508. doi:https://doi.org/10.1155/2015/487508.
- Schuster M, Sexton DJ, Diggle SP, et al. Acyl-homoserine lactone quorum sensing: from evolution to application. Annu Rev Microbiol. 2013;67:43–63.
- Jakobsen TH, Bjarnsholt T, Jensen PØ, et al. Targeting quorum sensing in Pseudomonas aeruginosa biofilms: current and emerging inhibitors. Future Microbiol. 2013;8:901–921.
- Lee J, Wu J, Deng Y, et al. A cell-cell communication signal integrates quorum sensing and stress response. Nat Chem Biol. 2013;9:339–343.
- Cornelis P. Putting an end to the Pseudomonas aeruginosa IQS controversy. Microbiology Open. 2020 Feb; 9(2):e962. Epub 2019 Oct 30.
- Holloway BW, Krishnapillai V, Morgan AF. Chromosomal genetics of Pseudomonas. Microbiol Rev. 1979;43:73–102.
- Kuang Z, Hao Y, Walling BE, et al. Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-A. PLoS One. 2011;6:e27091.
- Pinzon NM, Ju LK. Analysis of rhamnolipid biosurfactants by methylene blue complexation. Appl Microbiol Biotechnol. 2009;82:975–981.
- Morihara K. Production of elastase and proteinase by Pseudomonas aeruginosa. J Bacteriol. 1964;88:745–757.
- Azghani AO. Pseudomonas aeruginosa and epithelial permeability: role of virulence factors elastase and exotoxin A. Am J Respir Cell Mol Biol. 1996;15:132–140.
- McIver KS, Kessler E, Ohman DE. Identification of residues in the Pseudomonas aeruginosa elastase propeptide required for chaperone and secretion activities. Microbiology. 2004;150:3969–3977.
- Ochsner UA, Koch AK, Fiechter A, et al. Isolation and characterization of a regulatory gene affecting rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. J Bacteriol. 1994;176:2044–2054.
- Zhu K, Rock CO. RhlA converts beta-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the beta-hydroxydecanoyl-beta-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J Bacteriol. 2008;190:3147–3154.
- Bartlett JA, Fischer AJ, McCray PB Jr. Innate immune functions of the airway epithelium. Contrib Microbiol. 2008;15:147–163.
- Ryu JH, Kim CH, Yoon JH. Innate immune responses of the airway epithelium. Mol Cells. 2010;30:173–183.
- Holweg A, Schnare M, Gessner A. The bactericidal/permeability-increasing protein (BPI) in the innate defense of the lower airways. Biochem Soc Trans. 2011;39:1045–1050.
- Seiler F, Lepper PM, Bals R, et al. Regulation and function of antimicrobial peptides in immunity and diseases of the lung. Protein Pept Lett. 2014;21:341–351.
- Werner JL, Steele C. Innate receptors and cellular defense against pulmonary infections. J Immunol. 2014;193:3842–3850.
- Alcorn JF, Wright JR. Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J Biol Chem. 2004;279:30871–30879.
- Mariencheck WI, Alcorn JF, Palmer SM, et al. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol. 2003;28:528–537.
- Horvat RT, Clabaugh M, Duval-Jobe C, et al. Inactivation of human gamma interferon by Pseudomonas aeruginosa proteases: elastase augments the effects of alkaline protease despite the presence of alpha 2-macroglobulin. Infect Immun. 1989;57:1668–1674.
- Kevin GL, Munson KL, Johnson MC, et al. Metalloproteases from Pseudomonas aeruginosa degrade human RANTES, MCP-1, and ENA-78. J Interferon Cytokine Res. 2003;23:307–318.
- Parmely M, Gale A, Clabaugh M, et al. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect Immun. 1990;58:3009–3014.
- Theander TG, Kharazmi A, Pedersen BK, et al. Inhibition of human lymphocyte proliferation and cleavage of interleukin-2 by Pseudomonas aeruginosa proteases. Infect Immun. 1988;56:1673–1677.
- LaBauve AE, Wargo MJ. Detection of host-derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog. 2014;10:e1003889.
- Teichgräber V, Ulrich M, Endlich N, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–391.
- Becker KA, Riethmüller J, Lüth A, et al. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. 2010;42:716–724.
- Brodlie M, McKean MC, Johnson GE, et al. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am J Respir Crit Care Med. 2010;182:369–375.
- Bodas M, Min T, Mazur S, et al. Critical modifier role of membrane‐cystic fibrosis transmembrane conductance regulator‐dependent ceramide signaling in lung injury and emphysema. J Immunol. 2011;186:602–613.
- Pewzner-Jung Y, Tavakoli Tabazavareh S, Grassmé H, et al. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol Med. 2014;6:1205–1214.
- Dekimpe V, Déziel E. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology. 2009;155(Pt3):712–723.
- Dandekar AA, Greenberg EP. Microbiology: plan B for quorum sensing. Nat Chem Biol. 2013;9:292–293.
- Zimmermann S, Wagner C, Müller W, et al. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Infect Immun. 2006;74(10):5687–5692.
- Karlsson T, Musse F, Magnusson KE, et al. N-Acylhomoserine lactones are potent neutrophil chemoattractants that act via calcium mobilization and actin remodeling. J Leukoc Biol. 2012;91:15–26.
- Wagner C, Zimmermann S, Brenner-Weiss G, et al. The quorum-sensing molecule N-3-oxododecanoyl homoserine lactone (3OC12-HSL) enhances the host defence by activating human polymorphonuclear neutrophils (PMN). Anal Bioanal. Chem. 2007;387(2):481–487.
- Vikström E, Magnusson KE, Pivoriunas A. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone stimulates phagocytic activity in human macrophages through the p38 MAPK pathway. Microbes Infect. 2005;7:1512–1518.
- Holm A, Magnusson KE, Vikström E. Pseudomonas aeruginosa N-3-oxo-dodecanoyl-homoserine lactone elicits changes in cell volume, morphology, and AQP9 characteristics in macrophages. Front Cell Infect Microbiol. 2016;6:32.
- Vikström E, Tafazoli F, Magnusson KE. Pseudomonas aeruginosa quorum sensing molecule N-(3 oxododecanoyl)-l-homoserine lactone disrupts epithelial barrier integrity of Caco-2 cells. FEBS Lett. 2006;580:6921–6928.
- Vikström E, Bui L, Konradsson P, et al. Role of calcium signalling and phosphorylations in disruption of the epithelial junctions by Pseudomonas aeruginosa quorum sensing molecule. Eur J Cell Biol. 2010;89:584–597.
- Karlsson T, Turkina MV, Yakymenko O, et al. The Pseudomonas aeruginosa N-acylhomoserine lactone quorum sensing molecules target IQGAP1 and modulate epithelial cell migration. PLoS Pathog. 2012;8(10):e1002953.
- Tateda K, Ishii Y, Horikawa M, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71:5785–5793.
- Josephson H, Ntzouni M, Skoglund C, et al. Pseudomonas aeruginosa N-3-oxo-dodecanoyl-homoserine lactone impacts mitochondrial networks morphology, energetics, and proteome in host cells. Front Microbiol. 2020;11:1069.
- Smith RS, Fedyk ER, Springer TA, et al. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-kappa B and activator protein-2. J Immunol. 2001;167(1):366–374.
- Smith RS, Harris SG, Phipps R, et al. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)homoserine lactone contributes to virulence and induces inflammation in vivo. J Bacteriol. 2002;184(4):1132–1139.
- Hooi DS, Bycroft BW, Chhabra SR, et al. Differential immune modulatory activity of Pseudomonas aeruginosa quorum-sensing signal molecules. Infect Immun. 2004;72(11):6463–6470.
- Telford G, Wheeler D, Williams P, et al. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-L-homoserine lactone has immunomodulatory activity. Infect Immun. 1998;66(1):36–42.
- Ritchie AJ, Yam AO, Tanabe KM, et al. Modification of in vivo and in vitro T- and B-cell-mediated immune responses by the Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Infect Immun. 2003;71(8):4421–4431.
- Ritchie AJ, Jansson A, Stallberg J, et al. The Pseudomonas aeruginosa quorum-sensing molecule N-3-(oxododecanoyl)-L-homoserine lactone inhibits T-cell differentiation and cytokine production by a mechanism involving an early step in T-cell activation. Infect Immun. 2005;73(3):1648–1655.
- Charlton TS, de Nys R, Netting A, et al. A novel and sensitive method for the quantification of N-3-oxoacyl homoserine lactones using gas chromatography-mass spectrometry: application to a model bacterial biofilm. Environ Microbiol. 2000;2(5):530–541.
- Singh PK, Schaefer AL, Parsek MR, et al. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764.
- Erickson DL, Endersby R, Kirkham A, et al. Pseudomonas aeruginosa quorum-sensing systems may control virulence factor expression in the lungs of patients with cystic fibrosis. Infect Immun. 2002;70:1783–1790.
- Chambers CE, Visser MB, Schwab U, et al. Identification of N-acylhomoserine lactones in mucopurulent respiratory secretions from cystic fibrosis patients. FEMS Microbiol Lett. 2005;244:297–304.
- Jesaitis AJ, Franklin MJ, Berglund D, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–4339.
- Jensen PO, Bjarnsholt T, Phipps R, et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology. 2007;153:1329–1338.
- Turkina MV, Vikström E. Bacteria-host crosstalk: sensing of the quorum in the context of pseudomonas aeruginosa infections. J Innate Immun. 2018;11(3):263–279.
- Kariminik A, Baseri-Salehi M, Kheirkhah B. Pseudomonas aeruginosa quorum sensing modulates immune responses: an updated review article. Immunol Lett. 2017;190:1–6.