
ABSTRACT
Bacterial genotoxins are peptide or protein virulence factors produced by several pathogens, which make single-strand breaks (SSBs) and/or double-strand DNA breaks (DSBs) in the target host cells. If host DNA inflictions are not resolved on time, host cell apoptosis, cell senescence, and/or even bacterial pathogen-related cancer may occur. Two multi-protein AB toxins, cytolethal distending toxin (CDT) produced by over 30 bacterial pathogens and typhoid toxin from Salmonella Typhi, as well as small polyketide-peptides named colibactin that causes the DNA interstrand cross-linking and subsequent DSBs is the most well-characterized bacterial genotoxins. Using these three examples, this review discusses the mechanisms by which these toxins deliver themselves into the nucleus of the target host cells and exert their genotoxic functions at the structural and functional levels.
Introduction
Bacterial genotoxins are polyketide-peptide metabolites or multi-protein virulent factors manipulating host DNAs in the target mammalian cells [Citation1] (). Genotoxin-mediated cellular effects include cell nuclear distension, DNA repair response, cell cycle arrest in G2/M, and/or cell death [Citation2,Citation3]. When the cellular DNA repair system was overwhelmed by the DNA damage triggered by the action of bacterial genotoxins, the host cell may enter a senescence state or apoptosis [Citation1]. There are two well-characterized multi-protein AB-type genotoxins, cytolethal distending toxins (CDTs) produced by over 30 Gram-negative bacteria species, and typhoid toxin from Salmonella enterica serovar Typhi (S. Typhi) [Citation4,Citation5] (). Colibactins, small polyketide-peptide metabolites produced primarily by Escherichia coli (E. coli) strains in the phylogenetic group B2, are another well-characterized genotoxin [Citation6] (). This review utilizes these three genotoxins as examples in discussing their delivery, structure, and function.
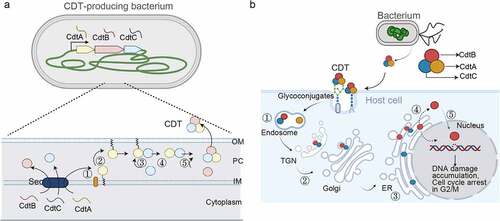
Figure 1. Multi-Protein and small peptide bacterial genotoxins. (a) Schematic diagram depicting the intoxication processes of multi-protein and small peptide bacterial genotoxins. (b) CDT structure. CdtB is the enzymatic ‘A’ subunit possessing DNase I-like activity. CdtA and CdtC are receptor-binding ‘B’ subunits of CDTs. (c) Typhoid toxin structure. CdtB is one of the ‘A’ subunits of typhoid toxin, possessing DNase I-like activity. PltA is the other ‘A’ subunit, while PltB forms the homopentameric receptor-binding ‘B’ subunits of typhoid toxin. (d) Representative colibactin structures. According to colibactin nomenclature, the numbers behind the word colibactin generally match their molecular weight [Citation94]. (b and c) panels are prepared using PyMOL. AaCDT, Aggregatibacter actinomycetemcomitans CDT (PDB: 2F2F). StyCdtb, Salmonella enterica subsp. enterica serovar Typhi CdtB (PDB: 4K6L). Created with BioRender.Com.
![Figure 1. Multi-Protein and small peptide bacterial genotoxins. (a) Schematic diagram depicting the intoxication processes of multi-protein and small peptide bacterial genotoxins. (b) CDT structure. CdtB is the enzymatic ‘A’ subunit possessing DNase I-like activity. CdtA and CdtC are receptor-binding ‘B’ subunits of CDTs. (c) Typhoid toxin structure. CdtB is one of the ‘A’ subunits of typhoid toxin, possessing DNase I-like activity. PltA is the other ‘A’ subunit, while PltB forms the homopentameric receptor-binding ‘B’ subunits of typhoid toxin. (d) Representative colibactin structures. According to colibactin nomenclature, the numbers behind the word colibactin generally match their molecular weight [Citation94]. (b and c) panels are prepared using PyMOL. AaCDT, Aggregatibacter actinomycetemcomitans CDT (PDB: 2F2F). StyCdtb, Salmonella enterica subsp. enterica serovar Typhi CdtB (PDB: 4K6L). Created with BioRender.Com.](/cms/asset/1a5579e5-6914-45ec-8a93-5f0861469867/kvir_a_2097417_f0001_oc.jpg)
AB toxins are protein virulence factors secreted by many bacterial pathogens, contributing to the pathogenicity of the cognate bacteria. AB toxins consist of two functionally distinct components: the enzymatic ‘A’ fragment (for single chain AB toxins) or subunit (for multi-protein AB toxins) for pathogenicity and the receptor-binding ‘B’ fragment or subunit for toxin delivery (). AB toxins are called by several different names, based on their structure-, A-component-, and B-component-mediated categorizations.
From the structural standpoint, CDTs are AB2 toxins, consisting of one enzymatic ‘A’ subunit, CdtB, and two receptor-binding ‘B’ subunits, CdtA and CdtC (). Their alias, genotoxin, is based on their A-component-mediated action. CDTs are produced by more than 30 γ- and ε-Proteobacteria, which are mostly Gram-negative bacterial pathogens, such as AaCDT from Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), CjCDT from Campylobacter jejuni (C. jejuni), HdCDT from Haemophilus ducreyi (H. ducreyi), and SdCDT from Shigella dysenteriae (S. dysenteriae) [Citation4]. Typhoid toxin from S. Typhi is an A2B5 toxin, consisting of two enzymatic ‘A’ subunits, CdtB and PltA, and a homopentamer of receptor-binding subunit, PltB (). As reflected in its name, typhoid toxin CdtB shares similarities to the enzymatic subunit CdtB from CDTs. Many of the cognate bacteria producing CDTs or typhoid toxin are human pathogens. For instance, A. actinomycetemcomitans, H. ducreyi, E. coli, C. jejuni, and S. Typhi are the causative agents of oral, genital, gastrointestinal (GI) tract, or systemic diseases [Citation7–9].
Colibactins are polyketide, non-ribosomal peptide hybrid compounds mainly produced by E. coli strains of the phylogenetic group B2 and other bacteria encoding polyketide synthases (pks), such as Klebsiella pneumoniae, Klebsiella aerogenes (previously known as Enterobacter aerogenes), and Citrobacter koseri (). Colibactin-mediated genotoxic effects may be indistinguishable from CDT-mediated cytotoxicity, as the DNA damage repair responses and cell cycle arrests primarily in G2/M are also induced, among others [Citation6,Citation10]. However, unlike CdtB from CDTs and typhoid toxin, colibactin-mediated genotoxic effects occur presumably more efficiently when there is a direct interaction (e.g. adhesion or invasion in host cells) between the cognate bacteria and host cells, which is presumably due to the unstable nature of colibactins [Citation6] ().
This review is divided into two parts. The first part focuses on the assembly and/or delivery mechanisms of CDTs and typhoid toxin, as well as colibactins, to the nucleus of the target host cells, which are essential for their genotoxic activities. The second part of this review discusses notable structural and functional features of CdtB and colibactin.
Delivery of bacterial genotoxins
Cytolethal distending toxins
CDT was discovered in Campylobacter spp, including C. coli, C. foetus, C. jejuni, and C. lari, as well as E. coli and Shigella, all of which encode the cdt genes within the same operon [Citation2,Citation11–14]. The genome of C. jejuni is approximately half of the genomes of other enteric pathogens, such as Salmonella spp. Consistently, C. jejuni encodes a simpler set of virulent factors, including CDT, lipooligosaccharide (LOS), HtrA, CadF, and fT3SS [Citation15], and CjCDT is an only exotoxin produced by C. jejuni [Citation13]. CjCDT induces cell cycle arrest in G2/M and nucleus distending [Citation16]. Some but not all C. jejuni strains encode CDT [Citation14]. Compared to C. jejuni lacking CjCDT, the counterparts having CjCDT are associated with increased episodes of progressive gastritis, invasion into epithelial cells, and septicaemia, as shown among human clinical patients and animal models [Citation17–19].
Among CDTs from more than 30 γ- and ε-Proteobacteria, EcCDT, HdCDT, and AaCDT are perhaps the most well characterized. For instance, E. coli strains isolated from paediatric patients with diarrhoea encode one of the five EcCDTs identified thus far, EcCDT-I to V [Citation20]. EcCDTs show over 40% sequence identity, which are encoded within the same operon in each E. coli strain that is associated with watery diarrhoea [Citation20,Citation21], neonatal meningitis [Citation22], porcine or poultry septicaemia [Citation23], or human urinary tract infections (UTIs) [Citation24]. The cdt-I, cdt-III, cdt-IV, and cdt-V operons are located near prophage genes or in a conjugative plasmid, suggesting that horizontal gene transfers were likely involved in the acquisition of CDTs across those pathogenic E. coli strains [Citation25–28].
HdCDT is produced by H. ducreyi, the pathogen associated with chancroid lesions and chronic skin ulcerations [Citation29]. HdCDT has been identified in 64–89% of clinical cases [Citation30], suggesting its associated role in clinical manifestations. Like other CDTs, HdCDT genes are encoded within the same operon [Citation31].
AaCDT genes are encoded in the cdt gene locus within the same operon in the chromosome of A. actinomycetemcomitans, the pathogen associated with aggressive periodontitis, infectious endocarditis, and chest wall abscesses, among others [Citation32,Citation33]. AaCDT, lipopolysaccharide (LPS), and leukotoxin are the primary virulence factors of A. actinomycetemcomitans associated with clinical manifestations [Citation34]. For instance, the administration of wild-type AaCDT to a live rat animal model recapitulated many oral clinical manifestations, including junctional epithelial tissue abrasions, which were not seen when a catalytic mutant AaCDT was administered [Citation35]. AaCDT is known to show cell-type-dependent cellular toxicities, including cell distension and apoptosis in epithelial cells, rapid cell cycle arrest and apoptosis of T lymphocytes, and pro-inflammation responses in phagocytic cells through the activation of the NLRP3 inflammasome, which are beneficial to the cognate pathogen by facilitating bacterial colonization and subverting innate and adaptive immune responses [Citation36–41].
Production and assembly of CDT holotoxins
All CDTs are tripartite AB toxins, consisting of CdtA, CdtB, and CdtC at a 1:1:1 stoichiometry. The genes, cdtA, cdtB, and cdtC, are encoded within the same operon [Citation42] (). As phage remnants are often found near the cdt genes, CDTs are believed to be acquired via horizontal gene transfer and evolved separately in the context of the interaction between the cognate bacteria and the host [Citation43–45]. These three protein subunits are produced individually in the bacterial cytoplasm, translocated across the inner membrane (IM) to the periplasmic compartment (PC) of the cognate Gram-negative bacteria, and self-assembled in the PC [Citation46] (). The translocation of immature CdtA, CdtB, and CdtC subunits from the bacterial cytoplasm to the PC by crossing the IM is dependent on the bacterial secretion system (Sec) utilizing their N-terminal signal peptide, which is removed by the action of the signal peptidase I and II during the translocation process [Citation46] (). The holotoxin assembly in the PC occurs in a step-wise fashion (). CdtA crossed from the cytoplasm to the PC contains the conserved cysteine residue at its N-terminus to be lipidated in the PC by the action of a lipoprotein processing enzyme before it starts making a complex with CdtC [Citation47] (). This conserved cysteine residue is part of the lipobox consensus sequence: (L)–3-(A/S)–2-(G/A)–1-C+1, where amino acid residues at the −2 and −1 positions can be any amino acid. The conserved ‘L-V-A-C’ sequence is found in AaCdtA, HdCdtA, CjCdtA, and EcCdtA-I, II, III [Citation46]. The cysteine at the +1 position becomes the first amino acid of lipoCdtA located in the PC [Citation48] (). The lipoCdtA moves from the IM to the outer membrane (OM) within the PC, which recruits mature CdtC and forms a lipoCdtA and CdtC complex (). The lipoCdtA and CdtC complex is then freed from the OM via the N-terminal truncation of lipoCdtA, which forms a tripartite CDT holotoxin complex via its self-assembly with CdtB in the PC [Citation47] (). The fully assembled CdtA/CdtB/CdtC holotoxin in the PC is ready to be secreted out to the extracellular environment. Given toxin’s tropism to specific cells and tissues, which is primarily mediated by toxin binding to specific glycan receptor displayed on the target host cells, an active toxin secretion mechanism(s) plays a larger role in toxin secretion. The mechanisms involved remain to be characterized in detail. In contrast, a passive secretion mechanism of the toxin that is encased in the outer membrane vesicle (OMV) has been characterized, which is the process that can be stimulated by environmental signals, including sodium taurocholate (a bile salt component) in the case of C. jejuni [Citation49].
Figure 2. CDT production, assembly, and trafficking. (a) The cdtA, cdtB, and cdtC genes are encoded within the same operon and expressed in the cytoplasm. ①, CdtA, CdtB, and CdtC are transported to the periplasm compartment (PC) through the Sec secretion pathway. Unlike CdtB and CdtC, CdtA is lipidated in the inner membrane (IM). ②, The lipoCdtA was transported onto the outer membrane (OM). ③, LipoCdtA on the OM recruits CdtC in the PC to form a heterodimer. ④, CdtC-lipoCdtA recruits a protease on the OM, resulting in the CdtA/CdtC heterodimer complex. ⑤, CdtA/CdtC heterodimer recruits free CdtB in the PC and forms CDT holotoxin. (b) Schematic diagram depicting the expression, assembly, trafficking, and intoxication processes of CDT: ①, CdtB/CdtC may be dissociated from CdtA in the endosome. ②, CdtB/CdtC transport to the ER through the retrograde transport pathway. ③, CdtB is dissociated from CdtC in the ER. ④, CdtB leaves the ER to the cytoplasm through an ERAD or ERAD-related pathway. ⑤, CdtB utilizes an atypical NLS sequence(s) to enter the nucleus and plays its function as a genotoxin. Created with BioRender.com.
CDT trafficking pathway: Receptor-binding
Delivery of CDTs from the extracellular environment to the nucleus involves toxin binding to the receptor, retrograde endocytosis, and nuclear translocation (). In brief, the B subunits CdtA and CdtC are known to utilize specific glycan moieties displayed by ganglioside glycolipids and N-linked glycoproteins [Citation50–52]. Similar to several other multi-protein AB toxins, the binding of CDTs to their receptor initiates a retrograde receptor-mediated endocytosis process, which was demonstrated by N-glycosylation and sulphation of engineered HdCDTs, indicating its Golgi and ER localization during the trafficking process [Citation53]. CDTs, including AaCDT and EcCDT, may use the endoplasmic reticulum (ER)-associated degradation pathway (ERAD) to exit from the ER to the cytoplasm [Citation54]. From the cytoplasm, CdtB traffics to the nucleus, which involves their intrinsic nuclear localization sequences (NLS), as demonstrated by using AaCdtB [Citation55]. This section discusses this multi-step process required for CDT delivery to the site of action, the nucleus.
The binding between CDTs and host cell surface receptors are mainly relying on B components (CdtA and CdtC) that preferentially bind to N-linked glycoproteins or glycosphingolipids. For instance, a broad range of ganglioside glycans, including GM1, GM2, GM3, and/or Gb4 serve as receptors for AaCDT from A. actinomycetemcomitans [Citation52]. The glycan-binding preferences of AaCdtA and AaCdtC are similar but different; AaCdtA showed strong binding affinities to GM1, GM2, GM3, and Gb4, while CdtC preferentially binds to GM1 and GM2 [Citation52], which may contribute to higher affinities of the holotoxin to GM1 and GM2 when all gangliosides are comparably present on one cell, as well as the relative importance of CdtA and CdtC, depending on cell types with different ganglioside expression profiles. In support, AaCDT utilized GM3 to bind to human monocytes, as demonstrated by the inhibition of glycolipid synthesis and the inhibition of the toxin binding by GM3 enriched liposome [Citation52], while GM3-depleted Chinese hamster ovary (CHO) cells remained sensitive to CDTs [Citation56]. N-linked glycans with/without fucosylation are utilized by several CDTs, including EcCDT from E. coli, AaCDT, and HdCDT from H. ducreyi [Citation50,Citation51]. For instance, inhibition of N-glycosylation by a pan-N-glycosylation inhibitor, tunicamycin, abolished HdCDT, AaCDT, and EcCDT-III-mediated toxicities [Citation53]. Consistently, HdCdtA and HdCdtC subunits share 91.9% and 93.5% of amino acid sequences with AaCdtA and AaCdtC, which showed similar cell surface binding to lipid rafts and N-linked glycans [Citation56]. This is in contrast to CjCDT whose toxic effects were increased by a tunicamycin treatment, which may suggest that glycoprotein receptor plays little to no role in CjCDT-mediated toxicities and subsequently the lack of tall glycoprotein glycans helped CDT bind to its receptor, such as ganglioside glycolipid receptors on the plasma membrane [Citation56]. It is noteworthy that CjCDT has the most divergent amino acid sequence identity (less than 30%) compared with the other three CDTs, HdCDT, AaCDT, and EcCDT [Citation56].
Furthermore, a genetic screen identified several genes that encode protein binding partners for CDTs: Transmembrane protein 181 (TMEM181 for EcCDT-I), Synaptogyrin-2 (SYNGR2 for HdCDT and AaCDT), and three receptors, Transmembrane 9 superfamily member 4 (TM9SF4), Transmembrane protein 127 (TMEM127) and Protein GPR107 (for CjCDT) [Citation57]. Among them, GRP107 is a glycoprotein that contains three predicted N-glycosylation sites at residues 70, 169, and 211 and thus the interaction between CDT and GRP107 may be through N-liked glycans. Thus, it is conceivable that CDTs may use specific protein receptors on some occasions, while glycan moieties are the primary receptors for CDTs. Nonetheless, the use of ubiquitous glycan moieties by most CDTs is in line with a broad range of their target host cells, including epithelial cells, macrophages, and lymphocytes [Citation1]. It is worth noting that, compared to CdtB subunits, the receptor binding subunits, CdtA and CdtC, share lower similarities across CDTs [Citation58], which is consistent with the coevolution concept for niche-specific adaptations of AB toxins and the cognate bacteria.
The role of lipid rafts in the interactions between CDTs and gangliosides has been investigated by using several approaches, including cellular imaging [Citation59,Citation60] in conjunction with cholesterol/sphingomyelin depletions [Citation53,Citation56,Citation59,Citation61]. For instance, either methyl β-cyclodextrin-mediated depletion of cholesterol or a mutation of sphingomyelin synthase 1 significantly made HeLa and Jurkat cells resistant to AaCDT, CjCDT, EcCDT, and HdCDT [Citation53,Citation56,Citation59,Citation61]. Consistently, CdtC subunits from AaCDT, CjCDT, and HdCDT have the conventional cholesterol recognition/interaction amino acid consensus (CRAC) sequences, whose mutations diminished toxin binding to lipid rafts [Citation62]. It is noteworthy that the CRAC-mutation-mediated phenotype appears to be cell type-dependent; unlike HeLa and Jurkat cells, the cholesterol-depleted CHO cells showed little to no effects on this cell’s susceptibility to AaCDT [Citation56,Citation63].
CDT trafficking pathway: Retrograde endocytosis
While the tripartite stoichiometry of CDTs is important for their interaction with cell surface receptors, CdtA and CdtC may dissociate from the complex at the different stages in the vesicle trafficking pathway (). CDT endocytosis appears to be a rapid process, as supported by the presence of HdCdtA in the early endosome within 10 minutes of toxin treatment. The fate of CdtA has been controversial, which was thought to remain on the plasma membrane, but the data from HdCdtA indicate that it is also part of the toxin complex trafficked to the endosome where CdtA is believed to be degraded [Citation64]. Nonetheless, the consensus is that at least the CdtB/CdtC complex traffic to the trans-Golgi and the ER via a retrograde pathway [Citation64]. The retrograde transport pathway was demonstrated by detecting Golgi- and ER-specific posttranslational modifications – sulphation and N-linked glycosylation of HdCdtB, respectively [Citation53]. Gene screen results also support the retrograde vesicle trafficking pathway [Citation61]. CdtC was transported with CdtB throughout the retrograde trafficking pathway, after which CdtB leaves the ER for further trafficking to enter the nucleus [Citation63,Citation65]. Similarly, genome-wide CRISPR/Cas9-mediated screens using CjCDT revealed several genes that encode proteins involved in the retrograde vesicle trafficking pathway, including the Golgi-associated retrograde protein complex components, such as Vps51 and Vps54, and the transmembrane emp24 domain-containing protein 2 (TMED2). HEK293 cells deficient in those genes became resistant to CjCDT [Citation66].
CDT trafficking pathway: Nuclear localization
The mechanism by which CdtB leaves the ER lumen during the CDT trafficking process is incompletely understood, although, like many other multi-protein AB toxins, such as ricin and cholera toxin, the conventional ERAD or alternative pathway is believed to be utilized by CDTs (). Conventional ERAD pathway-related genes, such as Derl2, Hrd1, and p97, were shown to be important for the intoxication of CHO cells by AaCDT, HdCDT, EcCDT-III, presumably by providing energy for the ERAD pathway for translocation of the misfolded protein to the cytoplasm. Similarly, HEK293 cells deficient in ERAD components (e.g. SEL1 L and HRD1) showed resistance to CjCDT, indicating that CjCDT utilizes the ERAD machinery for its retrograde transport to the cytoplasm [Citation66]. An alternative mechanism has also been proposed, which is supported by (1) the high thermal stability of CdtB which appears to be not ideal for the conventional ERAD pathway that requires the protein to be unfolded for translocation [Citation67], and (2) the insensitivity of HdCdtB to Derl1 and Derl2 mutations that are essential for the conventional ERAD-mediated translocation of misfolded proteins [Citation53]. It is conceivable that the use of the conventional ERAD and/or alternative pathway may be toxin-dependent.
After leaving the ER to the cytoplasm via a conventional ERAD and/or alternative pathway, CdtB utilizes its intrinsic, atypical NLS to translocate itself into the cell nucleus, which likely exploits the host machinery for nuclear protein localization (). For instance, EcCdtB-II contains two NLSs, NLS1 and NLS2, in its C-terminus region. △NLS1 mutant toxin showed its perinuclear localization in the trans-Golgi network (TGN)/ER, but not in the cytoplasm, while △NLS2 mutant toxin was found in the cytoplasm [Citation68], suggesting their roles in EcCdtB nuclear translocation at the different stages from the ER to the cytoplasm to the nucleus. AaCdtB has a single NLS that plays an essential role in toxin translocation from the cytoplasm to the nucleus, which was demonstrated by a series of gain- and loss-of-function studies primarily through fluorescence microscopy. In particular, deletion of the 11 amino acid long NLS sequence made CdtB unable to translocate to the nucleus, whereas the addition of the SV40 T NLS to AaCdtB∆NLS enables the toxin to translocate to the nucleus and subsequent cytotoxicity [Citation55].
Typhoid toxin
Typhoid toxin is an A2B5 toxin, consisting of one each of two A subunits, CdtB (29 kDa) and PltA (27 kDa), and five of its receptor-binding B subunits, PltB (13 kDa each), which are secreted by the Gram-negative bacterium, Salmonella enterica serovar Typhi (S. Typhi), the cause of typhoid fever [Citation5,Citation69]. S. Typhi infects the GI tract, followed by systemic infection and ultimately transmission to other hosts by returning to and excreting from the GI tract, as S. Typhi primarily infects via an oral-faecal route. Consistently, typhoid toxin targets intestinal epithelial cells, immune cells, brain endothelial cells, and gallbladder epithelial cells [Citation70–72]. S. Typhi is an exclusive, human pathogen responsible for typhoid fever, a life-threatening systemic disease [Citation73]. Typhoid toxin CdtB is homologous to the A subunit of CDTs that target the host cell DNA ( (a–)). Similar to CDTs, intoxication by typhoid toxin CdtB results in host cell DNA damage repair responses, cell cycle arrests in G2/M, and/or cell death [Citation69,Citation74,Citation75]. PltA is homologous to the A subunit of pertussis toxin and E. coli ArtA, both of which target G proteins, although S. Typhi PltA seems to target another protein rather than G proteins [Citation69]. CdtB and PltA are linked in tandem primarily via a single intermolecular disulphide bond through two cysteine residues positioned in each A subunit [Citation5] ( (a–)). The C-terminus of PltA forms an α-helix that is inserted into a channel of the donut-shaped PltB homopentamer, contributing to its distinct, pyramid-shaped A2B5 holotoxin structure [Citation5]. Consistently, there is no direct interaction between CdtB and PltB (). Both A subunits, CdtB and PltA, are always assembled with the pentameric B subunits [Citation5], which can be either PltB5 or, in some cases, PltC5 [Citation76].
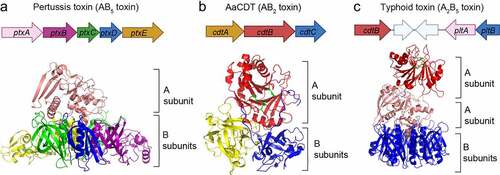
Figure 3. Typhoid toxin – two toxins become one. The gene organizations and 3D structures of pertussis toxin (a), AaCDT (b), and typhoid toxin (c). The enzymatic ‘A’ subunits and the receptor-binding ‘B’ subunits are indicated. Figures are prepared using PyMOL. Pertussis toxin (PDB: 1PRT). AaCDT, Aggregatibacter actinomycetemcomitans CDT (PDB: 2F2F). StyCdtB, Salmonella enterica serovar Typhi CdtB (PDB: 4K6 L).
Production and assembly of typhoid toxin
The three genes for typhoid holotoxin are encoded as two separate operons within the same islet in the chromosome (). This is atypical gene organization for multi-protein AB toxins that are organized frequently in the same operon, which, however, is consistent with the unique feature of typhoid toxin – two AB toxins evolved to become one toxin with two enzymatic activities [Citation5,Citation69] ( (a–)). Its unique A2B5 arrangement seems to be the outcome of the combination and adaptation events of CDT in the AB2 toxin family and pertussis toxin in the AB5 family. CdtB and PltA-PltB5 are linked primarily by a single disulphide bond between CdtBCys269 and PltACys214; These cysteine residues are unique to typhoid toxin, as corresponding residues to these two are not found in any of their homologs [Citation5] (). This event is consistent with the loss of additional B components, CdtA and CdtC, from the typhoid toxin complex, whose B subunit PltB underwent additional evolution in the context of S. Typhi infection [Citation70].
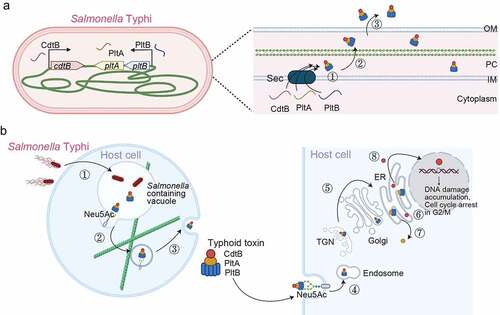
Nonetheless, similar to other AB toxins, typhoid toxin subunits are produced as separate proteins in the bacterial cytoplasm, each of which possesses an N-terminal signal peptide that makes each subunit individually cross the IM via the Sec system. However, unlike other AB toxins, typhoid toxin is produced exclusively by the intracellularly located S. Typhi by sensing specific cues presented in the vacuole environment within host cells [Citation69,Citation75] (). In the PC, mature A and B subunit proteins are assembled, which have 12 disulphide bonds to form [Citation5]. The assembled AB holotoxins are secreted from the bacteria to the lumen of the Salmonella-containing vacuole (SCV) via a mechanism involving specific enzymes located in the bacterial membrane [Citation77] (). Typhoid toxin in the SCV binds to N-acetylneuraminic acid (Neu5Ac)-bearing N-linked glycoprotein receptors on the SCV, which is cation-independent mannose 6-phosphate receptor (CI-M6PR) in Henle407 and HEK293 cells. This event subsequently coordinates downstream multistep vesicle export processes required to translocate typhoid toxin from the SCV to the extracellular environment, where host cell modifications by the Salmonella pathogenicity island 2 (SPI-2) type III secretion system (T3SS) play an essential role [Citation78]. Therefore, while the core mechanism for typhoid toxin gene expression, assembly, and secretion process in bacteria is shared with the mechanisms for other multi-protein AB toxins, including CDTs, there are two additional steps unique to typhoid toxin: (1) invasion into the host cells and (2) translocation from the SCV to outside infected cells ().
Figure 4. Typhoid toxin production, assembly, trafficking, and intoxication. (a) Typhoid toxin production, assembly, and secretion from S. Typhi. cdtB is transcribed from one operon, while pltB, pltA (and one additional gene, ttsA) are co-transcribed by another operon. ①, CdtB, PltA, PltB remains unstructured in the bacterial cytoplasm until they are transported into the PC through the Sec secretion pathway. The secreted CdtB, PltA, and PltB are self-assembled into typhoid toxin holotoxin. ②, Typhoid toxin enters the outer periplasm space by the action of the Tsa enzyme that breaks the peptidoglycan. ③, Typhoid toxin finally secretes to the lumen of the SCV. (b) Schematic diagram depicting typhoid toxin expression, assembly, trafficking, and intoxication. The steps involved are: ①, Salmonella invades host cells and forms the Salmonella-containing vacuole (SCV). ②, Salmonella in the SCV expresses and secretes the typhoid toxin under a specific SPI2-T3SS vacuole condition. The secreted typhoid toxin in the SCV binds to Neu5Ac-terminated glycoprotein receptors on the SCV and is sorted into anterograde vesicles. ③, the exocytosis of vesicles carrying typhoid toxin relies on the microtubule-based transport. Finally, typhoid toxin from S. Typhi-infected cells is released into the extracellular environment. ④, Typhoid toxin in the extracellular environment enters the target host cells through receptor-mediated endocytosis. Typhoid toxin utilizes multiantennary N-linked glycan receptors terminated in Neu5Ac. ⑤, the endocytosed typhoid toxin utilizes the retrograde transport pathway to enter the ER. ⑥, Typhoid toxin is disassembled into CdtB and PltA/pltB by a reductase. ⑦, PltA utilizes the ERAD pathway to translocate to the cytoplasm where it plays its ADP-ribosylation enzyme function. ⑧, CdtB utilizes an ERAD or ERAD-related pathway to leave the ER and ultimately be transported to the nucleus, where CdtB plays its genotoxic activity. Created with BioRender.Com.
Typhoid toxin trafficking pathway: Receptor-binding
Typhoid toxin preferentially binds to multiantennary N-linked glycoproteins as higher affinity binders, which is the basis for the tropism of typhoid toxin to certain target host cells in the infection site and located distantly from the primary infection site, as typhoid toxin circulates in the bloodstream and the lymphatic system [Citation70–72]. Typhoid toxin has tropism to a broad range of cells, including intestinal and gallbladder epithelial cells, immune cells, and brain endothelial cells, and intoxicates them, making this toxin enterotoxin, leukotoxin, and neurotoxin [Citation70,Citation72]. As we learned from typhoid toxin, AB toxin tropism is determined primarily by two factors: infection sites targeted by the cognate bacteria and toxin receptor expressions on mammalian cells [Citation70,Citation79]. Furthermore, the receptor-toxin interactions are affected by the numbers of receptor binding pockets on the B subunit monomer, B subunit stoichiometry of the assembled AB toxins, and asymmetry of the toxin’s 3D structure influenced by how their A subunit is linked to the B subunits, contributing to higher affinity multivalent bindings of AB toxins to their receptors on the target host cells [Citation70,Citation80].
The receptor-binding PltB pentamer of typhoid toxin has 15 glycan receptor binding pockets whose glycan preferences are similar yet different [Citation70]. The five pockets located on the lateral side of the pentamer preferentially bind to α2–3 and α2–6 sialosides displayed as part of N-linked glycoproteins with a higher affinity to α2–6 sialosides [Citation70]. The ten pockets located on the bottom of the pyramid-shaped typhoid toxin preferentially bind to α2–3 sialosides [Citation70]. 9-O-acetylation of the terminal sialic acids is known to further increase the affinity of PltB to glycans [Citation71]. It is worth noting that typhoid toxin orthologs encoded by some clade B nontyphoidal Salmonellae, collectively called Salmonella A2B5 toxins, contain the identical CdtB at the sequence level, while their receptor-binding subunits have variations, which are associated with an altered tropism of toxin variant [Citation70]. For instance, Javiana toxin from nontyphoidal Salmonella Javiana targets intestinal epithelial cells [Citation70], which is in line with the coevolution concept for niche-specific adaptations of AB toxins and the cognate bacteria [Citation81].
Typhoid toxin trafficking pathway: Retrograde endocytosis & nuclear localization
The binding of PltB to its glycan receptor initiates an extended endocytosis process of typhoid toxin involving the endosomes, the TGN, and the ER [Citation66] (). In particular, through a genome-wide CRISPR/Cas9-mediated screen, Chang et al demonstrated that typhoid toxin utilizes components of the retrograde transport cellular machinery to arrive in the ER, from where it is transported to the cell cytoplasm by the ERAD pathway [Citation66]. In particular, this screen identified several genes encoding components involved in the retrograde transport, including the Golgi-associated retrograde protein complex (GARP, Arl1, and VPS51–54), the conserved oligomeric Golgi (COG1–8) complex, TMED2, COP1, COP2 and various components of the core ERAD pathway. The two A subunits, CdtB and PltA, are tethered by a disulphide bond that is reduced by ER-resident reductases before their translocation from the ER to the cell cytoplasm, where PltA functions in host cells. CdtB further traffics from the cytoplasm to the nucleus presumably by a concerted mechanism involving its nuclear localization sequence and host proteins controlling nuclear trafficking of host proteins. In support, typhoid toxin CdtB contains an NLS-like sequence homologous to the NLS of AcCdtB.
Colibactin
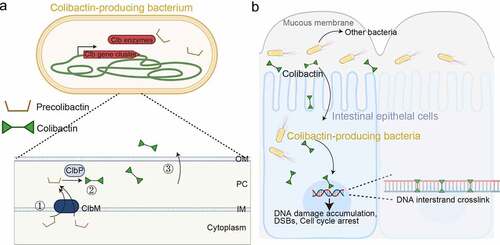
Colibactins are small secondary peptide metabolites synthesized by the actions of an enzyme complex consisting of a total of eight nonribosomal peptide synthetases and polyketide synthases (NRPSs-PKSs) [Citation6,Citation82] (). Colibactins induce DNA interstrand cross-linking and subsequently double-strand breaks (DSB), chromosome aberrations, and cell cycle arrests which primarily happen in G2/M but in some cases in S or G1 phase [Citation83,Citation84]. E. coli is the primary bacterial species carrying the 54 kb, pks genomic island encoding a total of 19 genes, clbA to clbS. These 19 genes encode for three NRPS (ClbH, ClbJ, and ClbN), three PKS (ClbC, ClbI, and ClbO), two of the hybrid NRPS-PKS megasynthases (ClbB and ClbK), and nine other enzymes for tailoring and editing [Citation6,Citation82]. A systemic mutagenesis study on the pks genes revealed that all NRPS-PKS enzymes and eight tailoring and editing enzymes are required for the synthesis of colibactins. The pks island was first identified in the E. coli strain isolated from neonatal bacterial meningitis, but such E. coli strains are found in gut microbiota, other infectious disease cases (e.g. septicaemia and urinary tract infections), and colorectal cancer cases [Citation85]. In addition, the majority of hypervirulent Klebsiella pneumonia (over 55%) [Citation86], as well as other members of the Enterobacteriaceae, such as Klebsiella aerogenes (Enterobacter aerogenes) and Citrobacter koseri are also known to encode the pks island.
Figure 5. Colibactin intoxication processes. (a) a diagram depicting the biogenesis of colibactin. Colibactins are peptide-polyketide metabolites synthesized by enzymes encoded in the colibactin (Clb) gene cluster. Through a coordinated action by Clb enzymes, precolibactin is synthesized in the bacterial cytoplasm. The lower panel indicates key steps in the synthesis of colibactin in the periplasm: ①, Precolibactins synthesized in the bacterial cytoplasm are inactive, and are transported into the periplasm through the ClbM transporter. ②, ClbP in the periplasm cuts the prodrug motif of precolibactin, resulting in the active colibactin. ③, Colibactins are secreted from the bacteria through a direct membrane diffusion or yet-to-be-identified mechanism. (b) Schematic diagram depicting an intoxication process of colibactin. It is still unclear, but two mechanisms are proposed: (1) colibactin produced extracellularly is diffused to enter host cells and (2) intracellular bacteria produce colibactin in the host cell cytoplasm. Created with BioRender.Com.
Procolibactin, a non-toxic precursor of colibactin, is made in the bacterial cytoplasm, which is transformed to genotoxic colibactin in the PC by the action of ClbP, a process that prevents self-DNA damage in colibactin-producing bacteria [Citation87,Citation88]. Precolibactin has the structure of colibactin but it has the N-myristoyl-D-Asn prodrug side chain [Citation89–91] (). Precolibactins are transported into the PC, a process resulting in the removal of the N-myristoyl-D-Asn side chain by ClbP [Citation87,Citation88] (). In the PC, a spontaneous cyclization of precolibactin occurs due to the removal of the N-myristoyl-D-Asn side chain from the compound, resulting in an active genotoxin colibactin [Citation92,Citation93] ().
Figure 6. Colibactin biogenesis. The N-myristoyl-D-Asn prodrug side chains in the Precolibactin are indicated in orange, which is later removed by ClbP in the periplasm. Once the N-myristoyl-D-Asn prodrug side chain is removed, precolibactin is four-fold cyclized to form the colibactin structure. The unstable α-aminoketone group in precolibactin is indicated in red, which is oxidated and hydrolysed. The oxidated and hydrolysed form in the colibactin structure is indicated in green. The α-diketone group found in colibactin is also unstable and easily cleaved under a mildly nucleophilic condition. The electrophilic cyclopropane groups and the DNA alkylation reaction groups are labelled in dark blue and light blue, respectively. The macrocyclic and 5-hydroxy-oxazole groups are shown in the structure of colibactin 645.
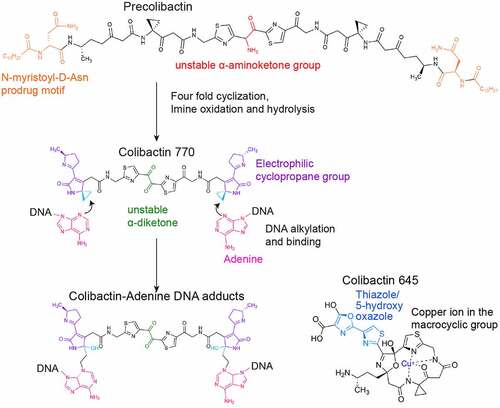
The translocation pathway of colibactin produced in the bacterial PC to the host cell nucleus is unclear (). However, previous studies indicated that a direct interaction between the cognate bacteria producing colibactin and host cells is linked to the efficient colibactin-mediated genotoxicity, which is supported by observations that the treatment of bacterial culture supernatants, cell lysates, or even the direct introduction of colibactin compounds to host cells did not cause host cell DNA damage [Citation6]. Consistently, previous studies revealed a rapid modification and/or decay of colibactin in many common environments that the bacteria encounter in the host [Citation94–96]. For instance, an α-aminoketone group found in colibactins is prone to be oxidated, hydrolysed, and spontaneously dissociated [Citation94] (). As such, colibactin is unstable in aerobic conditions, which is in line with the requirement for direct interaction between bacteria and host cells in such conditions [Citation6]. Given the largely anaerobic condition of the GI tract, this instability under aerobic conditions may not be a problem for colibactin-producing bacteria that inhabit the GI tract. Colibactins are poor in penetrating the mucin layer and the host cell membrane, which is another plausible explanation for the required direct surface interaction between the bacteria and host cells for colibactin-mediated genotoxicity [Citation6,Citation94,Citation97]. Furthermore, the inhibition of colibactin-producing bacterial invasion into host cells did not abolish its associated genotoxicity [Citation6]. Based on these results, we predict that bacterial invasion is not required, but those bacteria that can adhere to or invade host cells may cause more severe genotoxic damages due to their efficient toxin delivery mechanism to the target host cells. In support, E. coli B2 strains that produce colibactin that were isolated from colorectal cancer patients were able to induce carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) expression, which switches their poorly adherent phenotype to highly persistent colonization in the gut [Citation98].
In addition to its well-characterized trafficking to target host cell DNA, colibactin is able to traffic into other bacteria and trigger prophage induction, which required a direct interaction between colibactin-producing E. coli and prophage-containing bacteria, including E. coli, Salmonella, Staphylococcus, Citrobacter, and Enterococcus [Citation99].
Structure and function of bacterial genotoxins
CdtB
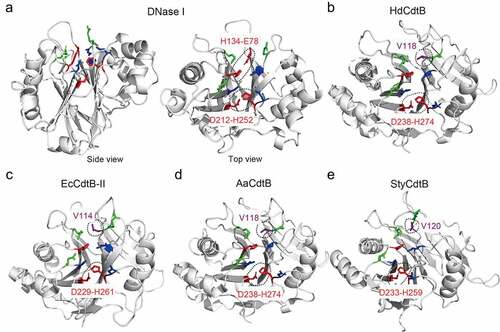
Four CdtB structures have been determined by X-ray crystallography: HdCdtB, EcCdtB, AaCdtB, and CdtB from S. Typhi typhoid toxin (StyCdtB), which share many structural features with metalloenzymes, such as DNase I and inositol polyphosphate-5-phosphatase (IP5P) [Citation5,Citation8,Citation58,Citation100–104] ( (a–)). The Root-Mean-Square Deviation (RMSD) value is 1.62 Å over 141 Cα atoms between bovine DNase I and HdCdtB, while 1.63 Å RMSD value over 121 Cα atoms is noted between IP5P and CdtB (reviewed in [Citation4]). CdtB appears to have both DNase I-like activity [Citation105–109] and IP5P activity [Citation110]. The nuclease activity of CdtB is particularly well characterized, which is approximately 100-fold weaker than the activity of human DNase I [Citation8], which is in line with the anticipated role of bacterial genotoxins in host cell manipulation to benefit the cognate pathogen during infection, rather than killing the host.
Figure 7. Key residues of DNaseI, HdCdtB, EcCdtB, AaCdtB, and StyCdtB. Structures of bovine DNase I (PDB: 2DNJ) (a), HdCdtb (PDB: 1SR4) (b), EcCdtb-II (PDB: 2F1N) (c), AaCdtb (PDB: 2F2F) (d), and StyCdtb (PDB: 4K6 L) (e). Catalytic (red), metal-binding (blue), and DNA binding (green) residues in their active site are indicated. Dotted circles are the catalytic histidine-partner amino acid hydrogen bonds, as well as the counterpart amino acid residues comparable to DNase I E78 that forms a hydrogen bond with H134. HdCdtb, Haemophilus ducreyi CdtB. EcCdtb. Escherichia coli CdtB. AaCdtb, Aggregatibacter actinomycetemcomitans. StyCdtb, Salmonella enterica subsp. enterica serovar Typhi CdtB. See also Supplemental . Figures are prepared using PyMOL. Bovine DNase I was used because of its structure complexed with DNA (PDB: 2DNJ).
CdtB vs. DNase I: Catalytic, metal binding, and DNA binding residues
Both CdtB and DNase I cut DNA, but the DNA nicking efficiency of CdtB is lower, which appears to be primarily due to differences in catalytic residue numbers and their associated hydrogen bonds between the two. Bovine DNase I has two conserved catalytic histidine residues H134 and H252 that form hydrogen bonds with residues E78 and D212, respectively [Citation103,Citation111] (). DNase IE78V introduced to disturb the hydrogen bond with H134 resulted in a decrease in DNase I activity, which was comparable to the CdtB-mediated activity [Citation111,Citation112]. E78 V was used to resemble HdCdtB, as CdtBH160 and CdtBV118 are located in the comparable positions to DNase IH134 and DNase IE78 within the catalytic sites but they do not form a hydrogen bond in CdtB ( (a–)).
Consistent with the importance of the catalytic histidine residue forming a hydrogen bond with its pair in the active site, EcCdtB-IID229A prevented the formation of a hydrogen bond with its catalytic residue H261. EcCdtB-II H261 and D229 are equivalent to the H252 and D212 interaction in DNase I (). EcCdtB-IID229A showed a decrease in the plasmid cleavage efficiency and abolished its ability to cause cell cycle arrests in G2/M [Citation111]. Similarly, CjCdtBD222A decreased its ability to nick the yeast chromosome and failed to induce a DNA damage repair response in both yeast and human foetal fibroblasts [Citation113,Citation114] (). These results are in line with the concept that catalytic histidine residues and their associated hydrogen bonds are indeed important for the DNA nicking activity and strength/efficiency ( (a–)).
As members of the Mg2+ metalloenzyme family, both CdtB and DNase I require Mg2+ for their enzyme activity. DNase I has three predicted metal-binding residues, E39, D168, and D251 [Citation111]. Matching residues are also found in CdtB sequences. The importance of Mg2+ for CdtB activity was demonstrated in AaCdtB [Citation110], CjCdtB [Citation2], EcCDT-II [Citation111,Citation115], HdCDT [Citation116,Citation117], StyCDT [Citation74] through in vivo chromatin condensation and/or in vitro plasmid degradation assays.
DNase I has three DNA binding residues, R41, R111, and N170 [Citation8,Citation58]. These residues are comparable to R117, R144, and N201 in HdCdtB. A single substitution of any of these three residues to alanine abolished CdtB activity, as demonstrated by the activity loss in the G2/M cell cycle. Residues critical for catalytic, metal binding, and DNA binding are conserved across CdtB (Supplemental Figure S1).
Colibactin
The genotoxicity of colibactin relies on the two electrophilic cyclopropane groups found on each side of the colibactin structure (). This electrophilic group makes a covalent interaction with adenine through DNA alkylation, resulting in interstrand crosslinked DNAs [Citation92,Citation96,Citation118]. Interstrand crosslinked (ICL) DNAs are also supported by colocalizations of the ICL DNA damage repair protein FANCD2 with phosphorylated γH2AX in colibactin-intoxicated Hela cells [Citation119]. ICL DNAs subsequently result in DSBs, which can occur during the ICL repair process and/or through spontaneous hydrolysis [Citation120,Citation121]. Consistent with colibactin-mediated ICL DNAs primarily through DNA alkylation with adenine, a recent study that determined colibactin-mediated DNA mutational signatures of human intestinal organoids treated for five months with colibactin-producing E. coli revealed its skewed “T to N” single base substitutions [Citation122]. These results also support the role of the ICL-associated nucleotide excision repair in the colibactin-induced accumulation of DNA mutations [Citation122].
Similar to CdtB-mediated genotoxicity, colibactins cause DNA damage in a dose-dependent manner. Colibactin produced after a low multiplicity of bacterial infection (m.o.i.) resulted in host cell DNA repair responses in intoxicated cells that still duplicated and passed the impaired DNAs to their daughter cells (e.g. anaphase bridging, micronuclei), while colibactin that was produced after a high m.o.i. infection caused cell senescence with clear signs of DSB and the distended nucleus [Citation10]. Instead of the electrophilic cyclopropane group, colibactin-mediated DSB formation relies on the macrocyclic and 5-hydroxy-oxazole groups [Citation83,Citation84] (). Colibactin-producing bacteria are associated with cancer due to the inheritable nature of damaged DNAs [Citation98,Citation123]. It is worthwhile noting that colibactin is produced not only by bacteria classified as pathogens but also by ones classified as beneficial bacteria/microbiome, which includes E. coli Nissle 1917 that is currently being used as a probiotic in human medicine in certain countries [Citation6,Citation99,Citation122,Citation124–126]; additional precautions should be taken particularly for prolonged treatments of such probiotics.
Concluding remarks
Genotoxins are the growing members of bacterial toxins, which cause DNA damage, nucleus distension, cell cycle arrests in G2/M, apoptosis, or senescence. When the damages are not too severe, the altered genome can be inherited in their daughter cells, which correlates with clinical manifestations associated with the cognate bacterial infection and, in some cases, cancer. The biogenesis, delivery, structure, and function of most well-characterized bacterial genotoxins are discussed in this review. Less well-known genotoxin examples include uropathogenic specific protein (Usp) found in phylogenetic E. coli group B2, Shigella VirA, and E. coli EspF. The Usp exerts its genotoxic activity through the C-terminal region that contains a DNase-like domain, which induces DSBs in HUVECs or HEK293 cell lines as demonstrated via comet assays [Citation127]. The Usp genotoxin is encoded by E. coli strains that are associated with prostatitis, pyelonephritis, bacteraemia, and colorectal cancer [Citation127–130]. Intriguingly, Usp-producing bacteria encode the associated antitoxin genes, imu1-3, which are required for self-protection from the Usp genotoxin [Citation131]. Shigella VirA and E. coli EspF also have genotoxicities, which intervene in the normal DNA repair activity in host cells. For instance, Shigella VirA promotes the degradation of p53 [Citation132], while E.coli strains isolated from colorectal cancer patients produce Type III secretion system effector EspF that targets the DNA mismatch repair enzyme [Citation133,Citation134], which as a result play genotoxin-like functions. Discovering additional members/homologs in these bacterial genotoxin family are perhaps a matter of further investigation. Nonetheless, one common feature across bacterial protein genotoxins appears to be causing milder DNA damage, compared to mammalian DNase I, critical for maintaining host cells survived and proliferating, thus benefiting the cognate bacteria. In contrast, colibactin, the small polyketide-peptide metabolite, has been suggested to induce cell senescence in intestinal epithelial cells, indicating higher level of DNA damage. The next chapter in the study of bacterial genotoxins would be using current knowledge for identifying and characterizing genotoxins in detail, which will offer critical insights into intervention strategies to prevent genotoxin-mediated diseases.
Supplemental Material
Download MS Word (24.3 KB)Acknowledgments
We acknowledge that we could not include and cite many other important papers in this article due to space constraints.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data that support this review article are available upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2097417.
Additional information
Funding
References
- Guerra L, Cortes-Bratti X, Guidi R, et al. The biology of the cytolethal distending toxins. Toxins (Basel). 2011;3:172–190.
- Lara-Tejero M, Galán JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290(5490):354–357.
- Lara-Tejero M, Galán JE. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002;10(3):147–152.
- Scuron MD, Boesze-Battaglia K, Dlakic M, et al. The cytolethal distending toxin contributes to microbial virulence and disease pathogenesis by acting as a tri-perditious toxin. Front Cell Infect Mi. 2016;6. DOI:10.3389/fcimb.2016.00168
- Song J, Gao X, Galan JE. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature. 2013;499(7458):350–354.
- Nougayrède JP, Homburg S, Taieb F, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–851.
- Klem F, Wadhwa A, Prokop LJ, et al. Prevalence, risk factors, and outcomes of irritable bowel syndrome after infectious enteritis: a systematic review and meta-analysis. Gastroenterology. 2017;152:1042–1054, e1041.
- Nesić D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433.
- Stanaway JD RR, Blacker BF, Goldberg EM, et al. The global burden of typhoid and paratyphoid fevers: a systematic analysis for the global burden of disease study 2017. Lancet Infect Dis. 2019;19(4):369–381.
- Cuevas-Ramos G, Petit CR, Marcq I, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci, USA. 2010;107(25):11537–11542.
- Johnson WM, Lior H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb Pathog. 1988a;4(2):115–126.
- Johnson WM, Lior H. A new heat-labile cytolethal distending toxin (CLDT) produced by Escherichia coli isolates from clinical material. Microb Pathog. 1988b;4(2):103–113.
- Lai CK, Chen YA, Lin CJ, et al. Molecular mechanisms and potential clinical applications of campylobacter jejuni cytolethal distending toxin. Front Cell Infect Microbiol. 2016;6:9. DOI:10.3389/fcimb.2016.00009
- Mortensen NP, Schiellerup P, Boisen N, et al. The role of Campylobacter jejuni cytolethal distending toxin in gastroenteritis: toxin detection, antibody production, and clinical outcome. Apmis. 2011;119:626–634.
- Tegtmeyer N, Sharafutdinov I, Harrer A, et al. Campylobacter virulence factors and molecular host-pathogen interactions. Curr Top Microbiol Immunol. 2021;431:169–202.
- Pickett CL, Pesci EC, Cottle DL, et al. Prevalence of cytolethal distending toxin production in Campylobacter jejuni and relatedness of Campylobacter sp. cdtB gene. Infect Immun. 1996;64(6):2070–2078.
- Fox JG, Rogers AB, Whary MT, et al. Gastroenteritis in NF-κB-deficient mice is produced with wild-type campylobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect Immun. 2004;72(2):1116–1125.
- Jain D, Prasad KN, Sinha S, et al. Differences in virulence attributes between cytolethal distending toxin positive and negative Campylobacter jejuni strains. J Med Microbiol. 2008;57(Pt 3):267–272.
- Purdy D, Buswell CM, Hodgson AE, et al. Characterisation of cytolethal distending toxin (CDT) mutants of Campylobacter jejuni. J Med Microbiol. 2000;49(5):473–479.
- Hinenoya A, Naigita A, Ninomiya K, et al. Prevalence and characteristics of cytolethal distending toxin-producing Escherichia coli from children with diarrhea in Japan. Microbiol Immunol. 2009;53(4):206–215.
- Meza-Segura M, Zaidi MB, Maldonado-Puga S, et al. Cytolethal distending toxin-producing Escherichia coli strains causing severe diarrhoea in young Mexican children. JMM Case Rep. 2017;4(2):e005079.
- Scorza FB, Doro F, Rodriguez-Ortega MJ, et al. Proteomics characterization of outer membrane vesicles from the extraintestinal pathogenic Escherichia coli δtolr IHE3034 mutant. Mol Cell Proteomics. 2008;7(3):473–485. DOI:10.1074/mcp.M700295-MCP200
- Taieb F, Svab D, Watrin C, et al. Cytolethal distending toxin A, B and C subunit proteins are necessary for the genotoxic effect of Escherichia coli CDT-V. Acta Vet Hung. 2015;63(1):1–10.
- Toth I, Herault F, Beutin L, et al. Production of cytolethal distending toxins by pathogenic Escherichia coli Strains isolated from human and animal sources: establishment of the existence of a new cdt variant (type IV). J Clin Microbiol. 2003;41(9):4285–4291.
- Johnson TJ, Kariyawasam S, Wannemuehler Y, et al. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J Bacteriol. 2007;189(8):3228–3236.
- Peres SY, Marches O, Daigle F, et al. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol Microbiol. 1997;24(5):1095–1107.
- Svab D, Horvath B, Maroti G, et al. Sequence variability of P2-like prophage genomes carrying the cytolethal distending toxin V operon in Escherichia coli O157. Appl Environ Microbiol. 2013;79(16):4958–4964.
- Toth I, Nougayrede JP, Dobrindt U, et al. cytolethal distending toxin type i and type iv genes are framed with lambdoid prophage genes in extraintestinal pathogenic Escherichia coli. Infect Immun. 2009;77(1):492–500.
- de Paula Coelho C, Motta PD, Petrillo M, et al. Homeopathic medicine Cantharis modulates uropathogenic E. coli (UPEC)-induced cystitis in susceptible mice. Cytokine. 2017;92:103–109.
- Ahmed HJ, Svensson LA, Cope LD, et al. Prevalence of cdtABC genes encoding cytolethal distending toxin among Haemophilus ducreyi and Actinobacillus actinomycetemcomitans strains. J Med Microbiol. 2001;50(10):860–864.
- Lewis DA, Stevens MK, Latimer JL, et al. Characterization of Haemophilus ducreyi cdtA, cdtB , and cdtC Mutants in in vitro and in vivo systems. Infect Immun. 2001;69(9):5626–5634.
- Kaplan AH, Weber DJ, Oddone EZ, et al. Infection due to Actinobacillus actinomycetemcomitans: 15 cases and review. Rev Infect Dis. 1989;11(1):46–63.
- Mayer MP, Bueno LC, Hansen EJ, et al. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. 1999;67(3):1227–1237.
- Herbert BA, Novince CM, Kirkwood KL. Aggregatibacter actinomycetemcomitans , a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol Oral Microbiol. 2016;31(3):207–227.
- Ohara M, Miyauchi M, Tsuruda K, et al. Topical application of Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces cell cycle arrest in the rat gingival epithelium in vivo. J Periodontal Res. 2011;46(3):389–395.
- Akifusa S, Poole S, Lewthwaite J, et al. Recombinant actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect Immun. 2001;69(9):5925–5930.
- Ando-Suguimoto ES, da Silva MP, Kawamoto D, et al. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine. 2014;66(1):46–53.
- Belibasakis GN, Johansson A. Aggregatibacter actinomycetemcomitans targets NLRP3 and NLRP6 inflammasome expression in human mononuclear leukocytes. Cytokine. 2012;59:124–130.
- Belibasakis GN, Johansson A, Wang Y, et al. Cytokine responses of human gingival fibroblasts to Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cytokine. 2005;30:56–63.
- Shenker BJ, Boesze-Battaglia K, Scuron MD, et al. The toxicity of the Aggregatibacter actinomycetemcomitans cytolethal distending toxin correlates with its phosphatidylinositol-3,4,5-triphosphate phosphatase activity. Cell Microbiol. 2016a;18(2):223–243.
- Shenker BJ, Walker LP, Zekavat A, et al. Lymphoid susceptibility to the Aggregatibacter actinomycetemcomitans cytolethal distending toxin is dependent upon baseline levels of the signaling lipid, phosphatidylinositol-3,4,5-triphosphate. Mol Oral Microbiol. 2016b;31(31):33–42.
- Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun. 1994;62(1):244–251.
- Tóth I, Hérault F, Beutin L, et al. Production of cytolethal distending toxins by pathogenic Escherichia coli strains isolated from human and animal sources: establishment of the existence of a new cdt variant (type iV). J Clin Microbiol. 2003;41(9):4285–4291.
- Tóth I, Nougayrède JP, Dobrindt U, et al. Cytolethal distending toxin type i and type iv genes are framed with lambdoid prophage genes in extraintestinal pathogenic Escherichia coli. Infect Immun. 2009;77(1):492–500.
- Wilson BA, Winkler ME, Ho BT. Bacterial pathogenesis: a molecular approach. 4th ed. Washington (DC): ASM Press(Chapter 12); 2019a.
- Ueno Y, Ohara M, Kawamoto T, et al. Biogenesis of the Actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect Immun. 2006;74(6):3480–3487.
- Tsuruda K, Matangkasombut O, Ohara M, et al. CdtC-Induced processing of membrane-bound CdtA is a crucial step in aggregatibacter actinomycetemcomitans cytolethal distending toxin holotoxin formation. Infect Immun. 2018;86(3). DOI:10.1128/IAI.00731-17
- Buddelmeijer N. The molecular mechanism of bacterial lipoprotein modification—how, when and why? FEMS Microbiol Rev. 2015;39(2):246–261.
- Davies C, Taylor AJ, Elmi A, et al. Sodium taurocholate stimulates campylobacter jejuni outer membrane vesicle production via down-regulation of the maintenance of lipid asymmetry pathway. Front Cell Infect Microbiol. 2019;9:177.
- Cao L, Volgina A, Huang CM, et al. Characterization of point mutations in the cdtA gene of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Mol Microbiol. 2005;58(5):1303–1321.
- McSweeney LA, Dreyfus LA. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect Immun. 2005;73(4):2051–2060.
- Mise K, Akifusa S, Watarai S, et al. Involvement of ganglioside GM3 in G 2 /M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect Immun. 2005;73(8):4846–4852.
- Guerra L, Teter K, Lilley BN, et al. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell Microbiol. 2005;7(7):921–934.
- Eshraghi A, Dixon SD, Tamilselvam B, et al. Cytolethal distending toxins require components of the ER-associated degradation pathway for host cell entry. PLoS Pathog. 2014;10(7):e1004295.
- Nishikubo S, Ohara M, Ueno Y, et al. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J Biol Chem. 2003;278:50671–50681.
- Eshraghi A, Maldonado-Arocho FJ, Gargi A, et al. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J Biol Chem. 2010;285:18199–18207.
- Frisan T. Bacterial genotoxins: the long journey to the nucleus of mammalian cells. Biochim Biophys Acta. 2016;1858(3):567–575.
- Hu X, Nesic D, Stebbins CE. Comparative structure-function analysis of cytolethal distending toxins. Proteins. 2006;62(2):421–434.
- Boesze-Battaglia K, Besack D, McKay T, et al. Cholesterol-Rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell Microbiol. 2006;8(5):823–836.
- Lin CD, Lai CK, Lin YH, et al. Cholesterol depletion reduces entry of Campylobacter jejuni cytolethal distending toxin and attenuates intoxication of host cells. Infect Immun. 2011;79(9):3563–3575.
- Carette JE, Guimaraes CP, Wuethrich I, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol. 2011;29(6):542–546. DOI:10.1038/nbt.1857
- Boesze-Battaglia K, Brown A, Walker L, et al. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J Biol Chem. 2009;284:10650–10658.
- Damek-Poprawa M, Jang JY, Volgina A, et al. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect Immun. 2012;80(8):2761–2770.
- Robb Huhn G 3rd, Torres-Mangual N, Clore J, et al. Endocytosis of the CdtA subunit from the Haemophilus ducreyi cytolethal distending toxin. Cell Microbiol. 2021;23(11):e13380.
- Gargi A, Tamilselvam B, Powers B, et al. Cellular interactions of the cytolethal distending toxins from Escherichia coli and Haemophilus ducreyi. J Biol Chem. 2013;288(11):7492–7505.
- Chang SJ, Jin SC, Jiao X, et al. Unique features in the intracellular transport of typhoid toxin revealed by a genome-wide screen. PLoS Pathog. 2019;15(4):e1007704.
- Guerra L, Nemec KN, Massey S, et al. A novel mode of translocation for cytolethal distending toxin. Biochim Biophys Acta. 2009;1793(3):489–495.
- McSweeney LA, Dreyfus LA. Nuclear localization of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell Microbiol. 2004;6:447–458.
- Spanò S, Ugalde JE, Galán JE. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe. 2008;3(1):30–38.
- Lee S, Yang YA, Milano SK, et al. Salmonella typhoid toxin PltB subunit and its non-typhoidal salmonella ortholog confer differential host adaptation and virulence. Cell Host Microbe. 2020;27(6):937–949. DOI:10.1016/j.chom.2020.04.005
- Nguyen T, Lee S, Yang YA, et al. The role of 9-O-acetylated glycan receptor moieties in the typhoid toxin binding and intoxication. PLoS Pathog. 2020;16(2):e1008336. DOI:10.1371/journal.ppat.1008336
- Yang YA, Lee S, Zhao J, et al. In vivo tropism of Salmonella Typhi toxin to cells expressing a multiantennal glycan receptor. Nat Microbiol. 2018;3(2):155–163.
- Bhutta ZA, Gaffey MF, Crump JA, et al. Typhoid fever: way forward. Am J Trop Med Hyg. 2018;99(3_Suppl):89–96.
- Ahn C, Yang Y-A, Neupane DP, et al. Mechanisms of typhoid toxin neutralization by antibodies targeting glycan receptor binding and nuclease subunits. iScience. 2021;24:102454.
- Haghjoo E, Galán JE. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc Natl Acad Sci, USA. 2004;101(13):4614–4619.
- Fowler CC, Stack G, Jiao X, et al. Alternate subunit assembly diversifies the function of a bacterial toxin. Nat Commun. 2019;10(1):3684.
- Geiger T, Lara-Tejero M, and Xiong Y, et al. Mechanisms of substrate recognition by a typhoid toxin secretion-associated muramidase. Elife. 2020;9:e53473. doi:10.7554/eLife.53473.
- Chang SJ, Song J, Galan JE. Receptor-mediated sorting of typhoid toxin during its export from salmonella typhi-infected cells. Cell Host Microbe. 2016;20(5):682–689.
- Petersen E, Miller SI. Toxin glycan binding: lectin keys unlocking host and tissue specificity. Cell Host Microbe. 2020;27(6):851–853.
- Nguyen T, Richards AF, Neupane DP, et al. The structural basis of Salmonella A(2)B(5) toxin neutralization by antibodies targeting the glycan-receptor binding subunits. Cell Rep. 2021;36(10):109654. DOI:10.1016/j.celrep.2021.109654
- Lee S, Inzerillo S, Lee GY, et al. Glycan-Mediated molecular interactions in bacterial pathogenesis. Trends Microbiol. 2021;30(3):254–267.
- Putze J, Hennequin C, Nougayrède JP, et al. Genetic structure and distribution of the colibactin genomic island among members of the family enterobacteriaceae. Infect Immun. 2009;77(11):4696–4703. DOI:10.1128/IAI.00522-09
- Colis LC, Woo CM, Hegan DC, et al. The cytotoxicity of (−)-lomaiviticin a arises from induction of double-strand breaks in DNA. Nat Chem. 2014;6(6):504–510.
- Woo CM, Li Z, Paulson EK, et al. Structural basis for DNA cleavage by the potent antiproliferative agent (–)-lomaiviticin a. Proc Natl Acad Sci, USA. 2016;113(11):2851–2856.
- Faïs T, Delmas J, Barnich N, et al. Colibactin: more than a new bacterial toxin. Toxins (Basel). 2018;10(4):151.
- Lan Y, Zhou M, Jian Z, et al. Prevalence of pks gene cluster and characteristics of Klebsiella pneumoniae -induced bloodstream infections. J Clin Lab Anal. 2019;33(4):e22838.
- Cougnoux A, Gibold L, Robin F, et al. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J Mol Biol. 2012;424(3–4):203–214.
- Dubois D, Baron O, Cougnoux A, et al. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J Biol Chem. 2011;286(41):35562–35570.
- Bian X, Fu J, Plaza A, et al. In vivo evidence for a prodrug activation mechanism during colibactin maturation. Chembiochem. 2013;14:1194–1197.
- Brotherton CA, Balskus EP. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J Am Chem Soc. 2013;135(9):3359–3362.
- Wernke KM, Xue M, Tirla A, et al. Structure and bioactivity of colibactin. Bioorg Med Chem Lett. 2020;30(15):127280.
- Healy AR, Nikolayevskiy H, Patel JR, et al. A mechanistic model for colibactin-induced genotoxicity. J Am Chem Soc. 2016;138(48):15563–15570.
- Trautman EP, Healy AR, Shine EE, et al. Domain-targeted metabolomics delineates the heterocycle assembly steps of colibactin biosynthesis. J Am Chem Soc. 2017;139(11):4195–4201.
- Healy AR, Wernke KM, Kim CS, et al. Synthesis and reactivity of precolibactin 886. Nat Chem. 2019;11(10):890–898.
- Taieb F, Petit C, Nougayrede JP, et al. The enterobacterial genotoxins: cytolethal distending toxin and colibactin. EcoSal Plus. 2016;7(1). DOI:10.1128/ecosalplus.ESP-0008-2016
- Wilson MR, Jiang YD, Villalta PW, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science. 2019b;363:709±. DOI:10.1126/science.aar7785
- Shine EE, Xue M, Patel JR, et al. Model colibactins exhibit human cell genotoxicity in the absence of host bacteria. ACS Chem Biol. 2018;13(12):3286–3293.
- Raisch J, Buc E, Bonnet M, et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 2014;20(21):6560–6572. DOI:10.3748/wjg.v20.i21.6560
- Silpe JE, Wong JWH, Owen SV, et al. The bacterial toxin colibactin triggers prophage induction. Nature. 2022;603(7900):315–320.
- Dlakic M. Functionally unrelated signalling proteins contain a fold similar to Mg2±dependent endonucleases. Trends Biochem Sci. 2000;25(6):272–273.
- Dlakic M. Is CdtB a nuclease or a phosphatase? Science. 2001;291(5504):547.
- Hontz JS, Villar-Lecumberri MT, Potter BM, et al. Differences in crystal and solution structures of the cytolethal distending toxin B subunit: relevance to nuclear translocation and functional activation. J Biol Chem. 2006;281(35):25365–25372.
- Lahm A, Suck D. Dnase I-induced DNA conformation. 2 a structure of a DNase I-octamer complex. J Mol Biol. 1991;222(3):645–667.
- Yamada T, Komoto J, Saiki K, et al. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci. 2006;15(2):362–372.
- Bezine E, Malaise Y, Loeuillet A, et al. Cell resistance to the cytolethal distending toxin involves an association of DNA repair mechanisms. Sci Rep. 2016;6(1):36022.
- Blazkova H, Krejcikova K, Moudry P, et al. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J Cell Mol Med. 2010;14(1–2):357–367.
- Fahrer J, Huelsenbeck J, Jaurich H, et al. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces, persistent levels of DNA.Double-strand breaks in human fibroblasts. DNA Repair (Amst). 2014;18:31–43.
- Fedor Y, Vignard J, Nicolau-Travers ML, et al. From single-strand breaks to double-strand breaks during S-phase: a new mode of action of the Escherichia coli cytolethal distending toxin. Cell Microbiol. 2013;15(1):1–15.
- Li L, Sharipo A, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell Microbiol. 2002;4(2):87–99.
- Shenker BJ, Dlakic M, Walker LP, et al. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J Immunol. 2007;178(8):5099–5108.
- Elwell CA, Dreyfus LA. Dnase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37(4):952–963.
- Pan CQ, Ulmer JS, Herzka A, et al. Mutational analysis of human DNase I at the DNA binding interface: implications for DNA recognition, catalysis, and metal ion dependence. Protein Sci. 1998;7(3):628–636.
- Hassane DC, Lee RB, Mendenhall MD, et al. Cytolethal distending toxin demonstrates genotoxic activity in a yeast model. Infect Immun. 2001;69(9):5752–5759.
- Hassane DC, Lee RB, Pickett CL. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect Immun. 2003;71(1):541–545.
- Elwell C, Chao K, Patel K, et al. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun. 2001;69(5):3418–3422.
- Frisan T, Nagy N, Chioureas D, et al. A bacterial genotoxin causes virus reactivation and genomic instability in Epstein-Barr virus infected epithelial cells pointing to a role of co-infection in viral oncogenesis. Int J Cancer. 2019;144(1):98–109.
- Seiwert N, Neitzel C, Stroh S, et al. AKT2 suppresses pro-survival autophagy triggered by DNA double-strand breaks in colorectal cancer cells. Cell Death Dis. 2017;8(8):e3019.
- Xue M, Shine E, Wang W, et al. Characterization of natural colibactin–nucleobase adducts by tandem mass spectrometry and isotopic labeling. support for DNA alkylation by cyclopropane ring opening. Biochemistry. 2018;57(45):6391–6394.
- Bossuet-Greif N, Vignard J, Taieb F, et al. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio. 2018;9(2). DOI:10.1128/mBio.02393-17
- Clauson C, Schärer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol. 2013;5(10):a012732.
- Xue M, Wernke KM, Herzon SB. Depurination of colibactin-derived interstrand cross-links. Biochemistry. 2020;59:892–900.
- Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature. 2020;580(7802):269–273. DOI:10.1038/s41586-020-2080-8
- Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–123. DOI:10.1126/science.1224820
- Nougayrède JP, Chagneau CV, Motta JP, et al. A toxic friend: genotoxic and mutagenic activity of the probiotic strain Escherichia coli Nissle 1917. mSphere. 2021;6:e0062421.
- Olier M, Marcq I, Salvador-Cartier C, et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes. 2012;3(6):501–509. DOI:10.4161/gmic.21737
- Tronnet S, Floch P, Lucarelli L, et al. The genotoxin colibactin shapes gut microbiota in mice. mSphere. 2020;5(4). DOI:10.1128/mSphere.00589-20
- Nipič D, Podlesek Z, Budič M, et al. Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J Infect Dis. 2013;208(10):1545–1552.
- Lloyd AL, Rasko DA, Mobley HL. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J Bacteriol. 2007;189(9):3532–3546.
- Roche-Lima A, Carrasquillo-Carrión K, Gómez-Moreno R, et al. The presence of genotoxic and/or pro-inflammatory bacterial genes in gut metagenomic databases and their possible link with inflammatory bowel diseases. Front Genet. 2018;9:116.
- Yamamoto S, Nakano M, Terai A, et al. The presence of the virulence island containing the usp gene in uropathogenic Escherichia coli is associated with urinary tract infection in an experimental mouse model. J Urol. 2001;165(4):1347–1351.
- Rihtar E, Zgur Bertok D, Podlesek Z. The uropathogenic specific protein gene usp from Escherichia coli and salmonella bongori is a novel member of the TyrR and H-NS regulons. Microorganisms. 2020;8(3):8.
- Bergounioux J, Elisee R, Prunier AL, et al. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium’s epithelial niche. Cell Host Microbe. 2012;11(3):240–252.
- Maddocks OD, Scanlon KM, Donnenberg MS. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. Mbio. 2013;4:e00152–00113. DOI:10.1128/mBio.00152-13.
- Maddocks OD, Short AJ, Donnenberg MS, et al. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One. 2009;4(5):e5517.