
ABSTRACT
Aeromonas caviae, an important food-borne pathogen, induces serious invasive infections and inflammation. The pro-inflammatory IL-1β functions against pathogenic infections and is elevated in various Aeromonas infection cases. However, the molecular mechanism of A. caviae-mediated IL-1β secretion remains unknown. In this study, mouse macrophages (PMs) were used to establish A. caviae infection model and multiple strategies were utilized to explore the mechanism of IL-1β secretion. IL-1β was elevated in A. caviae infected murine serum, PMs lysates or supernatants. This process triggered NLRP3 levels upregulation, ASC oligomerization, as well as dot gathering of NLRP3 and speck-like signals of ASC in the cytoplasm. MCC950 blocked A. caviae mediated IL-1β release. Meanwhile, NLRP3 inflammasome mediated the release of IL-1β in dose- and time-dependent manners, and the release of IL-1β was dependent on active caspase-1, as well as NLRP3 inflammasome was activated by potassium efflux and cathepsin B release ways. A. caviae also enhanced TLR2 levels, and deletion of TLR2 obviously decreased IL-1β secretion. What’s more, A. caviae resulted in NF-κB p65 nuclear translocation partly in a TLR2-dependent manner. Blocking NF-κB using BAY 11-7082 almost completely inhibited NLRP3 inflammasome first signal pro-IL-1β expression. Blocking TLR2, NF-κB, NLRP3 inflammasome significantly downregulated IL-1β release and TNF-α and IL-6 levels. These data illustrate that A. caviae caused IL-1β secretion in PMs is controlled by NLRP3 inflammasome, of which is mediated by NF-κB pathway and is partially dependent on TLR2, providing basis for drugs against A. caviae.
Introduction
Aeromonas caviae (A. caviae), an aero-anaerobic gram-negative bacteria, is classified into family Aeromonadaceae and genus Aeromonas [Citation1]. It is widely distributed, commonly found in fresh water and soil [Citation2]. As an emerging conditional pathogen to humans, A. caviae infection mainly occurs in elderly people or children and causes gastroenteritis, sepsis, meningitis, and wound infections [Citation3]. Statistically, among the Aeromonas spp. related clinical cases, 96.5% infections were triggered by A. caviae, A. hydrophila, A. veronii, A. dhakensis, and A. caviae infection accounts for the highest proportion of 29.9% [Citation1]. Antibiotic therapy strategy played a vital role against A. caviae infection [Citation4], however, the emergence of drug-resistant strains restricted treatment effects and such therapy would also bring serious side effects to patients [Citation5]. Therefore, the search for effective treat and control of the current A. caviae infection situation is needed to be investigated.
Studies so far revealed that the infection with A. hydrophila [Citation6], A. veronii [Citation7], A. sobria [Citation8], A. salmonicida [Citation9] and A. dhakensis [Citation10] would induce a series of inflammatory response with elevated IL-1β, TNF-α, IL-6, IL-8 and so on. IL-1β was a potent cytokine that functioned in host-response and the resistance to pathogens [Citation11]. Streptococcus pneumoniae infection induced the meningitis in mice with increased IL-1β levels [Citation12]. The release of IL-1β also participated the Staphylococcus aureus induced sepsis on mice model [Citation13]. Once the innate immunity system sensed the invading pathogenic bacteria through pattern-recognition receptors (PRRs), pro-inflammatory cytokines of IL-1β would be secreted to regulate the inflammatory response and recruit immune cells [Citation14]. Macrophages played a vital role in the resistance to the pathogenic infection [Citation15]. A. dhakensis infection enhanced pro-IL-1β transcription in the spleen as well as head kidney of South American fish Piaractus mesopotamicus [Citation10]. A. hydrophila induced IL-1β release and disturbed head kidney macrophages mitochondrial dysfunction and apoptosis [Citation16]. Moreover, IL-1β would promote TNF-α, IL-6 and IL-12 secretion to aggravate inflammation and cell death in mice macrophages infected with Vibrio alginolyticus and Vibrio harveyi [Citation17,Citation18]. However, whether A. caviae infection induces IL-1β release and the underlying molecular mechanism is needed to be further explored. The secretion of IL-1β is controlled by two signal pathways. Stimulants of flagellin, profilin, lipoprotein activates TLRs, transmits the signal to activate NF-κB, and subsequently accelerates the transcription level of pro-IL-1β as the first signal. The activation of inflammasomes within cytoplasm will recruit and cleave the pro-caspase-1 to matured caspase-1 form. Mature caspase-1 will cleave the pro-IL-1β to IL-1β p17 and serves as the second signal. Previous study reported that A. sobria led to NLRP3 inflammasome assembling and IL-1β enhancement in PMs [Citation8]. IL-1β and NLRP3 inflammasome were weakened in Hepatitis B virus infection through NF-κB and ROS in Kupffer cells [Citation19]. A. veronii-induced IL-1β release was mediated by the activation of NLRP3 and NLRC4 inflammasomes in bone marrow-derived macrophages [Citation7]. TLR2 and NLRP3 participated in regulating the maturation and secretion of IL-1β induced by Prevotella nigrescens-infected dendritic cells [Citation20]. Based on the known complex regulation ways of IL-1β secretion, it should be determined which kind of TLRs and NLRs may be involved in A. caviae infection and what’s the key inflammasome functioning in IL-1β release.
The IL-1β secretion during Aeromonas spp. infection is well studied in A. hydrophila [Citation6], A. veronii [Citation7] and A. sobria [Citation8], and less reports focused on the IL-1β secretion in A. caviae. Macrophages played vital roles in host innate immunity in response to pathogens invasion. Once bacteria invading into organisms, the chemokines would induce the massive recruitment of macrophages at the region of infection, then the macrophages would function in bacterial clearance and caused inflammation [Citation21,Citation22]. Therefore, we established A. caviae infection model based on mice peritoneal macrophages (PMs) and explored the IL-1β secretion mechanism. In our study, we analysed the IL-1β levels in murine serum, as well as the lysates and supernatants of PMs stimulated with A. caviae. Then, we screened the NLRs family, determined the activated inflammasome, and explored its effects on IL-1β. Meanwhile, we also measured the activated TLRs and explored its roles in IL-1β production. The NF-κB signal was investigated and further verified whether its activation was dependent on the key TLR and regulated pro-IL-1β. Finally, the roles of key TLR, NF-κB, and NLR were assessed in regulating A. caviae infection induced inflammatory response in PMs, intending to elucidate the molecular mechanism of IL-1β secretion in A. caviae infection.
Materials and methods
Bacteria strains, chemicals, antibodies, and kits
Aeromonas caviae (ATCC 15,468) was kept in our lab. The V X765 (specific caspase-1 inhibitor), CA074-Me (specific cathepsin B inhibitor) and BAY11–7082 (specific NF-κB inhibitor) were all obtained from Selleck (Shanghai). The Glyburide (specific K+ efflux inhibitor) and MCC950 (effective selective inhibitor of NLRP3) were aquired from MedChemExpress (Monmouth Junction). The gentamicin was obtained from Biological Industries (Israel). Antibodies of NLRP3 and caspase-1 were from Adipogen (San Diego). IL-1β was from R&D (Minneapolis). ASC was from Proteintech (Wuhan). The uncoated commercial kits for measuring mouse TNF-α, IL-6 and IL-1β were from Invitrogen (California).
Mice and cell culture
The female wild type (WT) C57BL/6 mice, TLR2-deficient (TLR2−/−) mice, NLRP3-deficient (NLRP3−/−) mice, caspase-1/11-deicient (caspase-1/11−/−) mice were kept in our lab, housed in the pathogen-free equipment and fed with sterile diet. The mouse peritoneal macrophages (PMs) were prepared referring to previous methods [Citation17,Citation18]. The cell purity was analysed by flow cytometry using CD11b (BioLegend).
Mice infection
Ten WT mice were randomized into control group and A. caviae infection group. When the OD600 nm value reached to 0.6, the bacterial suspension was collected by centrifugation and resuspended with sterile PBS. Mice were infected with A. cavaie by gavage at a dose of 8 × 109 CFU/mice once a week for 2 weeks and the control group was given the same volume of sterile PBS [Citation23,Citation24]. They were euthanized on day 14. Blood was harvested from the retro-orbital sinus and serum was collected for ELISA analysis.
Stimulation of macrophages with A. caviae
A. caviae was cultured in the Nutrient agar for 4–5 h to reach the logarithmic growth stage. The bacterial solution was collected and washed with sterile PBS. After three times, bacterial precipitation was resuspended with RPMI 1640 containing 1% FBS. Prior to infection, the number of bacteria was count by microscope. Then, the resuspended bacterial solution was inoculated into the macrophages at different multiples of infection (MOI) and incubated for 90 min. Then, cells were treated with a high concentration of gentamicin (150 μg/mL) for 90 min to remove the remaining A. caviae, and then incubation was continued with a low concentration of gentamicin (20 μg/mL) for 12 h. To detect the protein expression levels of p-p65 and p-IKBα, the cells were co-cultured with A. caviae at a MOI of 40:1 for 4 h and infected cells were collected for immunological detection.
Quantitative real-time PCR (qPRC)
RNA in A. caviae infected cells was obtained using Trizol (Monad, Wuhan) and cDNA was prepared using the reverse transcription kit (Monad). The qPCR assays were performed on an Stepone Plus machine (ThermoFisher, Waltham). The reaction program and system were set according to the instruction manual operation steps of SYBR Green qPCR reagent (Monad). The mRNA fold change was calculated by the 2−ΔΔCT method. The primers were shown in supplementary Table 1.
Western blot analysis
After infection with A. caviae for 12 h or 4 h, the cell precipitation was lysed with a mix containing 40 µL of RIPA lysis buffer, 0.4 µL of protease inhibitor 100 × PMSF (Solarbio, Beijing), and 0.4 µL of 100 × phosphatase inhibitor (Sangon Biotech, Shanghai). The protein concentration was assessed by the BCA method. The protein samples from cell culture supernatant was extracted using chloroform and methanol [Citation17,Citation18], and dissolved in 1% SDS buffer. Twenty microgram of cell lysates or 10 µL of supernatant were used for western blot analysis with respectively antibodies (Abs) of IL-1β, caspase-1, NLRP3, ASC, and β-actin. The immunoreactive bands were developed with moderate chemiluminescence substrate. The protein densitometric quantification was normalized to β-actin values using ImageJ software.
Elisa
The cell culture supernatant stimulated with A. caviae was harvested and used for measuring TNF-α, IL-6 and IL-1β levels with the established standard curve at OD450 nm value referring to recommended procedures.
Confocal immunofluorescence microscopy
For immunofluorescence detection, PMs were infected with A. caviae at a MOI of 40 for 12 h or 4 h followed by fixation, permeation, and sealing treatment [Citation17,Citation18]. Then, PMs were co-incubated with 200 times diluted Abs of ASC, NLRP3 or p-NF-κB p65, and 400 times diluted Abs of FITC labelled rabbit IgG or mouse IgG. Nucleus were stained by Hoechst (UE, Suzhou). Representive images were captured on a confocal microscope (Olympus, Japan).
Inhibitor studies
Inhibitors of VX765, CA074-Me, Glyburide, MCC950, and BAY11–7082 were dissolved in the corresponding solvent and stored at −80℃ according to the protocol of manufacturer. Inhibitors were co-incubated with PMs for 1 h prior to A. caviae stimulation. The working concentrations of V × 765, MCC950, and BAY11–7082 were 10 µM, whereas CA074-Me and Glyburide were 25 µM and 50 µM. After stimulation of 12 h or 4 h with A. caviae, samples were used for Western Blot assays, qPCR analysis, or ELISA assays.
Asc oligomerization
After A. caviae stimulation at a MOI of 40 for 12 h, the cells precipitation was disrupted in DSS crosslinking buffer, and the obtained supernatants were crosslinked with 2 mM DSS (Aladdin, Shanghai). The cross-link pellet was analysed by Western Blot [Citation17,Citation18].
Statistical analysis
The differences were conducted by t-test (two groups) or one-way ANOVA followed by Tukey’s post hoc testing (three or more groups) using Graphpad software. Results are exhibited as the mean ± SD and each experimental group was done at least three times. ns represented no significance. When P < 0.05 (* or #), the data was considered statistically significant.
Results
A. caviae infection triggers pro-inflammatory IL-1β release in mice serum and murine macrophages
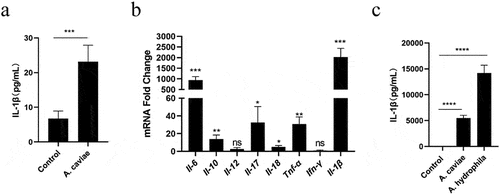
A. caviae infection in human occurs mainly through contaminated food and wounds in the clinical cases. The IL-1β levels in serum of mice injected with A. caviae increased significantly (P < 0.001) compared to those in healthy individuals (). It suggested that A. caviae infection was accompanied by the release of IL-1β. The correlation of IL-1β with A. caviae infection was further evaluated using A. caviae infected PMs model. Various inflammatory cytokines transcription levels were preliminary screened and the results suggested that most cytokines were changed in response to A. caviae infection and Il-1β was the highest (P < 0.001) (). A. hydrophila, a foodborne pathogen that was able to stimulate the secretion of IL-1β [Citation16], was used as a positive control here. The PMs was stimulated for 12 h with A. caviae at a MOI of 40 and A. hydrophila at a MOI of 1. ELISA results suggested the A. caviae obviously induced the secretion of IL-1β in PMs (P < 0.0001) (), which was consistent with that in A. hydrophila (P < 0.0001) [Citation25]. Furthermore, we observed the cell morphology after A. caviae infection (MOI = 40) at different timescales of 0 h, 4 h, 8 h, and 12 h. The results suggested that A. caviae was cytotoxic and could induce obvious macrophages death (supplementary Figure S1). Altogether, A. cavie infection causes the release of IL-1β in mice.
Figure 1. Effects of A. caviae infection on IL-1β secretion. (a) Mice were infected with A. cavaie by gavage at a dose of 8 × 109 CFU/mice once a week for 2 weeks and the IL-1β levels in serum from healthy individuals (n = 5) and A. caviae infection mice (n = 5) were measured by ELISA. (b) PMs were inoculated with A. caviae (MOI = 40) for 12 h and the transcript levels of cytokines were determined by qPCR. Differences represented between the A. caviae infection group and unstimulated control group. (c) the IL-1β levels in the culturing supernatant from PMs inoculated with A. caviae at MOI of 40 for 12 h was detected by ELISA. A. hydrophila stimulation (MOI = 1 and 12 h) was set as the positive control.
A. caviae infection activates NLRP3 inflammasome to induce pro-inflammatory IL-1β release in murine macrophages
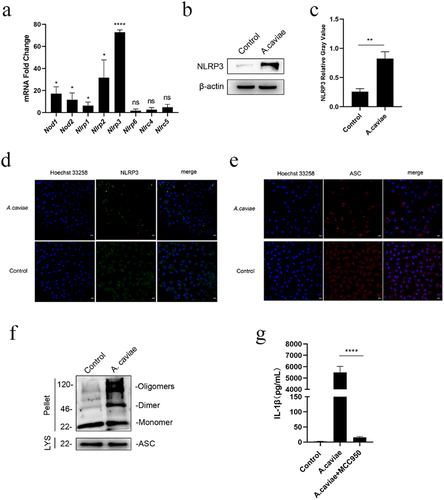
IL-1β, a key mediator in inflammation, is released by host in response to pathogens infection. Different from conventional endoplasmic reticulum-Golgi secretion route, more and more studies indicate that IL-1β release is associated with inflammasomes activation and assembly [Citation11]. In order to determine which intracellular NLR play a vital role during the A. caviae infection, PMs were treated with A. caviae and qPCR data showed that there were five Nlrs significantly up-regulated, namely Nod1, Nod2, Nlrp1, Nlrp2, and Nlrp3, whereas the Nlrp3 transcript level reached the highest (P < 0.0001) among the changed Nlrs (). To validate the activated NLR, NLRP3 protein was also detected in A. caviae infected PMs. In combination with the relative gray analysis to β-actin, A. caviae stimulation up-regulated the protein expressing level of NLRP3 in macrophages (P < 0.01, ). The activation of NLRP3 would bind with ASC and recruited immature caspase-1 to assemble NLRP3 inflammasome [Citation26]. The increase of protein expressing level of NLRP3 implies that NLRP3 inflammasome might be assembled in A. caviae infection. To verify our hypothesis, immunofluorescence staining of NLRP3 was conducted. The results showed that NLRP3 protein (green) aggregated into a speck and distributed around the nucleus after A. caviae infection, in contrast, no obvious speck was found in the untreated control (). Then, the ASC oligomerization and immunofluorescence of ASC protein were further studied. Macrophages were exposed to A. caviae for 12 h and the cell precipitation was collected for DSS cross-link experiment. There was clear oligomerization formation in response to A. caviae treatment by Western Blot analysis (). As expected, immunofluorescence results revealed that A. caviae infection induced the formation of specks of ASC around the nucleus, while ASC was widely dispersed in the nucleus and cytoplasm in the control (). These results imply that NLRP3 inflammasome is activated in PMs infected by A. caviae. MCC950 was a specific NLRP3 inflammasome inhibitor by directly interacting with Walker B motif in NATCH domain to inhibit ATP hydrolysis [Citation27]. To determine the effect of NLRP3 inflammasome in A. caviae infection mediated IL-1β release, PMs were pre-incubated with MCCP50 to disrupt NLRP3 inflammasome assembly. Pretreatment with MCC950 significantly inhibited A. caviae infection triggered the secretion of IL-1β (P < 0.0001) compared to single A. caviae infection group (). Collectively, NLRP3 inflammasome is assembled against A. caviae stimulation and participated in the production of IL-1β.
Figure 2. NLRP3 inflammasome mediated the A. caviae-induced IL-1β secretion. PMs were inoculated with A. caviae at a MOI of 40 for 12 h. (a) the mRNA expressing levels of Nlrs were evaluated by qPCR. Differences represented between the A. caviae infection group and unstimulated control group. (b, c) the NLRP3 protein level was explored by Western Blot and combined with relative grey value to β-actin. (d, e) Immunofluorescence observation of NLRP3 protein (green) and ASC protein (red), and the nucleus were stained in blue. (f) DSS cross link assays showed the ASC oligomerization with anti-ASC antibody during A. caviae infection. (g) PMs were pretreated with MCC950 for 1 h before A. caviae inoculation (MOI = 40) for 12 h. The IL-1β levels was determined by ELISA.
A. caviae activates NLRP3 inflammasome and releases IL-1β in time and dose dependent manners
PMs were, respectively, treated with different doses of A. caviae. The results showed that A. caviae infection led to the release of caspase-1 p20 and IL-1β p17. Moreover, these two inflammasome activation markers were both dose-dependent enhancement except for caspase-1 p20 activation between MOIs of 10 and 20. A. caviae infection did not affect pro-caspase-1 protein expression but elevate both pro-IL-1β and NLRP3 protein levels. Different A. caviae treatment doses exhibited no significant difference in these three proteins expressing levels except for an increase (P < 0.001) of NLRP3 at MOIs of 20 or 40 comparing with that of 10 (.
Figure 3. A. caviae activated NLRP3 inflammasome and induced IL-1β secretion in dose- and time-dependence. (a, b) PMs were stimulated with A. caviae at different MOIs of 10, 20, 40 for 12 h. The protein levels of caspase-1 p20 and IL-1β in the supernatant (sn), as well as pro-caspase-1, pro-IL-1β, nlrp3 and β-actin in lysates (lys) was detected and grey analysis was carried out. (c, d) A. caviae infected PMs for different timescales of 4 h, 8 h, 12 h (moi = 20). The caspase-1 p20 and IL-1β in the SN, as well as pro-caspase-1, pro-IL-1β and NLRP3 in the LYS were detected and grey analysis was carried out.
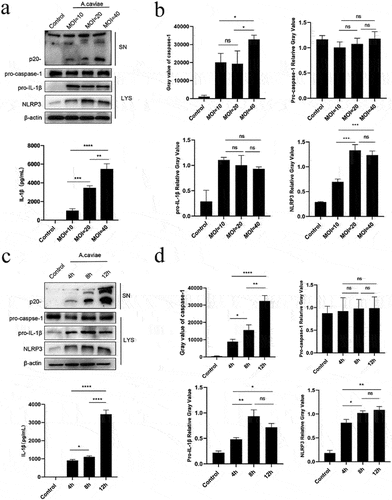
PMs were, respectively, treated with A. caviae for different duration time. A. caviae infection-induced caspase-1 p20 and IL-1β p17 release in time-dependent tendency within 4–12 h (). Pro-caspase-1 relative gray analysis indicated no noticeable change trend without or with A. caviae treatment for different time (). While the pro-IL-1β levels were presented remarkable increase at 8 h and maintained stable at 12 h (). The NLRP3 levels were enhanced along with the increasing infection time, however, not significant changed at 12 h compared with that of 8 h. (). Generally, these results illustrate that NLRP3 inflammasome mediated IL-1β release in A. caviae infected PMs are time and dose-dependent.
A. caviae mediated IL-1β secretion is regulated by caspase-1 p20 and NLRP3 inflammasome
Pre-incubation with MCC950 () indicated that blocking NLRP3 inhibited IL-1β secretion. NLRP3 inflammasome activation is controlled by various approaches and depends on signals provided by stimulants. Moreover, assembly of NLRP3 inflammasome with caspase-1 or 11 mediated canonical inflammasomes or not [Citation28,Citation29]. To determine the IL-1β secretion mechanisms in PMs induced by A. caviae, PMs were first pretreated withVX765, a caspase-1 specific inhibitor [Citation30], before A. caviae infection, and measured the NLRP3 inflammasome biomarkers. The results showed that caspase-1 in V X 765 pretreatment group was inhibited obviously (P < 0.01) (), but there was no obvious change in pro-caspase-1 (). Blocking caspase-1 highly decreased A. caviae induced IL-1β secretion (). Generally, these data suggest that IL-1β is regulated by caspase-1 p20 in A. caviae infection.
Figure 4. IL-1β was mediated by active caspase-1 and NLRP3 inflammasome dependent on K+ efflux and cathepsin B release. (a-d) PMs were pre-treated with VX765 for 1 h or without pretreatment and then stimulated with A. caviae at a MOI of 40 for 12 h. The protein levels of caspase-1 p20 and IL-1β in the SN, as well as pro-caspase-1 and β-actin in the LYS were measured, and grey analysis was made. (e-g) After 1 h pretreatment with MCC950, CA074-Me and Glyburide (Gly), PMs were infected with A. caviae for 12 h, the protein levels of caspase-1 p20 and IL-1β in the SN, as well as pro-caspase-1 and β-actin in the LYS were measured, and relative grey values were analysed.
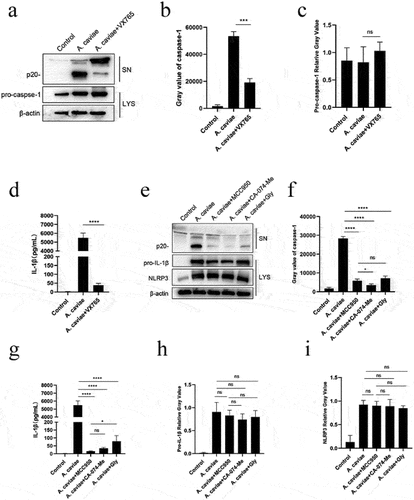
Approaches of K+ efflux and release of cathepsin B both enable NLRP3 inflammasome activation [Citation31]. To assess the signals released by A. caviae in PMs, cells were incubated with CA074-Me or Glyburide prior to A. caviae infection, and MCC950 pretreatment was used as a positive control here. Caspase-1 p20 declined significantly in PMs pre-treated by CA-074-Me and Glyburide compared to the infection group without inhibitor pretreatment (P < 0.0001), and there was a significant decrease (P < 0.05) in CA-074-Me pretreatment group compared to MCC950 pretreatment group while there was no significant change in Glyburide pretreatment group. (). As expected, decrease of functional caspase-1 reduced the concentration of IL-1β in the supernatant from PMs pre-treated by CA-074-Me (P < 0.0001) and Glyburide (P < 0.0001) compared to infection group without inhibitors treatment. In addition, there was no obvious distinction in IL-1β from CA-074-Me pretreatment group and MCC950 pretreatment group. But the use of Glyburide has a significant increase (P < 0.05) in IL-1β levels compared to MCC950, indicating that the IL-1β was partly dependent on K+ efflux approach and more sensitive to lysosome damage way (). Meanwhile, pro-IL-1β and NLRP3 stayed unchanged in the existence of these two inhibitors (). Therefore, IL-1β release is regulated by active caspase-1/11 and NLRP3 inflammasome activation mediated by K+ efflux approach as well as lysosome damage way.
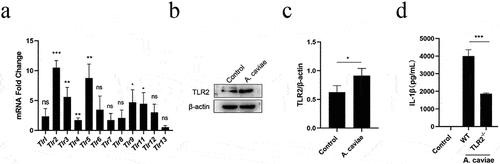
A. caviae infection activates TLR2 and regulates pro-inflammatory IL-1β production in murine macrophages
TLRs could recognize bacteria associated ligands and play fundamental roles in innate immune responses [Citation32]. In our study, many Tlrs were elevated after A. caviae stimulation and the mRNA expression level of Tlr2 had a significant increase (P < 0.001) in . Consistent with the transcriptional results, protein expression of TLR2 was significantly enhanced (P < 0.05) (). These results led us to further determine the role of TLR2 in IL-1β secretion. ELISA results showed that A. caviae-induced IL-1β secretion was significantly down-regulated (P < 0.001) when deletion of TLR2 (). Therefore, we can conclude that A. caviae infection activate TLR2 in PMs and the secretion of IL-1β is partly mediated through TLR2.
Figure 5. A. caviae-induced IL-1β secretion was partly dependent on TLR2. (a) After A. caviae infection for 12 h at a moi of 40, the cell lysates were prepared and subjected to qPCR assay for determining the Tlrs expression. (b, c) the TLR2 protein level in cell lysates was measured by Western Blot and quantified through relative grey value analysis to β-actin. (d) PMs from wild type (wt) or TLR2−/− mice were stimulated with A. caviae for 12 h (moi = 40) and the supernatant was collected for il-1β secretion level determination by elisa.
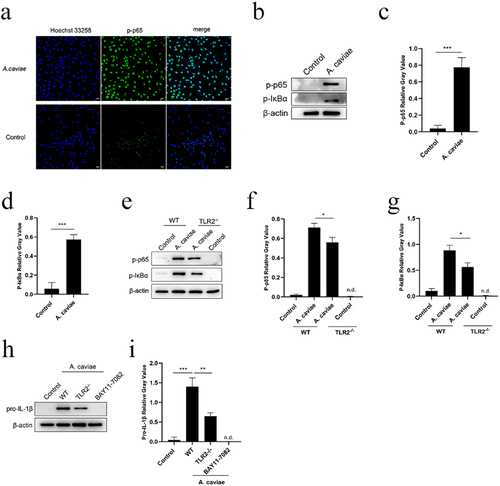
A. caviae infection activates NF-κB partly regulated by TLR2 and accelerates NLRP3 inflammasome first signal pro-IL-1β expression
NF-κB p65 mediated pro-inflammatory cytokines expression [Citation33]. Immunofluorescence results showed that A. caviae stimulation triggered NF-κB p65 subunit (green) translocation from cytoplasm to cell nuclear (). Furthermore, p-p65 and p-IκBα were both obviously up-regulated in A. caviae infection group compared to the control (P < 0.001, ), indicating that NF-κB was activated in PMs during A. caviae infection. Then, the relationship between TLR2 and NF-κB was further studied on the model of A. caviae infected PMs for 4 h. A. caviae-induced p-p65 and p-IKBα levels in PMs was significantly decreased in the absence of TLR2−/− (P < 0.05, ). This finding indicated that deficient of TLR2 significantly reduced NF-κB signal but not completely inhibited. Hence, the activation of NF-κB was partly dependent on TLR2. Pro-IL-1β, the first signal of NLRP3 inflammasome, was a potent inflammatory mediator for IL-1β secretion. The effect of TLR2 and NF-κB signaling pathway on pro-IL-1β expression was evaluated using TLR2 deletion PMs or BAY11–7082 (block IκBα phosphorylation) [Citation34]. The pro-IL-1β was significantly restricted in PMs prepared from TLR2−/− mice (P < 0.01). Meanwhile, the pro-IL-1β expression was absent in BAY11–7082 pre-treatment group (). Taken together, these results indicate that A. caviae infection activates NF-κB pathway partly regulated by TLR2 and promotes the expression of pro-IL-1β, which serves as the first signal for NLRP3 inflammasome.
Figure 6. NF-κB was activated during A. caviae infection and regulated the pro-IL-1β production partly dependent on TLR2. (a) PMs were incubated with A. caviae at a MOI of 40 for 4 h. Subcellular localization of p-p65 (green) and nucleus (blue) was detected by immunofluorescence assay. (b-d) the protein levels of p-p65, p-IKBα and β-actin were detected, and qualified through relative grey value to β-actin. (e-g) WT or TLR2−/− PMs were infected with A. caviae at a MOI of 40 for 4 h followed by the protein level analysis of p-p65, p-IκBα and β-actin. (h, i) PMs from WT (pretreatment with BAY11–7082 for 1 h) or TLR2−/− mice were infected with A. caviae for 4 h and the protein levels of pro-IL-1β were detected, and quantified by relative grey value analysis.
Roles of TLR2, NF-κB, and NLRP3 inflammasome in A. caviae mediated IL-1β and other cytokines production
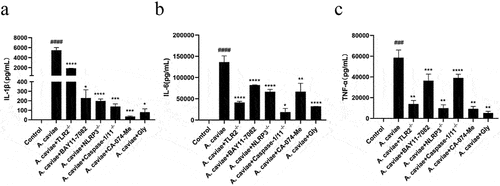
To determine the roles of TLR2, NF-κB, NLRP3 inflammasome in inflammatory cytokines, PMs were pre-treated with various inhibitors of BAY11–7082, CA-074-Me, and Glyburide, followed by A. caviae infection in PMs from WT, TLR2−/−, NLRP3−/−, and caspase-1/11−/− mice. Blocking NLRP3 inflammasome with CA-074Me or Glyburiden, deletion of NLRP3 and TLR2, inhibition of NF-κB, all reduced IL-1β secretion as well as other proinflammatory cytokines production (), indicating that blocking NLRP3 inflammasome and its upstream TLR2 and NF-κB signaling pathway exhibits positive roles in IL-1β and other pro-inflammatory cytokines release.
Figure 7. The roles of TLR2, NF-κB, and NLRP3 inflammasome in IL-1β release and cytokines of TNF-α and IL-6 secretion. PMs from TLR2−/−, NLRP3−/− and Caspase-1/11−/− mice as well as WT mice pretreated by BAY11–7082, CA-074 Me or Glyburide (Gly) were infected with A. caviae at a MOI of 40 for 12 h and ELISA assays were utilized to analyse the levels of IL-1β (a), TNF-α (b) and IL-6 (c) in the supernatant.
Discussion
Aeromonas genus members are widely distributed in the aquatic system, and infect fish [Citation35], other aquatic animals as well as human [Citation36]. Ingestion of contaminated food or direct contact with wounds both cause clinical infection cases, especially for immunocompromised population [Citation37], leading to gastroenteritis [Citation38], wound infections [Citation39], bacteraemia [Citation40], septicaemia [Citation41], urinary tract infections [Citation42], respiratory tract infections [Citation43]. Most human-associated pathogenic strains are focused on A. caviae, A. dhakensis, A. veronii and A. hydrophila. Statistically, A. caviae infection becomes the highest prevalent up to 37.26% among 1852 strains from clinical cases [Citation1]. The main treatment for Aeromonas infection is by antibiotics threapy, but most Aeromonas members exhibit resistance to ampicillin, penicillin, and the first generation of cephalosporins [Citation44]. Fortunately, carbapenems and updated cephalosporins have some effects against Aeromonas [Citation45]. However, the extensive use of antibiotics as well as its unique biofilms restricted these antibiotics effects against Aeromonas infection, which bring out huge challenge of emerging large numbers of drug resistance strains and threaten patients’ health currently [Citation46]. The current A. caviae infection situation pushes us to highlight our research on A. caviae infection mechanism.
Innate immunity functions in host resistance to bacteria. Macrophages are responsible for the recognition and clearance of foreign pathogens. This progress is accompanied by the production of inflammatory cytokines, and excessive release will bring damage to organism. A. hydrophila infection activated macrophages caspase-1/IL-1β inflammatory axis and led to head kidney macrophages apoptosis [Citation6]. A. hydrophila caused mitochondrial dysfunction, produced mtROS release, translocated mtDNA to cytoplasm, activated caspase-1/IL-1β, and induced IL-1β production [Citation16]. IL-1β also functioned against A. dhakensis infection in the spleen and head kidney of Piaractus mesopotamicus at the initial stage [Citation10], as well as A. sobria infection in primary mouse macrophages via activating NLRP3 inflammasome [Citation8]. In our study, mice in vivo infection assays demonstrated that A. caviae infection-induced IL-1β release in murine serum. In vitro experiment, A. caviae increased the transcription and expression of IL-1β in primary mouse macrophages. These data highlighted that IL-1β should be a key marker in response to Aeromonas and A. caviae infection.
Proinflammatory cytokine IL-1β is one major cytokine in inflammatory-associated diseases and played important roles in innate immune response of host against pathogens infection or other physiological and pathological progresses [Citation47]. Hence, there is an urgent need to explore in depth how A. caviae regulate IL-1β production in PMs. Generally, IL-1β is regulated by two signals, one is through the activation of NF-κB, thereby promoting pro-IL-1β expression. The second signal is regulated by the inflammasome complex, which promotes the cleavage of pro-caspase-1 and thus mediates the release of IL-1β. A. veronii utilized its type III secretion system to activate NLRP3 and NLRC4 inflammasomes, and induce caspase-1 p10 cleavage of pro-IL-1β to IL-1β p17 in macrophages [Citation7]. A. hydrophila cytotoxins triggered mouse macrophages caspase-1 activation and IL-1β release, which was regulated by the NLRP3 inflammasome [Citation48]. These cytosolic multiprotein complexes constructed inflammasomes are emerging regulators in innate immune defence and its associated inflammatory caspases are key mediators in the IL-1β release [Citation49]. In our research, to determine the role of NLRs in A. caviae-mediated IL-1β generation, the mRNA transcript levels of multiple NLRs were screened and the NLRP3 exhibited the highest level compared to other NLRs. Meanwhile, immunofluorescence and oligomerization assays verified that NLRP3 inflammasome was activated, which was similar to A. hydrophila infection [Citation48]. The ELISA results suggested that inhibitor of MCC950 pretreatment sharply weakened IL-1β, but not entirely inhibited, meanwhile this result was also verified in NLRP3−/− mice. Hence, A. caviae-induced IL-1β generation is controlled by the NLRP3 inflammasome. However, the exact PAMPs in A. caviae that participate in the assembly of NLRP3 inflammasomes require further invested afterwards.
Assembly of NLRP3 inflammasome is mediated by canonical or non-canonical ways in Gram-negative bacteria infection [Citation50]. V × 765 specially blocked caspase-1 [Citation30,Citation51]. Pre-treatment of V × 765 diminished active caspase and IL-1β. Similarly, A. caviae-induced IL-1β level was also declined in murine caspase-1/11−/− PMs. Hence, A. caviae-induced IL-1β release is also controlled by active caspase-1. NLRP3 inflammasome is regulated by cellular signaling events, including ionic flux, ROS production, lysosomal damage released cathepsin B, mitochondrial damage [Citation52]. Previous studies reported that bacterial toxin activated NLRP3 inflammasome was commonly realized through K+ efflux [Citation53]. Shigella sonnei activated NLRP3 inflammasome in macrophages through approaches of ROS, mitochondrial dysfunction, K+ efflux, and decreased bacterial activity [Citation53]. Mycobacterium kansasii infection-induced NLRP3 inflammasome assembly in THP-1 macrophages, thereby limiting its proliferation within cellular [Citation54]. Here, we found that K+ efflux and lysosomal damage induced by A. caviae signals both involved in activating NLRP3 inflammasome and releasing IL-1β by introducing inhibitors of CA-074-Me and Glyburide, which was consistent with that in common reported bacterial toxins, Shigella sonnei, and Mycobacterium kansasii. Hence, A. caviae-mediated IL-1β release is controlled by K+ efflux and lysosomal damage mediated NLRP3 inflammasome assembly.
IL-1β release not only depended on active caspase-1 and NLRP3 inflammasome but also needed indispensable first signal, namely pro-IL-1β production [Citation55]. NF-κB, a member of nuclear transcription factor family, functioned as a mediator in accelerating the expression of pro-inflammatory cytokines and chemokines [Citation56], being an inflammatory-associated therapeutic target [Citation57]. Canonical NF-κB signal pathway was triggered by TLRs mediated signal transduction to activate IκB kinase and led to phosphorylation and degradation, releasing NF-κB dimer [Citation56]. Streptococcus pyogenes resulted in NLRP3 inflammasome-mediated IL-1β secretion was regulated by NF-κB signal pathway [Citation58]. TLRs was type I transmembrane proteins and recognized the bacterial products during the infection [Citation59]. TLR2 could recognize lipopeptides from Gram-negative bacteria [Citation60,Citation61]. The activation of TLR2 activated the NF-κB p65 and delivered it to the nucleus, which subsequently facilitated the transcription of inflammatory cytokines [Citation62,Citation63]. In our study, TLR2 was activated after A. caviae stimulation and deletion of TLR2 plays a decreasing but incomplete role in IL-1β production. Meanwhile, NF-κB was activated via phosphorylating IκBα and triggered nuclear shift from cytoplasma. Deletion of TLR2 obviously disrupted IκBα phosphorylation and further significantly declined NLRP3 inflammasome first signal pro-IL-1β expression. Altogether, our results fully illustrate that the first signal that leads to A. caviae-caused IL-1β secretion is regulated by NF-κB signal pathway partly in a TLR2-dependent manner. Not only TLR2 but also other TLRs may be involved in A. caviae-induced NF-κB activation and production of pro-IL-1β, which need to be furthered explored.
The release of IL-1β would recruit the aggregation of macrophages in pathogenic bacteria infection. Once IL-1 R receptor that distributed on the surface of macrophages received IL-1β stimulation [Citation64], it would feedback the signal to the NF-κB pathway to promote TNF-α and IL-6. As expected, blocking NLRP3 inflammasome activation with utilization of NLRP3−/− mice, Caspase-1−/− mice or introducing inhibitors of MCC950, CA-074-Me, and Glyburide all decreased IL-1β, thereby down-regulating TNF-α and IL-6 production in A. caviae-stimulated cell culturing supernatant simultaneously. Therefore, NLRP3 inflammasome functions in IL-1β and other inflammatory cytokines production in PMs during A. caviae infection.
Kinds of virulence factors were involved in the Aeromonas infection progress. It was reported that there were multiple virulence factors in Aeromonas including heat-stable cytotonic toxins (ast), aerolysin-related cytotoxic enterotoxin (act), heat-labile cytotonic enterotoxin (alt), haemolysin (hlyA), aerolysin (aerA), type III secretion system (TTSS), elastase (ela), lipase (lip), polar flagellum (fla), and lateral flagella (laf), which would damage healthy body and induced inflammatory response during infection [Citation65]. In A. hydrophila, virulence factors of aerolysin, haemolysin and multifunctional repeat-in-toxin were reported to activate the NLRP3 inflammasome and play a vital role in the secretion of IL-1β [Citation23]. In A. veronii, virulence factors of aerolysin and type III secretion system were verified to take part in the secretion of IL-1β in macrophages [Citation7]. Meanwhile, different Aeromonas carry various combination of virulence factors [Citation65]. Hence, inflammatory cytokines, such as IL-1β and IL-6, released by macrophages during different Aeromonas infections will reach different levels of secretion or transcription.
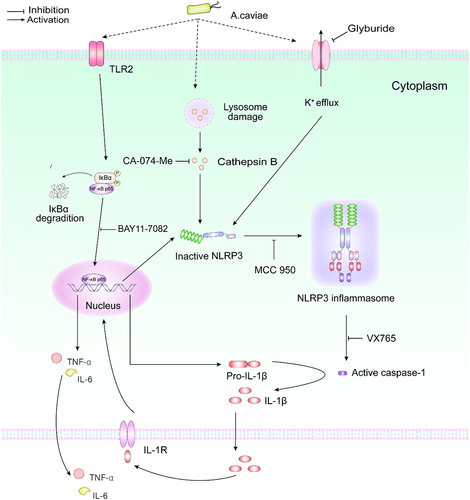
In conclusion, our study for the first time illustrated that A. caviae infection induced proinflammatory cytokine IL-1β release in PMs, which was dependent on caspase-1 p20 and NLRP3 inflammasome triggered by K+ efflux and lysosomal damage. Moreover, activation NF-κB partly regulated by TLR2 further accelerated pro-IL-1β generation, which served as the first signal for IL-1β release. Inhibition of IL-1β could down-regulate other proinflammatory cytokines of TNF-α and IL-6 (). Therefore, our study revealed a potential therapeutic target for clinical infection induced by A. caviae.
Figure 8. The molecular mechanism of A. caviae infection induced IL-1β release in PMs.
Author contribution
QY conducted the assays, collected related papers, and wrote the manuscript. JZ conducted the assays, organized the results, and prepared the figures. FL conducted the assays and wrote the manuscript. HC, WZ, and HY conducted the assays. NH performed data analysis. JD and PZ contributed to conception of the study, provided the guidance throughout the program, and reviewed the manuscript. All authors approved the submitted version.
Ethics Statement
Animal experiments steps were permitted by the Experimental Animal Ethics Committee of Jiangsu Ocean University.
Supplemental Material
Download MS Word (203.2 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data are available from the corresponding author Panpan Zhao.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2116169.
Additional information
Funding
References
- Fernández-Bravo A, Figueras MJ. An update on the genus aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms. 2020;8(1):129.
- Parker JL, Shaw JG. Aeromonas spp. clinical microbiology and disease. J Infect. 2011;62(2):109–118.
- Pund RP, Theegarten D. The importance of aeromonads as a human pathogen. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2008;51(5):569–576.
- Maeda A, Nakajima Y, Yanagisawa K, et al. Combination therapy with levofloxacin and cefepime to treat severe respiratory infection due to aeromonas caviae after a near-drowning accident in river water. Acute Med Surg. 2021;8(1):e708. DOI:10.1002/ams2.708
- Zinner SH. Antibiotic use: present and future. New Microbiol. 2007;30(3):321–325.
- Kumar M, Kumar J, Sharma S, et al. TLR22-Mediated activation of TNF-α-caspase-1/IL-1β inflammatory axis leads to apoptosis of aeromonas hydrophila-infected macrophages. Mol Immunol. 2021;137:114–123.
- McCoy AJ, Koizumi Y, Higa N, et al. Differential regulation of caspase-1 activation via NLRP3/NLRC4 inflammasomes mediated by aerolysin and type III secretion system during aeromonas veronii infection. J Immunol. 2010;185(11):7077–7084. DOI:10.4049/jimmunol.1002165
- Zhang W, Li Z, Yang H, et al. Aeromonas sobria Induces proinflammatory cytokines production in mouse macrophages via activating nlrp3 inflammasome signaling pathways. Front Cell Infect Microbiol. 2021;11:691445.
- Xu Z, Jin P, Zhou X, et al. Isolation of a virulent aeromonas salmonicida subsp. masoucida bacteriophage and its application in phage therapy in turbot (scophthalmus maximus). Appl Environ Microbiol. 2021;87(21):e0146821. DOI:10.1128/AEM.01468-21
- Carriero MM, Henrique-Silva F, Meira CM, et al. Molecular characterization and gene expression analysis of the pro-inflammatory cytokines IL-1β and IL-8 in the South American fish piaractus mesopotamicus challenged with Aeromonas dhakensis. Genet Mol Biol. 2020;43(4):e20200006. DOI:10.1590/1678-4685-gmb-2020-0006
- Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011;22(4):189–195.
- Barichello T, dos Santos I, Savi GD, et al. TNF-Alpha, IL-1beta, IL-6, and cinc-1 levels in rat brain after meningitis induced by streptococcus pneumoniae. J Neuroimmunol. 2010;221(1–2):42–45. DOI:10.1016/j.jneuroim.2010.02.009
- Zuelli FM, Cárnio EC, Saia RS. Cholecystokinin protects rats against sepsis induced by staphylococcus aureus. Med Microbiol Immunol. 2014;203(3):165–176.
- Kaplanov I, Carmi Y, Kornetsky R, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116(4):1361–1369. DOI:10.1073/pnas.1812266115
- Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol. 2020;15:123–147.
- Kumar M, Shelly A, Dahiya P, et al. Aeromonas hydrophila inhibits autophagy triggering cytosolic translocation of mtDNA which activates the pro-apoptotic caspase-1/IL-1β-nitric oxide axis in headkidney macrophages. Virulence. 2022;13(1):60–76. DOI:10.1080/21505594.2021.2018767
- Wang J, Ding Q, Yang Q, et al. Vibrio alginolyticus triggers inflammatory response in mouse peritoneal macrophages via activation of Nlrp3 inflammasome. Front Cell Infect Microbiol. 2021;11:769777.
- Yu G, Wang J, Zhang W, et al. NLRP3 inflammasome signal pathway involves in vibrio harveyi-induced inflammatory response in murine peritoneal macrophages in vitro. Acta Biochim Biophys Sin (Shanghai). 2021;53(12):1590–1601. DOI:10.1093/abbs/gmab137
- Yu X, Lan P, Hou X, et al. HBV inhibits lps-induced nlrp3 inflammasome activation and il-1β production via suppressing the nf-κB pathway and ros production. J Hepatol. 2017;66(4):693–702. DOI:10.1016/j.jhep.2016.12.018
- Jang HM, Park JY, Lee YJ, et al. TLR2 and the NLRP3 inflammasome mediate IL-1β production in prevotella nigrescens-infected dendritic cells. Int J Med Sci. 2021;18(2):432–440. DOI:10.7150/ijms.47197
- Matsushima K. Chemokines. introduction. Springer Semin Immunopathol. 2000;22(4):321–328.
- Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5):a016303.
- Abuelsaad AS, Mohamed I, Allam G, et al. Antimicrobial and immunomodulating activities of hesperidin and ellagic acid against diarrheic aeromonas hydrophila in a murine model. Life Sci. 2013;93(20):714–722. DOI:10.1016/j.lfs.2013.09.019
- Vitovec J, Aldova E, Vladik P, et al. Enteropathogenicity of plesiomonas shigelloides and aeromonas spp. In experimental mono- and coinfection with cryptosporidium parvum in the intestine of neonatal BALB/c mice. Comp Immunol Microbiol Infect Dis. 2001;24(1):39–55. DOI:10.1016/S0147-9571(00)00012-6
- Krzymińska S, Kaznowski A, Chodysz M. Aeromonas spp. human isolates induce apoptosis of murine macrophages. Curr Microbiol. 2009;58(3):252–257.
- Shao BZ, Xu ZQ, Han BZ, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262.
- Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15(6):556–559. DOI:10.1038/s41589-019-0277-7
- Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3(7):e338.
- Kayagaki N, Warming S, Lamkanfi M, et al. Non-Canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. DOI:10.1038/nature10558
- Wu S, Li Z, Ye M, et al. VX765, a specific caspase-1 inhibitor, alleviates lung ischemia reperfusion injury by suppressing endothelial pyroptosis and barrier dysfunction. Biomed Res Int. 2021;2021:4525988.
- He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 iNflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021.
- Kawai T, Akira S. Tlr signaling. Semin Immunol. 2007;19(1):24–32.
- Mitchell JP, Carmody RJ. NF-κB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. 2018;335:41–84.
- Irrera N, Vaccaro M, Bitto A, et al. Bay 11-7082 inhibits the nf-κB and nlrp3 inflammasome pathways and protects against imq-induced psoriasis. Clin Sci (Lond). 2017;131(6):487–498. DOI:10.1042/CS20160645
- Lee HJ, Hoel S, Lunestad BT, et al. Aeromonas spp. isolated from ready-to-eat seafood on the Norwegian market: prevalence, putative virulence factors and antimicrobial resistance. J Appl Microbiol. 2021;130(4):1380–1393. DOI:10.1111/jam.14865
- Kienzle N, Muller M, Pegg S. Aeromonas wound infection in burns. Burns. 2000;26(5):478–482.
- Fernández-Bravo A, Fort-Gallifa I, Ballester F, et al. A case of aeromonas trota in an immunocompromised patient with diarrhea. Microorganisms. 2020;8(3): DOI:10.3390/microorganisms8030399
- Teunis P, Figueras MJ. Reassessment of the enteropathogenicity of mesophilic aeromonas species. Front Microbiol. 2016;7:1395.
- Tena D, Aspiroz C, Figueras MJ, et al. Surgical site infection due to aeromonas species: report of nine cases and literature review. Scand J Infect Dis. 2009;41(3):164–170. DOI:10.1080/00365540802660492
- Tang HJ, Lai CC, Lin HL, et al. Clinical manifestations of bacteremia caused by aeromonas species in southern Taiwan. PLoS One. 2014;9(3):e91642. DOI:10.1371/journal.pone.0091642
- Makino I, Tajima H, Kitagawa H, et al. A case of severe sepsis presenting marked decrease of neutrophils and interesting findings on dynamic ct. Am J Case Rep. 2015;16:322–327.
- Mandal J, Dhodapkar R, Acharya NS, et al. Urinary tract infection due to aeromonas spp., a lesser known causative bacterium. J Infect Dev Ctries. 2010;4(10):679–681. DOI:10.3855/jidc.1052
- Kao HT, Huang YC, Lin TY. Fatal bacteremic pneumonia caused by aeromonas hydrophila in a previously healthy child. J Microbiol Immunol Infect. 2003;36(3):209–211.
- Vila J, Ruiz J, Gallardo F, et al. Aeromonas spp. And traveler’s diarrhea: clinical features and antimicrobial resistance. Emerg Infect Dis. 2003;9(5):552–555. DOI:10.3201/eid0905.020451
- Batra P, Mathur P, Misra MC. Aeromonas spp.: an emerging nosocomial pathogen. J Lab Physicians. 2016;8(1):1–4.
- Liu J, Gao S, Dong Y, et al. Isolation and characterization of bacteriophages against virulent aeromonas hydrophila. BMC Microbiol. 2020;20(1):141. DOI:10.1186/s12866-020-01811-w
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232–241.
- McCoy AJ, Koizumi Y, Toma C, et al. Cytotoxins of the human pathogen aeromonas hydrophila trigger, via the NLRP3 inflammasome, caspase-1 activation in macrophages. Eur J Immunol. 2010;40(10):2797–2803. DOI:10.1002/eji.201040490
- Ta A, Vanaja SK. Inflammasome activation and evasion by bacterial pathogens. Curr Opin Immunol. 2021;68:125–133.
- Rathinam VA, Vanaja SK, Waggoner L, et al. Trif licenses caspase-11-dependent nlrp3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. DOI:10.1016/j.cell.2012.07.007
- Acuña G, Ortiz-Riaño E, Vinagre J, et al. Application of capillary electrophoresis for the identification of Atlantic salmon and rainbow trout under raw and heat treatment. J Capill Electrophor Microchip Technol. 2008;10(5–6):93–99.
- Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. DOI:10.3390/ijms20133328
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. DOI:10.1016/j.immuni.2013.05.016
- Chen CC, Tsai SH, Lu CC, et al. Activation of an NLRP3 inflammasome restricts mycobacterium kansasii infection. PLoS One. 2012;7(4):e36292. DOI:10.1371/journal.pone.0036292
- Hirano S, Zhou Q, Furuyama A, et al. Differential regulation of IL-1β and IL-6 release in murine macrophages. Inflammation. 2017;40(6):1933–1943. DOI:10.1007/s10753-017-0634-1
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651.
- Sehnert B, Burkhardt H, Dübel S, et al. Cell-type targeted NF-kappaB inhibition for the treatment of inflammatory diseases. Cells. 2020;9(7): DOI:10.3390/cells9071627
- Harder J, Franchi L, Muñoz-Planillo R, et al. Activation of the Nlrp3 inflammasome by streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183(9):5823–5829. DOI:10.4049/jimmunol.0900444
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384.
- Jin MS, Kim SE, Heo JY, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130(6):1071–1082. DOI:10.1016/j.cell.2007.09.008
- Kang JY, Nan X, Jin MS, et al. Recognition of lipopeptide patterns by toll-like receptor 2-toll-like receptor 6 heterodimer. Immunity. 2009;31(6):873–884. DOI:10.1016/j.immuni.2009.09.018
- Ma SQ, Wei HL, Zhang X. TLR2 regulates allergic airway inflammation through NF-κB and MAPK signaling pathways in asthmatic mice. Eur Rev Med Pharmacol Sci. 2018;22(10):3138–3146.
- Wang M, Wang L, Fang L, et al. NLRC5 negatively regulates LTA-induced inflammation via TLR2/NF-κB and participates in TLR2-mediated allergic airway inflammation. J Cell Physiol. 2019;234(11):19990–20001. DOI:10.1002/jcp.28596
- Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27.
- Zhou Y, Yu L, Nan Z, et al. Taxonomy, virulence genes and antimicrobial resistance of aeromonas isolated from extra-intestinal and intestinal infections. BMC Infect Dis. 2019;19(1):158. DOI:10.1186/s12879-019-3766-0