
ABSTRACT
Although four Shigella species (S. flexneri, S. sonnei, S. dysenteriae, and S. boydii) have been reported, S. sp. PAMC 28760, an Antarctica isolate, is the only one with a complete genome deposited in NCBI database as an uncharacterized isolate. Because it is the world’s driest, windiest, and coldest continent, Antarctica provides an unfavourable environment for microorganisms. Computational analysis of genomic sequences of four Shigella species and our uncategorized Antarctica isolates Shigella sp. PAMC28760 was performed using MP3 (offline version) program to predict trehalase encoding genes as a pathogenic or non-pathogenic form. Additionally, we employed RAST and Prokka (offline version) annotation programs to determine locations of periplasmic (treA) and cytoplasmic (treF) trehalase genes in studied genomes. Our results showed that only 56 out of 134 Shigella strains had two different trehalase genes (treF and treA). It was revealed that the treF gene tends to be prevalent in Shigella species. In addition, both treA and treF genes were present in our strain S. sp. PAMC28760. The main objective of this study was to predict the prevalence of two different trehalase genes (treF and treA) in the complete genome of Shigella sp. PAMC28760 and other complete genomes of Shigella species. Till date, it is the first study to show that two types of trehalase genes are involved in Shigella species, which could offer insight on how the bacteria use accessible carbohydrate like glucose produced from the trehalose degradation pathway, and importance of periplasmic trehalase involvement in bacterial virulence.
Introduction
Shigella is a Gram-negative bacterium that is genetically related to Escherichia coli [Citation1]. It is a facultative anaerobe and a non-spore former. It belongs to non-motile and rod-shaped bacteria. Shigella are among common causes of diarrhoea worldwide. Shigella infection is one of the top four infections among African and South Asian children [Citation2]. Based on its serological features, Shigella genus can be differentiated into four species: S. dysenteriae (serogroup A), S. flexneri (serogroups B), S. boydii (serogroups C), and S. sonnei (serogroup D). Shigella species has a highly immunogenic O-antigen made of many oligosaccharides unit (O) repeats with a wide range of sugar components, number of repeats, arrangements, and linkages. Each Shigella species can be further differentiated into several serotypes based on O-antigen on its lipopolysaccharide layer: S. dysenteriae having 15 serotypes, S. flexneri having 6 serotypes with 15 subtypes, S. boydii having 18 serotypes, and S. sonnei having only 1 serotype [Citation3–5]. Although serogroups A, B, and C are physiologically identical, due to its positive beta-D-galactosidase and ornithine decarboxylase activity, S. sonnei is distinguished as a single serogroup D [Citation6]. A previous study has reported that 60% of all infections worldwide are caused by S. flexneri. Thus, S. flexneri has been intensively studied, which has enhanced our understanding of Shigella pathophysiology and the underlying “host-pathogen” communication [Citation7]. S. sp. PAMC28760 is a lichen-associated polar bacteria isolated from Antarctica. It has been deposited in the NCBI (National Center for Biotechnology Information) database (https://www.ncbi.nlm.nih.gov/) as an uncharacterized organism. Antarctica is a geographical mass covered with up to 13000 feet of ice and bare rock, with small mosses and lichens being its primary vegetation [Citation8].
Various microorganisms remain unknown in such a harsh environment since they have developed specific adaption abilities towards a wide range of extreme conditions to thrive in such habitat [Citation9]. Generally, Shigella species can grow in a temperature range from (6–8) °C to (45–47) °C [Citation10]. However, temperatures about 65 °C cause their rapid inactivation. Some Shigella species can survive for long durations when they are frozen at −20 °C or refrigerated at 4 °C [Citation11,Citation12]. Bacteria have developed a wide range of coping mechanisms to endure adverse environments such as food deprivation, biochemical and biological changes, and extreme temperatures. Temperature is one of the most crucial elements that can influence microbial protein expression. According to previous studies, expression levels of outer membrane proteins were analysed using proteome profiles of S. flexneri cells grown at 37, 38.5, and 40° C. Pathogens might use the overexpression of specific proteins (18.4, 25.6, and 57.0 kDa) to govern the expression of virulence-related proteins when cells were exposed to higher temperatures [Citation13]. Moreover, cold-adapted enzymes from organisms living in polar regions, deep oceans, and high altitudes have several benefits, they have been increasingly analysed in recent years.
Trehalose is also essential to organisms as a survival mechanism in a stress environment because of its unique physiochemical properties, which allow it to protect cell integrity against a different environmental damage and nutritional limitations [Citation14]. Also, trehalose and its derivatives have also been found to possess crucial functions in the pathogenicity of a wide range of organisms, including bacteria (Gram-positive and Gram-negative) and plants [Citation15] Also, trehalose metabolism could be employed as a target for novel pathogen-specific treatments. Trehalose is a disaccharide produced by various organisms. It can be degraded via several pathways. Among these pathways, the trehalose-6-phosphate pathway (TPP) is used by many bacteria to degrade trehalose. This pathway has been investigated under conditions of low osmolarity in both Gram-positive and Gram-negative bacteria [Citation16,Citation17]. It was reported in E. coli K-12 that under different osmolarity conditions, it may survive on trehalose as its sole carbon source and uses different pathways for its breakdown. Likewise, the external trehalose is hydrolysed by periplasmic trehalase (TreA) at high osmotic conditions. At that moment, the glucose PTS then transports the produced glucose molecules back into the cytoplasm [Citation17,Citation18]. During the transition between high and low osmolarity, a second trehalase, cytoplasmic trehalase (TreF), is active which removes the internal pool of trehalose as the cells alter their metabolism to low osmolarity. TreF’s low enzymatic activity is low enough not to interfere with trehalose biosynthesis during high osmolarity, but high enough to breakdown the accumulated trehalose during the return to normal conditions, when no more biosynthesis proceeds [Citation19].
Several prokaryotes and eukaryotes can degrade trehalose to glucose through the enzyme trehalase [EC 3.2.1.28] [Citation20,Citation21]. It has been reported that E. coli has two trehalases, including cytoplasmic trehalase (TreF) and periplasmic trehalase (TreA). The periplasm is a small space between the outer and inner membranes of Gram-negative bacteria. Trehalases from E. coli, such as periplasmic TreA (Tre37A), have an extra C-terminal region, whereas TreF has an extended N-terminal region. Both enzymes are monomeric and have a 47% similarity [Citation22]. Neutral trehalase (L72) is a protein found in Klebsiella oxytoca that has been linked to several functions, including energy sources and stress protection [Citation23]. Experimental evidence of periplasmic treA gene in needed for optimal development of type 1 fimbriae for cell invasion and colonization in extraintestinal pathogenic E. coli (ExPEC) strain MT78 has been addressed in the previous study [Citation24]. Similarly, in Burkholderia pseudomallei, a single trehalase-encoding gene, identical to E. coli TreA, which is involved in stress tolerance and virulence in mouse and insect infection models, plays a role in stress tolerance and virulence [Citation25]. Despite its tiny size, the periplasm contains many important proteins required for a variety of physiological activities and bacterial survival under stress. Periplasmic proteins aid in the defence against different stresses, making it easier for bacteria such as S. Typhimurium to colonize the host [Citation26]. However, there has been no complete analysis of the expression of many periplasmic proteins, especially periplasmic trehalase (TreA), in Shigella strains. The goal of this study was to determine the prevalence of two different trehalase genes (treF and treA) in 134 complete Shigella genomes, including lichen-associated S. sp. PAMC28760 isolated from the Antarctica region. Additionally, we would like to determine which trehalase genes (treF or treA) might contribute to virulence. It is thought that analysis of pathogenic and non-pathogenic trehalase might provide a new direction to understand bacterial pathogenic mechanism at the genetic level and to provide a new insight on drug development for the treatment of bacterial infections. The use of a bioinformatics tools such as MP3 can allow the study of virulence genes involved in respective strains without the need to perform hazardous laboratory experiments.
Materials and methods
Data sources
The complete genome and amino acid sequences of Shigella species were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/) [Citation27]. A total of 134 Shigella strains deposited in NCBI by September 2021 were analysed, including our Antarctica isolate S. sp. PAMC28760, whose genome size was 4,558,287 bp [Citation28].
Phylogenetic tree construction and average nucleotide identity (ANI) analysis
To compare 16S rRNA sequences of S. sp. PAMC28760 with those in other complete genomes of Shigella strains (133 strains), phylogenetic analysis was performed using the ClustalW alignment tool and the Molecular Evolutionary Genetic Analysis (MEGA X) (https://www.megasoftware.net/) tools [Citation29]. MEGAX was used to create the phylogenetic tree, which was built on a neighbour-joining tree method [Citation30] and 1,000 bootstrap replications [Citation31]. The online software Interactive Tree life (iTOL) v6 (https://itol.embl.de/) was used to analyse phylogenetic trees [Citation32]. Orthologous Average Nucleotide Identity Software Tool (OAT) [Citation33] was used to determine the average nucleotide identity (ANI) of 16S rRNA from closely related species acquired from EziBio Cloud (www.ezibiocloud.net) [Citation34]. To determine if the strain PAMC28760 belonged to Shigella or Escherichia, EziBio cloud 16S rRNA sequencing was used. Secondary data was used to identify the cytoplasmic trehalase or periplasmic trehalase from the characterized strains E. coli strain K-12 substrain MG1655 (NC 000913.3) as a reference for the construction of a phylogenetic tree for trehalase genes (treA and treF) in those studied strains who possess both trehalase genes. NCBI, RAST, and Prokka were used to find the cytoplasmic and periplasmic genes. MUSCLE [Citation35,Citation36] was used to align amino acid sequences, and maximum-likelihood and neighbour-joining methods were used to build a phylogenetic tree.
Comparative genomic analysis and, prediction of periplasmic trehalase and cytoplasmic trehalase
The prevalence of trehalase genes in the studied genome, as well as to predict pathogenic and non-pathogenic factors, were carried out using the MP3 (offline version) program (http://metagenomics.iiserb.ac.in/mp3/index.php) [Citation37]. This program uses two modules including Support Vector Model (SVM) and Hidden Markov Model (HMM) to predict pathogenic and non-pathogenic proteins in the genome. Furthermore, Rapid Annotations utilizing Subsystems Technology (RAST, https://rast.nmpdr.org/rast.cgi) [Citation38] and Prokka annotation (Prokka 1.14.6 offline version) [Citation39] were used to locate predicted trehalase genes. CGView ServerBETA (www.cgview.ca) was used to better visualization of location predicted trehalase genes [Citation40].
Results and discussion
Phylogenetic tree analysis of S. sp. PAMC28760
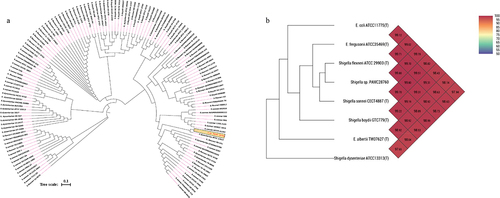
Phylogenomic analysis revealed that S. sp. PAMC28760 and S. dysenteriae ATCC12037 belonged to the same branch (). MEGA X program was used to construct phylogenetic tree to analyse their evolutionary history using the neighbour-joining method [Citation41] with 1,000-replicate bootstrap. Furthermore, ANI value revealed that S. sp. PAMC28760 had a close relationship with strains S. flexneri ATCC29903(T) (99.80%), S. sonnei CECT4887(T) (99.70%), E. coli ATCC11775(T) and S. boydii GTC779(T) (99.19%), E. fergusonii ATCC35469(T) (99.70%), S. dysenteriae ATCC13313 (T) (98.99%), and E. albertii TW07627 (T) (98.89%) (). These results suggest that the S. sp. PAMC28760 strain is closely related to Escherichia strain as both belong to the same family Enterobacteriaceae.
Figure 1. (a) Circular phylogenetic analysis of the complete genomes of Shigella: Phylogenetic tree showing the relationships of genomes of a total 134 Shigella strains including an Antarctica isolate Shigella sp. PAMC28760 (represented in red text), and their phylogenetic position. This analysis was prepared using MEGA X based on 16S rRNA sequences with neighbour-joining method with 1,000-replicate bootstrap. (b) Heatmap generated with OrthoANI values calculated using the OAT software to determine the close relationship of strain S. sp. PAMC28760 with S. flexneri ATCC29903(T), S. sonnei CECT4887(T), E. coli ATCC11775(T), S. boydii GTC779(T), E. fergusonii ATCC35469(T), S. dysenteriae ATCC13313(T), and E. albertii TW07627(T).
Trehalase gene and its phylogeny
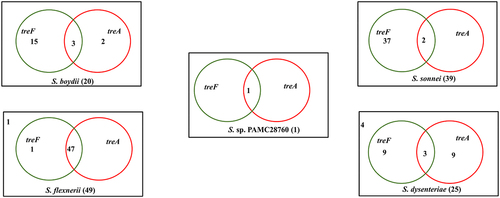
When complete genomes of 134 Shigella strains including our strain PAMC28760 were studied, only 56 strains were found to have two types of trehalase (treF and treA) genes. Furthermore, we employed RAST annotation database and, Prokka annotation to differentiate cytoplasmic (treF) and periplasmic (treA) trehalase. In addition, the CGview online server () visualize the predicted trehalase genes in S. sp. PAMC28760. When we aligned them with characterized trehalase genes (treF and treA) of E. coli K-12 substrain MG655, S. sp. PAMC28760 was found to also encode the same genes involved in trehalose degradation (). While 48, 47, and 47 of S. flexneri’s strains had treF, treA, and both treF and treA genes, respectively, 39, 2, and 2 of S. sonnei’s strains had treF, treA, and both treF and treA genes, respectively. In addition, of a total of 20 S. boydii strains, 18, 5, and 3 strains had treF, treA, and both treF and treA genes, respectively. For a total of 25 S. dysenteriae strains, 12,12, and 3 strains had treF, treA, and both treF and treA genes, respectively (). Results showed that S. sp. PAMC28760 had both trehalase genes treF (cytoplasmic trehalase) and treA (periplasmic trehalase).
Figure 2. Circular genome comparison using CGView ServerBETA (http://cgview.Ca/) tool for the representation of genome and features of the S. sp. PAMC28760. The contents of the featured rings (starting with the outermost ring to the centre) are as follows. Ring 1, combined ORFs in forward and reverse strands; Ring 2, trehalose degradative genes, combined forward and reverse strand, and CDS (including tRNA and rRNA) in forward and reverse strands; Ring 3, GC skew plot, values above average are depicted in green, and below average in purple; Ring 4, GC content plot; and Ring 5, Sequence ruler.
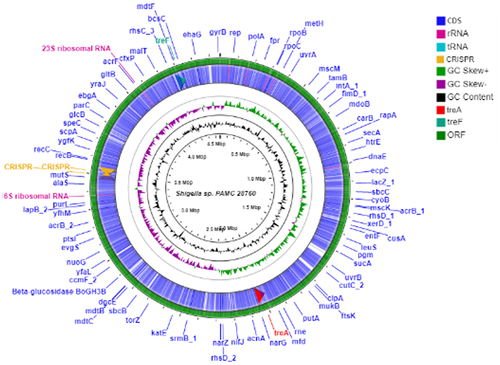
Figure 3. Cytoplasmic trehalase (TreF) amino acid sequence alignment with a characterized trehalase (TreF). TreF (GH37) from E. coli K-12 substr. MG1655, trehalase from S. flexneri C32, trehalase from Shigella sp. PAMC28760, and trehalase from S. boydii ATCC49812. The signature motif 1 and signature motif 2 represent two highly conserved sequence segments that belong to the GH37 family. The “#” symbol denotes the catalytic sites of Asp312 and Glu496. the three black boxes represent conserved regions (CR3–CR5).

Figure 4. Venn diagram categorizes trehalase genes involved in the complete genomes of four Shigella species along with uncategorized Shigella sp. PAMC28760. Green circle represents the cytoplasmic trehalase (treF), whereas red circle represents the periplasmic trehalase (treA). The number outside the circles represents the absence of both trehalase genes.
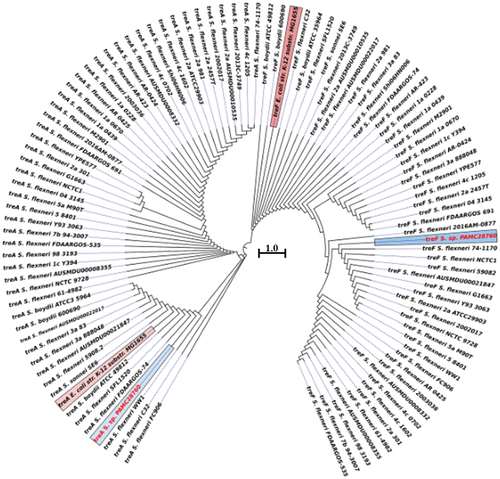
Phylogenetic tree analysis of trehalase genes (treF and treA) with a characterized E. coli K-12 substrain MG 1655 revealed that treA of S. sp. PAMC28760 and E. coli K-12 substrain MG1655 shared the same clade with 100% sequence identity, whereas S. sp. PAMC28760 did not share the same clade as E. coli K-12 substrain MG1655, although both shared 99.82% sequence identity (). This shows that trehalase genes (treA and treF) of S. sp. PAMC28760 could be distinctly divided into two major clades. It was found that treA and treF genes from studied genome clustered together more closely with both genes of S. flexneri. The treA gene is clustered with S. flexneri FDAARGOS-74 and S. flexneri WW1 whereas treF is clustered with S. flexneri 2016AM–0877 and S. flexneri 74–1170.
Figure 5. Circular phylogenetic tree based on trehalase genes (treF/treA) sequence in the complete genomes of Shigella strains with reference to the characterized trehalase of E. coli strain K-12 substrain MG165 using a neighbour-joining tree method with 1,000-replicate bootstrap. The pink highlighted boxes represent the characterized trehalase genes (treF and treA), whereas the red text indicates the strain (Shigella sp. PAMC28760) under study.
These results suggest that S. sp. PAMC28760 might have a trehalose degradation pathway like that of E. coli. Also, it has been reported that TreA in E. coli is a trehalase found in the periplasmic area of cells that hydrolyzes trehalose glucose under high osmolarity, whereas TreF is a cytoplasmic isoform of TreA trehalase that plays important role in trehalose breakdown produced within bacterial cells under high osmolarity conditions [Citation42,Citation43]. Similarly, in the case of cytoplasmic trehalase (TreF), it becomes active during the transition between high and low osmolarity. TreF can deplete the internal trehalose pool as the cell metabolism shifts to a low osmolarity state. TreF has a low enzymatic activity that is low enough not to interfere with trehalose production under high osmolarity, but high enough to degrade the accumulated trehalose once the environment returns to normal [Citation19].
Trehalose degradative pathway
Six routes of trehalose degradation pathways (trehalose degradation I, II, III, IV, V, and VI) have been found in organisms depending on their subcellular locations. These pathways have been reported in the MetaCyc pathway database [Citation44]. They are summarized in (). Depending on the organism, trehalose might enter cells via a permease where it remains unmodified, or it gets transformed to phosphorylated trehalose 6-phosphate forms via a phosphotransferase system (PTS). Trehalose that cannot be modified might get degraded by a hydrolysing trehalase (EC 3.2.1.28) or might be split by trehalose phosphorylase (EC 2.4.1.64, and EC 2.4.1.231) (). It was revealed that our Antarctica isolate S. sp. PAMC28760 had the trehalase gene based on the prediction of trehalose degradative pathway. The result is summarized in . Trehalose is broken down into two molecules of glucose and water by the trehalase enzyme that utilizes glucose as a carbon source. Trehalase is classified into glucoside hydrolase (GH) families such as GH37, GH65, and GH15 in the CAZy (Carbohydrate-Active Enzyme) database (http://www.cazy.org/) [Citation45]. The GH37 family possesses only trehalase enzymes, whereas GH65 and GH15 families possess other enzymes along with trehalase enzymes. In 2007, it was reported that Mycobacterium smegmatis and Mycobacterium tuberculosis possessed trehalase that belonged to the GH15 family [Citation46].
Figure 6. Trehalose degradative pathways. Six different trehalose degradative pathways are found in organisms (bacteria, fungi, yeast, Arthropoda, and plants). Among them, only two degradation pathways (Trehalose degradation pathway II (cytosolic) and VI (periplasmic)) are found in Shigella species.
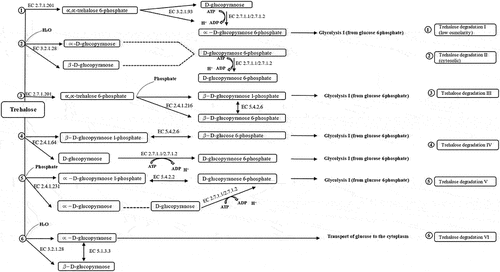
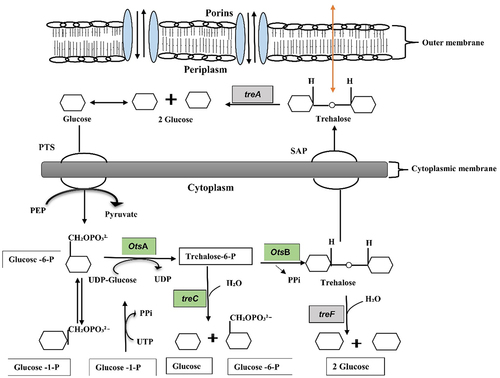
Figure 7. Schematic diagram of the trehalose metabolism pathway in Gram-negative bacteria is formulated from Kosciow et al., 2014 and Purvis et al., 2005. The green boxes represent the trehalose synthesis genes (otsA, trehalose-6-phosphate phosphatase; otsB, trehalose-6-phosphate synthase; and treC, trehalose-6-phosphate hydrolase), whereas grey boxes represent the trehalose degrading genes (treA, periplasmic trehalase; and treF, cytoplasmic trehalase). At cytoplasm, trehalose is degraded by cytoplasmic trehalase gene (treF). The plasma membrane, stretch-activated proteins (SAP) facilitate the exit of trehalose under hypotonic conditions to the periplasm where it further degraded by periplasmic trehalase gene (treA).
Trehalase belonging to the GH37 family can hydrolyse a molecule of ∝,∝-trehalose into two molecules of glucose by inverting the anomeric orientation. Trehalase belonging to the GH37 family have been found in different species, including bacteria, fungi, yeasts, plants, insects, and vertebrates [Citation22]. GH family has been divided into “clans” in the CAZy database, where enzymes are regarded to have a common evolutionary origin. Clan GH-G was ascribed to GH37 enzymes, while clan GH-L was ascribed to GH65 and GH15 enzymes. Although clans GH-G and GH-L share only a low amount of sequence homology, such finding is significant. GH37 trehalase has two catalytic residues, Asp and Glu, in their CDs (catalytic domains). Asp and Glu residues tend to be involved in the function of GH65 and GH15 trehalases. These amino acid residues are most likely to be involved in a common inverting mechanism during catalysis [Citation47]. Structures of these trehalases are comprised of conserved regions (CRs), which include catalytic residues. These CRs can form active sites that usually have loops. CDs of GH enzymes contain well-known trehalase signature motifs, motif 1 (PGGRFXEXY[G/Y] D[S/T] Y] and motif 2 (QWD[Y/F]PN/Y) [G/A] W[P/A] P), whereas GH65 and GH15 trehalases do not [Citation48,Citation49]. Our Antarctica isolate S. sp. PAMC28760 possesses GH37 trehalase with two signature motifs (motifs 1 and 2) as well as highly conserved regions (CR3-CR5), which have also been found in E. coli. Further study confirms that S. sp. PAMC28760 possesses trehalase enzyme, a member of the GH37 CAZyme family (). The Gram-positive bacteria like Bacillus subtilis (non-pathogenic) and Clostridioidess difficile (pathogenic) share a pathway in which exogenous trehalose can be imported by a PTS to produce glucose and glucose-6-phosphate via the phosphotreahalose TerA (analogous to the PTS-TreC system in pathogenic E. coli). Due to the acquisition of an additional cluster of trehalose metabolism genes, namely a second PTS that mediates high-efficiency trehalose uptake from the environment, epidemic C. difficile strains can also grow on low trehalose. By increasing toxin levels, both modified trehalose utilization systems contributed to the growth and toxicity of these epidemic C. difficile strains [Citation49]. There have been no previous papers on the function of the trehalose degradation pathway in virulence in Antarctic isolates till date. However, in Variovorax sp. PAMC28711 [Citation50], the presence of trehalose metabolic pathway was mentioned.
Prediction of pathogenic and non-pathogenic proteins
MP3 (standalone program) can predict the presence of pathogenic and non-pathogenic proteins in a complete genome of a microbe based on two models, SVM and HMM, and their hybrids (integrated SVM and HMM models). To predict pathogenic and non-pathogenic trehalase, we retrieved complete genomes of 134 Shigella species (strains) from the NCBI database along with our S. sp. PAMC28760 isolates from Antarctica. Our strain S. sp. PAMC28760 showed pathogenic proteins of 1,136 (based on SVM model) out of 4329 total proteins (), with periplasmic trehalase as a pathogenic trehalase (data not shown). MP3 tool can be used to compare numbers of pathogenic proteins in healthy and infected samples by precisely identifying pathogenic protein fragments (based on amino acid composition and dipeptide composition) commonly found in metagenomic data without needing a time-consuming homology-based alignment [Citation37]. In comparison with other publicly available bioinformatic tools, this program can predict pathogenic proteins with improved accuracy (95.06%), sensitivity (85.59%), and specificity (96.64%) as it employs both SVM and HMM models. Also, it is essential to analyse complete genome sequences of pathogenic and non-pathogenic bacteria of closely related species to determine if any significant genomic changes have occurred. It has been proposed that both pathogenic and non-pathogenic strains have virulence factors/genes. They can be distinguished based on gene content. When other genes suppress the virulence factors/genes, the bacterium becomes non-pathogenic. However, when suppressing genes are lost, a commensal can become pathogenic [Citation51].
Table 1. MP3 prediction of the total proteins, pathogenic protein, and non-pathogenic proteins in all the complete genomes of Shigella strains including Shigella sp. PAMC28760, which is indicated as a asterisk symbol. Hybrid: predictions from both HMM and SVM models.
In addition, the detection of transposon mutants in extraintestinal pathogenic E. coli (ExPEC) that are defective in binding to non-phagocytic cells is an unexpected finding on the probable role of periplasmic trehalase (treA) in virulence [Citation24]. Furthermore, while trehalase enzymes are known to have a role in virulence of some fungal species, the occurrence of multiple enzymes can inhibit their potential as an antifungal drug target. Because the trehalose pathway and its enzymes are not found in mammals (including humans), fungi-specific inhibitors of the trehalose pathway and their enzymes should be generally non-toxic to mammals [Citation52,Citation53]. Likewise, a previous study has reported that inactivating trehalose biosynthesis pathways does not reduce resistance to oxidative stress in many bacteria, but a periplasmic trehalase gene (treA) mutant in Burkholderia pseudomallei shows increased sensitivity to oxidative stress despite elevated trehalose levels in the mutant, which is expected to protect against this stress [Citation25]. Another study also reported that validmycin A was ineffective against Clostridioides difficile TreA, whereas trehalose derivatives such as epimers containing hydroxyl groups (2- and 4-positions), and thiotrehalose derivatives showed promise as TreA inhibitors with a larger spectrum. The efficacy of these drugs in treating specific bacterial infections is currently being studied [Citation54]. It has also been reported that the PTS route for trehalose uptake (trehalose degradation I, low osmolarity) is inhibited when the osmolarity is high. Thus, trehalase (TreA) in the periplasm can allow cells to utilize trehalose at a high osmolarity by breaking it down into glucose molecules, which can be subsequently transported by phosphotransferase mediated system [Citation55]. Genome of Shigella strains were analysed for pathogenic and non-pathogenic trehalase genes in this study for the first time. It is assumed that studying trehalase in one pathogenic bacterium like Shigella species could be important for further studies. Trehalase (TreA) from the pathogenic strain of extraintestinal E. coli known as MT78 has also been identified as a member of glycoside hydrolase 37 (GH37). Similarly, deletion of these genes in the meningoencephalitis-causing yeast Crytococcus neoformans resulted in severe defects in spore production, a decrease in spore germination, and an increase in the production of alternative development structures, which spores forms are plausible infectious particles [Citation56]. Trehalose does not have to solely play a role in osmoregulation. According to Lee et al., it has stated that if glucose is present in the cytoplasm, molecules like trehalose are produced at levels approaching 400 mM in the cytoplasm [Citation57]. Glycine betaine and L-proline often accumulate in the cytoplasm (around 700 and 400 mM, respectively) and can replace trehalose [Citation58]. Many species utilize these osmolytes, which appear to be well-adapted to cellular functions. The electro-neutral solutes trehalose, glycine betaine, and L-proline, as well as potassium glutamate, have various chemical characteristics that may suit their functions in cell survival during osmotic shock.
Conclusions
Although there are many studies on trehalase, it was not studied in Shigella species based on two different trehalase genes (treF and treA) and pathogenicity. Most Shigella species (S. flexneri, S. boydii, S. dysenteriae, and S. sonnei), as well as our strain S. sp. PAMC28760, have cytoplasmic trehalase, and all periplasmic trehalase predicted in the studied strains showed up as pathogenic proteins using MP3, RAST, and Prokka tools. Notably, treF was detected in all strains of S. sonnei, but treA was identified in only two strains. This sort of research on pathogenic and non-pathogenic trehalase could help researchers to elucidate how and why Shigella species have certain traits. Furthermore, before performing any kinds of wet lab work, these bioinformatics tools are important in determining the nature of proteins present in a complete genome of bacteria.
Acknowledgment
The authors would like to thank Ms. Phataratah Sa-nguannarm for her help in downloading the offline version of MP3 and Prokka programs.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data used in this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Yabuuchi E. Bacillus dysentericus (sic) 1897 was the first taxonomic rather than Bacillus dysenteriae 1898. Int J Syst Evol Microbiol. 2002;3:1041. doi:10.1099/00207713-52-3-1041.
- Kotloff KL, Nataro JP, Blackwelder WC, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382(9888):209–222. DOI:10.1016/S0140-6736(13)60844-2.
- Muthuirulandi Sethuvel DP, Devanga Ragupathi NK, Anandan S, et al. Update on: shigella new serogroups/serotypes and their antimicrobial resistance. Lett Appl Microbiol. 2017;64(1):8–18.
- Lampel KA, Maurelli AT. Shigella species. Ch 11. In: Miliotis M and J Bier, editors. International handbook of foodborne pathogens. Boca Raton: CRC Press. 2003:167–180. doi:10.1201/9780203912065.
- Levine MM, Kotloff KL, Barry EM, et al. Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat Rev Microbiol. 2007;5(7):540–553.
- Hale TL, Keusch GT. Med Microbiol. Galveston (TX): University of Texas Medical Branch at Galveston. 1996:303–310.
- Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21(1):134–156.
- Bargagli R. Environmental contamination in Antarctic ecosystems. Sci Total Environ. 2008;400(1–3):212–226.
- Stan-Lotter H, Fendrihan S. Adaptation of microbial life to environmental extremes: novel research results and application. second edition. (Switzerland: Springer Cham). 2017.
- Stewart SE, Parker MD, Amézquita A, et al. Microbiological risk assessment for personal care products. Int J Cosmet Sci. 2016;38(6):634–645.
- Coates K. Foodborne microorganisms of public health significance. Aust Vet J. 1999;77(1):54.
- Warren BR, Parish ME, Schneider KR. Shigella as a foodborne pathogen and current methods for detection in food. Crit Rev Food Sci Nutr. 2006;46(7):551–567.
- Harikrishnan H, Ismail A, Banga Singh KK. Temperature-Regulated expression of outer membrane proteins in Shigella flexneri. Gut Pathog. 2013;5(1). DOI:10.1186/1757-4749-5-38
- Luyckx J, Baudouin C. Trehalose: an intriguing disaccharide with potential for medical application in ophthalmology. Clin Ophthalmol. 2011;5:577–581.
- Bhumiratana A, Anderson RL, Costilow RN. Trehalose metabolism by Bacillus popilliae. J Bacteriol. 1974;119(2):484–493.
- Helfert C, Gotsche S, Dahl MK. Cleavage of trehalose‐phosphate in Bacillus subtilis is catalysed by a phospho‐α‐(1–1)‐glucosidase encoded by the treA gene. Mol Microbiol. 1995;16(1):111–120.
- Boos W, Ehmann U, Bremer E, et al. Trehalase of Escherichia coli. Mapping and cloning of its structural gene and identification of the enzyme as a periplasmic protein induced under high osmolarity growth conditions. J Biol Chem. 1987;262(27):13212–13218.
- Styrvold OB, Strom AR. Synthesis, accumulation, and excretion of trehalose in osmotically stressed Escherichia coli K-12 strains: influence of amber suppressors and function of the periplasmic trehalase. J Bacteriol. 1991;173(3):1187–1192.
- Horlacher R, Uhland K, Klein W, et al. Characterization of a cytoplasmic trehalase of Escherichia coli. J Bacteriol. 1996;178(21):6250–6257.
- Zhou Y, Li X, Katsuma S, et al. Duplication, and diversification of trehalase confers evolutionary advantages on lepidopteran insects. Mol Ecol. 2019;28(24):5282–5298.
- Shukla E, Thorat L, Bendre AD, et al. Cloning, and characterization of trehalase: a conserved glycosidase from oriental midge. Chironomus Ramosus. 3 Biotech. 2018;8:1–7.
- Sakaguchi M. Diverse and common features of trehalases and their contributions to microbial trehalose metabolism. Appl Microbiol Biotechnol. 2020;104(5):1837–1847.
- Tang P, Hseu YC, Chou HH, et al. Proteomic analysis of the effect of cyanide on klebsiella oxytoca. Curr Microbiol. 2010;60(3):224–228.
- Pavanelo DB, Houle S, Matter LB, et al. The periplasmic trehalase affects type 1 fimbria production and virulence of extraintestinal pathogenic Escherichia coli strain MT78. Infect Immun. 2018;86(8). DOI:10.1128/IAI.00241-18
- Vanaporn M, Sarkar-Tyson M, Kovacs-Simon A, et al. Trehalase plays a role in macrophage colonization and virulence of Burkholderia pseudomallei in insect and mammalian hosts. Virulence. 2017;8(1):30–40.
- Shome A, Kumawat M, Pesingi PK, et al. Isolation and identification of periplasmic proteins in Salmonella typhimurium. Int J Curr Microbiol Appl Sci. 2020;9(6):1923–1936.
- Sayers EW, Beck J, Bolton EE, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021;49(D1):D10–D17. DOI:10.1093/nar/gkaa892.
- Han SR, Kim DW, Kim B, et al. Complete genome sequencing of Shigella sp. PAMC 28760: identification of CAZyme genes and analysis of their potential role in glycogen metabolism for cold survival adaptation. Microb Pathog. 2019;137:103759.
- Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425.
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. N Y: Evolution; 1985.
- Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments | Nucleic Acids Res | Oxford Academic. Nucleic Acids Res. 2019;47(W1):W256–W259.
- Lee I, Kim YO, Park SC, et al. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66(2): 1100–1103.
- Yoon SH, Ha SM, Kwon S, et al. Introducing EzBiocloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017;67(5):1613–1617.
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5(1):113.
- Rc E. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5): 1792–1797.
- Gupta A, Kapil R, Dhakan DB, et al. MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLoS One. 2014;9(4):e93907.
- Aziz RK, Bartels D, Best A, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1): DOI:10.1186/1471-2164-9-75.
- Seemann T. Prokka: rapid prokaryotic genome annotation | Bioinformatics | Oxford Academic. Bioinformatics. 2014;30(14):2068–2069.
- Grant JR, Stothard P. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(Web Server):W181–W184.
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101(30):11030–11035.
- Tourinho-dos-Santos CF, Bachinski N, Paschoalin VM, et al. Periplasmic trehalase from Escherichia coli--characterization and immobilization on spherisorb. Braz J Med Biol Res. 1994;27(3):627–636.
- Uhland K, Mondigler M, Spiess C, et al. Determinants of translocation and folding of TreF, a trehalase of Escherichia coli. J Biol Chem. 2000;275(31):23439–23445.
- Caspi R, Billington R, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes-a 2019 update. Nucleic Acids Res. 2020;48(D1):D445–D453.
- Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–D495.
- Carroll JD, Pastuszak I, Edavana VK, et al. A novel trehalase from Mycobacterium smegmatis − purification, properties, requirements. Febs J. 2007;274(7):1701–1714.
- Maicas S, Guirao-Abad JP, Argüelles JC. Yeast trehalases: two enzymes, one catalytic mission. Biochim Biophys Acta - Gen Subj. 2016;1860(10):2249–2254.
- Barraza A, Sánchez F. Trehalases: a neglected carbon metabolism regulator? Plant Signal Behav. 2013;8(7):e24778.
- Kalera K, Stothard AI, Woodruff PJ, et al. The role of chemoenzymatic synthesis in advancing trehalose analogues as tools for combatting bacterial pathogens. Chem Commun. 2020;56(78):11528–11547.
- Shrestha P, Kim MS, Elbasani E, et al. Prediction of trehalose-metabolic pathway and comparative analysis of KEGG, MetaCyc, and RAST databases based on complete genome of Variovorax sp. PAMC28711. BMC Genom. 2022;23(1). DOI:10.1186/s12863-021-01020-y
- Loren B, Ali N, Bocklitz T, et al. Discrimination between pathogenic and non-pathogenic E. coli strains by means of Raman microspectroscopy. Anal Bioanal Chem. 2020;412(30): 8241–8247.
- Perfect JR, Tenor JL, Miao Y, et al. Trehalose pathway as an antifungal target. Virulence. 2017;8(2):143–149.
- Argüelles JC. Trehalose as antifungal target: the picture is still incomplete. Virulence. 2017;8(2):237–238.
- Danielson ND, Collins J, Stothard AI, et al. Degradation-Resistant trehalose analogues block utilization of trehalose by hypervirulent Clostridioides difficile. Chem Commun. 2019;55(34):5009–5012.
- Gutierrez C, Ardourel M, Bremer E, et al. Analysis and DNA sequence of the osmoregulated treA gene encoding the periplasmic trehalase of Escherichia coli K12. MGG Mol Gen Genet. 1989;217(2–3):347–354.
- Botts MR, Huang M, Borchardt RK, et al. Developmental cell fate and virulence are linked to trehalose homeostasis in Cryptococcus neoformans. Eukaryot Cell. 2014;13(9):1158–1168.
- Lee SJ, Gralla JD. Osmoregulation of bacterial transcription via poised RNA polymerase. Mol Cell. 2004;14(2):153–162.
- Larsen PI, Sydnes LK, Landfald B, et al. Osmoregulation in Escherichia coli by accumulation of organic osmolytes: betaines, glutamic acid, and trehalose. Arch Microbiol. 1987;147(1):1–7.