
ABSTRACT
The emergence of antibiotic-resistant Aeromonas strains in clinical settings has presented an escalating burden on human and public health. The dissemination of antibiotic resistance in Aeromonas is predominantly facilitated by chromosome-borne accessory genetic elements, although the existing literature on this subject remains limited. Hence, the primary objective of this study is to comprehensively investigate the genomic characteristics of chromosome-borne accessory genetic elements in Aeromonas. Moreover, the study aims to uncover novel genetic environments associated with antibiotic resistance on these elements. Aeromonas were screened from nonduplicated strains collected from two tertiary hospitals in China. Complete sequencing and population genetics analysis were performed. BLAST analysis was employed to identify related elements. All newly identified elements were subjected to detailed sequence annotation, dissection, and comparison. We identified and newly designated 19 chromosomal elements, including 18 integrative and mobilizable elements (IMEs) that could be classified into four categories: Tn6737-related, Tn6836-related, Tn6840-related, and Tn6844a-related IMEs. Each class exhibited a distinct pattern in the types of resistance genes carried by the IMEs. Several novel antibiotic resistance genetic environments were uncovered in these elements. Notably, we report the first identification of the blaOXA-10 gene and blaVEB-1 gene in clinical A. veronii genome, the first presence of a tetA(E)–tetR(E) resistance gene environment within the backbone region in IMEs, and a new mcr-3.15 resistance gene environment. The implications of these findings are substantial, as they provide new insights into the evolution, structure, and dissemination of chromosomal-borne accessory elements.
Introduction
Aeromonas, a Gram-negative bacterium belonging to the Enterobacteriaceae family, is commonly found in diverse environmental habitats, including aquatic and soil ecosystems [Citation1,Citation2]. However, in recent years, Aeromonas has increasingly been isolated from clinical settings, with a particular emphasis on the four commonly occurring species A. caviae, A. dhakensis, A. veronii, and A. hydrophila [Citation1,Citation3–5]. Unfortunately, the concomitant use of antibiotics and natural selection pressures have resulted in a rapid escalation of antibiotic resistance in Aeromonas. Clinical isolates of Aeromonas are becoming increasingly resistant to commonly used antibiotics, including β-lactams and carbapenems [Citation6,Citation7]. Furthermore, Aeromonas serves as a reservoir of antibiotic resistance genes and has the potential to disseminate resistance to other bacterial species [Citation6,Citation8]. Therefore, antibiotic-resistant Aeromonas poses an ever-growing burden on human and public health, presenting a serious public safety threat that warrants immediate attention.
Mobile genetic elements (MGEs) are transmissible DNA fragments that often carry multiple antibiotic resistance genes. They are frequently involved in the transfer and dissemination of antibiotic-resistance genes, serving as essential vehicles for the spread of antibiotic resistance [Citation9,Citation10]. Integrative and mobilizable elements (IMEs) are a class of MGEs that are often located on the chromosome [Citation11]. IMEs are distinguished by their capacity to integrate and exploit the conjugative apparatus of unrelated co-resident conjugative elements to promote their transfer [Citation11]. IMEs are typically comprised of several critical components, including int (integrase), rlx (relaxase), oriT (origin of conjugative replication), and attL/attR (attachment site at the left/right end of the IME). Previous research has demonstrated that IMEs are highly diverse and widely distributed [Citation11]. IMEs have been shown to play a crucial role in the acquisition and accumulation of antibiotic resistance genes, thereby contributing to the emergence of novel genetic environments for antibiotic resistance [Citation12,Citation13]. Despite the crucial role in the dissemination of antibiotic resistance, IMEs and other chromosome-borne accessory genetic elements have received little attention, with few studies conducting detailed genomic analyses of the types, structures, and antibiotic resistance gene environments. To better understand the bacterial resistance adaption and evolution in Aeromonas, there is a pressing need for comprehensive genetic dissection and in-depth analysis of these chromosome-borne elements.
In this study, we screened clinical isolates of Aeromonas from two Chinese tertiary hospitals in Beijing and Zhengzhou. A total of five isolates (A. caviae 710029, A. veronii 829,120, A. dhakensis 628330, A. veronii 183,026, and A. caviae 21006) were collected. Their genomes were sequenced and finely annotated. Combined with the results of BLAST correlation analysis in the NCBI, a total of 18 new IMEs (Tn6737, Tn6836-Tn6843, Tn6844a-e, Tn6845, and Tn7400-Tn7402) and one inserted region (Inserted regionorf1293) were identified and newly named in Aeromonas. All the antibiotic-resistance genes carried on these 19 chromosome-borne accessory genetic elements were identified. Following a comparative genomics analysis, it was found that although the IMEs originated from diverse sources, certain IME structures displayed notable homology. The 18 IMEs can be classified into four categories based on the integrase gene and backbone regions: Tn6737-related, Tn6836-related, Tn6840-related, and Tn6844a-related IMEs. Within the Tn6844a-related IMEs, the IME backbone region was found to carry the tetA(E) gene, which is responsible for conferring tetracycline resistance phenotype [Citation14]. Notably, this is the first report of an Aeromonas harboring an IME with backbone carrying the tetA(E) gene. Simultaneously, Tn6844b, a member of the Tn6844a-related IMEs, containing the tetA(E) gene within its backbone, was also integrated into Tn6841, which belongs to the Tn6840-related IMEs. This resulted in the creation of a novel tetA(E) genetic environment. Moreover, the β-lactamase gene blaOXA-10 was identified on Tn7402, a gene that was previously exclusively detected in A. caviae during clinical isolation within the genus Aeromonas [Citation15]. The strain 183026 where Tn7402 is located is the first clinically isolated A. veronii carrying the blaOXA-10 gene. Furthermore, the newly discovered integron In1832 on Tn6737 harbors the extended-spectrum β-lactamase gene blaVEB-1 [Citation16], and strain 829120, where Tn6737 was identified, represents the first reported instance of A. veronii carrying the blaVEB-1 gene. Additionally, within the Inserted regionorf1293 on the chromosome of A. caviae 21006, the genetic environment of the mcr-3.15 gene was reported for the first time, which is involved in the expression of resistance to colistin (regarded as the few last line of antibiotics) [Citation7].
In this work, five strains of clinical Aeromonas were bioinformatically characterized newly identified, and 18 IMEs of four types on the chromosome were identified and newly assigned named. This study provides evidence that chromosome-borne accessory genetic elements are distributed among multiple species of Aeromonas. Due to their carriage of antibiotic-resistance genes, these elements are vital in the bacterial adaptation and may play a significant role in the persistence of bacterial populations. In addition, they can be a critical evolutionary force in bacterial genomes. Additionally, a series of novel antibiotic resistance genetic environments have been discovered on these elements, and these first reports may serve as a warning regarding the emerging situation of Aeromonas drug resistance. Furthermore, we anticipate that this research will yield new insights into the structure, evolution, and dissemination of antibiotic-resistant chromosome-borne accessory genetic elements of Aeromonas, as well as the novel genetic context of antibiotic resistance.
Materials and Methods
Sample collection and bacterial isolation
Five clinical isolates of Aeromonas were screened from nonduplicate strains collected from two tertiary hospitals located in Beijing and Zhengzhou, China (). Among these strains, A. caviae 710029, A. veronii 829120, and A. dhakensis 628330 were sourced from Hospital A in Beijing, while A. veronii 183026 and A. caviae 21006 were sourced from Hospital B in Zhengzhou. A. caviae 710029 was isolated in 2016 from the bile drainage fluid sample of a 43-year-old female patient with cholangitis. A. veronii 829120 was isolated in 2017 from the bile sample of a 58-year-old female patient with multiple intra- and extrahepatic bile duct stones. A. dhakensis 628330 was isolated in 2017 from the bile drainage fluid sample of a 28-year-old female patient who had undergone biliary tract surgery. A. veronii 183026 was isolated in 2019 from the blood sample of a 14-year-old female patient who had been diagnosed with acute B-cell lymphoblastic leukemia. Finally, A. caviae 21006 was isolated in 2019 from the sputum sample of a 53-year-old female patient with lymphoma. To identify the bacterial species of the five clinical Aeromonas isolates described above, average nucleotide identity (ANI) analysis (http://www.ezbiocloud.net/tools/ani) were performed using the genome sequences, respectively [Citation17].
Table 1. Background information on the five Aeromonas isolates.
Antibiotic susceptibility test
The drug minimum inhibitory concentration (MIC) of these five Aeromonas strains were determined by bioMérieux VITEK2 (Supplementary Table S1). The antibiotic susceptibility test results were determined by the Clinical and Laboratory Standards Institute (CLSI) guidelines [Citation18].
Sequencing and sequence assembly
Bacterial genomic DNA of five clinical isolates of Aeromonas were isolated using the UltraClean Microbial Kit from Qiagen (Hilden, Germany). The genomes were subsequently sequenced using an Illumina HiSeq sequencer from Illumina Inc. (CA, USA) with a paired-end library and an average insert size of 350 bp (range from 150 to 600 bp). Additionally, a shared DNA library with an average size of 15 kb (range from 10 to 20 kb) was sequenced using a PacBio RSII sequencer from Pacific Biosciences (CA, USA) [Citation19,Citation20]. Proovread software was utilized to correct the long PacBio reads with the paired-end short Illumina reads [Citation21]. The corrected PacBio reads were then assembled de novo using SMARdenovo (https://github.com/ruanjue/smartdenovo). To ensure high-quality sequencing data, NanoPack29 and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc) were employed [Citation22].
Phylogenetic Reconstruction and Analysis
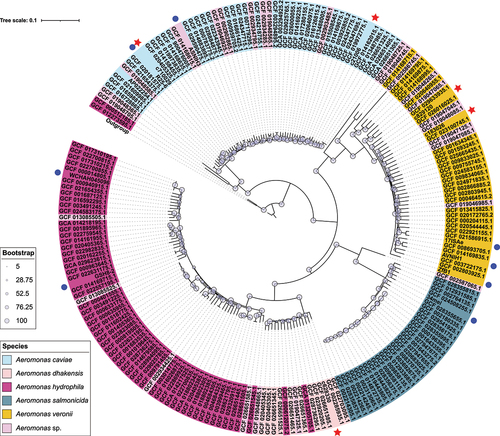
A total of 207 complete sequences of Aeromonas strains, which were identified as A. caviae, A. dhakensis, A. media, A. salmonicida, A. veronii, and Aeromonas sp., were downloaded from RefSeq or GenBank of NCBI (Supplementary Table S2). These sequences were submitted from various sources between 2006 and 2023. MUMmer v3.1 was used for alignments against the reference genome to create a core genome alignment [Citation23]. A total of 208,058 single nucleotide polymorphisms (SNPs) in the backbone regions were identified and extracted. A maximum-likelihood phylogenetic tree was constructed using IQ-TREE 2 [Citation24], which was based on the SNPs’ dataset with an ultrafast bootstrap iteration of 1000 (). The resulting phylogenetic tree, along with relevant information such as leaf name and species, was displayed using the iTOL programs [Citation25].
Figure 1. Population distribution of the five Aeromonas isolates with 207 Aeromonas genomes. The phylogenetic tree was generated using the maximum-likelihood method and accompanied by bootstrap analysis, which assigned a degree of support (percentage) to each associated taxa cluster. The bootstrap values were represented using a bootstrap bar with different-sized circles for each branch. The tree scale bar corresponded to the scale of sequence divergence. The species in NCBI of each branch were denoted with different background colours. The newly sequenced Aeromonas isolates were marked with red stars, while blue dots were used to indicate other isolates carrying chromosomal borne elements identified in this study.
Sequence annotation and comparison
Open reading frames (ORFs) and pseudogenes from the genomes of the five clinical isolates of Aeromonas were predicted using RAST 2.0 [Citation26]. The 19 chromosome-borne accessory genetic elements were identified, detailed dissected, and manually annotated using BLASTP/BLASTN [Citation27]. Online databases such as CARD [Citation28], ResFinder [Citation29], ISfinder [Citation30], INTEGRALL [Citation31], the Tn Number Registry [Citation32], and DANMEL [Citation10] were employed for the annotation of resistance genes, MGEs, and other features. MUSCLE 3.8.31 and BLASTN were used to perform multiple and pairwise sequence comparisons [Citation33]. Comparison gene diagrams of chromosome-borne accessory genetic elements were drawn using Inkscape 1.1 (https://inkscape.org/en).
Nucleotide sequence accession numbers
The chromosome sequences of the five clinical Aeromonas isolates, including A. caviae 710029 (chromosome c710029), A. veronii 829120 (chromosome c829120), A. dhakensis 628330 (chromosome c628330), A. veronii 183026 (chromosome c183026), and A. caviae 21006 (chromosome c21006), were submitted to the GenBank database. These sequences can be accessed through the following accession numbers: CP047981, CP054855, CP054854, CP072325, and CP072326, respectively.
Results
Overview of the five clinical Aeromonas isolates
Five clinical Aeromonas isolates (A. caviae 710029, A. veronii 829120, A. dhakensis 628330, A. veronii 183026, and A. caviae 21006) were obtained from clinical patients in tertiary hospitals in China (). A. caviae 710029, A. veronii 829120, and A. dhakensis 628330 were isolated from Hospital A in Beijing, while A. veronii 183026 and A. caviae 21006 were isolated from Hospital B in Zhengzhou. A. caviae 710029 was isolated in 2016, A. veronii 829120 and A. dhakensis 628330 were isolated in 2017, and A. veronii 183026 and A. caviae 21006 were isolated in 2019. Antimicrobial susceptibility tests on these five strains were performed (Supplementary Table S1), and the whole genome sequences of these five clinical Aeromonas isolates were obtained.
A. caviae 710029 had a chromosome (c710029, accession number CP047981) with a length of 4,520,672 bp, and the mean G+C content of c710029 was 61.6%. Additionally, a new plasmid was identified in the genome of A. caviae 710029 and was designated as p710029-qnrS. A. veronii 829120 had only one chromosome (c829120, accession number CP054855) with a length of 4,500,467 bp, and the mean G+C content of c829120 was 58.9%. A. dhakensis 628330 also had only one chromosome (c628330, accession number CP054854) with a length of 4,933,619 bp, and the mean G+C content of c628330 was 61.5%. A. veronii 183026 had a chromosome (c183026, accession number CP072325) with a length of 4,565,950 bp, and the mean G+C content of c183026 was 58.8%. Moreover, a new plasmid was identified in the genome of A. veronii 183026 and was designated as p183026–1. Lastly, A. caviae 21006 had a chromosome (c21006, accession number CP072326) with a length of 4,573,307 bp, and the mean G+C content of c21006 was 61.4%. Furthermore, a new plasmid was identified in the genome of A. caviae 21006 and was designated as p21006–1.
Phylogenetic analysis of the five clinical Aeromonas isolates
Phylogenetic analysis was conducted to investigate the diversity and evolutionary relationships among Aeromonas strains, using the genome sequences of the five isolates newly sequenced and all strains identified as A. caviae, A. dhakensis, A. salmonicida, A. veronii, and Aeromonas sp. whose assembly levels are complete available from GenBank or RefSeq (last accessed on 22 April 2023). A total of 212 Aeromonas strains were included in the analysis, with the chromosome sequence of A. caviae WP8-S18-ESBL-04 (accession number AP022254, GCF_014169735.1, the standard strain of A. caviae) serving as the reference. An outgroup was also selected using strain TR3_1 (GCF_017310215.1, the standard strain of A. media). After multiple sequence alignments, 208,058 SNPs located in the core genome regions were identified and extracted to construct a maximum likelihood (ML) phylogenetic tree. Supplementary Table S2 contains additional information about the strains included in the analysis.
Based on the phylogenetic tree, each species forms a unique cluster with distinct differences from other species (). The newly sequenced five strains of Aeromonas in this study are located on different branches of the tree according to their respective species, which is consistent with the previous species identification results. Notably, c710029, R25–6 and GCF_020181575.1 are relatively close on the phylogenetic tree, sharing a common branch. In 2018, R25–6, previously isolated from wastewater in China, was identified as A. caviae [Citation34]. In subsequent study, two types of IMEs were identified in the R25–6 genome, which are similar to those carried by strain 710,029. GCF_020181575.1 was isolated from a clinical setting at a tertiary hospital in Zhejiang, China in 2020, and was reported to carry the blaNDM resistance gene on its chromosome [Citation35]. Furthermore, strain 710,029 was obtained from the clinical setting, raising the possibility of a transmission link between clinical and environmental strains, although this relationship is yet to be fully elucidated. GCF_029633935.1, which is the most closely related strain to 829,120, was isolated from zebrafish, and GCF_002285935.1, which is the most closely related strain to 628,330, was isolated from Myocastor coypus [Citation36]. Both of these strains were isolated from animals in natural environments, which may suggest a possible association with clinical strains. The background information of GCF_019048085.1, which is the most closely related strain to 183,026, has not been reported. Strain 21,006 has a relatively large evolutionary distance from other strains, making it difficult to infer its transmission and evolutionary relationships.
In addition, two strains of A. hydrophila (GCA_013205705.1 and GCF_000612075.2) have been mistakenly classified within the A. dhakensis clade, likely due to the recent recognition of A. dhakensis as a distinct species [Citation4]. Misidentification of A. dhakensis as A. hydrophila or A. caviae using phenotypic methods was common prior to its recognition as a separate entity [Citation4]. Despite the population structure indicating that these strains may be more accurately classified as A. dhakensis, they have been designated as A. hydrophila in NCBI. Furthermore, as the precise classification of strains within the Aeromonas sp. remains unknown, they are distributed across all branches of the phylogenetic tree.
Overview of the 19 chromosome-borne accessory genetic elements
The five clinical Aeromonas isolates were subjected to detailed genome annotation and analysis to identify the location and genetic context of chromosome-borne accessory genetic elements. A total of eight chromosomal-borne elements were identified and named (seven IMEs and one inserted region, ) through manual curation. Among these strains, c710029 carried two IMEs (Tn6838 and Tn6844c), c829120 carried three IMEs (Tn6737, Tn6837, and Tn6844b), c628330 carried two IMEs, with one of them being Tn6844b, which was located within another IME, Tn6841. c183026 carried only one IME, Tn7402. Notably, c21006 did not carry any IMEs, but an accessory inserted region was identified and named as Inserted regionorf1293.
Table 2. Major features of the 19 chromosome-borne accessory genetic elements.
In order to conduct a systematic structural analysis of the IMEs carried by Aeromonas chromosome and explore their evolutionary patterns, BLAST analysis was performed in GenBank (sharing >90% coverage and > 70% identity) based on the int genes of seven non-redundant IMEs. After manual verification and in combination with relevant literature reports, 11 IMEs in GenBank were newly identified and named, which exhibit high similarity and homology with the aforementioned seven IMEs. These 18 IMEs are all located on the chromosome and range in length from 14,031 bp to 78,805 bp (). The 18 IMEs are hosted by various Aeromonas species, which are indicated on the phylogenetic tree presented in . Based on the int gene and backbone structure, these 18 IMEs can be divided into four categories: Tn6737-related IMEs, Tn6836-related IMEs, Tn6840-related IMEs, and Tn6844a-related IMEs. The Tn6737-related IMEs contain only one IME, Tn6737. The Tn6836-related IMEs include seven IMEs, named Tn6836-Tn6839, and Tn7400-Tn7402. The Tn6840-related IMEs comprise four IMEs, named Tn6840-Tn6843. The Tn6844a-related IMEs consist of six IMEs, named Tn6844a-e, and Tn6845. Within each category of IMEs, the majority display a high level of consistency in their backbone region structure, with variations predominantly observed in the accessory region.
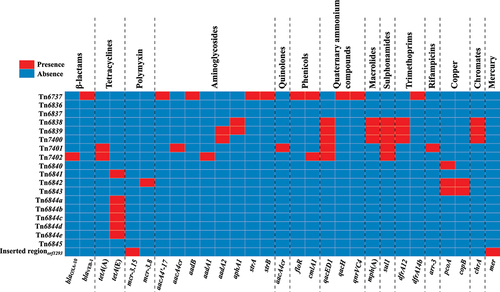
In this study, 29 antibiotic resistance genes belonging to 14 different categories, including β-lactam, tetracycline, polymyxin, aminoglycoside, quinolone, phenicol, quaternary ammonium compound, macrolide, sulfonamide, trimethoprim, rifampicin, copper, chromate, and mercury, were identified on 19 chromosomal-borne elements, except for Tn6836, Tn6837, and Tn6845 ( and Supplementary Table S3). Tn6737 harbored the most resistance genes, including 10 genes of five different types (blaVEB-1, aacA4’−17, aadB, strA, strB, floR, cmlA1, qacH, qnrVC4, and dfrA14b). A total of 14 resistance genes belonging to 11 different types were detected in Tn6836-related IMEs. Tn6840-related IMEs contained relatively fewer resistance genes, with only four genes of three different types. Tn6844a-related IMEs harbored only one tetracycline resistance gene, tetA(E), and its corresponding regulatory gene tetR(E), without any other types of resistance genes. In addition, two resistance genes, mcr-3.15 and mer, were identified on the Inserted regionorf1293. These results suggest a correlation between the distribution of resistance genes and the type of IME.
Figure 2. Heatmap of the prevalence of drug resistance genes. Each row represents a unique element, while each column represents a resistance gene. Resistance genes are categorized by dashed lines for clarity. The heatmap utilizes a colour scheme in which red squares indicate gene presence and blue squares indicate gene absence.
To gain insight into the evolution and transmission of the 18 IMEs, we undertook a meticulous analysis of each element through detailed manual annotation, characterization, and precise comparison. Through this approach, we were able to explore the similarities and differences in gene structure among the four categories of IMEs. Furthermore, we subjected the Inserted regionorf1293 to the same rigorous analysis.
Genetic Characterization of Tn6836-related IMEs
Tn6836-related IMEs comprise Tn6836-Tn6839 and Tn7400-Tn7402, as shown in . The Tn6836-related IMEs range in length from 14,936 bp to 35,738 bp and all of them are integrated into the Aeromonas chromosome (). Tn6836 (from cWL1843), Tn6839 (from cR25–6), Tn7400 (from cASNIH3), and Tn7401 (from c17ISAe) were derived from sequenced Aeromonas strains available in GenBank [Citation34,Citation37–39], while Tn6837 (from c829120), Tn6838 (from c710029), and Tn7402 (from c183026) were obtained from newly sequenced strains in this study. Among these, Tn6837 and Tn6838 were found in Aeromonas strains 829,120 and 710,029, respectively, which were isolated from patients in the same hospital located in Beijing. In contrast, Tn7402 was detected in strain 183,026, which was isolated from a patient in a hospital in Zhengzhou. The hosts of these IMEs belong to multiple species, including A. schubertii, A. veronii, and A. caviae.
Figure 3. Comparison of Tn6836 and six related IMEs. Genes were visually depicted as arrows, while mobile genetic elements and other features were distinguished by their assigned colour based on their functional classification. Regions of homology, defined by nucleotide identity greater than 95%, were highlighted with a shaded background. Pseudogenes were denoted by a single quotation mark preceding their name.
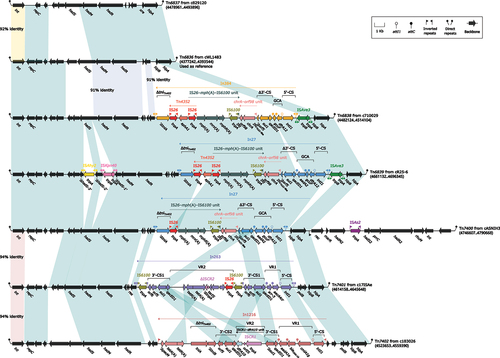
The IME Tn6836 is 16,303 bp in length and is located on the chromosome of A. schubertii WL1483 between 4,377,242 bp and 4,393,544 bp. Tn6836 is flanked by a pair of 20 bp attL/attR sites. Functionally, Tn6836 contains only the backbone region without any accessory region (). The backbone region carries genes such as int (integrase), repC (replication), hsdS–hsdM–hsdR (type I restriction-modification system), hipA–hipB (type II toxin-antitoxin system), and other genes assigned in black in . Unlike Tn6836, other IMEs have a pair of 18 bp attL/attR sites at both ends. The backbone regions of other IMEs show high homology with Tn6836 and have similar gene structures. Tn6837, containing only the backbone region, has the most similar gene structure to Tn6836. However, accessory regions have been identified in other IMEs except Tn6836 and Tn6837. Tn6838, Tn7401, and Tn7402 contain only one accessory region that mainly consists of an integron. In Tn6839 and Tn7400, apart from a longer accessory region with integron as the main element, some scattered small accessory regions resulting from the insertion of insertion sequences (IS) into their backbone regions were also identified. In Tn6839, the insertion of IS elements such as ISAhy2 and ISKpn40 splits the hsdS gene in the backbone region into three parts, whereas in Tn7400, ISAse2 is inserted between the hsdM2 and hsdS2 genes. The integron in the accessory regions of both Tn6838 and Tn6839 are followed by an ISAve3.
In Tn6839 and Tn7400, In27 is the main component in the accessory region, as shown in and Supplementary Table S3. In27 in Tn6839 and Tn7400 is a concise class 1 integron that contains the primary components of the 5’-conserved region (5‘−CS), 3’-conserved region (3‘−CS), and a gene cassette array (GCA) consisting of dfrA12–gcuF–aadA2 [Citation43,Citation44]. Downstream of the 3’-CS, there is a putative resistance unit chrA–orf98 unit. In the downstream region of this unit is a reverse putative resistance unit IS26–mph(A) – IS6100 unit, which carries the mph(A) gene and is involved in conferring resistance to macrolides [Citation9]. Additionally, in Tn6839, downstream of the IS26–mph(A) – IS6100 unit, there is a composite transposon Tn4352, which shares an IS26 with the IS26–mph(A) – IS6100 unit. In Tn6839 and Tn7400, In27 ends with a truncated tniA encoding DD(35)E transposase TniA. In27 is flanked by a pair of 25-bp IRi (inverted repeat at the integrase end) and IRt (inverted repeat at the tni end). Outside of IRi and IRt, there are 5-bp DRs (direct repeats, target site duplication signals for transposition) on either side. In Tn6838, the accessory region is similar to that of Tn6839, with the exception that In27 is replaced by In384. In384 is also a concise class 1 integron, and it has a similar structure with In27 in Tn6839, except that the GCA content is dfrA12–gcuF. In263 and In1216 are two integrons located in the accessory regions of Tn7401 and Tn7402, respectively, and they are both complex class 1 integrons. In263 and In1216 have a pair of 25-bp IRi/IRt on both sides. In the case of In263, there are also 5-bp DRs on the outside of IRi/IRt, while no DRs were discovered on either side of In1216. In addition to the 5’−CS and 3’-CS backbone regions, In263 contains two variable regions (VRs): VR1, which consists of arr-3–aacA4cr, and VR2, which consists of IS26–ΔISCR2–tetA(A)–tetR(A) and is associated with tetracycline resistance [Citation14]. VR1 of In1216 is composed of cmlA1g–blaOXA-10–aadA1e, while VR2 is composed of ISCR1–dfrA10 unit. ISCR is a type of element that is bounded by an origin (oriIS) downstream and a terminus (terIS) upstream, which are responsible for capturing resistance genes [Citation9,Citation44]. At the end of the backbone region of In1216, there is also a gene structure tetA(A)–tetR(A) similar to that in In263. It is worth mentioning that the β-lactam resistance gene blaOXA-10, located in In1216, was reported for the first time in clinically isolated A. veronii, which had previously only been reported in A. caviae within Aeromonas genus [Citation15]. This is a newly emerged resistance gene environment in clinical A. veronii, which is located on the GCA of In1216, suggesting strong potential for dissemination.
Genetic Characterization of Tn6844a-related IMEs
Tn6844a-related IMEs, including Tn6844a-e and Tn6845 (), have been identified in various Aeromonas species, such as A. veronii, A. dhakensis, A. caviae, and A. salmonicida. Tn6844a (from cAVNIH1), Tn6844d (from cR25–6), Tn6844e (from cZfB1), and Tn6845 (from c34mel) were obtained from previously submitted sequences in GenBank [Citation34,Citation41,Citation42], while Tn6844b (from c628330 and c829120) and Tn6844c (from c710029) were identified in newly sequenced strains in this study. Notably, Tn6844b was found in the chromosomes of both strains 628,330 and 829,120.
Figure 4. Comparison of Tn6844a and five related IMEs. Genes were illustrated as arrows, while mobile genetic elements and other features were designated with distinct colours corresponding to their functional classification. Regions of homology, which were defined by nucleotide identity greater than 95%, were highlighted with a shaded background. Pseudogenes were indicated by a single quotation mark preceding their name.
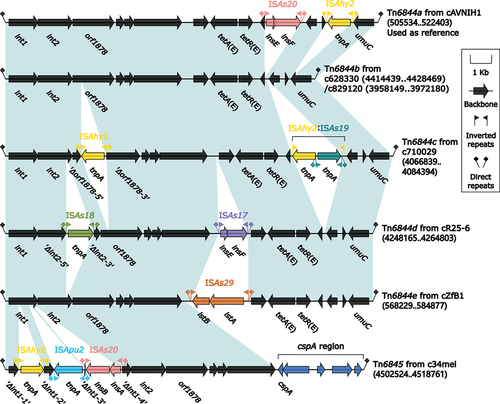
Tn6844a-related IMEs are located on the chromosome of Aeromonas, and their lengths vary from 14,031 bp to 17,556 bp (). Each of these IMEs has a pair of attL/attR sites that are 16 bp in length on both sides. With the exception of Tn6844b, these IMEs have accessory regions, but it is only a small portion of the overall structure, as the majority of the sequence is composed of the backbone region. The backbone region of these IMEs, except for Tn6845, consists of int1–int2–orf1878–tetA(E)–tetR(E)–umuC. Tn6845 has a shorter backbone region, which is comprised of int1–int2–orf1878. Accessory regions of these IMEs are mostly composed of scattered IS elements. Insertions of ISAs20 and ISAhy2 are present in Tn6844a, while ISAhy2 and ISAs19 are found in Tn6844c. Tn6844d contains ISAs18 and ISAs17 insertions, and Tn6844e shows the presence of ISAs29 insertion. Additionally, Tn6845 exhibits the insertions of ISAhy2, ISApu2, and ISAs20. Furthermore, a cspA region is located at the end of Tn6845, which is related to the expression of cold shock protein CspA.
Tn6844a-related IMEs have caught the attention of researchers due to the presence of tetA(E) – tetR(E) resistance genes within their backbone region. These genes are responsible for conferring resistance to tetracycline [Citation14]. These novel antibiotic resistance gene environments are discovered within the backbone region of Tn6844a-e, marking the first report of tetA(E)–tetR(E) resistance gene environments carried within the backbone region of IMEs in Aeromonas. The emergence and dissemination of these IMEs may contribute to the spread and amplification of tetA(E) – tetR(E) resistance genes, posing a potential threat to public health.
Genetic characterization of Tn6840-related IMEs
Tn6840-related IMEs comprise four IMEs, namely Tn6840 from cWCHAH045096, Tn6841 from c628330 (a novel Aeromonas strain identified in this study), Tn6842 from c17ISAe, and Tn6843 from cGSH8–2 (). Tn6840, Tn6842, and Tn6843 sequences were sourced from the GenBank [Citation39,Citation40]. The lengths of these IMEs vary from 15,792 bp to 78,805 bp (). The host range of these IMEs includes multiple species such as A. hydrophila, A. dhakensis, and A. veronii.
Figure 5. Comparison of Tn6840 and three related IMEs. The figure utilizes arrows to represent genes, while mobile genetic elements and other features are distinguished with distinct colours based on their functional classification. Regions of homology, which are defined by nucleotide identity greater than 95%, are highlighted with a shaded background. Pseudogenes are identified by a single quotation mark preceding their name.
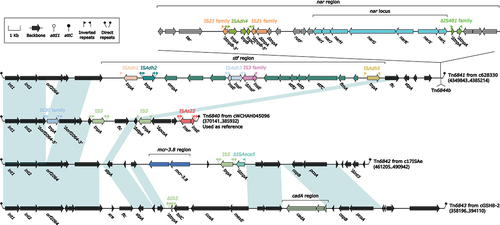
The Tn6840 element is situated on the A. hydrophila WCHAH045096 chromosome, spanning a length of 15,792 bp from positions 370,141 bp to 385,932 bp and flanked by a pair of attL/attR sites, each 10 bp long. It can be subdivided into two regions based on gene function, the backbone region and the accessory region. The backbone region comprises the int1–int2–orf2064–stpA genes, which are interrupted by several IS elements. Among these, an IS element belonging to the IS30 family disrupts the orf2064 gene into two segments. Additionally, IS5 elements are present upstream of the flc gene and downstream of the stpA gene, while an insertion sequence ISAs22 is found at the end of Tn6840. Other IMEs related to Tn6840 also contain attL/attR sites and share a similar backbone structure. However, the accessory regions differ. In Tn6842, the accessory region harbors IS5 and ISAeca5 elements, as well as an mcr-3.8 region that carries the mcr-3.8 gene (Supplementary Table S3), which is involved in the expression of phosphoethanolamine, playing a crucial role in colistin resistance [Citation7]. Another related IME, Tn6843, contains a cadA region, which participates in the expression of a cadmium-translocating P-type ATPase.
Tn6841 is distinct from Tn6840, Tn6842, and Tn6843, as it carries longer accessory regions (Supplementary Table S3). The stf region, which is located in the accessory region of Tn6841, contains ompA–stfG–stfD–stfC–fimA genes that are responsible for encoding fimbrial-related proteins. This region also contains several ISs, such as ISAdh1, ISAdh2, ISAdh3, and ISAdh5. Another region in Tn6841, the nar region, contains the core gene structure narI–narJ–narH–narG–narK– narX–narL, which encodes respiratory nitrate reductase. Interestingly, Tn6844b (a Tn6844a-related IME mentioned above) is inserted at the end of Tn6841. The Tn6844b backbone carries the tetA(E)– tetR(E) resistance gene environment, making Tn6841 a large genetic environment that includes tetA(E)–tetR(E) resistance genes. This nested arrangement of resistance genes may accelerate their spread to some extent.
Genetic characterization of Tn6737-related IMEs
Tn6737 is the sole member of the Tn6737-related IMEs. It spans 34,888 bp and is situated within the chromosome of A. veronii 829120, specifically at positions 2,207,485 bp to 2,242,372 bp (). Tn6737 is flanked by a pair of 18 bp attL/attR sequences, although its attL/attR sequences differ by three positions. Tn6737 can be divided into backbone region and accessory region based on gene structure (). The backbone region contains the genes int–zot–rstB–rep. Interestingly, a newly discovered unit transposon, Tn6738, is inserted within the backbone region. Tn6738 is bounded by a pair of 38 bp IRL/IRR (inverted repeat left/right), and a pair of 5 bp DRs serves as the boundary outside the IRL/IRR. Tn6738 shares a similar structure with Tn1722, as it also carries the genes tnpA–tnpR–res–mcp, and belongs to the Tn21 subfamily of the Tn3 family [Citation45]. However, Tn6738 has truncated portions of the mcp gene and the res site due to the insertion of a long gene structure, Tn5393c–virD2–floR–lysR–traG–copG–In1832. Tn5393c is a previously identified unit transposon that belongs to the Tn163 subfamily in the Tn3 family [Citation46,Citation47]. It is flanked by an 81 bp IRL/IRR sequence, and the backbone region contains the genes tnpA–res–tnpR–strA–strB. The virD2–floR–lysR–traG–copG sequence that follows is related to conjugative transfer. At the end of the structure, a new integron, In1832, is identified.
Figure 6. Organization of Tn6737. The figure utilizes arrows to represent genes, while transposons, integrons, and other features are distinguished with distinct colours based on their functional classification. Regions of homology, which are defined by nucleotide identity greater than 95%, are highlighted with a shaded background. Pseudogenes are identified by a single quotation mark preceding their name.
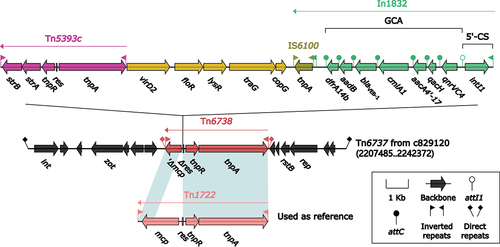
In1832, a novel concise class 1 integron was identified that consists of three main components: 5’-CS, GCA, and IS6100. The 5’-CS includes the integrase gene int1, while the GCA carries a plethora of resistance genes with a structural configuration of qnrVC4–qacH–aacA4’−17–cmlA1–blaVEB-1–aadB–dfrA14b. Downstream of GCA is a 25-bp IRt, which is repeated directly to the IRi outside of the upstream of In1832. IS6100, serving as the end of In1832, is inserted downstream of IRt. It is worth noting that the extended-spectrum β-lactamase gene blaVEB-1 is carried in the gene cassette of In1832, probably due to the capture of the integron [Citation16]. Prior to this discovery, blaVEB-1 had not been reported in A. veronii, making this the first report of its presence in this species. Furthermore, the fact that it is located in a new integron, In1832, suggests a novel mechanism for resistance gene dissemination, potentially accelerating its spread in this novel drug-resistant environment.
Genetic characterization of inserted regionorf1293
Inserted regionorf1293 is a 58,974 bp sequence situated on the chromosome of A. caviae 21,006, spanning positions 1,220,204 bp and 1,279,177 bp ( and ). This region lacks core backbone genes of MGEs and is therefore difficult to define as an MGE such as a transposon, integron, or IME. Nonetheless, the presence of 15 bp DRs at both ends of Inserted regionorf1293 serves as boundaries for the Inserted regionorf1293 and indicates that it should be considered as a discrete entity. Notably, the upstream DR is located within an ORF called orf1293 on the A. caviae 21,006 chromosome, from which the name Inserted regionorf1293 is derived.
Figure 7. Organization of Inserted regionorf1293. Arrows are utilized to represent genes. Transposons and other features are distinguished with distinct colours based on their functional classification. Pseudogenes are identified by a single quotation mark preceding their name.
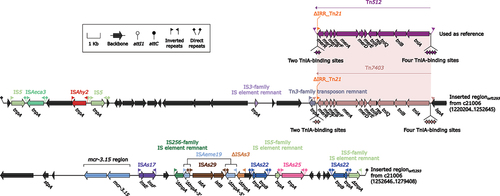
In the Inserted regionorf1293, multiple MGEs were discovered, including a novel transposon named Tn7403. This transposon is 8,445 bp in length and is delimited by a pair of 25 bp IRL/IRR without any DRs found outside of the IRL/IRR. Tn7403 was classified as a unit transposon of the Tn7 family [Citation48,Citation49], and contains four repeat regions serving as TniA-binding sites upstream, as well as two repeat regions serving as TniA-binding sites downstream of Tn7403. Like other unit transposons in the Tn7 family, Tn7403 contains the essential transposition determinants, including tniA (transposase), tniB (transposition regulator), tniQ (target-site selection protein), and tniR (resolvase) [Citation48,Citation49]. Additionally, a mer locus, including merE–merD–merA–merF–merP–merT–merR, was identified on Tn7403, which may be associated with mercury resistance expression. Comparative genomics analysis revealed a high similarity between Tn7403 and Tn512, with an identity value of up to 94%. However, a comparison of the core genes tniA and tniB in Tn7403 showed that they had 86% and 90% identity with tniA and tniB in Tn512, respectively. Hence, despite the high similarity between Tn7403 and Tn512, they should be regarded as distinct transposons.
Downstream of Tn7403, a region containing the mcr-3.15 gene has been identified. The mcr-3.15 gene is associated with polymyxin resistance, which is recognized as a last-resort antibiotic treatment for multidrug-resistant Gram-negative infections [Citation7]. Notably, this is the first report of the mcr-3.15 gene in the genetic context of A. caviae. Additionally, several ISs have been identified on the Inserted regionorf1293, such as IS5, ISAeca3, ISAhy2, ISAs17, ISAeme19, ISAs29, ISAs22, and ISAs25. The high frequency of these ISs suggests that multiple horizontal gene transfer (HGT) events may have occurred in this region, highlighting the potential for rapid evolution and antibiotic resistance development.
Discussion
Chromosome-borne accessory genetic elements of Aeromonas are widespread in different species within the genus, creating hotspots for HGT and facilitating the emergence of antibiotic resistant genes [Citation11,Citation13]. Despite the abundance of literature on MGEs in Aeromonas, research on chromosomal elements has been limited and imprecise [Citation8,Citation12,Citation13]. To better understand the diversity and classification of these elements and their role in facilitating antibiotic resistance, more comprehensive gene dissection and comparative genomics analyses are needed. Such insights would be invaluable for controlling and preventing this opportunistic pathogen, and are therefore of great importance to the scientific community.
In this study, we screened five Aeromonas strains isolated from clinical samples in two hospitals located in Beijing and Zhengzhou (). Whole-genome sequencing and population genetic analysis were performed, in conjunction with the Aeromonas sequences available in GenBank. Our findings revealed that these five strains belonged to different species within the Aeromonas genus ( and Supplementary Table S2). One of the notable observations was the close genetic distance between c710029, R25–6, and GCF_020181575.1 in the phylogenetic tree, despite their different host origins. While c710029 originated from a clinical patient in a Beijing hospital, GCF_020181575.1 was isolated from a clinical patient in a hospital in Zhejiang Province, China [Citation35], and R25–6 was obtained from a wastewater environment [Citation34]. Furthermore, R25–6 harbored several IMEs similar to those in c710029, suggesting a possible transmission and dissemination of Aeromonas strains between clinical and natural environments. Similarly, we observed close genetic distances between c829120 and GCF_029633935.1, and between c628330 and GCF_002285935.1. Notably, both GCF_029633935.1 and GCF_002285935.1 were obtained from animal hosts in natural environments [Citation36], indicating a potential transmission and dissemination of Aeromonas between animal and human clinical settings. Given the One Health concept in public health [Citation50], it is crucial to monitor and analyze environmental strains of Aeromonas for potential transmission and dissemination between natural, animal, and clinical environments to prevent potential public health issues. Therefore, rigorous monitoring and analysis of environmental strains of Aeromonas are necessary to prepare for potential challenges.
From the analysis of five newly sequenced strains and strains in GenBank, 18 IMEs and one inserted region were identified from the Aeromonas chromosomes (). Based on a comparative genomics analysis, the 18 IMEs were classified into four categories, each characterized by distinct structural features. Interestingly, these four types of IMEs were found in multiple species of the genus Aeromonas, indicating their wide distribution. At the same time, the high degree of structural similarity observed in different regions for the IMEs suggests that it has spread across multiple regions of bacterial populations. This indicates an overall increase in the resistance levels of Aeromonas, and the possibility of more pan-resistant Aeromonas emerging in the future, which will pose a greater challenge for resistance prevention and control measures.
After careful identification, it was observed that the distribution of drug-resistant genes on these chromosome elements was closely correlated with their categories ( and Supplementary Table S3). Remarkably, each class of IME contained only specific drug-resistant genes. For instance, Tn6844a-related IMEs were found to exclusively carry tetracycline resistance genes, while Tn6840-related IMEs were found to harbor resistance genes for tetracycline, polymyxin, and copper. This intriguing finding suggests a potential tendency between IMEs and drug-resistant genes in Aeromonas. In future studies, it would be valuable to collect and analyze more IMEs in Aeromonas to explore in greater detail the association between the presence of drug-resistant genes and the categories of IMEs.
In the Tn6836-related IMEs (), the blaOXA-10 gene is carried in the VR1 region of the In1216 integron on Tn7402. This is the first report of blaOXA-10 in a clinical A. veronii genome, and the In1216 integron, along with the IME Tn7402, constitutes a novel resistance environment for blaOXA-10. The Tn6844-related IME contains a tetA(E)–tetR(E) resistance gene environment in its backbone (). Notably, this resistance environment is not dependent on foreign genetic elements but rather is part of the IME backbone genes that move with the entire IME. The dissemination of such IMEs can provide a selective advantage to their host by enabling faster adaptation to environmental pressures. The spread of these IMEs in Aeromonas poses increasing challenges for clinical treatment and prevention. In Tn6840-related IME, the resistance environment on Tn6841 is more complex (), with a Tn6844b insertion at the end. Because the Tn6844b backbone carries the tetA(E)–tetR(E) resistance gene environment, the insertion of Tn6844b marks Tn6841 as an IME carrying this resistance gene environment as well. The nested arrangement of different types of IMEs can facilitate the spread of resistance genes across multiple IMEs, accelerating their dissemination. This arrangement also suggests that different IMEs are not independent of each other but are interconnected, with HGT and gene exchange between them. In the Tn6737-related IMEs, only Tn6737 has been identified (), and no other IMEs have been identified. Notably, a newly discovered integron, In1832, is carried on Tn6737, with several genes, including the blaVEB-1 resistance gene, within its gene cassette. This is the first report of blaVEB-1 in A. veronii. In1832 provides a novel resistance environment for blaVEB-1 in Tn6737, which highlights the importance of this complex resistance environment. Additionally, a novel mcr-3.15 resistance gene environment is present on the Inserted regionorf1293 (). This is the first report of mcr-3.15 genetic environment isolated from clinical strains of Aeromonas, and previous studies have shown that strains carrying mcr have higher resistance to colistin, the last line of useful antibiotics for treating bacterial infections [Citation7]. Therefore, this study provides a detailed description of the mcr-3.15 resistance gene environment and contributes to the field of bacterial resistance infection treatment, providing a foundation for further research on this resistance gene.
Chromosomal accessory elements are prevalent in the genus Aeromonas and encompass multiple types, each with varying resistance environments. Despite their significance in the dissemination of resistance genes, the formation of new resistant gene environments, and the adaptation and evolution of Aeromonas, they have not received adequate attention to date. In this study, we conducted an in-depth analysis of the genetic structure of chromosomal accessory elements in Aeromonas from two monitored hospitals and identified several common types of chromosomal elements. We also provided accurate annotations of novel resistance gene environments that emerged on these elements. Given the potential for these elements to continue appearing in Aeromonas in the future, they represent a hidden threat that requires sufficient attention and vigilance. Thus, we hope that our study will provide essential information on the chromosomal accessory element situation in Aeromonas, support the evolution of resistance elements in Aeromonas, and provide guidance for better prevention and control of resistance dissemination and proliferation.
Conclusion
In this study, we performed genomic sequencing and population genetics analysis on five strains of Aeromonas and characterized and deciphered the genomic features of 19 elements carried on their chromosomes. These elements, newly discovered and named, are widely distributed in various Aeromonas strains and play an important role in the adaptation and evolution of these opportunistic pathogens under natural selection pressure. Moreover, these elements carry a complex array of novel resistance gene environments. Notably, this study reports for the first time the presence of blaOXA-10 in a clinical A. veronii genome, the presence of a tetA(E)–tetR(E) resistance gene environment carried by backbone region in a category of IME, and the occurrence of blaVEB-1 gene in clinical A. veronii. Furthermore, we have characterized the mcr-3.15 resistance gene environment in clinical A. caviae. Overall, our findings have significant implications for understanding resistance chromosomal borne accessory elements and their novel resistance gene environments in Aeromonas, and could provide important insights into the control and prevention of these important opportunistic pathogens.
List of abbreviations
IME: integrative and mobilizable element; MGE: mobile genetic element; ANI: average nucleotide identity; SNP: single nucleotide polymorphism; ORF: open reading frame; ML: maximum likelihood; IS: insertion sequence; 5’-CS: 5‘-conserved region; 3’-CS: 3’-conserved region; GCA: gene cassette array; IRi: inverted repeat at the integrase end; IRt: inverted repeat at the tni end; DR: direct repeat; VR: variable region; IRL: inverted repeat left; IRR: inverted repeat right; HGT: horizontal gene transfer.
Author contributions
Conceptualization, F.C., K.M., B.G., and T.Y.; methodology, Z.Y., P.W., B.G., and D.Z.; data analysis, F.C., T.Y., X.L., J.H., and Y.Z.; resources, T.Y., Z.Y., K.M. and D.Z.; writing – original draft, F.C. and T.Y.; writing – review and editing, K.M. and B.G. All authors have read and agreed to the published version of the manuscript.
Consent for Publication
All the authors of the manuscript have read and agreed to its content.
Ethics Statement
The specimens were collected with the patient’s informed consent, and all experimental protocols involving human specimens were reviewed and approved by the Ethics Committee of the institutions involved in the study, in compliance with the medical research regulations of the Ministry of Health in China. Similarly, research procedures involving biohazardous materials were approved by the Biosafety Committee of the institutions involved. This study was conducted in China and adhered to all relevant ethical and safety guidelines.
Supplemental Material
Download Zip (65.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data Availability statement
The data presented in this study are available on request from the corresponding author. The sequences analyzed in this study can be found in the public GenBank database. The accession numbers were provided in this article.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2023.2271688
Additional information
Funding
References
- Fernandez-Bravo A, Figueras MJ. An update on the genus Aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms. 2020;8(1):129. doi: 10.3390/microorganisms8010129
- Pessoa RBG, de Oliveira WF, Correia M, et al. Aeromonas and human health disorders: clinical approaches. Front Microbiol. 2022;13:868890. doi: 10.3389/fmicb.2022.868890
- Song Y, Wang LF, Zhou K, et al. Epidemiological characteristics, virulence potential, antimicrobial resistance profiles, and phylogenetic analysis of Aeromonas caviae isolated from extra-intestinal infections. Front Cell Infect Microbiol. 2023;13:1084352. doi: 10.3389/fcimb.2023.1084352
- Puah SM, Khor WC, Aung KT, et al. Aeromonas dhakensis: clinical isolates with high carbapenem resistance. Pathogens. 2022;11(8):833. doi: 10.3390/pathogens11080833
- Xu Z, Shen W, Zhang R, et al. Clonal dissemination of Aeromonas hydrophila with binary carriage of bla (KPC-2)-bearing plasmids in a Chinese Hospital. Front Microbiol. 2022;13:918561. doi: 10.3389/fmicb.2022.918561
- Wu Y, Dong N, Cai C, et al. Aeromonas spp. From hospital sewage act as a reservoir of genes resistant to last-line antibiotics. Drug Resist Updat. 2023;67:100925. doi: 10.1016/j.drup.2023.100925
- Gonzalez-Avila LU, Loyola-Cruz MA, Hernandez-Cortez C, et al. Colistin resistance in Aeromonas spp. Int J Mol Sci. 2021;22(11):5974. doi: 10.3390/ijms22115974
- Piotrowska M, Popowska M. Insight into the mobilome of Aeromonas strains. Front Microbiol. 2015;6:494. doi: 10.3389/fmicb.2015.00494
- Partridge SR, Kwong SM, Firth N, et al. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31(4). doi: 10.1128/CMR.00088-17
- Wang P, Jiang X, Mu K, et al. DANMEL: a manually curated reference database for analyzing mobile genetic elements associated with bacterial drug resistance. mLife. 2022;1(4):460–16. doi: 10.1002/mlf2.12046
- Guedon G, Libante V, Coluzzi C, et al. The obscure world of integrative and mobilizable elements, highly widespread elements that Pirate bacterial conjugative systems. genes (Basel). Genes. 2017;8(11):337. doi: 10.3390/genes8110337
- Michaelis C, Grohmann E. Horizontal gene transfer of antibiotic resistance genes in biofilms. Antibiotics. 2023;12(2):328. doi: 10.3390/antibiotics12020328
- Bello-Lopez JM, Cabrero-Martinez OA, Ibanez-Cervantes G, et al. Horizontal gene transfer and its association with antibiotic resistance in the genus Aeromonas spp. Microorganisms. 2019;7(9):363. doi: 10.3390/microorganisms7090363
- Urban-Chmiel R, Marek A, Stepien-Pysniak D, et al. Antibiotic resistance in bacteria—A review. Antibiotics. 2022;11(8):1079. doi: 10.3390/antibiotics11081079
- Tang L, Huang J, She J, et al. Co-occurrence of the bla KPC-2 and mcr-3.3 gene in Aeromonas caviae SCAc2001 isolated from patients with diarrheal disease. Infect Drug Resist. 2020;13:1527–1536. doi: 10.2147/IDR.S245553
- Messaoudi A, Belguith H, Ben Hamida J. Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theor Biol Med Model. 2013;10(1):22. doi: 10.1186/1742-4682-10-22
- Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–19131. doi: 10.1073/pnas.0906412106
- CLSI, Methods for antimicrobial dilution and disk susceptibility testing of infrequently isolated or fastidious bacteria. 3rd. ( CLSI guideline M45). Clinical and Laboratory Standards Institute: Wayne, PA: 2016.
- Fu J, Zhang J, Yang L, et al. Precision methylome and in vivo methylation kinetics characterization of Klebsiella pneumoniae. Int J Genomics Proteomics. 2022;20(2):418–434. doi: 10.1016/j.gpb.2021.04.002
- Li C, Jiang X, Yang T, et al. Genomic epidemiology of carbapenemase-producing Klebsiella pneumoniae in china. Genomics Proteomics Bioinf. 2022;20(6):1154–1167. doi: 10.1016/j.gpb.2022.02.005
- Hackl T, Hedrich R, Schultz J, et al. Proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics. 2014;30(21):3004–3011. doi: 10.1093/bioinformatics/btu392
- De Coster W, D’Hert S, Schultz DT, et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34(15):2666–2669. doi: 10.1093/bioinformatics/bty149
- Delcher AL, Salzberg SL, Phillippy AM. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics. 2003;(1). Chapter 10:Unit 10 13. doi: 10.1002/0471250953.bi1003s00
- Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic Era. Mol Biol Evol. 2020;37(5):1530–1534. doi: 10.1093/molbev/msaa015
- Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi: 10.1093/nar/gkab301
- Brettin T, Davis JJ, Disz T, et al. Rasttk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5(1):8365. doi: 10.1038/srep08365
- Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41(Web Server issue):W29–33. doi: 10.1093/nar/gkt282
- Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48(D1):D517–D525. doi: 10.1093/nar/gkz935
- Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi: 10.1093/jac/dks261
- Siguier P, Perochon J, Lestrade L, et al. Isfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(Database issue):D32–36. doi: 10.1093/nar/gkj014
- Moura A, Soares M, Pereira C, et al. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009;25(8):1096–1098. doi: 10.1093/bioinformatics/btp105
- Roberts AP, Chandler M, Courvalin P, et al. Revised nomenclature for transposable genetic elements. Plasmid. 2008;60(3):167–173. doi: 10.1016/j.plasmid.2008.08.001
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004;5(1):113. doi: 10.1186/1471-2105-5-113
- Shi Y, Tian Z, Leclercq SO, et al. Genetic characterization and potential molecular dissemination mechanism of tet(31) gene in Aeromonas caviae from an oxytetracycline wastewater treatment system. J Environ Sci. 2019;76:259–266. doi: 10.1016/j.jes.2018.05.008
- Luo X, Mu K, Zhao Y, et al. Emergence of bla NDM- 1-Carrying Aeromonas caviae K433 isolated from patient with community-acquired pneumonia. Front Microbiol. 2022;13:825389. doi: 10.3389/fmicb.2022.825389
- Lim SR, Lee DH, Park SY, et al. Wild nutria (Myocastor coypus) is a potential reservoir of carbapenem-resistant and zoonotic Aeromonas spp. In Korea. Microorganisms. 2019;7(8):224. doi: 10.3390/microorganisms7080224
- Liu L, Li N, Zhang D, et al. Complete genome sequence of the highly virulent Aeromonas schubertii strain WL1483, isolated from Diseased Snakehead Fish (Channa argus) in China. Genome Announc. 2016;4(1). doi: 10.1128/genomeA.01567-15
- Weingarten RA, Johnson RC, Conlan S, et al. Genomic analysis of Hospital plumbing reveals diverse reservoir of bacterial plasmids conferring carbapenem resistance. MBio. 2018;9(1). doi: 10.1128/mBio.02011-17
- Roh HJ, Kim BS, Kim A, et al. Whole-genome analysis of multi-drug-resistant Aeromonas veronii isolated from diseased discus (symphysodon discus) imported to Korea. J Fish Dis. 2019;42(1):147–153. doi: 10.1111/jfd.12908
- Sekizuka T, Inamine Y, Segawa T, et al. Potential KPC-2 carbapenemase reservoir of environmental Aeromonas hydrophila and Aeromonas caviae isolates from the effluent of an urban wastewater treatment plant in Japan. Environ Microbiol Rep. 2019;11(4):589–597. doi: 10.1111/1758-2229.12772
- Hughes HY, Conlan SP, Lau AF, et al. Detection and Whole-genome sequencing of carbapenemase-producing Aeromonas hydrophila isolates from routine perirectal surveillance culture. J Clin Microbiol. 2016;54(4):1167–1170. doi: 10.1128/JCM.03229-15
- Pfeiffer F, Zamora-Lagos MA, Blettinger M, et al. The complete and fully assembled genome sequence of Aeromonas salmonicida subsp. pectinolytica and its comparative analysis with other Aeromonas species: investigation of the mobilome in environmental and pathogenic strains. BMC Genomics. 2018;19(1):20. doi: 10.1186/s12864-017-4301-6
- Partridge SR, Tsafnat G, Coiera E, et al. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev. 2009;33(4):757–784. doi: 10.1111/j.1574-6976.2009.00175.x
- Partridge SR. Analysis of antibiotic resistance regions in Gram-negative bacteria. FEMS Microbiol Rev. 2011;35(5):820–855. doi: 10.1111/j.1574-6976.2011.00277.x
- Nicolas E, Lambin M, Dandoy D, et al. The Tn3-family of replicative transposons. Microbiol Spectr. 2015;3(4). doi: 10.1128/microbiolspec.MDNA3-0060-2014
- Stokes HW, Elbourne LD, Hall RM. Tn1403, a multiple-antibiotic resistance transposon made up of three distinct transposons. Antimicrob Agents Chemother. 2007;51(5):1827–1829. doi: 10.1128/AAC.01279-06
- L’Abee-Lund TM, Sorum H. Functional Tn5393-like transposon in the R plasmid pRAS2 from the fish pathogen Aeromonas salmonicida subspecies salmonicida isolated in Norway. Appl Environ Microbiol. 2000;66(12):5533–5535. doi: 10.1128/AEM.66.12.5533-5535.2000
- Peters JE, Fricker AD, Kapili BJ, et al. Heteromeric transposase elements: generators of genomic islands across diverse bacteria. Mol Microbiol. 2014;93(6):1084–1092. doi: 10.1111/mmi.12740
- Peters JE. Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol Microbiol. 2019;112(6):1635–1644. doi: 10.1111/mmi.14383
- Hernando-Amado S, Coque TM, Baquero F, et al. Defining and combating antibiotic resistance from one health and Global health perspectives. Nat Microbiol. 2019;4(9):1432–1442. doi: 10.1038/s41564-019-0503-9