
ABSTRACT
Pasteurella multocida (P. multocida) is a bacterial pathogen responsible for a range of infections in humans and various animal hosts, causing significant economic losses in farming. Integrative and conjugative elements (ICEs) are important horizontal gene transfer elements, potentially enabling host bacteria to enhance adaptability by acquiring multiple functional genes. However, the understanding of ICEs in P. multocida and their impact on the transmission of this pathogen remains limited. In this study, 42 poultry-sourced P. multocida genomes obtained by high-throughput sequencing together with 393 publicly available P. multocida genomes were used to analyse the horizontal transfer of ICEs. Eighty-two ICEs were identified in P. multocida, including SXT/R391 and Tn916 subtypes, as well as three subtypes of ICEHin1056 family, with the latter being widely prevalent in P. multocida and carrying multiple resistance genes. The correlations between insertion sequences and resistant genes in ICEs were also identified, and some ICEs introduced the carbapenem gene blaOXA-2 and the bleomycin gene bleO to P. multocida. Phylogenetic and collinearity analyses of these bioinformatics found that ICEs in P. multocida were transmitted vertically and horizontally and have evolved with host specialization. These findings provide insight into the transmission and evolution mode of ICEs in P. multocida and highlight the importance of understanding these elements for controlling the spread of antibiotic resistance.
Introduction
Pasteurella multocida (P. multocida) is one of the most common opportunistic pathogens that have been responsible for causing multi-host diseases such as avian cholera, porcine atrophic rhinitis, bovine haemorrhagic sepsis [Citation1,Citation2], and human infections [Citation3] since its discovery in 1880 [Citation4]. The adaptation and evolution through mobile genetic elements, such as integrative and conjugative elements (ICEs), is a major area of interest in the study of widespread bacteria [Citation5–7]. ICEs have been shown to be an important means for bacteria to obtain foreign genes and enhance their environmental adaptability [Citation8,Citation9]. Previous research has identified ICEs in many bacteria phyla, including Proteobacteria, Actinobacteria, and Firmicutes [Citation5,Citation7,Citation10,Citation11]. Some ICEs have been associated with high virulence phenotypes [Citation12], adaptability [Citation11,Citation13], invasion, environmental stresses [Citation14] and multiple resistance to antimicrobials [Citation15]. However, there have been few studies on ICEs in P. multocida, only reported one transferable resistant ICEPmu1 from American bovine-sourced strain P. multocida 36950 [Citation16] and some resistant ICEs from European and American bovine, and Asian swine P. multocida identified by bioinformation analysis [Citation17–20], which have not yet been involved in correlation analysis and holistic studies of ICEs for P. multocida population. Meanwhile, ICEs could be transmitted horizontally and vertically to a variety of host bacteria, and the genetic plasticity of ICEs could be enhanced by insertion sequences (ISs) carrying resistance genes, but the transmission mode of ICEs in P. multocida has not been discussed, and the association between ISs and resistance genes in P. multocida has not been systematically studied [Citation21–23]. Therefore, this study aims to contribute to this growing area of research by exploring the ICEs in 435 P. multocida isolated from 13 hosts, from 1889 to 2022, to explore transmission mode of ICEs in P. multocida, assess the extent to which ICEs affect adaptability of the clinical strains, and seek to holistic study data by the association analysis between ICEs and P. multocida population.
Materials and methods
Genome sequences of Pasteurella multocida
A total of 435 P. multocida clinical isolates were used in this study, of which 42 were laboratory-sequenced strains (Table S1, Table S2). Among the 42 sequenced strains, 35 were isolated from duck livers and swollen feet with thick juice in China, and the remaining isolates comprised six poultry-sourced P. multocida acquired from the American Type Culture Collection (ATCC) and one P. multocida CVCC44801 (C48–1) acquired from the China Veterinary Culture Collection Center (CVCC). High-quality genomic DNA was extracted with a TIANamp Bacteria DNA Kit (Tiangen Biotech Co, Ltd., Beijing, China). The paired-end (PE350) library was prepared using the NEBNext Ultra IIkit (all New England Biolabs) according to the manufacturer’s instructions [Citation24]. Sequencing of the 42 isolates was conducted using an Illumina HiSeq 2500. Illumina reads were corrected by Quake [Citation25]. The raw reads were trimmed and quality controlled by Fastp (https://github.com/OpenGene/fastp). De Novo assembly was performed using SPAdes version 3.1.0 with the recommended parameters (-k 21,33,55,77,99,127 -careful) [Citation26]. The whole genome sequences of the remaining 393 P. multocida strains were retrieved from data published by GenBank at NCBI before December 2022 (Table S2). To assess genome completeness of all strains, CheckM14 was used as a genome quality estimator with “–taxonomy_wf” parameter; only genome with contamination <5% and completeness >95% were used for further analysis [Citation27].
MLST and serotype analysis
MLST was used to determine the STs of P. multocida strains based on the RIRDC MLST scheme [Citation28]. Each ST was uniquely determined by the seven MLST loci each with a different allelic profile, and a clonal complexes (CCs) type was formed by multiple different STs with a common ancestor (https://pubmlst.org/bigsdb?db=pubmlst_pmultocida_seqdef&page=query&scheme_id=2&submit=1) [Citation29]. The STs and CCs of all P. multocida were determined using the MLST online service at the Center for Genomic Epidemiology with “Pasteurella multocida#1” parameter (https://cge.food.dtu.dk/services/MLST/) [Citation30].
The serotype information of 393 PM strains obtained from NCBI was statistically analysed according to the database records, while the serotype information of 42 clinical strains was obtained by simulating amplification using primer sequences [Citation31,Citation32] combined with Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome).
Identification of ICEs
The whole-genome sequences of all strains were uploaded to IslandViewer 4 [Citation33], where the potential GIs were predicted with default parameters. The GIs with ICEs elements were screened based on the “Product” in the excel sheet from IslandViewer 4 [Citation8] and then these P. multocida genomes with ICE elements was uploaded to ICEBerg 2.0 for reanalysis to determine the existence of ICEs [Citation34]. The software Mauve 2015-2-26 was used as a multi-collinearity tool to compare the ICEs and the reference strain P. multocida Pm70 one by one in 82 P. multocida genomes. The software was utilized to analyse transmutation and development among strains, such as inversion, rearrangement, insertion and deletion in the form of locally collinear blocks [Citation35,Citation36], particularly focusing on the initiation and termination sites of ICEs based on the unique genomic characteristics of ICEs [Citation37] as shown the example illustration Figure S3.
Infrastructure and variable region analysis of ICEs
The genetic structure of 82 ICEs was precisely determined based on various characteristics, including the functions of ICEs, abnormal length and GC content, ISs and other mobility-related genes, such as essential functional integrase, recombinase, relaxase or helicase, and T4SS [Citation38,Citation39]. The specific methods were as follows, hypothetical proteins in ICEs were validated the functional categories by performing a BlastP search and utilizing the Conserved Domains Database (CDD) of NCBI [Citation40]; ISs analysis was performed using ISfinder [Citation41]; resistance genes were identified using Resfinder [Citation42] and T4SS was identified using oriTfinfer [Citation43]. Important elements within differential ICEs in multiple strains were compared by SWISSProt to determine gene level divergence. The detailed statistical data of the 82 ICEs is available in Table S4.
The collinearity analysis of 42 ICEHin1056-like ICEs families of in P. multocida using the software Mauve 2015-2-26 [Citation37] (). The gene structures and collinear alignments of SXT/R391 family ICEs and Tn916 family ICEs were plotted using the software Easyfig 2.2.5 [Citation44] to visualize the conserved and variable regions of the ICEs (). The similarity of T4SS (tfc1-tfc24) in each ICEs were compared using blastn, and the data were uploaded to the BIC online mapping platform (http://www.bic.ac.cn/BIC/#/) to form heatmap to visualize these results.
Figure 1. Comparison analysis of 42 ICEHin1056 family ICEs.
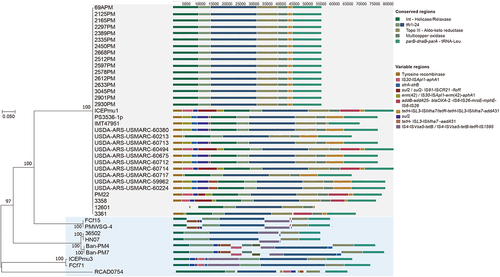
Conservative structural and gene sets of all P. multocida and ICEs
7Roary, a large-scale pan-genome analysis tool, was used to analyse the related genomes of all strains [Citation45]. The cut-off of BLAST was set to 50% identity without paralogous clustering. The nucleotide sequences corresponding to the whole genome of 435 P. multocida strains and the core genes of 42 ICEs of ICEHin1056 family, respectively, were concatenated to construct a core genome concatamer. The core genome concatamer was aligned using the MAFFT online service [Citation46] with default parameters, and the neighbour-joining trees of 435 strains, the maximum likelihood trees of 42 strains with ICEs and the maximum likelihood trees of 42 ICEs were constructed using IQtree-1.6.12 programme with a p-distance of less than 0.219 and a JC distance of less than 1, with 2000 bootstrapping replications [Citation47].
For Bayesian tree, TempEst v1.5.3 [Citation48] was used to detect the temporal signals for 42 ICEs and 42 strains with ICEs. And then BEAST v1.10.4 [Citation49] was used for Bayesian time-scaled phylogenetic analysis, using ML phylogeny as starting tree, the uncorrelated relaxed as clock model [Citation50], “Speciation: Yule Process” as tree prior [Citation51], and implementing the “GTR + Gamma” substitution on model. Markov chain Monte Carlo duplicate runs of 10,000,000 states each, sampling every 1000 steps was computed. The trees were obtained using TreeAnnotator (http://beast.community/index.html) after 10% burn‐in and visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Prevalence analysis
The software Origin 2022 was used to perform statistical analysis on the separation time and geographical location among ICEs and P. multocida, and to obtain the correlation between the ICEs in P. multocida and the timeline and host sources. The prevalence analysis results were visualized using Origin 2022.
Results
General features of P. multocida
A total of 435 P. multocida were collected in this study, of which 393 were from NCBI and 42 from local sequencing (Figure S1). The strains genomic data analysis and their information based on NCBI revealed that 17 of 393 strains from NCBI were duplicates uploaded by different institutions (Figure S2), 27 had genome quality assessment with completeness <95% and contamination >5% (Table S1). Therefore, a total of 418 strains were used for basic information statistics (Table S2), and 391 strains were further bioinformatically analysed (Figure S1).
From 1889 to 2022, a total of 418 isolates of P. multocida were isolated from Europe, the Americas, Asia, Africa, Oceania, and some unknown geographical regions, originating from 13 different host sources (Table S3). The initial isolation time of P. multocida from Europe and the Americas was earlier than that from the Asian-sourced only based on these available strains collected in this study. Clinical strains from Europe and the Americas were mainly isolated in large quantities from 2010 to 2013, while those from Asian-sourced were mainly obtained from 2015 to 2018. The serotypes of P. multocida are usually represented by capsular type (ABDEF) [Citation32,Citation52,Citation53]: lipopolysaccharide type (L1-L8) [Citation32]: Multi-locus sequence typing (MLST) genotype [Citation28], in which the MLST types are obtained by screening for the presence of 7 unique conserved genes in genomic data of P. multocida. For these strains in this study, their serotypes were mostly incomplete. The largest number of the available MLST types for P. multocida was ST13 (93 strains), which was mainly derived from the A:L3 (56 strains) of Europe and the Americas bovine-sourced strains. This was followed by ST122 (48 strains), mainly B:L2 (33 strains) of Asia bovine-sourced strains. The main available serotypes of swine-sourced P. multocda were A:L3:ST13 (17 strains) and D:L6:ST50 (20 strains) from Asia; the poultry-sourced were mainly A:L1:ST129 (31 strains) from Asia; the Alpacas-sourced were mainly A:L6:ST203 (2 strains) from the Europe and the Americas. The rabbit -sourced were distributed in Europe, the Americas and Asia, mainly A/F:L3:ST9 (12 strains) from Europe. The available MLST types of the remaining host sources strains had no obvious characteristics.
Integrative and conjugative elements in 391 P. multocida
The whole genomes of 391 P. multocida strains were analysed to identify possible GIs (Genomic Islands) containing ICEs. Using ISlanderViewer 4 and ICEberg 2.0 tools, 82 ICEs were detected among the P. multocida stains; Mauve 2015-2-26 software was used to compared the 82 ICEs with the reference strain Pm70, respectively, enabling identification of the complete initiation and termination sites of ICEs structures in each strain. To check the accuracy of the analyses method, the sequence error of initiation and termination sites between the predicted ICE region in P. multocida 36950 and ICEPmu1 in P. multocida 36950, one and only experimentally validated transferable ICE in P. multocida currently, was less than 50 bp, which verified the feasibility of the ICEs acquisition method in this study (Figure S3). And out of the 42 clinical isolates in our laboratory, only P. multocida HN141014 contains ICE.
Among the 391 P. multocida isolates, 302 was known their geographical location, host and isolation time, while these information of 89 were incomplete. The 89 strains did not contain ICEs according to the above analysis. Out of the 302 P. multocida isolates analysed, 189 were from Asia, and 37 contained ICEs; 113 were from Europe and the Americas, and 45 contained ICEs. The 82 ICEs in 391 isolates were found in cattle, poultry, swine, sheep and human-sourced strain, suggesting that more than 21% of P. multocida in this study contained ICEs. The functions and structures of these ICEs above were analysed based on their gene sequence. Of the 82 ICEs, 64 were identified as resistance-related ICEs, while two were likely metabolism-related, one was likely virulence-related and one was likely both virulence and resistance-related (Table S4). However, the completeness of genome sequencing varied among the 391 strains, because some ICEs structures located at the start or end of a “contig” were possibly incomplete, and some ICEs structures across multiple “contig” region were potentially flawed in the middle of these ICEs. Therefore, the analysis and collation of genome sequence of the strains and important functional genes of ICEs resulted in 26 complete ICEs located within a “contig,” 19 suspected complete ICEs located at a contig endpoint but with a largely complete ICE structure, and 39 incomplete ICEs across multiple contigs or with non-ICE structures (Table S4). Subsequent analyses were based on 45 ICEs that is 26 complete and 19 suspected complete ICEs.
ICEs of the ICEHin1056 family in P. multocida
This study identified 45 complete and suspected complete ICEs in P. multocida, of which 42 ICEs were found to belong to the ICEHin1056 family, and the remaining ICEs were two SXT/R391 family and one Tn916 family (Table S4). The ICEHin1056 family is the most widespread in P. multocida and was found to be present in all 42 resistant ICEs with resistance genes. These ICEs ranged in size from 36-108kb and had a GC% of 36–48%, with tRNA-Leu at both ends of the ICEs, except for P. multocida HN141014 and Fcf71, which had tRNA-Asn at both ends of the ICEs. The conserved structures and variable regions of 42 ICEs were compared and analysed. The collinear alignment of the complete structures with variable regions and the phylogenetic tree of their conserved structures revealed consistent phylogenetic relationship between their variable regions and the conserved structures (). Almost all ICEs in P. multocida genomes from Asia contained IS4-ISVsa5-tetB in the variable regions, while ICEs in P. multocida genomes from Europe and the Americas exhibited a variety of antimicrobial resistance genes but all contained ISL3-ISMha7-tetH with 100% sequence identity. The conserved structures of all these ICEs contained relaxase/helicase/TraI, DNA breaking-rejoining enzyme of tyrosine recombinase/integrase family, and almost complete T4SS (Tfc1-Tfc24), without Tfc1, Tfc18, Tfc20 or Tfc21–24 (, Table S5).
Figure 2. T4SS of 42 ICEHin1056 family ICEs in P. multocida.
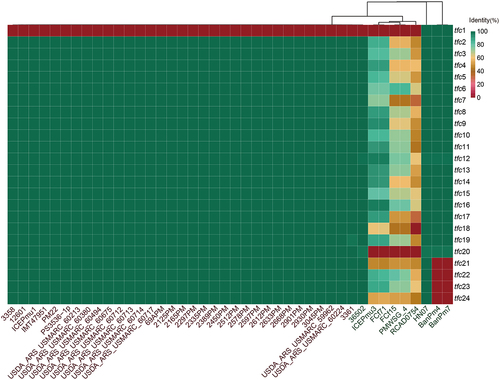
For the T4SSs, one of the conservative structures of the ICEHin1056 family, they were aligned and collated, and the alignment scores were recorded (Table S5). The phylogenetic analysis of the conserved region of T4SS was carried out, and the presence of Tcf1-Tcf24 in ICEs was visualized to determine the difference of T4SS. As shown in , the phylogenetic tree based on T4SS is still classified according to geographical location and host source. The Europe and the Americas P. multocida containing ICEs lacked Tfc1, of which ICEPmu1 without Tfc1 has been verified to be transferable, indicating that Tfc1 is not necessary for ICEs transfer [Citation16]. The Asia P. multocida containing ICEs lacked Tfc1, Tfc18, Tfc20, or Tfc21–24, with only strain HN07 has intact Tfc1–24. Previous unpublished studies in our laboratory have verified the transferability of ICEPmu3 from Asian P. multocida HN141014, indicating that Tfc1 and Tfc20 are not necessary for ICE transfer, and the nucleotide sequence differences (>60%) of Tfc18, Tfc21 and Tfc24 have not significantly affected the integrative and conjugative transfer.
ICEs of the SXT/R391 family and Tn916 family in P. multocida
In P. multocida, two ICEs of SXT/R391 family (Indian cattle-sourced P. multocida CUL-TANUVAS 2020, NZ_CP092967.1; Chinese pig-sourced P. multocida HND11, NZ_PPWN00000000.1) and one ICE of the Tn916 family (Pakistan cattle-sourced P. multocida TX1, JQAH00000000) have been identified. The ICEs in strain CUL-TANUVAS 2020 and HND11 belong to the typical SXT/R391 family and possess the insertion sites prfC-5,’ both contain T4SS-related elements which are clearly distinct from the ICEHin1056 family. Otherwise, strain HND11 lacks relaxase/helicase/TraI, CUL-TANUVAS 2020 contains related structures; HND11 contain a resistant ICE with resistant genes and IS4-ISVsa5, and CUL-TANUVAS 2020 is suspected to contain a metabolic ICE without resistance-related genes or ISs. They are most similar to ICEPmiCHN3300 from Vibrio cholerae and ICEVchBan9 from Bacillus mirabilis in the SXT/R391 family according to the blast alignment score value of ICEberg databases, but lack the partial resistant structure strB-strA-sul2 in the variable region ().
Figure 3. Collinear alignment of SXT/R39 family ICEs and Tn916 family ICEs.
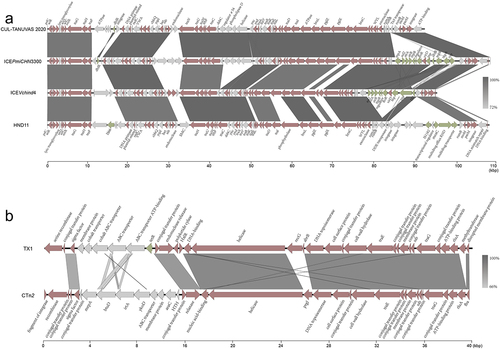
On the other hand, P. multocida T×1contains the ICE structure of the Tn916 family with an intergenic region as its insertion site. The ICE has an integrase of Serine recombinase family that different from the integrase of all other ICEs in P. muloticda, but does not contain T4SS, resistant genes, or ISs. It has the highest similarity to CTn2 of Clostridium difficile in the Tn916 family according to ICEberg databases, without obvious functional genes besides a hypothetical virulent protein ().
Resistance gene transfer of ICEs in P. multocida
Resistance genes analysis revealed that 45 ICEs might have different multiple resistant phenotypes, for example aminoglycosamines, sulphonamides, macrolides, chloramphenicol, amidols, penicillin, cephalosporins, and glycopeptides. Sequence blast alignment of the resistance genes of all ICEs in the SWISSProt database was used to determined amino acid differences, and these differences in amino acid coverage and identify were counted (Table S6). The resistance genes tetH, strB, aadA and sul2 showed almost consistent amino acid changes in multiple strains, while aph(3’)-Ia, strA, and floR had amino acid differences in strain 3358 in Table S6, and erm(42) had amino acid differences in strain USDA-ARS-USMARC-60494 due to incomplete sequencing with a large number of Ns instead of bases. And the polypeptide antibiotic bleomycin (bleO) in strain PMWSG-4 had a few amino acid differences, while the bleO in strain Fcf15 matched completely.
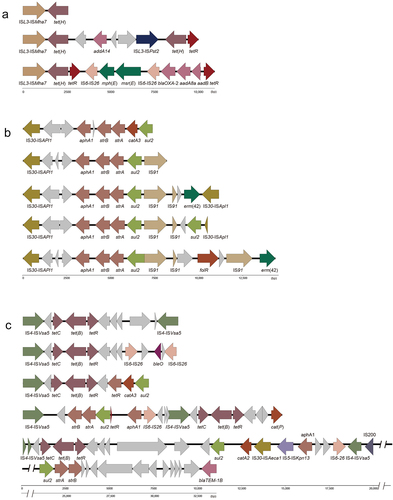
ISs are associated with the spread of resistance genes [Citation22]. ICEs in P. multocida contain several ISs (Table S7, Table S8), where ISL3-ISMha7 is the most frequently occurring and 100% consistent with ISL3-ISMha7 in Mannheimia haemolytica (28 ICEs, 62%). Others ISs in P. multocida include ISL3-ISPst2 100% consistent with ISL3-ISPst2 in Pseudomonas stutzeri, IS30-ISApl1 100% consistent with Actinobacillus pleuropneumoniae, IS91-ISVsa3 100% consistent with Vibrio Salmonicida, IS6-IS26 100% consistent with Proteus vulgaris, IS6-IS15DIV/IS10 R 100% consistent with Salmonella typhimurium, IS4-ISVsa5 100% consistent with Aliivibrio salmonicida, IS1182-ISCfr-1 100% consistent with Citrobacter freundii, IS5-ISKpn13 100% consistent with Klebsiella pneumoniae, IS30-ISAeca1 100% consistent with Aeromonas caviae, and IS91 100% consistent with the IS91-ISCR21 in ICEPmu1 marked as IS91-ISCR21 in this paper (Table S8). In addition, there are IS66, IS200/IS605, IS3, IS1595 with no exact match (Table S7). Theses ISs and resistance genes in ICEs showed certain patterns, among which ISL3-ISMha7/ISPst2 is immediately adjacent to the coordinated transfer of tetracycline gene tet(H); IS4-ISVsa5 is immediately adjacent to the tetracycline gene tet(B); IS30-ISApl1 is immediately adjacent to the aminoglycoside gene aphA1-strB-strA and sulphonamide gene sul2; IS6 is immediately adjacent to the macrolide gene mph(E)-msr(E), the β-lactam gene blaOXA-2-aadA8b-aadB and the polypeptide antibiotic gene bleO; IS91 is immediately adjacent to the macrolide gene erm(42) and acylaminol gene floR ().
Figure 4. Module structure of resistance genes of ICEs in P. multocida.
Prevalence of integrative and conjugative elements in P. multocida
Due to the horizontal gene transfer properties of ICEs carrying cargo genes and their widespread transmission in bacteria [Citation5,Citation8], ICEs genome sequences could partially demonstrate changes in ecological niches of P. multocida. Analysis of 302 P. multocida strains (Figure S1) revealed a significant temporal bias from 2010 to 2018, with the spread of ICEs, particularly resistant ICEs, increasing significantly during these years (). Although the isolation time of P. multocida strains in Europe and the Americas is earlier than that of Asia, the occurrence of the isolates with ICEs was later in Europe and the Americas than in Asia (). The P. multocida strains with ICEs in Europe and the Americas exhibited a rapid transmission trend mainly concentrated in 2011 and after this the P. multocida strains with ICEs was decreased year by year. The limited number of ICEs in Asia P. multocida strains appeared earlier than those in the Europe and the Americas, mainly concentrated in 2017, and after this the resistant ICEs in Asian strains decreased year by year. As shown in , P. multocida chronically exists in cattle and poultry-sourced hosts (), among which the cattle-sourced strains containing ICEs have also been found to be already present since 2003 and poultry-sourced P. multocida since 2004 (). The first human-sourced P. multocida containing ICEs was discovered in 2014, and another human-source P. multocida containing ICEs was found in 2016. At present, only one sheep-sourced P. multocida containing ICE has been discovered in 2016. After 2010, ICEs were found in swine-source P. multocida, with the highest occurrence of swine-sourced P. multocida containing ICEs in 2017. The highest occurrence of cattle-sourced P. multocida containing ICEs was in 2011, and the highest occurrence of poultry-sourced P. multocida containing ICEs was in 2017. Then, the number of P. multocida containing ICEs decreased as the number of clinical P. multocida decreased after about 2017.
Figure 5. Prevalence analysis of ICEs in P. multocida.
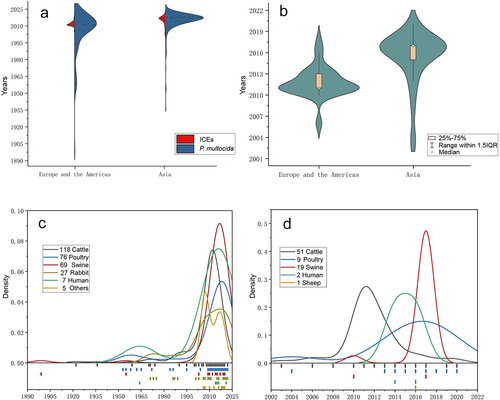
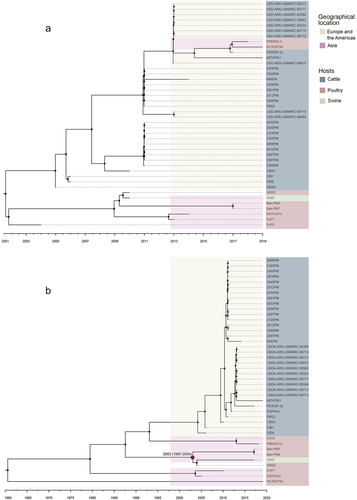
The evolutionary relationship among 45 complete and suspected complete ICEs was analysed using Bayesian tree construction. Sequence alignment of ICEs revealed that the strains HND11, TX1, CUL_TANUVAS 2020 had a less conserved structure compared to ICEs in other P. multocida strains. The Bayesian tree was constructed based on the remaining 42 ICEHin1056-like ICEs and 42 whole genomes of P. multocida with time signal (, Figure. S4). The ICEs of all cattle-sourced strains from Europe and the Americas were clustered into a clade, while all poultry-sourced and one swine-sourced strains from Asia were clustered into another clade (). The whole genome of these strains had a phylogenetic relationship consistent with the conserved structure of ICEs except for Asian poultry-sourced strains PMWSG-4 and RCAD0754 (). The results showed that the prevalence of different ICEs was correlated with the geographical locations and host sources of P. multocida. The different phylogenetic relationships observed between the whole genomes of P. multocida PMWSG-4 and RCAD0754 and the ICEs in them suggested that the ICEs in these two strains may be acquired through horizontal transfer of existing Asian ICEs after their introduction from Europe and the Americas into Asia. On the other hand, the whole genome of the remaining strains had a consistent phylogenetic relationship with their ICEs, indicating that the ICEs in these strains may have prevailed through vertical transmission.
Figure 6. The Bayesian tree of ICEs and P. multocida.
Discussion
ICEs are known to be highly problematic due to their broad host range and genetic plasticity since their discovery in 1991 [Citation5,Citation23,Citation54]. However, there has been limited comprehensive research on ICEs in P. multocida bacterial populations with limited available data. In this study, a total of 435 P. multocida strains were selected including the published all available data in NCBI before December 2022 and 42 strains isolated from our laboratory, and the correlation between 391 strains and 82 ICEs was analysed after screening (). Only 302 of the 391 strains had detailed and complete isolation information. Out of the 302 P. multocida strains analysed, 113 were from Europe and the Americas and 45 contained ICEs. These clinical isolates were observed in large quantities from 2010 to 2015 and the ICEs mainly concentrated from 2010 to 2013. That is to say, the frequency of ICEs increased during the mass transmission of P. multocida, and the main functions of the ICEs in P. multocida at that time were most likely required by P. multocida to resist environmental stress. Europe and the Americas P. multocida strains mainly contain resistant ICEs, which decreased after the concentration appeared in 2011, possibly that the spread of resistant ICEs was affected during the period of the widespread use of antibiotic, until the transmission of resistant ICEs weakened after European strategic action plan on antibiotic resistance 2011–2016 [Citation55–57]. In addition, 189 of the 302 P. multocida strains were from Asia, and 37 contained ICEs. These clinical isolates were observed in large quantities from 2015 to 2018, and the ICEs also mainly concentrated from 2015 to 2018. The existing limited number ICEs in Asia P. multocida is mostly associated with resistance and appeared earlier than in Europe and the Americas but decreased later than in Europe and the Americas. This discrepancy might be attributed to the fact that antibiotic abuse has not been effectively suppressed until more relevant national policies were implemented, such as the Ministry of Agriculture and Rural Affairs issued the National Action Plan for Restraining Bacteria of Animal Origin and the National Five-Year Action Plan for Comprehensive Management of Veterinary Drugs (2016–2020) in China [Citation58]. The frequency of resistant ICEs transmission in P. multocida roughly coincides with the implementation of global policies to curb antibiotic abuse, perhaps indicating the effectiveness of these policies from the side. Due to variations in the availability of data, particularly regarding the number of laboratory strains versus publicly accessible information from the NCBI, Jiang et al. conducted an analysis of A:L1:ST129 strains among 546 P. multocida strains and determined the adaptability of this serotype strains to ecological niches through ICEs harbouring resistance gene clusters [Citation59]. Our findings align with their conclusions. In general, the widespread distribution of ICEs in P. multocida improve adaptive resistance to harsh living environment, and the significance reduction of ICEs indicated that ICEs themselves has a fitness cost and burden for P. multocida, as with ICEs in other bacteria [Citation13]. Thus, there is a highly likely loss of ICEs in non-resistant environments to maintain a more suitable survival state [Citation13,Citation22,Citation60]. Furthermore, we have conducted an expanded analysis to investigate the dissemination of ICEs in P. multocida and the transmission patterns of resistance genes within these ICEs.
For instance, the ability of ICEs to transmit vertically and horizontally was demonstrated in this study. Collinear analysis of 82 ICEs based on whole genome sequences of 391 P. multocida isolates revealed at least five ICEs subtypes, including three subtypes of the ICEHin1056 family (), one subtype of the SXT/R391 family and one subtype of the Tn916 family. Among these, 33 P. multocida isolates from Europe and the Americas contained two subtypes of ICEHin1056 family (), one of which was provided with ISL3-ISMha7-tet(H), while the other was provided with ISL3-ISMha7-tet(H), ISApl1-aphA1-strB-strA-sul2, and IS91-ISCR21 (Table S8). Additionally, 8 P. multocida isolates from Asia contained a subtype of ICEHin1056 family (), which could almost exclusively be provided with IS4-ISVsa5-tetB (Table S8). One subtype of Tn916 family, which lacked obvious functional genes, and one subtype of SXT/R391 family were found to be provided with strB-strA-sul2 (). The discovery of Tn916 family further supported the finding of Moustafa et al. that P. multocida T×1contains an ICE [Citation19]. However, the failure of the Tn916 family to continue transmission in P. multocida, apart from the strain TX1 May 2001be due to its inability to introduce adaptive genes or due to the greater burden on the host’s normal growth and reproduction that this type of ICEs imposes [Citation61]. Furthermore, the study also found that the SXT/R391 family was not widely transmission in P. multocida, occurring in only two Asia P. multocida isolates, possibly also due to the adaptive burden on different species [Citation22]. The ICEHin1056 family was identified as the predominant circulating type of ICEs in P. multocida, likely because Haemophilus influenzae and P. multocida are of the same species, of which this family may be more easily acquired by P. multocida at a lower compensatory cost. The phylogeny of ICEHin1056-like ICEs is consistent with the whole genome of the respective strains (), indicating that the transmission of such ICEs in P. multocida is an early event. The ICEHin1056-like ICEs already existed in P. multocida before the pre-differentiation of the ICEs in Europe and the Americas, and Asia P. multocida. Subsequently, the ICEs were transmitted both vertically and horizontally in P. multocida, but the ICEs mainly spread through vertical transmission and evolved with the evolution of the P. multocida genomes, eventually presenting the phylogenetic relationship between ICEs and strain genomes. Among the P. multocida isolates studied, the Bangladesh poultry-sourced strain Ban-PM4 (NZ_CP052764.1), Ban-PM7 (NZ_CP052765.1) and the China swine-sourced strain HN07 (CP007040.1), diverged in 1997–2004 and then clustered into one branch. This pattern was roughly consistent with the time of strengthening agricultural cooperation between China and Bangladesh after 1983 [Citation62]. Notably, although the swine-sourced strain HN07 was clustered with the poultry-source, it was observed that the T4SS of ICEs in HN07 was noticeably different from that of the poultry-sourced strains based on the sequence alignment and phylogenetic tree of the conserved structure T4SS of ICEHin1056-like ICEs (). Hence, there are consistent differences between the host sources across the whole genomes of P. multocida and the conserved structures of ICEs. Overall, the conserved structure of ICEs in bovine and poultry-sourced P. multocida demonstrated more obvious host differences than other features of P. multocida genome, might suggesting that the ICEs in P. multocida were highly specialized to their host.
Moreover, the impact of ICEs on P. multocida primarily involves the acquisition of multiple resistance genes, and ISs has a significant correlation with the resistance phenotypes of ICEs [Citation63,Citation64]. Previous research has shown that IS91–floR [Citation65], IS4–tetB [Citation66,Citation67], and IS6–mph(E)–msr(E) [Citation68] exhibit synergistic transfers correlation. Additionally, ICEs containing IS91–erm(42), IS6–blaOXA-2–aadA8b–aadB, ISL3–tetH, IS30–aphA1–strB–strA, and IS30-sul2 have been identified, of which the correlation of these ISs and resistance were not previously mentioned in Pasteuraceae or any other species. Literature reports have revealed clinical P. multocida strains resistant to tetracyclines, aminoglycosides (kanamycin, streptomycin), sulphonamides, acylaminosol (florfenicol, chloramphenicol), macrolides (erythromycin) and lincomamides (clindamycin) [Citation69–71], and the resistance genes related to these antibiotics were identified in the ICEs elements of P. multocida in this study. These findings suggested that ICEs were closely related to the clinically resistant phenotypes of P. multocida. In addition, a β-lactam blaOXA-2 gene was found in the ICEs of P. multocida. This gene is 100% identical to the blaOXA-2 gene in Acinetobacter baumannii, Pseudomonas aeruginosa and a plasmid of Klebsiella pneumoniae, which have been associated with the carbapenemase phenotype in Acinetobacter baumannii [Citation72] and Pseudomonas aeruginosa [Citation73], and etapenem and ceftazidime phenotypes in a plasmid of Klebsiella pneumoniae [Citation74]. Moreover, this study identified that ICEs introduced the aminoglycopeptide resistance gene bleomycin bleO to P. multocida. This gene is predominantly found in the plasmids of Salmonella enterica [Citation75], E. coli [Citation76], Klebsiella pneumoniae [Citation77] Staphylococcus aureus [Citation78], and other bacteria, with small amounts presence in the genomes of Proteus mirabilis and Aliarcobacter butzleri, as well as in the ICEs such as ICEPmiChn-HBRJC2 and ICEPmiChnPm-SC1111, as reported by NCBI Blast. However, the gene bleO has not been previously identified in Pasteureliaceae.
To the best of our knowledge, this study is the first to shed light on the transmission and evolution patterns of ICEs in P. multocida based on the integrality of ICEs in the P. multocida population. Nevertheless, an additional uncontrolled factor is the possibility that although the number of genomes of P. multocida has increased significantly, there are still many laboratories with a large amount of P. multocida data that are not publicly available, so this study could only analyse the data of the publicly available part to explore some of the biological laws of ICEs in P. multocida population.
Summary
This project comprehensively investigated 82 ICEs, mainly associated with resistance among the 435 strains of 13 host-sourced P. multocida. The results indicated that the prevalence of ICEs in P. multocida is an early event, mainly through vertical transmission and partially through horizontal transmission and the conserved structure of ICEs showed distinct host characteristics at the genetic level for cattle and poultry-sourced P. multocida. ICEs were mainly classified into five types: three subtypes of ICEHin1056 family, one subtype of SXT/R391 family and one subtype of Tn916 family, of which the SXT/R391 family and Tn916 family were not previously reported in P. multocida. Additionally, the presence of multi-resistant ICEs in P. multocida contributes to the emergence of common resistance phenotypes, such as carbapenems and aminoglycopeptides, in host strains. This makes the clinical prevention and control of P. multocida more challenging, and highlights the need for continuous monitoring of the spread of ICEs in P. multocida to obtain important data for clinical rational use of antibiotics.
Authors contribution statement
DZ and AC conceived and designed the project; JH, XO, DS, BT, YH and ZW collected bacteria; JH, ZY, MW, RJ, SC and ML extracted genomic DNA and assembled and annotated the genomes; JH, ZY, J Huang and SZ performed MLST and serotype analysis; JH, ZY, XZ, QY and YW performed the construction of the phylogenetic tree and statistical analysis; JH and DZ performed the genome and their ICEs analysis in P. multocida; JH and DZ drafted and revised the manuscript. All authors have read and approved the final version manuscript.
Availability of data and materials
The data supporting the results of this study are available in the NCBI WGS database at https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/912/. The accession numbers of P. multocida genomes are included in Table S2 and Figure S2.
Supplemental Material
Download Zip (2.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2024.2359467
Additional information
Funding
References
- Wilkie IW, Harper M, Boyce JD, et al. Pasteurella multocida: diseases and pathogenesis. Curr Top Microbiol Immunol. 2012;361:1–16.
- Boyce JD, Seemann T, Adler B, et al. Pathogenomics of Pasteurella multocida. Curr Top Microbiol Immunol. 2012;361:23.
- Dziva F, Muhairwa AP, Bisgaard M, et al. Diagnostic and typing options for investigating diseases associated with Pasteurella multocida. Vet Microbiol. 2008;128(1–2):1–22. doi: 10.1016/j.vetmic.2007.10.018
- Kubatzky KF. Pasteurella multocida and immune cells. Curr Top Microbiol Immunol. 2012;361(361):53–72.
- Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nature Rev Microbiol. 2010;8(8):552–563. doi: 10.1038/nrmicro2382
- Bellanger X, Payot S, Leblond-Bourget N, et al. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev. 2014;38(4):720–760. doi: 10.1111/1574-6976.12058
- Ryan MP, Carraro N, Slattery S, et al. Integrative Conjugative Elements (ICEs) of the SXT/R391 family drive adaptation and evolution in γ-Proteobacteria. Critical Reviews In Microbiology. 2023(1):1–22. doi: 10.1080/1040841X.2022.2161870
- Johnson CM, Grossman AD. Integrative and conjugative elements (ICEs): what they do and how they work. Ann Rev Genet. 2015;49(1):577–601. doi: 10.1146/annurev-genet-112414-055018
- Botelho J, Schulenburg H. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 2021;29(1):8–18. doi: 10.1016/j.tim.2020.05.011
- Te Poele EM, Bolhuis H, Dijkhuizen L. Actinomycete integrative and conjugative elements. Antonie Van Leeuwenhoek. 2008;94(1):127–143. doi: 10.1007/s10482-008-9255-x
- Haenni M, Lupo A, Madec JY, et al. Antimicrobial Resistance in Streptococcus spp. Microbiol Spectr. 2018;6(2). doi: 10.1128/microbiolspec.ARBA-0008-2017
- Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3). doi: 10.1128/CMR.00001-19
- Botelho J, Grosso F, Peixe L. Antibiotic resistance in Pseudomonas aeruginosa - Mechanisms, epidemiology and evolution. Drug Resist Updat. 2019;44:100640. doi: 10.1016/j.drup.2019.07.002
- Weaver KE, Fischetti VA, Novick RP, et al. Enterococcal Genetics. Microbiol Spectr. 2019;7(2). doi: 10.1128/microbiolspec.GPP3-0055-2018
- Spagnoletti M, Ceccarelli D, Rieux A, et al. Acquisition and evolution of SXT-R391 integrative conjugative elements in the seventh-pandemic Vibrio cholerae lineage. MBio. 2014;5(4). doi: 10.1128/mBio.01356-14
- Michael GB, Kadlec K, Sweeney MT, et al. ICEPmu1, an integrative conjugative element (ICE) of Pasteurella multocida: structure and transfer. J Antimicrob Chemother. 2012;67(1):91–100. doi: 10.1093/jac/dkr411
- Schink AK, Hanke D, Semmler T, et al. Novel multiresistance-mediating integrative and conjugative elements carrying unusual antimicrobial resistance genes in Mannheimia haemolytica and Pasteurella multocida. J Antimicrob Chemother. 2022;77(7):2033–2035. doi: 10.1093/jac/dkac116
- Klima CL, Zaheer R, Cook SR, et al. Pathogens of bovine respiratory disease in North American feedlots conferring multidrug resistance via integrative conjugative elements. J Clin Microbiol. 2014;52(2):438. doi: 10.1128/JCM.02485-13
- Moustafa AM, Seemann T, Gladman S, et al. Comparative genomic analysis of Asian haemorrhagic septicaemia-associated strains of Pasteurella multocida identifies more than 90 haemorrhagic Septicaemia-Specific Genes. PLOS ONE. 2015;10(7):1–16. doi: 10.1371/journal.pone.0130296
- Peng Z, Liang W, Wang Y, et al. Experimental pathogenicity and complete genome characterization of a pig origin Pasteurella multocida serogroup F isolate HN07. Vet Microbiol. 2017;198:23–33. doi: 10.1016/j.vetmic.2016.11.028
- Delavat F, Miyazaki R, Carraro N, et al. The hidden life of integrative and conjugative elements. FEMS Microbiol Rev. 2017;41(4):512–537. doi: 10.1093/femsre/fux008
- He J, Sun L, Zhang L, et al. A novel SXT/R391 Integrative and conjugative element carries two copies of the bla NDM-1 gene in Proteus mirabilis. mSphere. 2021;6(4):e0058821. doi: 10.1128/mSphere.00588-21
- Cury J, Oliveira PH, De La Cruz F, et al. Host range and genetic plasticity explain the coexistence of integrative and extrachromosomal mobile genetic elements. Mol Biol Evol. 2018;35(9):2230–2239. doi: 10.1093/molbev/msy123
- Korostin D, Kulemin N, Naumov V, et al. Comparative analysis of novel MGISEQ-2000 sequencing platform vs Illumina HiSeq 2500 for whole-genome sequencing. PLoS One. 2020;15(3):e0230301. doi: 10.1371/journal.pone.0230301
- Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Bio. 2010;11(11):R116. doi: 10.1186/gb-2010-11-11-r116
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021
- Parks DH, Imelfort M, Skennerton CT, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114
- Subaaharan S, Blackall LL, Blackall PJ. Development of a multi-locus sequence typing scheme for avian isolates of Pasteurella multocida. Veterinary Microbiology. 2010;141(3–4):354–361. doi: 10.1016/j.vetmic.2010.01.017
- Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.Org website and their applications. Wellcome Open Res. 2018;3:124. doi: 10.12688/wellcomeopenres.14826.1
- Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–1361. doi: 10.1128/JCM.06094-11
- Townsend KM, Boyce JD, Jing YC, et al. Genetic organization of Pasteurella multocida cap Loci and development of a multiplex capsular PCR typing system. J Clin Microbiol. 2001;39(3):924–929. doi: 10.1128/JCM.39.3.924-929.2001
- Harper M, John M, Turni C, et al. Development of a rapid multiplex PCR assay to genotype Pasteurella multocida strains by use of the lipopolysaccharide outer core biosynthesis locus. J Clin Microbiol. 2015;53(2):477–485. doi: 10.1128/JCM.02824-14
- Bertelli C, Laird MR, Williams KP, et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45(W1):W30–W35. doi: 10.1093/nar/gkx343
- Liu M, Li X, Xie Y, et al. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019;47(D1):D660–D665. doi: 10.1093/nar/gky1123
- Darling AC, Mau B, Blattner FR, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704
- Ball P. Chemistry: Perkin, the mauve maker. Nature. 2006;440(7083):429. doi: 10.1038/440429a
- Darling AE, Mau B, Perna NT, et al. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147
- Dobrindt U, Hochhut B, Hentschel U, et al. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. 2004;2(5):414–424. doi: 10.1038/nrmicro884
- Burrus V, Pavlovic G, Decaris B, et al. Conjugative transposons: the tip of the iceberg. Mol Microbiol. 2002;46(3):601–610. doi: 10.1046/j.1365-2958.2002.03191.x
- Marchlerbauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Research. 2015;43(D1):D222–6. doi: 10.1093/nar/gku1221
- Siguier P, Perochon J, Lestrade L, et al. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(90001):D32–6. doi: 10.1093/nar/gkj014
- Florensa AF, Kaas RS, Clausen P, et al. ResFinder - an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb Genom. 2022;8(1):000748. doi: 10.1099/mgen.0.000748
- Li X, Xie Y, Liu M, et al. oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 2018;46(W1):W229–W34. doi: 10.1093/nar/gky352
- Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039
- Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics (Oxford, England). 2015;31(22):3691. doi: 10.1093/bioinformatics/btv421
- Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20(4):1160–1166. doi: 10.1093/bib/bbx108
- Nguyen LT, Schmidt HA, Von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. doi: 10.1093/molbev/msu300
- Rambaut A, Lam TT, Max Carvalho L, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2(1):vew007. doi: 10.1093/ve/vew007
- Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016. doi: 10.1093/ve/vey016
- Drummond AJ, Syw H, Phillips MJ, et al. Relaxed phylogenetics and dating with confidence - art. no. e88. PLOS Biol. 2006;4:4. doi: 10.1371/journal.pbio.0040088
- Gernhard T. The conditioned reconstructed process. J Theor Biol. 2008;253(4):769–778. doi: 10.1016/j.jtbi.2008.04.005
- Carter GR. Studies on Pasteurella multocida. I. A hemagglutination test for the identification of serological types. Am J Vet Res. 1955;16(60):481–484.
- Rimler RB, Rhoades KR, Serogroup F. A new capsule serogroup of Pasteurella multocida. J Clin Microbiol. 1987;25(4):615–618. doi: 10.1128/jcm.25.4.615-618.1987
- Flannagan SE, Clewell DB. Conjugative transfer of Tn916 in Enterococcus faecalis: trans activation of homologous transposons. J Bacteriol. 1991;173(22):7136–7141. doi: 10.1128/jb.173.22.7136-7141.1991
- Watson R. Europe launches 12 point plan to tackle antimicrobial resistance. BMJ. 2011;343(nov21 1):d7528. doi: 10.1136/bmj.d7528
- Smith E, Lichten CA, Taylor J, et al. Europe’s plan to tackle antimicrobial resistance: The success stories and challenges. Santa Monica, CA: RAND Corporation; 2016. doi: 10.7249/RB9930
- De Briyne N, Atkinson J, Pokludová L, et al. Antibiotics used most commonly to treat animals in Europe. Vet Rec. 2014;175(13):325. doi: 10.1136/vr.102462
- Rui Y, Qiu G. Analysis of gut microbial communities and resistance genes in pigs and chickens in Central China. Animals (Basel). 2022;12(23):3404. doi: 10.3390/ani12233404
- Jiang N, Chen H, Cheng L, et al. Genomic analysis reveals the population structure and antimicrobial resistance of avian Pasteurella multocida in China. J Antimicrob Chemother. 2024;79(1):186–194. doi: 10.1093/jac/dkad365
- Choufa C, Tidjani AR, Gauthier A, et al. Prevalence and mobility of integrative and conjugative elements within a Streptomyces natural population. Front Microbiol. 2022;13:970179. doi: 10.3389/fmicb.2022.970179
- Bean EL, Mclellan LK, Grossman AD, et al. Activation of the integrative and conjugative element Tn916 causes growth arrest and death of host bacteria. PLOS Genet. 2022;18(10):e1010467. doi: 10.1371/journal.pgen.1010467
- The Embassy of the People’s Republic of China in the People’s Republic of Bangladesh. The relations between China and Bangladesh [Z]. 2019. Available from: http://bd.china-embassy.gov.cn/chn/zmgx/gxgk/202003/t20200322_1780848.htm
- Partridge SR. Analysis of antibiotic resistance regions in gram-negative bacteria. FEMS Microbiol Rev. 2011;35(5):820–855. doi: 10.1111/j.1574-6976.2011.00277.x
- Partridge SR, Kwong SM, Firth N, et al. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31(4). doi: 10.1128/CMR.00088-17
- Li A, Yu R, Zhao W, et al. Characterization of a genomic Island carrying the tet(X4) gene in porcine Acinetobacter towneri co-harboring plasmid-borne bla (NDM-1) and bla (OXA-58) genes. Front Vet Sci. 2022;9:1002149. doi: 10.3389/fvets.2022.1002149
- Mbelle N, Osei Sekyere J, Feldman C, et al. Genomic analysis of two drug-resistant clinical Morganella morganii strains isolated from UTI patients in Pretoria, South Africa. Lett Appl Microbiol. 2020;70(1):21–28. doi: 10.1111/lam.13237
- Mbelle NM, Osei Sekyere J, Amoako DG, et al. Genomic analysis of a multidrug-resistant clinical Providencia rettgeri (PR002) strain with the novel integron ln1483 and an A/C plasmid replicon. Ann N Y Acad Sci. 2020;1462(1):92–103. doi: 10.1111/nyas.14237
- Nusrin S, Asad A, Hayat S, et al. Multiple mechanisms confer resistance to azithromycin in Shigella in Bangladesh: a comprehensive whole genome-based approach. Microbiol Spectr. 2022;10(4):e0074122. doi: 10.1128/spectrum.00741-22
- Nhung NT, Chansiripornchai N, Carrique-Mas JJ. Antimicrobial Resistance in Bacterial Poultry Pathogens: A Review. Front Vet Sci. 2017;4:126. doi: 10.3389/fvets.2017.00126
- Kehrenberg C, Schwarz S. Plasmid-borne florfenicol resistance in Pasteurella multocida. J Antimicrob Chemother. 2005;55(5):773–775. doi: 10.1093/jac/dki102
- Jamali H, Rezagholipour M, Fallah S, et al. Prevalence, characterization and antibiotic resistance of Pasteurella multocida isolated from bovine respiratory infection. Vet J (London, England: 1997). 2014;202(2):381–383. doi: 10.1016/j.tvjl.2014.07.024
- Asgin N, Otlu B, Cakmakliogullari EK, et al. High prevalence of TEM, VIM, and OXA-2 beta-lactamases and clonal diversity among Acinetobacter baumannii isolates in Turkey. J Infect Dev Ctries. 2019;13(9):794–801. doi: 10.3855/jidc.11684
- Lasarte-Monterrubio C, Fraile-Ribot PA, Vázquez-Ucha JC, et al. Activity of cefiderocol, imipenem/relebactam, cefepime/taniborbactam and cefepime/zidebactam against ceftolozane/tazobactam- and ceftazidime/avibactam-resistant Pseudomonas aeruginosa. J Antimicrob Chemother. 2022;77(10):2809–2815. doi: 10.1093/jac/dkac241
- Iovleva A, Mettus RT, Mcelheny CL, et al. Reduced ceftazidime and ertapenem susceptibility due to production of OXA-2 in Klebsiella pneumoniae ST258. J Antimicrob Chemother. 2019;74(8):2203–2208. doi: 10.1093/jac/dkz183
- Huang J, Deng S, Ren J, et al. Characterization of a blaNDM‑1‑harboring plasmid from a Salmonella enterica clinical isolate in China. Mol Med Rep. 2017;16(2):1087–1092. doi: 10.3892/mmr.2017.6733
- Bonnin RA, Poirel L, Carattoli A, et al. Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLOS ONE. 2012;7(4):e34752. doi: 10.1371/journal.pone.0034752
- Ahmad N, Ali SM, Khan AU. Molecular characterization of novel sequence type of carbapenem-resistant new delhi metallo-β-lactamase-1-producing Klebsiella pneumoniae in the neonatal intensive care unit of an Indian hospital. Int J Antimicrob Agents. 2019;53(4):525–529. doi: 10.1016/j.ijantimicag.2018.12.005
- Semon D, Movva NR, Smith TF, et al. Plasmid-determined bleomycin resistance in Staphylococcus aureus. Plasmid. 1987;17(1):46–53. doi: 10.1016/0147-619X(87)90007-2