
ABSTRACT
A GGGGCC hexanucleotide repeat expansion in the first intron of the C9orf72 gene is the most common genetic defect associated with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (C9ALS/FTD). Haploinsufficiency and a resulting loss of C9orf72 protein function has been suggested as a possible pathogenic mechanism in C9ALS/FTD. C9ALS/FTD patients exhibit specific ubiquitin and p62/sequestosome-1 positive but TDP-43 negative inclusions in the cerebellum and hippocampus, indicating possible autophagy deficits in these patients. In a recent study, we investigated this possibility by reducing expression of C9orf72 in cell lines and primary neurons and found that C9orf72 regulates the initiation of autophagy. C9orf72 interacts with Rab1a, preferentially in its GTP-bound state, as well as the ULK1 autophagy initiation complex. As an effector of Rab1a, C9orf72 controls the Rab1a-dependent trafficking of the ULK1 initiation complex prior to autophagosome formation. In line with this function, C9orf72 depletion in cell lines and primary neurons caused the accumulation of p62/sequestosome-1-positive inclusions. In support of a role in disease pathogenesis, C9ALS/FTD patient-derived iNeurons showed markedly reduced levels of autophagy. In this Commentary we summarise recent findings supporting the key role of C9orf72 in Rab GTPase-dependent regulation of autophagy and discuss autophagy dysregulation as a pathogenic mechanism in ALS/FTD.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common adult onset motor neuron disorder. ALS is caused by selective degeneration of upper and lower motor neurons, which leads to progressive muscle weakness, gait abnormalities, paralysis and ultimately death. Frontotemporal dementia (FTD) is the second most common form of dementia in the under 65s. FTD is characterized by loss of neurons predominantly in the frontal and temporal cortex, leading to changes in personality, behavior and cognitive function. ALS and FTD are now recognized to be at the extremes of a spectrum of the same disorder with up to 25% of ALS cases clinically diagnosed with FTD and approximately 50% of FTD cases displaying some degree of motor neuron involvement.Citation1
A GGGGCC hexanucleotide repeat expansion within the first intron of the C9orf72 gene was found to be the most common genetic defect associated with both ALS and FTD (referred to as C9ALS/FTD).Citation2,3 The pathogenic mechanism by which the repeat expansion causes disease may involve gain-of-toxic-function mechanisms, namely RNA toxicityCitation2,4-8 and protein toxicity by aberrant dipeptide repeat protein (DPR) accumulation.Citation9-13 Alternatively, reduced C9orf72 mRNA levels in a range of patient tissues and patient-derived cell linesCitation2,14-18 and reduced C9orf72 protein levels in the frontal cortex of C9ALS/FTD patientsCitation19,20 suggest loss-of-function by C9orf72 haploinsufficiency may also contribute to C9ALS/FTD. Mild neurodegeneration and cognitive impairment have been reported in some gain-of-toxic-function GGGGCC repeat C9orf72 BAC-transgenic miceCitation21,22 but not others,Citation23,24 while loss-of-function C9orf72 knockout mice exhibit an immunological rather than neuronal phenotype.Citation25-29 Thus neither loss- nor gain-of-function appears sufficient to model all aspects of disease in vivo, suggesting that all 3 mechanisms might be conspiring to cause C9ALS/FTD.
The alternatively spliced C9orf72 gene codes for 2 protein isoforms, a 481 amino acid (aa) “long” isoform (C9orf72L) and a 222 aa “short” isoform (C9orf72S). C9orf72 does not show obvious sequence homology to other proteins, but bioinformatics analysis revealed that the C9orf72 protein is structurally related to Differentially Expressed in Normal and Neoplasia (DENN) domain-containing proteins, which function as GDP/GTP exchange factors (GEFs) for the Rab family of GTPases.Citation30,31 Rab GTPases function as molecular switches that alternate between a GTP-bound active state and a GDP-bound inactive state. GEFs catalyze the exchange of GDP for GTP while GTPase activating proteins (GAPs) catalyze GTP hydrolysis to GDP. GTP-bound, active Rabs recruit and/or activate a variety of effector molecules and in doing so Rabs control a range of cellular trafficking events, including autophagy (reviewed in refs. Citation32, 33).
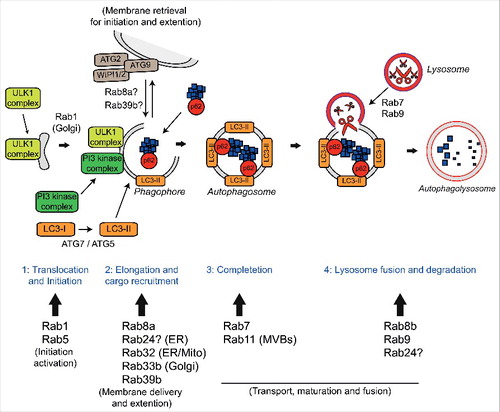
Autophagy (derived from the Greek and meaning “self-eating”) is a highly conserved catabolic process that involves the delivery of cytosolic components to the lysosome for degradation. Macroautophagy, herein referred to as autophagy, requires the formation of a double membrane structure called the autophagosome, which encapsulates cytosolic substrates such as misfolded proteins and damaged organelles, before fusing with the lysosome to allow bulk degradation and recycling. The autophagy process can be subdivided into 4 steps, (i) translocation and initiation, (ii) elongation and cargo recruitment, (iii) completion, and (iv) lysosome fusion and degradation (reviewed in ref. Citation34). A large number of Rab GTPases have been shown to be involved at each stage of autophagy (reviewed in ref. Citation32) ().
Figure 1. Autophagy and the Rab GTPases. The four stages of autophagy are indicated with the different Rabs involved at each stage. 1) Translocation of the ULK1 initiation complex to the phagophore is the first step in autophagy initiation. Rab1a mediates trafficking of the ULK1 complex to the phagophore and is involved in delivery of ATG9 positive membranes to the site of phagophore formation. Rab5 is involved in translocation of the Class III PI3 kinase complex and delivery of LC3-II. 2) Elongation of the phagophore membrane requires the Class III PI3 kinase complex. Rab8a, Rab24, Rab32, Rab33b and Rab39b are all involved in autophagosome formation and may aid in elongation by the delivery of additional membrane via ATG9/ATG2/WIPI1/2. Autophagy substrates are recruited to the growing phagophore by autophagy receptors such as p62/sequestosome-1 and optineurin. Autophagy receptors bind ubiquitin on the substrates and LC3-II on the nascent phagophore resulting in substrate delivery. 3) After completion and closure, autophagosomes are transported to allow fusion with the lysosome. Rab7 is involved in autophagosome transport while Rab11 delivers multi-vesicular bodies (MVBs) to the autophagosome, which appears to be required for maturation. 4) Autophagosome fusion with the lysosome allows degradation of the autophagic substrates. Rab7, Rab8b and Rab9 are involved in the fusion of autophagosomes and lysosomes, a process that may also require Rab24. Finally, autophagic substrates are degraded by the acid hydolases of the lysosome and recycled back to the cytoplasm.
Here we summarise the role of C9orf72 in Rab-mediated regulation of autophagy and discuss autophagy dysregulation as a pathogenic mechanism in ALS/FTD.
C9orf72 and autophagy induction
While the pathology associated with C9ALS/FTD is in many ways consistent with other forms of ALS/FTD, including the classical TDP-43 neuronal cytoplasmic inclusions, C9ALS/FTD cases are distinguished by the presence of specific ubiquitin and p62/sequestosome-1 positive, TDP-43 negative, neuronal cytoplasmic inclusions in the cerebellum, hippocampus and neocortex.Citation16,35,36 p62/sequestosome-1 is a well-characterized autophagy receptor that binds ubiquitinated substrates and recruits them to the nascent phagosome via interaction with phagosomal LC3-II (). Because p62/sequestosome-1 is itself degraded by autophagy in the process of delivering substrates, accumulations of p62/sequestosome-1 are indicative of defective autophagy.Citation37 Thus, the presence of p62/sequestosome-1-positive inclusions together with C9orf72 haploinsufficiency suggested the possibility of defective autophagy by loss of C9orf72 function in C9ALS/FTD.
To test for a role of C9orf72 in autophagy we depleted C9orf72 or, conversely, overexpressed C9orf72 in cell lines and primary neurons and quantified autophagy by monitoring LC3-II flux.Citation38 We found that loss of C9orf72 disrupted autophagy at the initiation stage whereas overexpression of C9orf72 induced autophagy.Citation39 These results corroborated other reports showing that loss of C9orf72 led to a disruption in autophagy in neuronal cell linesCitation40 and in vivo in C9orf72 knockout mice.Citation23,28
The initiation of autophagy is controlled by the ULK1 autophagy initiation complex, which is comprised of Unc-51-like kinase 1 (ULK1), FAK family kinase-interacting protein of 200 kDa (FIP200), autophagy-related 13 (ATG13) and ATG101.Citation41-44 When we disrupted the ULK1 complex by depletion of FIP200, C9orf72 overexpression was no longer able to induce autophagy indicating that C9orf72 acted at the level of the ULK1 complex.Citation39 Using immunoprecipitation, in vitro binding and proximity ligation assays we went on to show a direct interaction between C9orf72 and ULK1, FIP200 and ATG13.Citation39 The interaction with FIP200 confirmed a previously reported proteomics screen.Citation45 In support of our findings, C9orf72, in complex with SMCR8 and WDR41, was independently reported to interact with the ULK1 initiation complex.Citation28
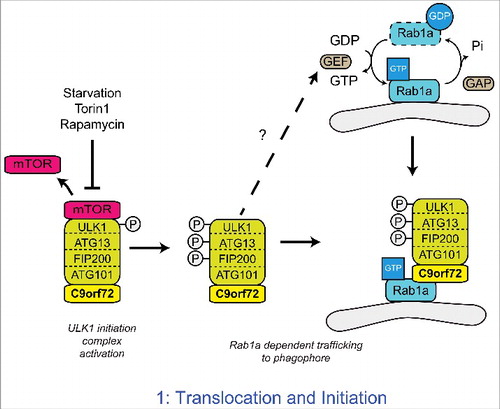
The ULK1 complex is kept inactive by mTOR, which phosphorylates ULK1 at serine 757.Citation46 Inactivation of mTOR leads to its release from the ULK1 complex and ULK1 phosphorylation is lost thereby enhancing ULK1 kinase activity. In turn, ULK1 phosphorylates FIP200 and ATG13 to activate the complexCitation41,46,47 (). The activated ULK1 complex translocates to the phagophore to initiate autophagosome formation.Citation42,48 In a series of experiments aimed at defining the role of C9orf72 in autophagy initiation we found that depletion of C9orf72 did not affect the ULK1 complex at the level of mTOR, but rather prevented the translocation of the activated ULK1 complex to the phagophore.Citation39
Figure 2. C9orf72 regulates the Rab1a dependent trafficking of the ULK1 complex. Upon inhibition of mTOR, the inhibitory phosphorylation of ULK1 is lost, leading to the activation of ULK1. Active ULK1 phosphorylates the other members of the initiation complex, including FIP200 and ATG13. Functioning as a Rab1a effector, C9orf72 mediates the interaction between the active ULK1 initiation complex and GTP-Rab1a within its target membrane. Thus C9orf72 controls the Rab1a dependent trafficking of the ULK1 initiation complex to the phagophore. How Rab1 is activated in response to autophagy induction is not yet known.
C9orf72 as a Rab1a effector
In yeast the translocation of Atg1, the yeast homolog of ULK1, is regulated by Ypt1, the yeast homolog of Rab1Citation49 and in mammalian cells Rab1 has been shown to regulate the early stages of autophagy.Citation50-52 We therefore investigated whether the C9orf72-mediated regulation of ULK1 complex translocation involved Rab1. In cells depleted of Rab1a by siRNA treatment or expressing constitutively GDP-bound, dominant negative Rab1a(S25N), overexpression of C9orf72 no longer induced translocation of the ULK1 initiation complex and autophagy,Citation39 indicating C9orf72 acted through Rab1a.
The structural homology of C9orf72 with DENN family Rab GEF proteinsCitation30,31 and the identification of C9orf72 as a potential Rab1a interaction partner in a yeast 2 hybrid screenCitation39 prompted us to further investigate the interaction of C9orf72 and Rab1a in immunoprecipitation and in-vitro binding assays. Rab1a interacted with C9orf72 in both assays, but interestingly C9orf72 preferentially bound to GTP-bound Rab1a.Citation39 Thus our data identified C9orf72 as an effector of Rab1a rather than a GEF as would be expected for a DENN family protein. As a Rab1a effector, C9orf72 was found to mediate the interaction between Rab1a and the ULK1 initiation complex and proved essential for Rab1a-mediated translocation of the ULK1 complex.Citation39 Thus our data support a model in which C9orf72 is an effector of Rab1a that facilitates trafficking of the ULK1 initiation complex to the phagophore ().
C9orf72 acts in a Rab cascade
While our work shows that C9orf72 is a Rab1a effector,Citation39 others have shown that C9orf72 forms a complex with SMCR8 and WDR41Citation28,40,53,54 and acts as a GEF for RAB8a and RAB39b in the autophagy pathway.Citation40 However, the interaction between the C9orf72/SMCR8/WDR41 complex and RAB8a or RAB39b appeared to be mainly through binding with SMCR8, and Rab GEF activity was only observed when SMCR8 was present.Citation40 As SMCR8 is itself a DENN domain protein,Citation31 it appears that SMCR8 rather than C9orf72 may mediate the GEF activity toward Rab8a and Rab39b.
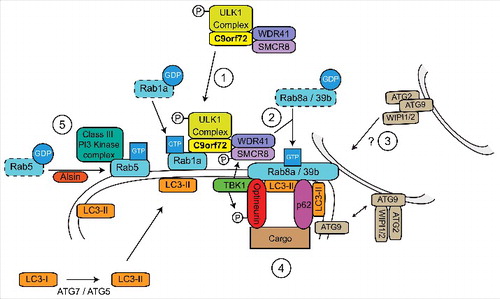
Sequential activation of Rab GTPases in a cascade-like fashion allows the correct spatial and temporal recruitment of successive Rabs in a signal transduction pathway. In these Rab cascades the upstream Rab and its effectors recruit the GEF of the next downstream Rab, while the downstream Rab recruits the GAP for the upstream Rab (reviewed in ref. Citation55). Thus a model emerges in which C9orf72 links autophagy initiation to downstream events via a Rab cascade, involving Rab1a upstream of Rab8a and Rab39b (). Consistent with this, Rab1 has been shown to be involved in autophagy initiationCitation39,50,52 whereas Rab8 and Rab39 are involved in autophagosome maturationCitation56,57 (). Furthermore Rab8a and Rab39b were shown to interact with the autophagy receptors optineurin and p62/sequestosome-1 suggesting the interesting possibility of C9orf72 regulated site-directed autophagy initiation.Citation40,56
Figure 3. C9orf72 is a central hub in a Rab cascade pathway during autophagy induction. 1) C9orf72 controls the Rab1a-dependent translocation of the ULK1 initiation complex to the site of phagophore formation by interacting with the ULK1 complex and active GTP-Rab1a. 2) At the site of phagophore formation, the C9orf72/SMCR8/WDR41 complex functions as a GEF for Rab8a and Rab39b. 3) Active Rab8a and Rab39b may be involved in delivery of additional membrane to the site of phagophore formation by retrieval of ATG9 positive membranes. Additional membrane allows elongation of the nascent phagophore and formation of an autophagosome. 4) The autophagy receptors p62/sequestosome-1 and optineurin interact with Rab8a and Rab39b, promoting nucleation and site specific recruitment of autophagic substrates to the growing phagophore. Furthermore, Rab8a also recruits TBK1, leading to phosphorylation of optineurin, SMCR8 and ULK1. The phosphorylation of optineurin enhances its interaction with LC3-II, facilitating substrate delivery and the progression of autophagy. 5) The Class III PI3 Kinase complex is also required for autophagosome formation. Rab5, activated by Alsin, delivers the PI3 Kinase complex to the phagophore and also facilitates the recruitment of the ATG7-ATG5 conjugation system required for LC3-II formation.
The role of C9orf72 in inflammation
Autophagy plays an important role in both innate and adaptive immunity. Thus, autophagy eliminates invading pathogens, controls pro-inflammatory signaling, regulates the secretion of immune mediators, and modulates MHC class II antigen presentation (reviewed in refs. Citation58, 59, 60).
C9orf72 knockout mice present with varying degrees of autoimmunity and inflammation, accompanied by splenomegaly and increased expression of inflammatory cytokines.Citation23,25-29 Our study into the regulation of autophagy initiation by C9orf72 indicates that these phenotypes could be a consequence of compromised autophagy. In support of this both ULK1 and AMPKα (an upstream regulator of ULK1) knockout mice show splenomegaly similar to the C9orf72 knockout mice.Citation61,62 Furthermore, C9orf72 knockout mice show increased secretion of IL-1ßCitation23 and increased numbers of activated CD69+ T-cells,Citation25 phenotypes that have previously been reported in autophagy-deficient ATG16L knockout miceCitation63 and ATG5 knockout mice,Citation64 respectively. Hence the phenotype of these C9orf72 knockout mice may relate to a specific role of C9orf72 in immune-related autophagy.
In agreement, TANK-binding kinase 1 (TBK1), a well-known regulator of inflammation and autophagy (reviewed in ref. Citation65) has been shown to phosphorylate SMCR8 in the C9orf72/SMCR8/WDR41 complex and this phosphorylation appears to be important for its function in autophagy.Citation40 Furthermore, the C9orf72/SMCR8/WDR41 complex has been shown to interact with optineurin, an autophagy substrate receptor involved in clearance of intracellular bacteria and itself a TBK1 substrate.Citation40,66 As mutations in both TBK1 and optineurin have also been associated with ALS/FTD,Citation67,68 a pattern of disrupted immune-related autophagy linked to the pathogenesis of ALS/FTD is emerging.
Defective autophagy and C9ALS/FTD
C9ALS/FTD patients show specific p62/sequestosome-1 pathology.Citation16,35,36 To directly test whether this C9ALS/FTD-associated pathology was linked to C9orf72 haploinsufficiency we investigated p62/sequestosome-1 distribution in cells and neurons depleted of C9orf72. Loss of C9orf72 resulted in the accumulation of p62/sequestosome-1 positive puncta in both HeLa cells and rat cortical neurons and this was specific to the loss of C9orf72 as reintroduction of C9orf72 in the rat cortical neurons was sufficient to rescue this accumulation.Citation39 In a similar experiment, Sellier et al. also demonstrated that reduced expression of C9orf72 led to the accumulation of p62/sequestosome-1.Citation40 We next went on to investigate autophagy in C9ALS/FTD patient-derived iNeurons and found markedly decreased levels of basal autophagy compared to their age and gender matched controls, lending further support to C9orf72 haploinsufficiency as a cause of autophagy deficits and p62/sequestosome-1 pathology in C9ALS/FTD.Citation39 If loss of C9orf72 causes p62/sequestosome-1 pathology in vivo is not yet clear as p62/sequestosome-1 levels have not yet been reported in full or neuronal specific C9orf72 knockout mice.Citation25-29 Interestingly neuronal specific ablation of C9orf72 did not lead to the accumulation of ubiquitin-positive inclusions in the spinal cord, hippocampus and frontal cortex, suggesting that at least in this case loss of neuronal C9orf72 is not enough to replicate disease associated ubiquitin pathology.Citation29 It will be of interest to further characterize the role of C9orf72 in autophagy in the CNS to establish its relationship with p62/sequestosome-1 and ubiquitin pathology in vivo.
The data from C9orf72 knockout mice show that loss of C9orf72 does not lead to the overt neurodegeneration that is typically associated with ALS/FTD.Citation23,25-29 Similarly, GGGGCC repeat C9orf72 BAC-transgenic mice that model toxic gain-of-function RNA and DPR pathology (but retain mouse C9orf72) show noCitation24,69 or mild neurodegeneration.Citation21,22 Hence it is plausible that C9orf72 haploinsufficiency may be a modifier of GGGGCC repeat-associated toxic gain-of-function mechanisms. An attractive possibility is a 2-hit model in which reduced autophagy by loss of C9orf72 function exacerbates DPR accumulation and toxicity. Alternatively, defective autophagy caused by C9orf72 haploinsufficiency in microglia and macrophages may lead to a failure in the support, and therefore survival, of (motor) neurons. Indeed, we know from studies of mutant SOD1-related ALS that ALS is a non-cell-autonomous process that critically involves glial cells.Citation70,71 Furthermore, the association of C9orf72 with immune-related autophagy discussed above may provide a direct link to the neuroinflammation that is observed in ALS/FTD. Future experiments should dissect any direct pathogenic effects of immune-related autophagy in ALS/FTD by, for example, genetic inhibition of autophagy in specific cell lineages such as myeloid cells.
Conclusions
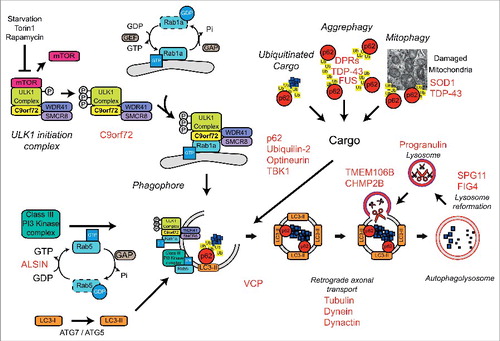
Autophagy deficits appear to have a central role in ALS/FTD. Indeed, in addition to C9orf72, TBK1 and optineurin many other genes implicated in the autophagy/lysosomal pathway have been associated with ALS/FTD, including Alsin (a Rab5 GEF), charged multivesicular body protein-2B (CHMP2B), p62/sequestosome-1, progranulin, ubiquilin-2, valosin-containing protein (VCP), Fig4, and TMEM106b (). Future research will have to elucidate the contribution of autophagy to the disease process and determine the effect of disease-associated mutations in neuronal and non-neuronal cells. Furthermore, as emerging evidence implicates multiple Rab GTPases in ALS/FTD and other neurodegenerative diseases, such as Parkinson disease, studying the specific functions of Rab GTPases in the CNS is warranted.
Figure 4. Autophagy and ALS/FTD. A number of genes, indicated in red, implicated in ALS/FTD have been linked to the autophagy/lysosomal pathway.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014; 10:661-70; PMID:25311585; https://doi.org/10.1038/nrneurol.2014.184
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 2011; 72:245-56; PMID:21944778; https://doi.org/10.1016/j.neuron.2011.09.011
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72:257-68; PMID:21944779; https://doi.org/10.1016/j.neuron.2011.09.010
- Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry Na, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013; 80:415-28; PMID:24139042; https://doi.org/10.1016/j.neuron.2013.10.015
- Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013; 110:E4530-9; PMID:24170860; https://doi.org/10.1073/pnas.1318835110
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 2013; 126:845-57; PMID:24170096; https://doi.org/10.1007/s00401-013-1200-z
- Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, et al. HnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 2013; 125:413-23; PMID:23381195; https://doi.org/10.1007/s00401-013-1088-7
- Sareen D, O'Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Translational Med 2013; 5:208ra149; PMID:24154603; https://doi.org/10.1126/scitranslmed.3007529
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013; 77:639-46; PMID:23415312; https://doi.org/10.1016/j.neuron.2013.02.004
- Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PEa, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013; 126:829-44; PMID:24129584; https://doi.org/10.1007/s00401-013-1192-8
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science (New York, NY) 2014; 16:1131-5
- Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 2013; 126:881-93; PMID:24132570; https://doi.org/10.1007/s00401-013-1189-3
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, NY) 2013; 339:1335-8; https://doi.org/10.1126/science.1232927
- Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 2013; 126:895-905; PMID:24166615; https://doi.org/10.1007/s00401-013-1199-1
- Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Annals Neurol 2013; 74:180-7; PMID:23720273
- Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012; 135:751-64; PMID:22366792; https://doi.org/10.1093/brain/awr365
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol 2012; 11:54-65; PMID:22154785; https://doi.org/10.1016/S1474-4422(11)70261-7
- Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genetics 2013; 92:981-9; PMID:23731538; https://doi.org/10.1016/j.ajhg.2013.04.017
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 2014; 35:1779.e5-1779.e13; PMID:24559645; https://doi.org/10.1016/j.neurobiolaging.2014.01.016
- Xiao S, Macnair L, Mcgoldrick P, Mckeever PM, Robertson J. Isoform specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 2015; 78(4):568-83; PMID:26174152
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, et al. Gain of toxicity from ALS/FTD-Linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 2016; 90:535-50; PMID:27112497; https://doi.org/10.1016/j.neuron.2016.04.006
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC mouse model with motor deficits and Neurodegenerative features of ALS/FTD. Neuron 2016; 90:521-34; PMID:27112499; https://doi.org/10.1016/j.neuron.2016.04.005
- O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AKMG, Ho R, Carmona S, Vit JP, Zarrow J, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016; 351:1324-9; PMID:Can't; https://doi.org/10.1126/science.aaf1064
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, et al. Human C9ORF72 Hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not Neurodegeneration in BAC transgenic mice. Neuron 2015; 88:902-9; PMID:26637797; https://doi.org/10.1016/j.neuron.2015.11.018
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Scientific Reports 2016; 6:23204; PMID:26979938; https://doi.org/10.1038/srep23204
- Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Translational Med 2016; 8:347ra93-ra93; PMID:27412785; https://doi.org/10.1126/scitranslmed.aaf6038
- Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CAC, Youssef SA, Harkema L, de Bruin A, Veldink JH, et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 2016; 132:145-7; PMID:27206760; https://doi.org/10.1007/s00401-016-1581-x
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathologica Commun 2016; 4:51; PMID:27193190; https://doi.org/10.1186/s40478-016-0324-5
- Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CaC, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals Neurol 2015; 78(3):426-38: n/a-n/a; PMID:26044557
- Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013; 29:499-503; PMID:23329412; https://doi.org/10.1093/bioinformatics/bts725
- Zhang D, Iyer LM, He F, Aravind L. Discovery of novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Frontiers Genetics 2012; 3:283
- Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differentiation 2014; 21:348-58; PMID:24440914; https://doi.org/10.1038/cdd.2013.187
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10:513-25; PMID:19603039; https://doi.org/10.1038/nrm2728
- Rubinsztein DC, Bento CF, Deretic V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med 2015; 212:979-90; PMID:26101267; https://doi.org/10.1084/jem.20150956
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011; 122:691-702; PMID:22101323; https://doi.org/10.1007/s00401-011-0911-2
- Mackenzie IRA, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 2014; 127:347-57; PMID:24356984; https://doi.org/10.1007/s00401-013-1232-4
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy*[S]. J Biol Chem 2007; 282:24131-45
- Kabeya Y. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19(21):5720-8; PMID:11060023; https://doi.org/10.1093/emboj/19.21.5720
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 2016; 35(15):1656-76; PMID:27334615
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 2016; 35:1276-97; PMID:27103069; https://doi.org/10.15252/embj.201593350
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; https://doi.org/10.1074/jbc.M900573200
- Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497-510; PMID:18443221; https://doi.org/10.1083/jcb.200712064
- Hosokawa N, Sasaki T, Iemura SI, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973-9; PMID:19597335; https://doi.org/10.4161/auto.5.7.9296
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649-62; PMID:19287211; https://doi.org/10.4161/auto.5.5.8249
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; https://doi.org/10.1038/nature09204
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; https://doi.org/10.1038/ncb2152
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; https://doi.org/10.1091/mbc.E08-12-1249
- Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, Ktistakis NT. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 2013; 126:5224-38; PMID:24013547; https://doi.org/10.1242/jcs.132415
- Wang J, Menon S, Yamasaki A, Chou HT, Walz T, Jiang Y, Ferro-Novick S. Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc Natl Acad Sci U S A 2013; 110:9800-5; PMID:23716696; https://doi.org/10.1073/pnas.1302337110
- Zoppino FCM, Militello RD, Slavin I, Álvarez C, Colombo MI. Autophagosome formation depends on the small GTPase rab1 and functional ER exit sites. Traffic 2010; 11:1246-61; PMID:20545908; https://doi.org/10.1111/j.1600-0854.2010.01086.x
- Meiling-Wesse K, Epple UD, Krick R, Barth H, Appelles A, Voss C, Eskelinen EL, Thumm M. Trs85 (Gsg1), a component of the TRAPP complexes, is required for the organization of the Preautophagosomal structure during selective autophagy via the Cvt pathway. J Biol Chem 2005; 280:33669-78; PMID:16079147; https://doi.org/10.1074/jbc.M501701200
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. α-Synuclein impairs macroautophagy: implications for Parkinson disease. J Cell Biol 2010; 190:1023-37; PMID:20855506; https://doi.org/10.1083/jcb.201003122
- Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes and regulates mTORC1 signaling. Mol Biol Cell 2016; PMID:27559131; https://doi.org/10.1091/mbc.E16-01-0003
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, Chen JF. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2016; 2:e1601167
- Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 2011; 91:119-49; PMID:21248164; https://doi.org/10.1152/physrev.00059.2009
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. TBK-1 Promotes Autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012; 37:223-34; PMID:22921120; https://doi.org/10.1016/j.immuni.2012.04.015
- Seto S, Sugaya K, Tsujimura K, Nagata T, Horii T, Koide Y. Rab39a interacts with Phosphatidylinositol 3-Kinase and negatively regulates autophagy induced by lipopolysaccharide stimulation in macrophages. PLoS One 2013; 8:e83324; PMID:24349490; https://doi.org/10.1371/journal.pone.0083324
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; https://doi.org/10.1038/nri3532
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; https://doi.org/10.1038/nature09782
- Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol 2012; 30:611-46; PMID:22449030; https://doi.org/10.1146/annurev-immunol-020711-074948
- Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 2014; 5:4004; https://doi.org/10.1038/ncomms5004
- Zhu H, Foretz M, Xie Z, Zhang M, Zhu Z, Xing J, Leclerc J, Gaudry M, Viollet B, Zou MH. PRKAA1/AMPKα1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy 2014; 10:1522-34; PMID:24988326; https://doi.org/10.4161/auto.29197
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; https://doi.org/10.1038/nature07383
- Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008; 455:396-400; PMID:18701890; https://doi.org/10.1038/nature07208
- Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: A gatekeeper of Neuroinflammation. Trends Mol Med 2016; 22:511-27; PMID:27211305; https://doi.org/10.1016/j.molmed.2016.04.006
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (New York, NY) 2011; 333:228-33; https://doi.org/10.1126/science.1205405
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18:631-6; PMID:25803835; https://doi.org/10.1038/nn.4000
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465:223-6; PMID:20428114; https://doi.org/10.1038/nature08971
- O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 2015; 88:892-901; PMID:26637796; https://doi.org/10.1016/j.neuron.2015.10.027
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003; 302:113-7; PMID:14526083; https://doi.org/10.1126/science.1086071
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014; 81:1009-23; PMID:24607225; https://doi.org/10.1016/j.neuron.2014.01.013