
ABSTRACT
SynGAP, encoded by SYNGAP1, is a Ras/Rap GTPase activator specifically expressed in the nervous systems. SynGAP is one of the most abundant proteins in the postsynaptic densities (PSDs) of excitatory synapses and acts as a critical synaptic activity brake by tuning down synaptic GTPase activities. Mutations of SYNGAP1 have been frequently linked to brain disorders including intellectual disability, autisms, and seizure. SynGAP has been shown to undergo fast dispersions from synapses in response to stimulations, a strategy that neurons use to control the specific activities of the enzyme within the tiny, semi-open compartments in dendritic spines. However, the mechanism governing the activity-dependent synaptic localization modulations of SynGAP is poorly understood. It has been shown recently that SynGAP α1, via specifically binding to PSD-95, can undergo liquid-liquid phase separation forming membraneless, condensed protein-rich sub-compartments. This phase transition-mediated, PSD-95-dependent synaptic enrichment of SynGAP α1 not only suggests a dynamic anchoring mechanism of the protein within the PSD, but also implies a new model for the PSD formation in living neurons.
Small G proteins regulate many aspects of cellular processes including cell adhesion and migration, growth and differentiation, vesicle trafficking and fusion by circling between their GTP-bound active forms and GDP-bound inactive forms. A wide variety of external and internal stimuli can activate small G proteins via converting GDP-bound form proteins to GTP-bound forms often aided by guanosine nucleotide exchange factors (GEFs). Conversely, GTPase activating proteins (GAPs) facilitate the reverse process via accelerating the GTP hydrolysis activity of the enzymes and converting them into inactive GDP-bound forms. Proper balance of the 2 opposite processes is of vital importance for physiological functions of small GTPase-mediated signaling events. Unlike many other enzymes that often contain intricate regulatory elements within their primary sequences, small G proteins contain minimal extra amino acid sequences other than the catalytic domains. The intricate enzymatic activities of small G proteins are, instead, largely regulated by a diverse collection of GEFs and GAPs, most of which are large proteins containing many regulatory elements in addition to catalytic GTPase exchanging or activating domainsCitation5,Citation8 As such, proper understanding of a GTPase signaling requires integrated studies of its specific GTPase/GEF/GAP trio.
Neurons are extremely polarized and with massive peripheral processes (in terms of volumes, physical lengths, and excessive branching of axons and dendrites). Additionally, physiological functions of neurons require the physical structures of dendrites and axons as well as molecular assemblies within these processes to be dynamically regulated in a very broad time span (from seconds to hours and even longer) both in developing and mature neurons. Therefore, small GTPase signaling in the nervous systems perhaps requires even more elaborated spatial and temporal regulations when compared with GTPases in cells from other tissues. Consistent with this reasoning, neuronal processes and their sub-compartments (e.g. synapses) are often highly enriched in small GTPases and various GEFs and GAPs.Citation9,Citation30 Mutations of many GEFs and GAPs are known to cause different forms of brain diseases ranging from brain mal-formations to psychiatric disorders in humans.Citation31,Citation4
This brief review focuses on a brain-specific GAP protein called SynGAP (encoded by SYNGAP1) which is a major component of the excitatory postsynaptic density (PSD) protein assemblies. Mutations of SYNGAP1 are frequently found in patients suffering brain disorders including intellectual disability (ID), autism spectrum disorders (ASDs), and epilepsy.Citation4,Citation6,Citation14,Citation15,Citation21,Citation25,Citation27,Citation28,Citation34 We first briefly summarize biochemical properties and cellular functions of SynGAP characterized to date. We then discuss a potentially novel spatial localization and concentration mechanism of SynGAP in synapses mediated by a physical process known as liquid-liquid phase separation (LLPS) that was discovered recently. Structural analysis indicates that some of the SYNGAP1 mutations identified in psychiatric disorder patients do not directly alter the GAP activity of SynGAP. Instead, these mutations may affect its PSD localization via mutation-induced impairment of PSD-95 binding or mutation-induced SynGAP mRNA decay resulting loss of one of the encoding gene.
Biochemical features of SynGAP
SynGAP is a brain specific GAP with no detectable expressions in other tissues tested. In neuron, SynGAP predominantly localizes in the PSD regions of excitatory synapses.Citation7,Citation17 There are evidences showing that SynGAP is also expressed in inhibitory synapsesCitation22 and even in growth cones of axons.Citation32 Quantitative proteomic studies of purified PSDs revealed that SynGAP, as with its binding partner PSD-95, is among the most abundant PSD components that can reach a concentration of ∼100 μM (calculated by detected copy numbers in molar terms and an average PSD volume).Citation30 Its extremely high concentration suggests that, in addition to function as a GTPase activator, SynGAP may also function as a scaffold in PSDs.
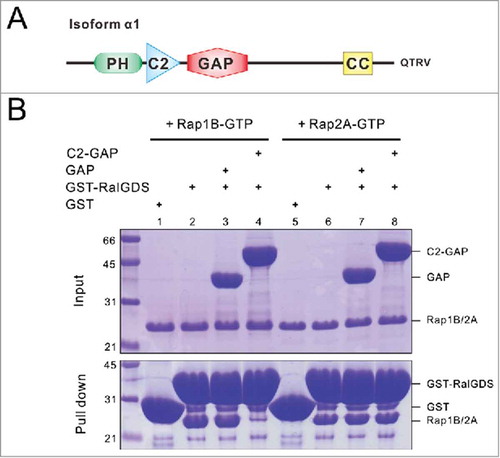
The full-length SynGAP is ∼140 kDa in its molecular mass and contains a PH (Pleckstrin homology) domain, a C2 domain and a GAP domain at its N-terminal half (). It was originally identified as a RasGAP because its GAP domain shows high sequence similarities to canonical RasGAPs such as p120GAP.Citation7 However, subsequent studies reported that SynGAP has higher catalytic activities toward Rap (another small G protein homologous to Ras but with different functions) than Ras.Citation19,Citation26 The RapGAP activity of SynGAP requires the direct coupling between the C2 and GAP domains, as the isolated GAP domain has little detectable GAP activity toward Rap.Citation26 In line with this study, we also confirmed that the C2 domain is essential for SynGAP's RapGAP activity toward Rap1B using highly purified recombinant proteins (). RalGDS specifically recruits GTP-bound Rap. Using GST-RalGDS-mediated binding to Rap-GTP as a readout, we demonstrate that the C2-GAP tandem, instead of the isolated GAP domain, catalyzes GTP hydrolysis of Rap1B-GTP. However, no obvious Rap2A GAP activity could be detected (). We also assayed Ras-GTP hydrolysis by the isolated SynGAP GAP domain using a spectrophotometry-based GAP assay,Citation38 and failed to detect obvious RasGAP activity using both M-Ras-GTP and R-Ras-GTP as substrates. In our controls, p120GAP can efficiently catalyze the GTP hydrolysis of both M-Ras-GTP and R-Ras-GTP (data not shown). Interestingly, a recent study indicated that SynGAP phosphorylation by different protein kinases can differentially modulate GAP activities of SynGAP toward Rap and Ras.Citation37 SynGAP phosphorylation by Ca2+/calmodulin-dependent protein kinase II (CaMKII) preferentially promotes its RapGAP activity; cyclin-dependent kinase 5 (Cdk5)-mediated phosphorylation instead only stimulated RasGAP activity of SynGAP.Citation37 This study provides a nice example showing that, in addition to protein-protein or protein-lipid interactions mediated by the well-defined domains in GAPs (and GEFs as well), post-translational modifications can also exert different regulatory roles on the activities of the enzymes.
Figure 1. The C2-GAP tandem is required for SynGAP's GAP activity toward Rap1B. (A) Schematic diagram showing the domain organization of SynGAP α1. (B) GST-RalGDS pull-down assay showing the C2-GAP tandem, instead of the isolated GAP domain, of SynGAP is required for its Rap1B GAP activity. It is noted that neither the GAP domain nor the C2-GAP tandem of SynGAP had detectable GAP activity toward Rap2A.
After the GAP domain, there is a long flexible region without defined domains or structures and followed by a coiled-coil domain near its C-terminus (). Recently, we reported the atomic structure of the SynGAP coiled-coil domain, showing that it forms a parallel trimeric coiled-coil structure.Citation40 The coiled-coil-mediated trimer formation is critical for concentrating SynGAP at PSDs by efficiently and specifically binding to PSD-95. Additionally, the coiled-coil domain may also play a role for forming SynGAP hetero-trimers, as all isoforms of SynGAP contain the identical coiled-coil sequence.
SynGAP is encoded by a single gene called SYNGAP1. SYNGAP1 is a complex gene that gives rise to multiple SynGAP isoforms via alternative promoter usage and alternative splicing.Citation20 Due to alternative promoter usage, SynGAP isoforms A, B and C are with distinct N-termini, in which the isoform C does not contain a PH domain. Alternative splicing generates additional SynGAP isoforms with variations in their C-termini. Among all isoforms, SynGAP α1 is abundant and also most studied. SynGAP α1 contains a PDZ binding motif (PBM, with “-QTRV” sequence) at its C-terminus (), and this PBM is responsible for binding to PDZ domain-containing scaffold proteins in excitatory PSD such as PSD-95. Earlier studies suggested that SynGAP α1 binds to all 3 PDZ domains of PSD-95 as well as a few other PDZ proteins.Citation17,Citation19 By carefully comparing quantitative binding affinities using purified recombinant proteins, we have demonstrated that SynGAP α1 binds to PDZ3 of PSD-95 with significantly higher affinity than to PDZ1 or PDZ2. The specific interaction between SynGAP α1 and PSD-95 PDZ3 originates from a second target binding site formed by the C-terminal α-helix extension and the βB-, βC-strands of PDZ3 in addition to the canonical target recognition groove of the PDZ domain. This new binding site accommodates a stretch of conserved hydrophobic residues at the upstream of the canonical SynGAP α1 PBM.Citation40 Additionally, the formation of the PDZ3-SH3-GK supramodule further enhances the interaction between PSD-95 PDZ3 with SynGAP α1 by another 10-fold or more. As such, the binding of SynGAP α1 to PSD-95 PDZ3 is several hundred times stronger than to PSD-95 PDZ1 or PDZ2 or to a few other reported PDZ domains.Citation40 Differing from α1, SynGAP α2 isoform does not contain a C-terminal PBM. Nonetheless, both SynGAP α1 and α2 have been shown to localize predominantly in PSDs of excitatory synapses by electron microscopy and mass spectrometry studies.Citation20,Citation39 As mentioned above, SynGAP α2 may localize to PSDs via forming hetero-trimer with the α1 isoform via the coiled-coil domain.
Cellular functions of SynGAP
Complete elimination of SYNGAP1 in mice is lethal at early post-natal stages,Citation16,Citation18 indicating that SynGAP plays critical roles for proper neuronal development. Heterozygous SYNGAP1+/− mice are viable but display obvious long-term potentiation (LTP) impairments as well as social and cognitive defects.Citation10,Citation13,Citation16,Citation18,Citation23 Restored expression of SYNGAP1 in young mutant, but not adult, mice can rescue social and cognitive defects caused by SYNGAP1 haploinsufficiency, supporting the critical roles of SynGAP for brain development.Citation1,Citation10 It is estimated that SYNGAP1 mutation is accounted for 0.7–1% of sporadic ID casesCitation21 and up to 1% of epileptic encephalopathies in humans.Citation6 Most of SYNGAP1 mutations lead to loss of functions of one copy of the gene, therefore most of the clinical phenotypes observed in patients are likely resulted from the loss of half of SynGAP due to haploinsufficiency. It is noted that, in addition to social and cognitive defects, patients with SYNGAP1 mutations can also have other symptoms including mild microcephaly, growth retardation, constipation, hirsutism, hypotonia, and limb problems,Citation4,Citation21,Citation25,Citation28 suggesting that SynGAP also plays functional roles beyond synaptic development and plasticity. In the following, we limit our discussion on the mechanistic roles of SynGAP in excitatory synapses.
SynGAP acts as a synaptic activity “brake” in excitatory synapses by tuning down local Rap/Ras signaling and inhibiting AMPA receptor (AMPAR) incorporation into synaptic membranes. Overexpression of SynGAP α1 in neuronal culture significantly reduces AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs) and results in a decreased AMPAR surface expression.Citation20,Citation29 Both its GAP activity and synaptic localization signal are necessary for SynGAP's function in regulating synaptic transmissions. Consistent with its inhibitory role in synaptic AMPAR expression, synaptic transmission is elevated in neurons derived from SYNGAP1−/− mice.Citation29,Citation33
It appears that modulating synaptic localization of SynGAP (and presumably the subsequent Rap or Ras activities in synapse) is a powerful means in regulating synaptic activities. Immunogold staining coupled with electron microscopic imaging as well as living imaging by fluorescence microscopy of rat hippocampal neuron cultures revealed that both SynGAP α1 and α2 are enriched at the PSD core under basal conditions, and the enzymes can move out of the PSD after NMDA receptor (NMDAR) activation.Citation39,Citation2 Mechanistically, CaMKII-mediated phosphorylation on SynGAP C-terminus may weaken its binding to PSD-95, and thus facilitate its dispersion from dendritic spines.Citation39,Citation2 SynGAP dispersion could have 2 possible effects on synapse activities. The most direct outcome is to release the negative “brake” from the synapse via overall GAP activity decrease in PSDs (i.e. via modulating the enzyme activity of SynGAP at PSDs).Citation2 Alternatively, activity-dependent dispersion of SynGAP out of the PSD core may free its occupied PDZ “slots” on PSD-95 or other Dlgs for glutamate receptors and adhesion molecules (i.e., via regulating the scaffolding role of SynGAP at PSDs).Citation36 Whatever the mechanisms are, the outcomes following SynGAP dispersions from synapses are enlarged spine volumes, more synaptic AMPAR insertions, and higher synaptic activities.Citation2 It is noted that the 2 mechanisms are not mutually exclusive, instead may act synergistically to modulate synaptic activities.
Phase transition of SynGAP and PSD-95
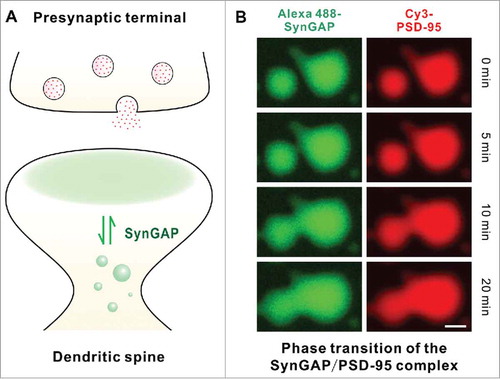
PSDs of excitatory synapses are one of the most complicated protein-rich assemblies in neurons. Thousands of proteins are enriched in this tiny semi-open compartment with plasma membrane on one side and dendritic spine cytoplasm on the other, forming detergent insoluble protein assemblies responsible for synaptic signal processing and storages. Yet, numerous studies have demonstrated that proteins in PSDs can undergo activity-dependent exchanges with those in cytoplasm. The disk-shaped, protein-rich assembly of PSD was discovered more than 60 y ago.Citation24 However, how such semi-open PSD compartments can autonomously form beneath the synaptic plasma membranes remains largely unknown (). The activity-dependent SynGAP dispersions from PSDs offer a good opportunity for studying the mechanisms governing the PSD enrichment as well as dynamic exchanges of SynGAP between PSD and cytoplasm.
We recently discovered that the SynGAP (the SynGAP α1 C-terminal CC-PBM fragment to be precise) and PSD-95 (the PDZ3-SH3-GK tandem to be exact) complex mixture can undergo a protein concentration-dependent liquid-liquid phase separation, forming a dynamic, membrane-lacking protein-rich phase both in vitro and in living cells.Citation40 Both components are highly enriched in the condensed liquid phase comparing to the surrounding aqueous solutions in vitro or in cytoplasm of living cells (i.e., a large concentration gradient of the SynGAP/PSD-95 complex is maintained between the protein-rich condensed phase and the low concentration aqueous phase) (). Importantly, proteins in the 2 phases constantly undergo rapid exchange. The phase transition of the SynGAP/PSD-95 complex requires the multivalent interaction between SynGAP and PSD-95. Both formation of the SynGAP trimer and weak, intermolecular PDZ3-SH3-GK-mediated multimerization capacity of PSD-95 are required for the SynGAP/PSD-95 complex to undergo phase transition. Additionally, specific SynGAP CC-PBM and PSD-95 PDZ3-SH3-GK interaction is also essential for phase transition to occur in living cells. Using hippocampal neuronal cultures as the model, we have demonstrated that the SynGAP α1 mutants (e.g., converting the SynGAP trimer into a monomer or weakening the SynGAP/PSD-95 interaction) that are defective in the SynGAP/PSD-95 phase transitions in vitro or in heterologous cells have been observed to be less well integrated into PSDs as well as to undergo more obvious chemical LTP-induced dispersions. Such mutant SynGAP expressing synapses contain higher amount of AMPA receptors and can be activated by low amplitude stimulations that are not sufficient to activate the wild type SynGAP containing synapses. These observations are consistent with the role of SynGAP as a synaptic activity “brake” in neurons, and mutations of SynGAP can lead to over-activation of synapses.Citation10,Citation2
The phase transition of the SynGAP/PSD-95 complex is highly protein concentration-dependent. Only when protein concentrations pass certain thresholds can phase transition occur. Regulating local SynGAP α1 concentration through protein synthesis/degradation or by isoform replacements might be other ways to modulate phase transitions in a longer timescale. This hypothesis coincides with the observation that overexpression of SynGAP α1 and α2 isoforms in hippocampal neurons have opposing effects on synaptic transmissions. Overexpression of PSD-95 binding SynGAP α1 isoform weakens synaptic strength, possibly by promoting more SynGAP α1 in the protein-rich PSD phase.Citation20,Citation29 Oppositely, overexpression PBM-lacking SynGAP α2 leads to elevated mean mEPSCs amplitudes, presumably by lowering its PSD localization due to lowering the concentration of the effective PSD-95 binding population of SynGAP.Citation20
The protein concentration dependence of the SynGAP/PSD-95 complex phase transition may also shed light on a long-lasting puzzle to us as to why SynGAP as an enzyme exists at such a high concentration in PSDs and why mammalian brain is so sensitive to the dosage of SynGAP. In rodents, SynGAP expression reaches its peak level around 14 d after birth, a period coinciding with the time window of substantial synaptogenesis.Citation10 Loss of both copies of SYNGAP1 in mice (SYNGAP1−/−) leads to postnatal death, indicating that the gene is indispensable for normal brain development. Loss of 50% of SynGAP in mice (i.e., SYNGAP1+/−) is sufficient to cause premature dendritic spine maturation, impaired LTP, exaggerated signal propagation and shifts of the balance of neural circuits toward hyper-excitable states.Citation10,Citation16,Citation18,Citation33 Altered dendritic spine development and neuronal circuits function are believed to be closely related to cognitive dysfunctions and behavioral disorders.Citation35 In agreement with this theory, lost one copy of SYNGAP1 during early development leads to impaired cognition and reduced seizure threshold in mice.Citation1,Citation3,Citation10,Citation23 Theoretically, the enzymatic activity decrease caused by a 50% reduction of SynGAP in SYNGAP1+/− animals should be easily buffered by the remaining high concentrations of the enzyme and by longer reaction time. However, under the scenario of phase transition-mediated incorporation of SynGAP into the PSD via binding to PSD-95, the concentration decrease of SynGAP in the PSD caused by loss of one copy of SYNGAP1 may be much more severe. It is possible that a 50% reduction of total enzyme expression in SYNGAP1+/− animals may lower the final concentration of SynGAP in the PSD by many folds due to the sharp concentration-dependent property of the SynGAP/PSD-95 complex phase transition.Citation40
The liquid-liquid phase separation of the SynGAP/PSD-95 complex in aqueous solution is reminiscent of the dynamic PSD assembly in living neurons in the following ways: protein assemblies in the PSD are highly concentrated compared with the same proteins in the cytoplasm of dendritic spines; proteins in the PSD and in dendritic spines can coexist without needing of physical membrane-like structure for separations; proteins within and outside the PSD can undergo dynamic exchanges in response to neuronal activity changes; PSD protein assemblies can undergo size changes in responding to neuronal activity. Therefore, there is a possibility that formation and dynamic assembly/disassembly of PSDs can also occur via the liquid-liquid phase separation (). This is certainly a bold hypothesis that requires further experimental tests. For example, the excitatory PSDs contain many abundant scaffold proteins and enzymes including PSD-95 (and other Dlgs), SAPAPs, Shank, Homer, CaMKII, and among others. These proteins can form a highly complicated protein-protein interaction network with many multivalent interactions. It remains to be tested whether these protein complexes can undergo liquid-liquid phase separation. If these protein complexes can also undergo phase transition, it will be important to characterize both the thermodynamic (e.g., the threshold concentrations, stoichiometric compositions, stabilities of the assemblies, etc.) and kinetic (e.g., the assembly and exchange rates of the components, possible enzyme-mediated regulations of the assemblies) properties of the phase transitions and to investigate functional implications of forming such self-organized PSD assemblies not delimited by membrane bilayers.
SYNGAP1 mutations in patients with neurodevelopmental disorders
Human genetic studies in the past have identified many mutations of SYNGAP1 in ASD and ID patients.Citation4,Citation6,Citation14,Citation15,Citation21,Citation25,Citation27,Citation28,Citation34 We have curated these mutations () and propose to divide these mutations into 2 major categories: one with mutations occur within or before the catalytic C2-GAP domain tandem, and these mutations are expected to impair the GAP activity of SynGAP; the mutations in the second category occur in the C-terminal half of the protein, and interestingly all these identified mutations are truncation mutations caused either by direct gain of stop codons or introduction of premature stop codons due to frameshifts.
Figure 2. Mutations of SynGAP1 identified in human patients. (A) Mapping of SYNGAP1 mutations found in ID or ASD human patients onto the amino acid sequence of the protein with accession code of NP_006763. K138*, R579* and L813RfsX are reported inCitation15; V348AfsX is reported inCitation34; K108VfsX, c.2294+1G>A (p.E706LfsX) and Q893RfsX are reported inCitation14; K418RfsX and T878DfsX are reported inCitation27; H95PfsX, W362R, P562L, N729TfsX and S738* are reported inCitation4; K114SfsX, c.389–2A>T, R143*, W267* and Q702* are reported inCitation6; c.3583–6G>A (p.V1195AfsX) is reported inCitation28; T144SfsX, R170Q, L327P, Y518NfsX, R922*, L925PfsX, Q928* and Q1093* are reported in.Citation25 Domains of SynGAP are defined as the PH domain (aa 150–253), C2 domain (aa 263–370), GAP domain (aa 408–734) and coiled-coil or CC domain (1188–1277). (B) Comparison of the Rap1B GAP activities of the WT or mutant SynGAP C2-GAP tandem. Data represent 3 batches of independent experiments and are presented as mean ± SEM.
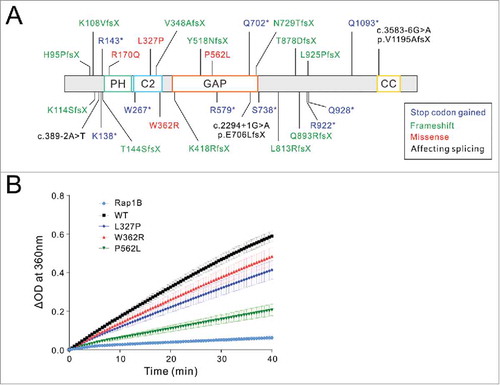
Figure 3. A schematic diagram of Phase transition-mediated SynGAP enrichment in PSD. (A) A Schematic diagram showing SynGAP enriched in PSD (shaded in pale green) can undergo dynamic exchange with SynGAP in dendritic shafts. (B) Co-localization of SynGAP and PSD-95 in the droplets with enriched concentrations. PSD-95 PSG and SynGAP CC-PBM labeled with Cy3 and Alexa488, respectively. A 1:1 mixture of Alexa488-SynGAP and Cy3-PSD-95 at a concentration of 50 μM. The time-lapse images are to demonstrate that small droplets can grow and fuse into larger ones.
The resulting SynGAP mutant proteins in the second category are defective in PSD-95 binding due to the loss of the coiled-coil domain and/or the extreme C-terminal PDZ binding motif, although these mutations may not directly impair the catalytic activity of SynGAP (). Since the PSD-95 binding-mediated PSD localization is critical for SynGAP's functions in synapses, the SynGAP mutations in the second category are expected to have major impacts on brains functions in these patients if the mutant transcripts are stably transcribed. It should be noted that mutations leading to premature truncations of SYNGAP1 can also cause nonsense-mediated RNA decay. Under such situations, the mutation will be more or less equivalent to the loss of one copy of SYNGAP1 and the phenotype can be attributed to the haploinsufficiency of the gene. The exact outcomes of the truncation mutations are difficult to predict, and should be experimentally investigated. For example, it was demonstrated recently that 2 different mutations in the middle region of Shank3 have very different impacts on the mRNA stability and consequent mutant Shank3 protein levels.Citation41 One leads to very unstable mRNA transcripts and thus very little mutant protein expression, and the other with stable expression of truncated protein likely acting as a dominant negative. As the results, the 2 Shank3 mutations display distinct phenotypes in patientsCitation11,Citation12 and in mice models.Citation41
For the first category, we selected 3 missense mutations occurring in the SynGAP C2-GAP tandem (L327P, W362R, and P562L; ) to test whether these mutations may alter the GAP activity of the enzyme. L327P localizes in the C2 domain and was found in a pair of monozygotic twins both with severe ID, autism and seizure.Citation25 W362R is another mutation in the C2 domain and was identified in an ID patient with both autism and seizure.Citation4 P562L is in the GAP domain and was identified in an ID patient with autism but no seizure.Citation4 We purified the wild type SynGAP C2-GAP or its mutants each bearing one of the above 3 mutations, and compared their GAP activities toward Rap1B using a spectrophotometry-based GAP assay (). Rap1B without addition of SynGAP shows very low intrinsic GAP activity. Comparing to the WT C2-GAP, all 3 mutants show decreased catalytic activities. L327P and W362R display moderate decreases in their GAP activities, whereas P562L has a more obvious decrease of the GAP activity of the enzyme. The different degrees of GAP activity changes caused by the 3 mutants observed in was also confirmed by a GST-RalGDS pull-down assay (data not shown). The results shown in suggest that many forms of mutations throughout SYNGAP1, either due to direct alterations of its GAP activities or because of disruptions of its synaptic localizations, can all lead to brain function impairments.
Methods
Plasmids, protein expression and purification
The full length rat SynGAP construct is a gift from Dr. Richard L. Huganir at Johns Hopkins University School of Medicine, Baltimore, MD, USA (reference sequence UniProt: Q9QUH6, 1308aa). The fragments encoding C2-GAP (aa 244E-740R) and GAP (aa 408T-740R) were PCR amplified and cloned into an in-house modified pETm3c vector with N-terminal His6 tag. Note that, the amplified C2-GAP sequence does not contain the residues corresponding to 375G-389S of the reference sequence. Mutations of SynGAP C2-GAP were made by standard PCR method and confirmed by DNA sequencing. Constructs encoding His6-tagged GAP domain of p120GAP and small GTPases including Rap1B, Rap2A, M-Ras and R-Ras as well as GST-tagged RalGDS are gifts from Dr. Xuewu Zhang at University of Texas Southwestern Medical Center, Dallas, TX, USA.Citation38 Recombinant proteins were expressed in Escherichia coli BL21 (DE3) cells in LB medium at 16°C and purified using Ni2+-NTA agarose (for His6-tagged proteins) or glutathione sepharose (for GST-tagged protein) affinity chromatography followed by size-exclusion chromatography with a column buffer containing 50 mM Tris pH7.8, 200 mM NaCl, 1 mM EDTA, 1 mM DTT (for C2-GAP) or 50 mM Tris pH7.8, 100 mM NaCl, 1 mM EDTA, 1 mM DTT (for GAP proteins or GST-tagged proteins). MgCl2 at a concentration of 4 mM was kept in all purification buffers for small GTPases.
RalGDS pull-down assay
GTP-bound Rap was prepared by incubating purified Rap with 50-fold excess fresh GTP in the presence of 5 mM EDTA on ice for 1 hour. The reaction was terminated by adding 10 mM MgCl2 and excess nucleotide was removed by a size-exclusion column. GTP-bound Rap (7.5 µM) was initially incubated with 15 µM SynGAP GAP or C2-GAP at room temperature for 30min. Pull down assay was performed by adding GST or GST-RalGDS (10 µM) and incubating in 4°C for 30min, and followed by adding glutathione-sepharose beads for another 30 min incubation in 4°C. After extensive wash by PBS, Rap-GTP was eluted and detected by SDS-PAGE with Coomassie blue staining.
Spectrophotometry-based GAP activity assay
Rap1B (40 µM) with or without C2-GAP (WT or mutants, 1µM) were added to the reaction system containing 50 mM Tris pH7.8, 120 mM NaCl, 4 mM EDTA, 100μM GTP, 1 U purine nucleoside phosphorylase (PNP), and 200μM 2-amino-6-mercapto-7-methylpurine riboside (MESG). PNP and MESG were from EnzChek Phosphate Assay Kit (E6646, ThermoFisher). Directly after incubation, the absorbance at 360 nm was monitored continuously in a 1-cm path-length cuvette at room temperature and each GAP-catalyzed reaction was monitored for 40 min.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from RGC of Hong Kong (664113, 16103614, and AoE-M09–12), and a 973 program grant from the Minister of Science and Technology of China (2014CB910204) to M Zhang. M Zhang is a Kerry Holdings Professor in Science and a Senior Fellow of IAS at HKUST.
References
- Aceti M, Creson TK, Vaissiere T, Rojas C, Huang WC, Wang YX, Petralia RS, Page DT, Miller CA, Rumbaugh G. Syngap1 haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol Psychiatry 2015; 77:805-15; PMID:25444158; https://doi.org/10.1016/j.biopsych.2014.08.001
- Araki Y, Zeng M, Zhang M, Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 2015; 85:173-89; PMID:25569349; https://doi.org/10.1016/j.neuron.2014.12.023
- Berryer MH, Chattopadhyaya B, Xing P, Riebe I, Bosoi C, Sanon N, Antoine-Bertrand J, Lévesque M, Avoli M, Hamdan FF. Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat Commun 2016; 7:133340; PMID:27827368; https://doi.org/10.1038/ncomms13340
- Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot‐Cerioli M. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Human Mutation 2013; 34:385-94; PMID:23161826; https://doi.org/10.1002/humu.22248
- Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007; 129:865-77; PMID:17540168; https://doi.org/10.1016/j.cell.2007.05.018
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genetics 2013; 45:825-30; PMID:23708187; https://doi.org/10.1038/ng.2646
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 1998; 20:895-904; PMID:9620694; https://doi.org/10.1016/S0896-6273(00)80471-7
- Cherfils J, Zeghouf M. Regulation of small gtpases by gefs, gaps, and gdis. Physiological Rev 2013; 93:269-309; PMID:23303910; https://doi.org/10.1152/physrev.00003.2012
- Chua J.J.E. Macromolecular complexes at active zones: integrated nano-machineries for neurotransmitter release. Cell Mol Life Sci 2014; 71:3903-16; PMID:24912984; https://doi.org/10.1007/s00018-014-1657-5
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting the maturation of dendritic spine synapses. Cell 2012; 151:709-23; PMID:23141534; https://doi.org/10.1016/j.cell.2012.08.045
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genetics 2007; 39:25-7; PMID:17173049; https://doi.org/10.1038/ng1933
- Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Côté M, Noreau A. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci 2010; 107:7863-8; https://doi.org/10.1073/pnas.0906232107
- Guo X, Hamilton PJ, Reish NJ, Sweatt JD, Miller CA, Rumbaugh G. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 2009; 34:1659-72; PMID:19145222; https://doi.org/10.1038/npp.2008.223
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 2011; 69:898-901; PMID:21237447; https://doi.org/10.1016/j.biopsych.2010.11.015
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Carmant L. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 2009; 360:599-605; PMID:19196676; https://doi.org/10.1056/NEJMoa0805392
- Kim JH, Lee HK, Takamiya K, Huganir RL. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 2003; 23:1119-24; PMID:12598599
- Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 1998; 20:683-91; PMID:9581761; https://doi.org/10.1016/S0896-6273(00)81008-9
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJC, O'Carroll CM, Martin SJ, Morris RGM. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 2002; 22:9721-32; PMID:12427827
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron 2004; 43:563-74; PMID:15312654; https://doi.org/10.1016/j.neuron.2004.08.003
- McMahon AC, Barnett MW, O'Leary TS, Stoney PN, Collins MO, Papadia S, Choudhary JS, Komiyama NH, Grant S.G.N, Hardingham GE. SynGAP isoforms exert opposing effects on synaptic strength. Nat Communications 2012; 3:900; PMID:22692543; https://doi.org/10.1038/ncomms1900
- Mignot C, Von Stülpnagel C, Nava C, Ville D, Sanlaville D, Lesca G, Rastetter A, Gachet B, Marie Y, Korenke GC. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Medical Genetics 2016; 5:511-22; PMID:26989088; https://doi.org/10.1136/jmedgenet-2015-103451
- Moon IS, Sakagami H, Nakayama J, Suzuki T. Differential distribution of synGAPα1 and synGAPβ isoforms in rat neurons. Brain Res 2008; 1241:62-75; PMID:18824155; https://doi.org/10.1016/j.brainres.2008.09.033
- Ozkan ED, Creson TK, Kramár EA, Rojas C, Seese RR, Babyan AH, Shi Y, Lucero R, Xu X, Noebels JL. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 2014; 82:1317-33; PMID:24945774; https://doi.org/10.1016/j.neuron.2014.05.015
- Palay SL. Synapses in the central nervous system. J Biophys Biochem Cytology 1956; 2:193; PMID:13357542; https://doi.org/10.1083/jcb.2.4.193
- Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park SM, Benoit V. De novo, heterozygous, loss‐of‐function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Medical Genetics Part A 2015; 167:2231-7; https://doi.org/10.1002/ajmg.a.37189
- Pena V, Hothorn M, Eberth A, Kaschau N, Parret A, Gremer L, Bonneau F, Ahmadian MR, Scheffzek K. The C2 domain of SynGAP is essential for stimulation of the Rap GTPase reaction. EMBO Reports 2008; 9:350-5; PMID:18323856; https://doi.org/10.1038/embor.2008.20
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012; 380:1674-82; https://doi.org/10.1016/S0140-6736(12)61480-9
- Redin C., Gérard B., Lauer J., Herenger Y., Muller J., Quartier A., Masurel-Paulet A., Willems M., Lesca G, El-Chehadeh S. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 2014; 51(11):724-36
- Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A 2006; 103:4344-51; PMID:16537406; https://doi.org/10.1073/pnas.0600084103
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 2007; 76:823-47; PMID:17243894; https://doi.org/10.1146/annurev.biochem.76.060805.160029
- Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, Boyle J, Shaw M, Gardner A, Proos A, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 2010; 42:486-8; PMID:20473311; https://doi.org/10.1038/ng.588
- Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51. 1 and its binding partners in CNS axon outgrowth. Genes Dev 2004; 18:541-58; https://doi.org/10.1101/gad.1151204
- Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J Neurosci 2004; 24:8862-72; PMID:15470153; https://doi.org/10.1523/JNEUROSCI.3213-04.2004
- Vissers L.E.L.M, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M. A de novo paradigm for mental retardation. Nat Genetics 2010; 42:1109-12; PMID:21076407; https://doi.org/10.1038/ng.712
- Volk L, Chiu SL, Sharma K, Huganir RL. Glutamate synapses in human cognitive disorders. Annual Rev Neurosci 2015; 38:127-49; PMID:25897873; https://doi.org/10.1146/annurev-neuro-071714-033821
- Walkup Iv WG, Mastro TL, Schenker LT, Vielmetter J, Hu R, Iancu A, Reghunathan M, Bannon BD, Kennedy MB. A model for regulation by SynGAP-α1 of binding of synaptic proteins to PDZ-domain'Slots' in the postsynaptic density. Elife 2016; 5:e16813; PMID:27623146
- Walkup WG, Washburn L, Sweredoski MJ, Carlisle HJ, Graham RL, Hess S, Kennedy MB. Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. J Biol Chem 2015; 290:4908-27; PMID:25533468; https://doi.org/10.1074/jbc.M114.614420
- Wang Y, He H, Srivastava N, Vikarunnessa S, Chen Yb, Jiang J, Cowan CW, Zhang X. Plexins are GTPase activating proteins for rap and are activated by induced dimerization. Sci Signal 2012; 5:ra6; PMID:22253263
- Yang Y, Tao-Cheng JH, Bayer KU, Reese TS, Dosemeci A. Camkii-Mediated Phosphorylation regulates distributions of Syngap-α1 and–α2 at the Postsynaptic density. PloS One 2013; 8:e71795; PMID:23967245; https://doi.org/10.1371/journal.pone.0071795
- Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, Zhang M. Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell 2016; 166:1163-75; PMID:27565345; https://doi.org/10.1016/j.cell.2016.07.008
- Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 2016; 89:147-62; PMID:26687841; https://doi.org/10.1016/j.neuron.2015.11.023