
Abstract
Recent studies on the processes that lead to the development of pancreatic cancer indicate that inflammatory macrophages have key functions in the initiation of pre-neoplastic lesions. Specifically, acquisition of an activating Kras mutation in pancreatic acinar cells leads to upregulation of intercellular adhesion molecule-1 (ICAM-1), which serves as a chemoattractant for M1-polarized macrophages. M1 macrophages then contribute to acinar cell metaplasia and development of precancerous lesions through inflammatory cytokines and secreted proteases.
Introduction
Metaplasia of pancreatic epithelial cells leads to a dedifferentiated, progenitor-like cell type that can develop to pancreatic intraepithelial neoplastic lesions (PanINs) or other types of pancreatic lesions.Citation1,2 In genetic animal models for pancreatic cancer the metaplasia of pancreatic acinar cells and further progression to PanINs can be induced by introduction of a Kras mutation,Citation3 which is also found in over 95% of all human pancreatic cancers. Although in the above mentioned animal models Kras mutations are expressed in all acinar cells, the occurrence of areas of acinar-to-ductal metaplasia (ADM) and PanINs is focal, indicating additional non-genetic factors involved in their formation.
Presence of pancreatic cancer goes along with desmoplasia and pancreatic inflammation (pancreatitis), and inducers of inflammatory responses such as obesity and smoking have been identified as environmental risk factors.Citation4 Moreover, a synergistic connection between oncogenic Kras mutations and presence of chronic pancreatitis was demonstrated in mice to abrogate the senescence barrier in low-grade PanINs and to accelerate the development of pancreatic ductal adenocarcinoma (PDA).Citation5 Similarly, a high-fat diet accelerates development of PDA in mice expressing mutant Kras by causing pancreatic inflammation and macrophage infiltration.Citation6
While in acinar cells Kras-initiated signaling pathways that drive development of pancreatic cancer are relatively well defined,Citation7 the crosstalk with immune cells that contribute to this process remains unclear. Our recent study for the first time reveals a direct cooperative mechanism between oncogenic Kras mutations and the inflammatory environment to drive the initiation of pancreatic cancer. We demonstrate that Kras-expressing pancreatic acinar cells initiate the chemoattraction of M1 macrophages to induce local inflammation. Once present in areas of ADM, M1 macrophages expedite Kras-driven transdifferentiation and formation of PanINs by providing enzymes that mediate extracellular matrix (ECM) degradation and cytokines that further drive the ADM process.Citation8
Macrophages are Needed for Initiation and Progression of Pancreatic Cancer
Precursor lesions to PDA often are associated with focal pancreatitis. By treating p48cre;LSL-KrasG12D mice with the macrophage toxin Gadolinium (III) chloride we found that macrophages contribute to formation and progression of mutant Kras-caused PanIN lesions.Citation8 Similar depletion of macrophages in a mouse model for acute pancreatitis completely protected acinar cells from undergoing metaplasia,Citation9 indicating that inflammation alone may be sufficient to initiate the transdifferentiation process. While in absence of oncogenic Kras this is reversible, in presence of mutant Kras it can accelerate formation of pancreatic cancer. Mechanistic insight of how inflammation may contribute to changes in the pancreas microenvironment was provided by demonstrating that macrophages secrete proteases that modulate the ECM and cytokines that can drive ADM. For example we show increased activity of matrix metalloproteinases (MMPs), mainly MMP-9, in regions of ADM and PanINs and also increased presence of the pro-inflammatory cytokine tumor necrosis factor (TNF). This, combined with Kras-caused changes in gene expression in acinar cells, can expedite the formation of precancerous lesions.
Kras-expressing Acinar Cells Upregulate ICAM-1 to Facilitate Microinflammation
By analyzing human patient tissue samples, we found that pancreatic regions of ADM express the ICAM-1. Immunohistochemical analysis of pancreata from p48cre;LSL-KrasG12D mice indicated that ICAM-1 expression is due to expression of mutant Kras. Additionally, transfection of oncogenic mutant Kras into normal acinar cells led to dramatic increase in ICAM-1 mRNA and protein expression. Moreover, ICAM-1 expressed by acinar cells was processed to a soluble version (sICAM-1), possibly functioning as chemoatractant for immune cells. Using transwell assays we showed that sICAM-1 indeed is a chemoattractant for primary mouse macrophages. A more detailed analysis of the subtype involved indicated that M1-, but not M2-polarized macrophages are attracted by sICAM-1. Next we showed in similar transwell assays that pancreatic acinar cells expressing mutant Kras can attract primary mouse M1-polarized macrophages; and this can be blocked with an ICAM-1 neutralizing antibody (ICAM-1 NAB). These exciting in vitro data were confirmed and strengthened by in vivo animal experiments in which p48cre;LSL-KrasG12D-expressing mice were treated with ICAM-1 neutralizing antibody or control antibody over a period of 11 weeks. The presence of the neutralizing antibody significantlyblocked the formation of precancerous lesions, almost identical to the data we had obtained by depleting macrophages in mice. Overall, our data describes a novel mechanism of how acinar cells that have acquired an oncogenic Kras mutation can crosstalk with macrophages to drive the initiation of precancerous lesions ().
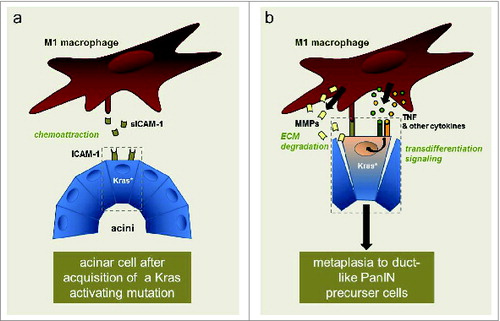
Figure 1. Crosstalk of macrophages with mutant Kras-expressing pancreatic acinar cells. Acquisition of an activating Kras mutation (Kras*) in acinar cells leads to expression of ICAM-1. A fraction of the ICAM-1 produced is shed and soluble (sICAM-1). Soluble ICAM-1 can act as a chemoattractant for M1-polarized macrophages (A). Macrophages then may directly interact with acinar cells via membrane ICAM-1. They provide enzymes that allow degradation of extracellular matrix (ECM) and inflammatory cytokines and chemokines that can drive transdifferentiation signaling. This leads to metaplasia of acinar cells to a duct-like phenotype that can give rise to pancreatic intraepithelial neoplastic (PanIN) lesions, which then can further progress to pancreatic cancer (B).
Conclusions
Two key problems prevent therapeutic efforts in pancreatic cancer. First, pancreatic cancer usually is diagnosed late, when it often is already metastatic. Second, once diagnosed PDA is difficult to treat because of its desmoplastic and inflammatory environment, which blocks drugs from reaching tumor cells.Citation10 The key to solve both issues is to develop methods for an early diagnosis and treatment. Moreover, at a certain age PanINs are present in almost every individual; and an additional difficulty is to determine which PanINs will progress to PDA. The characterization of the signaling events that drive formation and progression of PanINs will allow such development of novel methods for early diagnosis and for early treatment. Moreover, it will allow to reliably distinguish between individuals with high-grade pre-neoplastic lesions and individuals where it is “safe” to continue surveillance.
Experimentally-induced pancreatic inflammation enhances Kras-driven PanIN expansion and development of PDA in mice.Citation5 Our data now indicate that acquisition of a Kras mutation can also initiate microinflammation, which then leads to formation of PanINs.Citation8 Accompanying increased levels of sICAM-1 released by acinar cells or cytokines (e.g. TNF) produced by attracted macrophages, now could serve as markers for progression of disease. Both can be detected in pancreatic cystic fluid of pancreatitis and cancer patients. Moreover, we show that the use of neutralizing antibodies (e.g., ICAM-1 NAB) can reduce the progression of Kras-caused PanINs in vivo, by decreasing macrophage infiltration into the pancreas. Thus, we provide a proof-of-principle experiment for the use of such blocking antibodies for clinical use in humans.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Stanger BZ, Hebrok M. Control of cell identity in pancreas development and regeneration. Gastroenterology 2013; 144:1170-9; PMID:23622126; http://dx.doi.org/10.1053/j.gastro.2013.01.074
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2:897-909; PMID:12459728; http://dx.doi.org/10.1038/nrc949
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004; 5:375-87; PMID:15093544; http://dx.doi.org/10.1016/S1535-6108(04)00085-6
- Kolodecik T, Shugrue C, Ashat M, Thrower EC. Risk factors for pancreatic cancer: underlying mechanisms and potential targets. Front Physiol 2013; 4:415; PMID:24474939; http://dx.doi.org/10.3389/fphys.2013.00415
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M,Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011; 19:728-39; PMID:21665147; http://dx.doi.org/10.1016/j.ccr.2011.05.011
- Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, Lugea A, Gukovskaya AS, Li G et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 2013; 6:1064-73; PMID:23943783; http://dx.doi.org/10.1158/1940-6207.CAPR-13-0065
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012; 22:304-17; PMID:22975374; http://dx.doi.org/10.1016/j.ccr.2012.07.024
- Liou GY, Doppler H, Necela B, Edenfield B, Zhang L,Dawson DW, Storz P. Mutant Kras-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov 2015; 5:52-63; PMID:25361845; http://dx.doi.org/10.1158/2159-8290.CD-14-0474
- Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M, Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol 2013; 202:563-77; PMID:23918941; http://dx.doi.org/10.1083/jcb.201301001
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A,McIntyre D, Honess D, Madhu B, Goldgraben MA,Caldwell ME, Allard D et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457-61; PMID:19460966; http://dx.doi.org/10.1126/science.1171362