
Abstract
A key to improving cancer immunotherapy will be the identification of tumor-specific “neoantigens” that arise from mutations and augment the resultant host immune response. In this study we identified single nucleotide variants (SNVs) by RNA sequencing of asbestos-induced murine mesothelioma cell lines AB1 and AB1-HA. Using the NetMHCpan 2.8 algorithm, the theoretical binding affinity of predicted peptides arising from high-confidence, exonic, non-synonymous SNVs was determined for the BALB/c strain. The immunoreactivity to 20 candidate mutation-carrying peptides of increased affinity and the corresponding wild-type peptides was determined using interferon-γ ELISPOT assays and lymphoid organs of non-manipulated tumor-bearing mice. A strong endogenous immune response was demonstrated to one of the candidate neoantigens, Uqcrc2; this response was detected in the draining lymph node and spleen. Antigen reactive cells were not detected in non-tumor bearing mice. The magnitude of the response to the Uqcrc2 neoantigen was similar to that of the strong influenza hemagglutinin antigen, a model tumor neoantigen. This work confirms that the approach of RNAseq plus peptide prediction and ELISPOT testing is sufficient to identify natural tumor neoantigens.
Introduction
Although it has been known for many years that tumors express a large number of mutated proteins that could potentially elicit strong immune responses, the difficulty of detecting these antigens and quantifying their specific antitumor reactivity has meant that few such neoantigens have been identified.Citation1 This situation has changed recently with the development of high-throughput next-generation sequencing (NGS), which has enabled rapid delineation of the mutational profile of expressed cancer proteins, prediction of altered MHC binding of these mutated peptides, and testing of the peptides to identify those that induce specific CD8+ T-cell responses.
NGS of tumors, identification of non-synonymous mutations, and in silico epitope prediction have successfully been used to identify tumor neoantigens in several mouse cancers, including spontaneous melanomaCitation2 and methylcholanthrene (MCA)-induced sarcoma,Citation3,4 and in some human cancers, including melanoma,Citation5 cholangiocarcinoma,Citation6 ovarian cancer,Citation7 and chronic lymphocytic leukemiaCitation8 in a variety of treatment settings.
Despite this progress, it will not be possible to answer in humans some of the key outstanding questions addressing how best to use such reactivities to track responses and/or treat patients. Questions such as “What is the main anatomical compartment that the immune response occurs in?” cannot be answered in patients because of limited access to lymph nodes and tumors. Similarly, questions relating to the mechanisms of action and optimization of new checkpoint blockade-based treatments and the capacity of neoantigen vaccines to alter antitumor responses, especially when used in combination with other chemo- or immunotherapies, need to be addressed in relevant animal models.
Most immunocompetent animal models do not mimic human cancer in terms of how the cancer is initiated; most use artificial carcinogens, occur spontaneously, or are gene-targeted by transgenesis. One of the rare animal tumor models that is actually induced by a known human carcinogen and also faithfully mimics the human tumor counterpart is the asbestos-induced tumor mesothelioma.Citation9 We therefore conducted this study in mouse asbestos-induced mesothelioma to examine whether such a natural carcinogen induces tumor neoantigenic immune responses. As most published studies examined blood or spleen despite the finding that neoantigenic responses are preferentially localized to the lymph node draining the tumor (dLN),Citation10 we then compared neoantigen reactivities in dLN and the spleen. Finally, to investigate the strength of neoantigen reactivity, we compared endogenous neoantigen responses to those observed against a transduced strong tumor neoantigen, influenza hemagglutinin HA, in the same tumor cells, examining in the process any potential competition between HA and the “natural” neoantigen.Citation11
Results
Identification of non-synonymous mutations in expressed genes and predicted neoantigens in murine mesothelioma
The transcriptomes of the BALB/c mesothelioma cell lines AB1 and AB1-HA and the background BALB/c wild-type liver were sequenced. To increase the probability of identifying mutations that were expressed by the cancer, we used RNAseq. For each sample, more than 50 million sequenced reads with a mean length of 90 base pairs were generated. The concordant pair alignment was greater than 80%, and the average coverage depth was 133× in BALB/c liver, 77× in AB1, and 70× in AB1-HA. To improve confidence in the identification of the mutations called, the total exome of the cell lines and normal tissue was also sequenced such that a median of 40 million 150-nucleotide reads were generated with an average coverage depth of 70×.
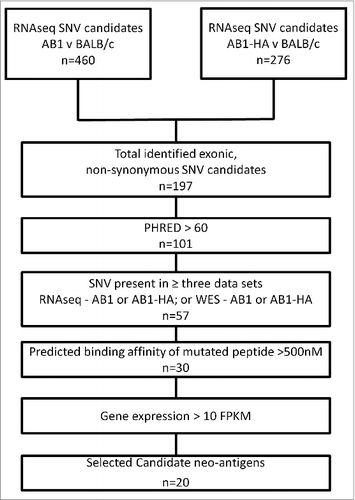
We identified 460 and 276 somatic single nucleotide variants (SNV) in the transcriptomes of AB1 and AB1-HA, respectively. A total of 197 SNVs that were predicted to result in non-synonymous protein changes were identified in exomic regions. Considering only high-quality data, a total of 57 mutations were detected in 3 of the 4 available sequence datasets; i.e., either RNAseq or exome sequencing of the 2 cell lines. In silico epitope prediction using netMHC pan 2.8 determined that 30 mutated peptides had the potential to bind to MHC class I of BALB/c mice: 21 with high affinity and 9 with weak affinity. Of these, 20 were selected for further study. PCR-based Sanger sequencing validated 18 of the 20 candidates; 2 candidates were not validated because of a failure to generate a clean amplicon by PCR , . Thus, mouse mesothelioma has a similar mutation rate to human mesothelioma (manuscript in preparation).
Table 1. Sequence and predicted binding characteristics of the 20 candidate peptide neoantigens
Figure 1. Flow diagram showing the selection process to identify candidate neoantigens for testing.
In vivo testing of predicted neoantigens
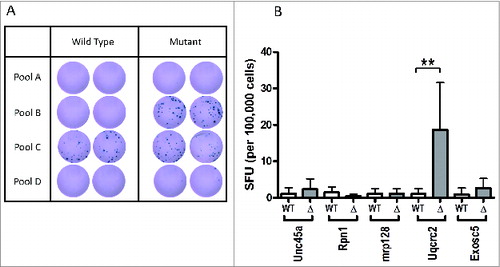
To determine whether an endogenous T-cell response was induced to any of these putative neoantigens we inoculated wild-type mice subcutaneously with the AB1 mesothelioma cell line. Tumors were grown to approximately 70 mm2 without intervention and then the dLNs were harvested. Twenty hours after recovery, dLN cells were tested for specific CD8+ T-cell reactivity using peptide-induced interferon γ production in an ELISPOT assay. We focused on CD8+ T-cell responses using short peptides and tested the peptides first in pools of 5 peptides/pool and then as separate peptides. All mice generated a CD8+ T-cell response that was restricted to the mutant peptides in pool B only (); no response was detected against peptides of pools A and D. Reactivity was demonstrated against the wild-type and mutant peptide C pool. Deconvolution of pool B revealed that the immune response was directed against only one of the 5 peptides, mutated Uqcrc2, with a median of 0.17 (interquartile range [IQR], 0–1.42) spot-forming units (SFU)/105 cells against the wild-type peptide detected in 9 mice, which was significantly less than the 14.38 (IQR 9.32–29.42) SFU/105 cells demonstrated against the mutant peptide (P < 0.01) . This finding demonstrates that spontaneous antigenic reactivity induced by this natural human carcinogen to at least one mutant peptide could readily be detected.
Figure 2. Endogenous immune responses to candidate neoantigens in mesothelioma tumor-bearing mice. (A) Representative duplicate wells from interferon-γ ELISPOT analysis of total dLN cell preparation from non-treated AB1 tumor-bearing mice against pools of peptides. (B) Summary of ELISPOT data showing mean ± SD for the deconvolution of peptide pool B. ELISPOT assays were performed in triplicate; **P < 0.01.
Comparison of responses in spleen versus tumor-draining lymph node
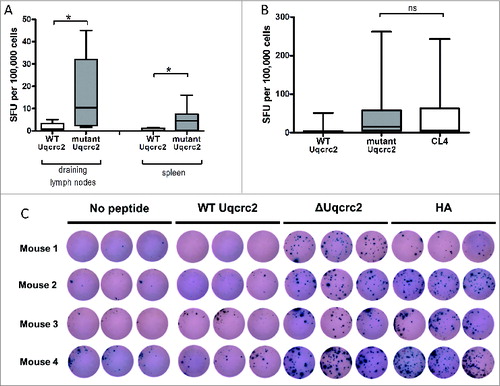
Because our previous studies using model antigens demonstrated that the immune response may be restricted to the tumor dLN, and because most human studies are not able to measure responses in this anatomical compartment, we were concerned that specific natural neoantigen reactivities might be missed when spleen and/or blood was studied. We therefore compared the response in spleen with that in dLN. Typically, responses were higher in the dLN but not restricted to that location. Although the total number of SFU/105 cells was lower in the spleen than in the dLN, there was still a significantly greater response against the mutant Uqcrc2 peptide (4.5 [IQR 0.34–7.5] SFU/105 cells) compared to the wild-type peptide (0.34 [IQR 0–1.12] SFU/105 cells; P < 0.05) in this compartment . Thus both dLN and spleen reveal detectable neoantigenic responses, at least to this mutant peptide.
Figure 3. (A) Endogenous immune responses to wild-type and mutant Urcqc2 in the dLN and spleen of 7 mesothelioma-bearing mice. ELISPOT assays were performed in triplicate; * P < 0.05. (B) Comparison of endogenous immune responses to wild-type and mutant Urcqc2, and the hemagglutinin peptide CL4 in the dLN of 15 AB1-HA tumor-bearing mice. ELISPOT assays were performed in triplicate; ns, not significant. (C) Representative triplicate wells from interferon-γ ELISPOT analysis of total dLN cell preparation from 4 non-treated AB1-HA tumor-bearing mice.
Comparison of the immune response to natural and model neoantigens
To determine whether the response to the natural neoantigen Uqcrc2 was similar to that of the strong transduced neoantigen HA, we compared the responses to these two peptides. Mice were inoculated with the AB1-HA mesothelioma cell line that expresses both the Uqcrc2 neoantigen and the HA antigen. There was no significant difference in the immune response in the dLN to the 2 neoantigens. The median response to the mutant Uqcrc2 peptide was 15.38 (IQR 4.90–58.50) SFU/105 cells, which is in the range of that seen with the HA CL4 peptide (5.04 [IQR 0.40–63.17] SFU/105 cells) . There was considerable intermouse variability in the response to the natural and artificial neoantigens in the same experiments with all conditions being identical; for example, one of the 12 mice tested (mouse 4) had a dramatic response to both peptides , whereas a second animal (mouse 1) had a significantly stronger response to the mutant Uqcrc2 peptide than to the HA peptide . Therefore, the response to Uqcrc2 is clearly not a weak response.
Discussion
The notion that the immune system is not ignorant of the presence of a cancer and mounts a measurable CD8+ T-cell response to the tumor has received a lot of attention in the past decade and is supported by studies showing that cross-presentation of tumor antigens is actually highly efficientCitation12 and cross-primes, albeit weakly, tumor-specific CD8+ T cells in the dLN.Citation13 Such observations using artificial model tumor antigens have reinvigorated the search for “natural” tumor-specific antigens that arise through the process of carcinogenesis. Most studies of tumor antigens have focused on “self” antigens such as cancer-testis antigens and overexpressed or cellular differentiation proteins.Citation14 Antigenic mutated tumor proteins have been much harder to study and characterize, although over the past 20 years a number have been described.Citation1 It has been assumed that such neoantigens would be stronger than self antigens because they would have escaped central tolerance, and such a notion has recently received support from human studies.Citation15,16
As neoantigens are almost always different in each individual patient, specific responses against them have been hard to study.Citation1 NGS has changed this situation by enabling the identification of the mutational profile of cancer proteins, which can then be coupled with peptide binding algorithms and in vitro studies. Identifying neoantigenic responses is potentially an important step in advancing cancer research and treatment. First, the immune response of the host to individual neoantigens can be tracked over time, enabling identification of the steps in the immunologic response to mutated proteins that are either effective or blocked during anticancer therapies. Second, immunoreactive neoepitopes could be exploited as vaccine candidates.
Although some neoantigenic reactivities have been described in patients,Citation7,15-17 many of the key outstanding questions relating to the use of such reactivities to track responses and/or treat patients cannot be answered in humans and will require relevant animal models. It is essential to study models that use a natural human carcinogen to determine whether such a carcinogen can induce any neoantigenic reactivities. Our data from this study of an asbestos-induced tumor, mesothelioma, in mice suggest that the number of mutations is similar to that seen in human mesothelioma (unpublished data). Although it is hard to compare DNA sequencing and analysis methodologies between studies, we found approximately 10-fold fewer non-synonymous transcribed exomic mutations in the murine AB1 mesothelioma cell line compared to the B16F10 melanoma cell lineCitation2 and at least 50-fold fewer than in the MCA-induced sarcoma model.Citation3
Importantly, when neoantigenic reactivities were examined, one strong neoantigen was detected among 20 candidates. Uqcrc2 is a component of the respiratory chain protein ubiquinol cytochrome c reductase that plays an important role in oxidative phosphorylation.Citation19 Although this molecule is involved in cellular respiration and its mutation might affect the altered glycolysis that is common in tumor biology, we do not yet know whether this mutation has any non-immunologic relevance in this model. The immune response in our study was not as broad as that seen to B16F10 tumors, in which 11 of 50 tested peptides elicited a mutant peptide-specific response,Citation2 but was similar to the oligoreactivity seen in the MCA model.Citation4
One of the main reasons for studying neoantigenic responses in mice is to gain access to different cellular compartments, which is hard to do in humans. For example, clinical priorities mean that at presentation only limited, if any, access to the dLN is possible. Furthermore, even if access to compartments such as dLN and tumors is possible at presentation, it is rarely possible afterwards. Such an issue becomes crucial when we consider that most human studies involve examination of blood and/or tumorsCitation7,15-17 yet mouse studies show that strong neoantigenic reactivities can be largely restricted to the dLN,Citation10,13,20,21 meaning that relevant reactivities might be missed if only blood or tumor is examined. Fortunately, we found that although the dLN has a higher level of response, neoantigenic responses were also readily detectable in the spleen. Thus, systemic studies in humans using blood as a source of responding T cells should be sufficient, rather than having to rely on obtaining dLNs to identify relevant neoantigenic reactivities.
The natural neoantigen reactivity discovered in this study was strong, as the level of response was similar to that to the artificial viral neoantigen HA used in this model. The notion that Uqcrc2 induced a strong rather than weak response is further supported by the fact that these responses were observed after 20 hours ex vivo rather than requiring prolonged ex vivo culture and T-cell amplification with peptide.
Importantly, although some strong antigens can compete with each other in viral systems,Citation11 there was no evidence of such competition between the natural neoantigen Uqcrc2 and the artificial neoantigen HA in this system. This suggests that oligoreactivity in response to multiple tumor mutations may not be hampered by competition, as has been seen in studies of tumors bearing strong poly-epitopes,Citation22 and raises the possibility that poly-epitope vaccines might be effective. Nevertheless, the response to the 2 strong neo-antigens Uqcrc2 and HA did not eradicate the tumor in tumor-bearing mice. This is consistent with other studies showing the presence of circulating antitumor CD8+ T cells with no discernible antitumor activity.Citation23 This implies a failure of some other essential component, such as CD4+ help,Citation20 inadequate lytic activity within the tumor,Citation24 or local suppression. These functions are all the subject of intense current study and are important to understand in the context of the current checkpoint blockade therapies, which broaden the CD8+ repertoire but may not be the only factor leading to tumor eradication.
In summary, studies of neoantigen-specific T-cell responses in this relevant animal model reveal a strong reactivity to a single mutated neoantigen, Uqcrc2, a protein important in oxidative phosphorylation. This response was greater in the dLN compared to spleen but detectable in both locations, and was not out-competed by the presence of a second strong neoantigen in the same tumor. This model allows examination of many aspects of the specific host antitumor responses to antigens induced by a natural human carcinogen, with direct applicability to the equivalent human disease. It also provides an ideal opportunity to study the effects of conventional cancer therapies such as chemotherapy, surgery, and radiation therapy on the specific antitumor response, to undertake preclinical testing of new immunotherapies such as new checkpoint blockade molecules, and to study the capacity of neoantigen vaccines to alter antitumor responses, especially in synergy with other chemo- or immunotherapies
Materials and Methods
Mice
Female BALB/c mice aged between 6 and 8 weeks were purchased from the Animal Resources Center Murdoch, Australia and maintained under standard specific pathogen-free housing conditions. Animal experiments were carried out in accordance with protocols approved by the University of Western Australia Animal Ethics Committee.
Cell lines
The murine mesothelioma cell line AB1 was previously generated by injecting crocidolite asbestos intraperitoneally into BALB/c mice;Citation9 the cell line was subsequently transfected with the PR8 influenza virus HA gene to produce AB1-HA.Citation25 Cell lines were maintained from original stocks in RPMI 1640 (Invitrogen, Mulgrave, Australia) supplemented with 20 mM HEPES, 0.05 mM 2-mercaptoethanol, 100 units/mL penicillin (CSL, Melbourne, Australia), 50 µg/mL gentamicin (David Bull Labs, Kewdale, Australia), and 10% FCS (Invitrogen). AB1-HA cells were maintained in media containing the neomycin analog geneticin (Invitrogen) at a final concentration of 400 µg/mL. Cell lines were used at below 20 passages for experiments and were regularly confirmed to be negative for Mycoplasma spp.
Next-generation sequencing
Total RNA was extracted from the mesothelioma cell lines AB1 and AB1-HA and from BALB/c liver tissue. RNA was sequenced using the Hiseq2000 sequencer (Illumina, CA) at Beijing Genomics Institute Hong Kong, China. Data were aligned to the murine reference genome mm9 (UCSC mm9 /NCBI build 37), derived from mice of the C57BL/6 strain using Bowtie 2.0Citation26 with quality analysis performed with Qualimap 2.0 software.Citation27
DNA was extracted from AB1 and AB1-HA cells and from BALB/c tail tissue. Library creation and exome capture were carried out with the SureSelectXT Mouse All Exon kit (Agilent, Mulgrave, Australia) following the manufacturer's instructions. Exomes were sequenced on the Hiseq2000 by the Australian Genome Research Facility (Parkville, Australia) and data were aligned to mm9 using the Burrows-Wheeler Alignment tool.Citation28 All exomes were sorted using SAMtoolsCitation29 and duplicate reads were marked using Picard. Local realignment and base quality score recalibration were performed using the Genome Analysis Toolkit.Citation30
Mutation selection and epitope prediction
From RNAseq data, somatic SNVs were identified using Varscan v2.3Citation31 and high-confidence calls were selected that had a minimum phred score of 60 or greater. Data were annotated using Annovar.Citation32 Homozygous SNVs were assumed to be germline mutations and excluded. For each high-confidence somatic, non-synonymous, exonic SNV identified in RNAseq data the corresponding data from exome data were examined using the Integrative Genomics Viewer.Citation33 Mutations that were present in 3 of the 4 sequenced data sets with a reads per kilobase of exon per million reads mapped (RPKM) expression score greater than 10 were selected. For validation, variants were amplified from cell lines and BALB/c DNA and subjected to Sanger sequencing (AGRF, Nedlands, Australia) and the results were visually examined. Protein sequences were retrieved from Genbank and for each of the mutations a set of peptides spanning the amino acid change and ranging from 8 to 11 amino acid in length was created in silico. For each peptide sequence, 2 sets of sequences were generated, one with the reference amino acid and one with the mutant variant. Matched sequences were analyzed using NetMHCpan 2.8 algorithmCitation34 and the potential class I binding affinity to murine MHC class I haplotype of H-2 Dd, H-2 Kd and H-2 Ld were determined. Strong binders, with a predicted binding affinity IC50 values less than 50 nM, and weak binders, with values of 50–500 nM, were selected as potential neoantigens.
Mouse mesothelioma model
Mice were inoculated subcutaneously with 5 × 105 AB1 or AB1-HA cells in a total volume of 100 μl PBS on the right flank. Mice were euthanized when tumors reached approximately 70 mm2 and the spleens and tumor-draining ipsilateral axillary and inguinal nodes lymph nodes (dLN) were harvested. Non-tumor bearing naïve mice were used as controls. Single-cell suspensions of spleen and dLN cells were prepared as previously describedCitation23 and used in ELISPOT assays.
ELISPOT
ELISPOT assays were performed as previously described.Citation35 Briefly, either 1 × 105 or 2 × 105 freshly isolated splenocytes or dLN cells were incubated in a microtiter plate pre-coated with 1 μg/mL anti-interferon-γ antibody (clone AN18; Mabtech, Preston, Australia) with 1 μg/mL test peptide (Mimtopes, Notting Hill, Australia), 1 μg/mL CL4 peptide from hemagglutinin of the Mt-Sinai strain of PR8 influenza virus (JPT, Berlin, Germany), 1 μg/mL anti-mouse CD3 monoclonal antibody (Raybiotech, GA), or media alone. After 20 h at 37°C, cytokine secretion was detected with 1 μg/mL biotinylated anti-interferon-γ antibody clone (R4–6A2; Mabtech), 1 μg/mL streptavidin horseradish peroxidase (Mabtech), and TMB substrate (Mabtech). Spots were visualized and enumerated using the Autoimmun Diagnostika AID ELISPOT reader system running count algorithm v.3.2.x. For each mouse the mean background number of SFU/105 cells was subtracted from the value for test wells.
Statistical analysis
Data were analyzed using Graph Pad Prism Software (Graph Pad Software, CA, USA). The paired Student's t test was used to compare responses of individual mice to different peptides. A P value < 0.05 using a 2-tailed test was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by the Insurance Commission of Western Australia and the Housing Industry of Australia Charitable Foundation.
References
- Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun 2013 13:15; PMID:23882160.
- Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012 72:1081-1091; PMID:22237626; http://dx.doi.org/10.1158/0008-5472.CAN-11-3722.
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012 482:400-404; PMID:22318521; http://dx.doi.org/10.1038/nature10755.
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014 515:577-581; PMID:25428507; http://dx.doi.org/10.1038/nature13988.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014 371:2189-2199; PMID:25409260; http://dx.doi.org/10.1056/NEJMoa1406498.
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014 344:641-645; PMID:24812403; http://dx.doi.org/10.1126/science.1251102.
- Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, Castellarin M, Twumasi-Boateng K, Watson PH, Holt RA, et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res 2014 20:1125-1134; PMID:24323902; http://dx.doi.org/10.1158/1078-0432.CCR-13-2147.
- Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014 124:453-462; PMID:24891321; http://dx.doi.org/10.1182/blood-2014-04-567933.
- Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW. Establishment of a murine model of malignant mesothelioma. Int J Cancer 1992 52:881-886; PMID:1459729; http://dx.doi.org/10.1002/ijc.2910520609.
- Marzo AL, Lake RA, Lo D, Sherman L, McWilliam A, Nelson D, Robinson BW, Scott B. Tumor antigens are constitutively presented in the draining lymph nodes. J Immunol 1999 162:5838-5845; PMID:10229818.
- Chen W, Anton LC, Bennink JR, Yewdell JW. Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity 2000 12:83-93; PMID:10661408; http://dx.doi.org/10.1016/S1074-76130080161-2.
- Anyaegbu CC, Lake RA, Heel K, Robinson BW, Fisher SA. Chemotherapy enhances cross-presentation of nuclear tumor antigens. PLoS One 2014 9:e107894; PMID:25243472; http://dx.doi.org/10.1371/journal.pone.0107894.
- Stumbles PA, Himbeck R, Frelinger JA, Collins EJ, Lake RA, Robinson BW. Cutting edge: tumor-specific CTL are constitutively cross-armed in draining lymph nodes and transiently disseminate to mediate tumor regression following systemic CD40 activation. J Immunol 2004 173:5923-5928; PMID:15528325; http://dx.doi.org/10.4049/jimmunol.173.10.5923.
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014 14:135-146; PMID:24457417; http://dx.doi.org/10.1038/nrc3670.
- Rosenberg SA, Kawakami Y, Robbins PF, Wang R. Identification of the genes encoding cancer antigens: implications for cancer immunotherapy. Adv Cancer Res 1996 70:145-177; PMID:8902056; http://dx.doi.org/10.1016/S0065-230X0860874-X.
- Kvistborg P, van Buuren MM, Schumacher TN. Human cancer regression antigens. Curr Opin Immunol 2013 25:284-290; PMID:23566921; http://dx.doi.org/10.1016/j.coi.2013.03.005.
- Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res 2014 2:522-529; PMID:24894089; http://dx.doi.org/10.1158/2326-6066.CIR-13-0227.
- Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015; 517:489-92; PMID:25363767.
- Duncan AM, Ozawa T, Suzuki H, Rozen R. Assignment of the gene for the core protein II UQCRC2 subunit of the mitochondrial cytochrome bc1 complex to human chromosome 16p12. Genomics 1993 18:455-456; PMID:8288258; http://dx.doi.org/10.1006/geno.1993.1500.
- Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, Scott B. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol 2000 165:6047-6055; PMID:11086036; http://dx.doi.org/10.4049/jimmunol.165.11.6047.
- McDonnell AM, Currie AJ, Brown M, Kania K, Wylie B, Cleaver A, Lake R, Robinson BW. Tumor cells, rather than dendritic cells, deliver antigen to the lymph node for cross-presentation. Oncoimmunology 2012 1:840-846; PMID:23162751; http://dx.doi.org/10.4161/onci.20493.
- Bundell CS, Jackaman C, Suhrbier A, Robinson BW, Nelson DJ. Functional endogenous cytotoxic T lymphocytes are generated to multiple antigens co-expressed by progressing tumors; after intra-tumoral IL-2 therapy these effector cells eradicate established tumors. Cancer Immunol Immunother 2006 55:933-947; PMID:16283304; http://dx.doi.org/10.1007/s00262-005-0086-6.
- Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003 63:4490-4496; PMID:12907622.
- Nelson DJ, Mukherjee S, Bundell C, Fisher S, van Hagen D, Robinson B. Tumor progression despite efficient tumor antigen cross-presentation and effective “arming” of tumor antigen-specific CTL. J Immunol 2001 166:5557-5566; PMID:11313395; http://dx.doi.org/10.4049/jimmunol.166.9.5557.
- Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res 1999 59:1071-1079; PMID:10070965.
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25.
- Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 2012 28:2678-2679; PMID:22914218; http://dx.doi.org/10.1093/bioinformatics/bts503.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009 25:1754-1760; PMID:19451168; http://dx.doi.org/10.1093/bioinformatics/btp324.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009 25:2078-2079; PMID:19505943; http://dx.doi.org/10.1093/bioinformatics/btp352.
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010 20:1297-1303; PMID:20644199; http://dx.doi.org/10.1101/gr.107524.110.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012 22:568-576; PMID:22300766; http://dx.doi.org/10.1101/gr.129684.111.
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010 38:e164; http://dx.doi.org/10.1093/nar/gkq603.
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer IGV: high-performance genomics data visualization and exploration. Brief Bioinform 2013 14:178-192; PMID:22517427; http://dx.doi.org/10.1093/bib/bbs017.
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009 61:1-13; PMID:19002680; http://dx.doi.org/10.1007/s00251-008-0341-z.
- Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Price P. Decreased IFNgamma production correlates with diminished production of cytokines by dendritic cells in patients infected with hepatitis C virus and receiving therapy. J Viral Hepat 2011 18:482-492; PMID:20529204; http://dx.doi.org/10.1111/j.1365-2893.2010.01331.x.