
Abstract
In a recent adoptive cell therapy (ACT) clinical trial using autologous tumor-infiltrating lymphocytes (TILs) in patients with metastatic melanoma, we found an association between CD8+ T cells expressing the inhibitory receptor B- and T-lymphocyte attenuator (BTLA) and clinical response. Here, we further characterized this CD8+BTLA+ TIL subset and their CD8+BTLA− counterparts. We found that the CD8+ BTLA+ TILs had an increased response to IL-2, were less-differentiated effector-memory (TEM) cells, and persisted longer in vivo after infusion. In contrast, CD8+BTLA− TILs failed to proliferate and expressed genes associated with T-cell deletion/tolerance. Paradoxically, activation of BTLA signaling by its ligand, herpes virus entry mediator (HVEM), inhibited T-cell division and cytokine production, but also activated the Akt/PKB pathway thus protecting CD8+BTLA+ TILs from apoptosis. Our results point to a new role of BTLA as a useful T-cell differentiation marker in ACT and a dual signaling molecule that curtails T-cell activation while also conferring a survival advantage for CD8+ T cells. These attributes may explain our previous observation that BTLA expression on CD8+ TILs correlates with clinical response to adoptive T-cell therapy in metastatic melanoma.
Abbreviations:
- AICD, activation-induced cell death
- ACT, adoptive cell therapy
- BTLA, B- and T-lymphocyte attenuator
- GB, granzyme B, Perf, perforin
- TIL, tumor-infiltrating lymphocyte
- ITIM, immunotyrosine-based inhibitory motif
- KIR, killer-cell immunoglobulin-like receptor
- TN, naïve T cell
- TCM, central memory T cell
- TEM, effector-memory T cell
- TEMRA, terminally-differentiated CD45RA-expressing T cell
Introduction
Adoptive T cell therapy (ACT) using tumor-infiltrating lymphocytes (TILs) has emerged as a promising treatment for metastatic melanoma. Multiple institutions, including our group at M.D. Anderson Cancer Center, are conducting Phase II studies for patients with Stage IIIc/IV metastatic melanoma with reported clinical response rates between 45% and 51%.Citation1-3 Identification of TIL subsets associated with clinical response is a critical biomarker for ACT. We previously reported that a positive clinical response to ACT was significantly related to the quantity and frequency of CD8+ cytotoxic T cells in the TILs infused into patients.Citation2 However, not all patients infused with a majority of CD8+ TIL were responders, therefore identification of more specific CD8+ subsets is critical, especially to further define the mechanism of action of CD8+ T cells during ACT. Moreover, identification of active CD8+ TIL subsets could lead to efforts to selectively expand these T cells prior to infusion into patients, which has a high potential to further improve the clinical efficacy of ACT.Citation4 Unexpectedly, we found that higher frequencies and numbers of CD8+ TILs expressing B- and T-lymphocyte attenuator (BTLA) were significantly correlated to positive clinical responses to ACT.Citation2 This was surprising as BTLA has been described as a coinhibitory receptor with an immunotyrosine-based inhibitory motif (ITIM) and an immunotyrosine-based switch motif (ITSM), similar to the well-known inhibitory receptors PD-1 and CTLA-4.Citation5 Like PD-1, BTLA has been shown to inhibit proliferation and cytokine production in melanoma tumor antigen-specific CD8+ T cells by recruitment of SHP1/2 phosphatases.Citation6,7 However, recent studies by other groups have found that BTLA may also promote cell survival by inducing a gene expression program similar to that induced by ICOS and CD28 costimulation.Citation8,9
Although BTLA has been characterized as a negative costimulatory molecule on T cells, a number of studies of human CD8+ T cells have shown that BTLA is constitutively expressed at a high level on naïve CD8+ T cells and gradually downregulated during T-cell expansion and cytotoxic T lymphocyte (CTL) differentiation.Citation6,10,11 A recent study suggested a close relationship between the expression of multiple inhibitory receptors (e.g., PD-1, TIM3, and LAG3) and antigen specificity, anatomic localization, and differentiation of CD8+ T cells in humans.Citation12 Therefore, it is likely that sustained BTLA expression marks a specific stage of differentiation in human CD8+ melanoma TILs.
In this study, we performed comprehensive phenotypic and functional characterization and gene expression profiling of CD8+BTLA+ TIL isolated and expanded from melanoma metastases in comparison with their CD8+BTLA− counterparts. We found that CD8+BTLA+ TIL were a less-differentiated, highly proliferative CD8+ effector memory (TEM) subset of TIL that were more acutely responsive to IL-2 and TCR signals and expressed genes reminiscent of early-stage effector-memory cells and central memory cells. In contrast, the CD8+BTLA− TIL subset consisted of more late-stage differentiated TEM and TEMRA TIL that were less responsive to IL-2 or TCR signals and exhibited a molecular signature of T-cell deletion.Citation13,14 We also found that the ligation of BTLA on CD8+BTLA+ TIL with its cognate receptor, herpes virus entry mediator (HVEM), exerted an inhibitory effect on proliferation and cytokine production but also promoted the phosphorylation of Akt and Bad, resulting in an increased expression of Bcl-xL and improved survival of TIL in the presence of apoptotic stimuli. Furthermore, in vivo tracking of TCR clones by high-throughput DNA sequencing of the TCR Vβ CDR3 region in patients after TIL ACT showed a greater number of persisting clones originating from the infused CD8+BTLA+ subset than from the BTLA− subset. Our findings provide a mechanistic basis for the positive clinical association with the CD8+BTLA+ TIL subset and support a new perspective of T-cell checkpoint molecules as inhibitory signaling systems that inhibit T-cell proliferation and effector function while promoting the survival of these inhibited cells so that critical antigen-specific clones are not depleted from the immune system. This dual functional role of BTLA may explain why CD8+ TEM cells accumulate in melanoma while being prevented from further cell division and differentiation.
Results
Differential gene expression profiles between CD8+BTLA+ and CD8+BTLA− melanoma TIL
To understand the differences between CD8+ BTLA+ and BTLA−TIL we performed expression gene profiling between 2 sorted subsets from 5 different melanoma patients using the Affymetrix® Human Gene 1.0 ST Array. The microarray data have been deposited into the NCBI Gene Expression Omnibus (GEO) database (Accession # GSE43260). An example of post-sort analysis of CD8+BTLA+ and CD8+ BTLA− TIL subsets and the expression intensity levels of CD3ϵ and CD3δ are shown in Figure S1. As an internal control, we confirmed that expression of the BTLA gene was significantly higher in the sorted CD8+BTLA+ subset (7.69-fold higher, P = 8.80×10−6 by paired t test) (, table). Using arbitrary selection criteria to assess genes that showed a significant difference between the 2 subsets (paired t test; fold change ≥1.45; P < 0.05), we identified 406 differentially expressed genes (DEGs). Among these, 171 genes were upregulated in the CD8+BTLA+ subset and 235 genes were upregulated in the CD8+BTLA− subset. In agreement with the previously described phenotype of CD8+BTLA+ T cells,Citation11,12 we found a significantly increased expression of early effector memory genes (CD28: 2.38-fold higher, P = 0.025; CD127 [also known as IL7R]: 2.08-fold higher, P = 0.03)] and differentiation genes (B3GAT1 [also known as CD57]: 1.79-fold higher, P = 0.0008)] (, table). On the other hand, the CD8+ BTLA− subset had significantly increased expression of 13 members of the killer-cell immunoglobulin-like (KIR) receptor gene family and the TYROBP gene (also known as DAP12, an adaptor protein for the KIR receptorsCitation15; 4.11-fold higher; P = 0.0002) (, table). KIR receptors are known to be expressed by natural killer (NK) cells,Citation16 highly differentiated CD8+PD-1−CD45RA+ TEMRA cells in lymphocytes from normal donors,Citation17,18 and senescent human CD8+CD28− T cells.Citation19,20 We confirmed the preferential protein expression of some KIR receptors in the CD8+BTLA− subset by flow cytometry (Fig. S2). We also found that the expression of other genes typically associated with NK cells, such as NCAM1 (also known as CD56; 3.42-fold higher, P = 0.01), LYN (2.41-fold higher, P = 0.025), and SYK (1.8-fold higher, P = 0.02)],Citation21,22 was significantly increased in the CD8+BTLA− subset (, table). Lastly, we observed that expression of genes associated with T-cell deletion and anergy,Citation14 such as IKAROS family zinc finger 2 (IKZF2; 5.08-fold higher, P = 0.00076)Citation13,23, early growth response 2 (EGR2; 1.82-fold higher, P = 0.0185)Citation13,14,24 and nuclear receptor subfamily, group A, member 3 (NR4A3; 1.8-fold higher, P = 0.0014)Citation13,24,25 was also significantly increased in the CD8+BTLA− TIL (, table).
Figure 1. CD8+BTLA+ TIL exhibit a less differentiated, more activated phenotype than their CD8+BTLA− counterparts. (A) Enrichment analysis of a global set of genes from CD8+ BLTA+ or BTLA− subsets. The top 50 genes that are immunologically relevant in either the BTLA+ or BTLA− subset are shown, ranked by enrichment scores using gene-set enrichment analysis (GSEA). Each row represents 1 gene and each column represents 1 sample of sorted CD8+ BTLA+ or BTLA− subset from 1 patient. TIL were isolated from melanoma metastases, cultured with 3,000 IU/mL IL-2 for 3 wks and stained for expression of CD8, BTLA, CD45RA, CCR7, CD28, CD27, PD-1, TIM3, granzyme B and perforin. Dead cells were excluded using Aqua® live/dead dye. (B, left panel) CD45RA and CCR7 expression on the CD8+BTLA+ (left dot plot) and CD8+BTLA− (right dot plot) populations on a representative TIL from patient 2360. (B, right panel) A summary of CD45RA and CCR7 expressions within each CD8+BTLA+ or CD8+BTLA− subset (n=11). Differentiation subsets are defined as follows: TN: CD45RA+CCR7+; TCM: CD45RA+CCR7+, TEM: CD45RA−CCR7−, TEMRA: CD45RA+CCR7−. The results are the percentages of TEM or TEMRA subsets within each gated CD8+ BTLA+ or BTLA− subset. TN and TCM are omitted because no significant populations were found. (C) Summary of the expression of surface markers CD28, PD-1, and TIM3 by the CD8+BTLA+ and CD8+BTLA− subsets (n ranges from 6–42 per marker). Statistical significance between the subsets was determined using Wilcoxon signed-rank test.
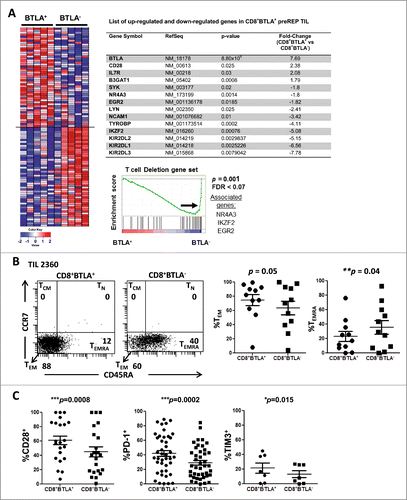
Enrichment of a molecular signature of T cell deletion in the CD8+BTLA− TIL subset
Gene set enrichment analysis (GSEA) was performed to look for enrichment of global or predefined sets of genes in TIL subsets. Given that BTLA has been described as a marker of exhaustion, we wanted to compare gene-set signatures derived from TIL with those derived from CD8+ T cells undergoing exhaustion,Citation26,27 deletion,Citation13 or anergy.Citation13 Interestingly, we did not find enrichment of an exhaustion gene set signature derived from Baitsch et al. (P = 0.31)Citation26 or Wherry et al. (P = 0.50)Citation27 in either TIL subset (Fig. S3). We also did not find enrichment of a T-cell anergy signature in any of the TIL subsets (P = 0.53; Fig. S3). However, we did discover enrichment of a molecular signature corresponding to T-cell deletion/tolerance (P = 0.00) in the CD8+BTLA− TIL subset (, bottom panel). These results suggest that BTLA does not mark exhausted CD8+ TIL; rather, they support the concept that CD8+BTLA+ TILs represent a less differentiated state of CD8+ TEM compared to CD8+BTLA− TILs.
Differentiation status of BTLA+ and BTLA− CD8+ TIL subsets
BTLA has been described as a marker that is progressively downregulated during CD8+ T-cell differentiation.Citation11,12 Furthermore, our microarray data suggested that there may be key differences in differentiation state between the BTLA+ and BTLA− subsets. Therefore, we analyzed TIL that were expanded ex vivo from metastatic tumors by flow cytometric analysis with a panel of differentiation and activation markers. The CD8+ T-cell differentiation subsets were broadly defined as follows: naïve T cells (TN: CD45RA+CCR7+), central memory T cells (TCM:CD45RA−CCR7+), effector-memory T cells (TEM: CD45RA−CCR7−), and terminally-differentiated effector T cells re-expressing CD45RA (TEMRA-int: CD45RAloCCR7−).Citation11,28 Analysis of a representative TIL line for expression of CCR7 versus CD45RA to delineate these types of memory cells is shown in . Overall, there was no statistically significant difference in the percentages of TEM cells within the CD8+BTLA+ and BTLA− subsets (, left graph); however, the BTLA− subset was significantly enriched for more differentiated TEMRA-int cells (, right graph) suggesting that these were more highly differentiated CTL. To further define the memory phenotype of the BTLA+ subset, TIL were stained for CD28 and CD27. Within the TEM subset, CD28 and CD27 are markers of early CD8+ effector-memory T (TEM) cells.Citation29 In concordance with the microarray analysis, we found significantly higher expression of CD28 in the BTLA+ subset compared to the BTLA− subset (61 ± 6.2% versus 45 ± 6.8%; P = 0.0008; ). However, the difference in the expression of CD27 between the 2 subsets was not statistically significant (33 ± 4.6% versus 36 ± 4.6%, respectively, P = 0.455; Fig. S4A). Surprisingly, 2 previously reported co-inhibitory markers, PD-1 and TIM3, were also found to be significantly expressed on the CD8+BTLA+ subset compared to the CD8+ BTLA− population; PD-1 expression was 42 ± 4.1% on the BTLA+ population versus 29 ± 3.4% on the BTLA− cells (P = 0.0002; n=42) () and TIM3 expression was 22 ± 7.1% on the BTLA+ subset compared to 13 ± 4.8% on the BTLA− subset (P = 0.015; n = 7) (). Finally, we examined expression of granzyme B (GB) and perforin (Perf) as markers associated with cytolytic activity. Among the 6 independent lines tested, there was no significant difference between the BTLA+ and BTLA− subsets for either cytolytic marker (GB: 95 ± 3.4% versus 95 ± 261 2.4%, p = 0.438; Perf: 50 ± 9% versus 55 ±10%, P = 0.688) (Fig. S4B and C). Overall, our phenotypic and microarray data suggest that the CD8+ BTLA+ TIL subset is composed mostly of effector-memory cells (CCR7−CD45RA−) with a less differentiated (CD28Hi) and more activated T-cell phenotype (PD-1+ and TIM3+) than the BTLA− subset. Interestingly, we found similar levels of GB and Perf expression between the 2 subsets, suggesting there may not be a difference in cytolytic ability between the 2 subsets.
CD8+BTLA+ TIL exhibit an enhanced proliferative response to IL-2
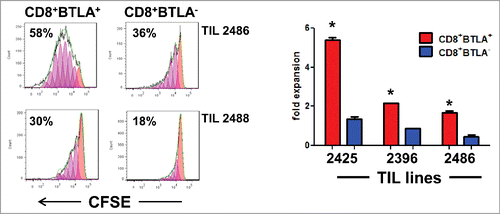
Our initial observation that BTLA+ TIL have a less-differentiated TEM phenotype suggested that there may be a significant difference in the response to IL-2 between the CD8+BTLA+ and CD8+BTLA− subsets. This is important because IL-2 is a key cytokine used in adoptive T-cell therapy to expand and promote the persistence of the TIL in vivo after infusion. Therefore, we examined the proliferative and cell survival responses between the 2 subsets. By manual cell counting (based on trypan blue exclusion) after 6 d of culture in the presence of IL-2, we found that CD8+BTLA+ TIL, on average, had a higher fold expansion than the CD8+ BTLA− TIL (TIL 2425: 5.4-fold versus 1.4-fold; TIL 2396: 2.2-fold versus 0.9-fold; TIL 2486: 1.7-fold versus 0.45-fold; , right graph and Fig. S5C). Next, we labeled sorted CD8+ BTLA+ and CD8+ BTLA− TIL with carboxyfluorescein succinimidyl ester (CFSE) and cultured the sorted cells with IL-2 for 3 d. Among all patients tested, the BLTA+ subset had higher fractions of proliferating cells and had undergone more cell divisions than the BTLA− subset (TIL 2486: 58% versus 36%; TIL 2488: 30% versus 18%; TIL 2605B: 84% versus 53%; TIL 2612: 73% versus 8%; , and Fig. S4B). Previous studies found that the quantity of the infused CD8+CD27+ TIL correlated with positive clinical response to ACT in metastatic melanoma.Citation30 As shown in Figure S4, CD27 was heterogeneously expressed in the BTLA+ and BTLA− subsets. Therefore, we examined whether a difference in IL-2 responsiveness was also found between the CD8+CD27+ and CD8+CD27− TIL subsets but did not find a significant difference in proliferation between the sorted CD27+ and CD27− subsets (Fig. S5). Together, our data suggest that the CD8+BTLA+ TIL proliferate better than CD8+BTLA− TIL in response to IL-2 and that the difference in CD27 expression did not account for this difference in proliferative potential.
Figure 2. CD8+BTLA+ TIL have enhanced proliferation in response to IL-2 stimulation compared to the CD8+BTLA− subset. Sorted CD8+BTLA+ and CD8+BTLA− TIL subsets were labeled with carboxyfluorescein succinimidyl ester (CFSE) and cultured at a density of 1 × 106/mL with high-dose IL-2 for 5 d (3,000 IU/mL) (n = 4). (A) Histograms showing percentages of proliferating cells from 2 lines (top). The absolute number of cells in each subset was determined using trypan blue exclusion and graphed as fold expansion (bottom). * indicates significance (P < 0.05) as determined by the Student t test.
Enhanced STAT5 activation by CD8+BTLA+ T-cells in expanding TIL cultures
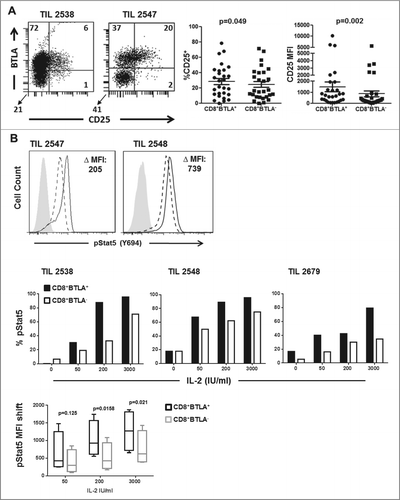
Because the CD8+BTLA+ subset expanded better with IL-2, we hypothesized that the CD8+BTLA+ subset might express a higher level of the high-affinity IL-2 receptor, CD25 (IL-2Rα) than the BTLA− subset. Indeed, we found that CD25 expression in TIL expanded with IL-2 tended to be associated with the CD8+ BTLA+ subset rather than the BTLA− TIL subset from the same patient (). Further analysis showed that there was a statistically significant difference in the level of cell surface CD25 expression between the CD8+BTLA+ and BTLA− subsets (, P = 0.002). Next, we asked whether the higher expression of CD25 in the CD8+BTLA+ subset was associated with stronger signaling induced by IL-2. To answer this question, we measured changes in STAT5 (Y694) phosphorylation, a marker of STAT5 activation downstream of the IL-2 receptor,Citation31 in sorted CD8+BTLA+ and BTLA− TIL. When treated with 200 IU/mL IL-2, CD8+BTLA+ TIL had higher levels of phospho-STAT5 (change in mean fluorescence intensity [MFI]: 205 for TIL 2547 and 739 for TIL 2548) (). We also performed an IL-2 dose–response study using different doses of IL-2 (0, 50, 200, or 3,000 U/mL) and found that the sorted CD8+BTLA+ subset had higher expression and MFI of pSTAT5 than the CD8+ BTLA− subset at all doses of IL-2 tested (, n = 4). Thus, IL-2 triggers a more robust signal activating STAT5 downstream of the IL-2R in the CD8+BTLA+ TIL subset, which may promote higher levels of CD25 expression in an autocrine loop.
Figure 3. Enhanced IL-2 responsiveness by CD8+BTLA+ TIL. (A) TIL were stained for expression of CD8, BTLA, and CD25 and with the Aqua® live/dead viability dye. The left dot plot shows representative staining of TIL 2538 and 2547. The numbers indicated percent expression within the AQUA−CD8+ population. The right graphs summarize the percentage CD25 expression and CD25 mean fluorescence intensity (MFI) for the CD8+BTLA+ and CD8+BTLA− subsets (n = 23). P value was calculated using Wilcoxon signed-rank test. (B) TIL were sorted into CD8+BTLA+ and CD8+BTLA− subsets and cultured with increasing concentrations of IL-2 for 20 min. Cells were fixed, permeabilized, and stained for pStat5 (Y694). The left histograms show the level of pSTAT5 (Y694) expression after treatment with 200 IU/mL IL-2 for 20 min. The change in MFI (ΔMFI) between the subsets is shown. The top graphs show the change in percent pSTAT5 levels after incubation with increasing concentrations of IL-2 for 3 independent TIL lines. The shift in pSTAT5 MFI was determined by subtracting the baseline levels for each subset (n = 4). Statistical significance was determined using a paired Student t test.
CD8+BTLA+ TIL have superior responses to TCR stimulation and produce more IL-2
The results above suggested that the BTLA+ TIL had a heightened response to IL-2. Next, we explored the relative capacity of the BTLA subsets to respond to TCR triggering. The proliferative response of CD8+BTLA+ and BTLA− T cell subsets after TCR stimulation was first analyzed. We also examined the induction of activation markers and the capacity of the 2 subsets to produce IL-2, a key feature of early-differentiated CD8+ memory T cells in humans.Citation32 We first compared the proliferation of the sorted CD8+BTLA+ and BTLA− subsets stimulated with plate-bound anti-CD3 plus anti-CD28 (OKT3 + CD28) in a 3[H]-thymidine incorporation assay in the absence of exogenous IL-2. As shown in , we consistently noted significantly higher proliferation in the CD8+BTLA+ subset in 4 different TIL lines. Next, we assessed the induction of the key activation markers CD25, PD-1, and LIGHT in response to the same TCR stimulation. Although both CD8+BTLA+ and CD8+BTLA− subsets could induce expression of PD-1 upon stimulation, expression of LIGHT was generally unchanged in BTLA− cells but modestly upregulated in BTLA+ cells (). However, the most dramatic and consistent difference was in the markedly greater increase in CD25 expression by CD8+BTLA+ cells (). Finally, in sorted CD8+ BTLA+ and BTLA− subsets from 2 different patients, we detected more intracellular IL-2 in CD8+BTLA+ TIL than in the CD8+BTLA− subset when stimulated with PMA and ionomycin (TIL 2547 ΔMFI: 274; TIL 2538 ΔMFI: 652) (). However, there was no difference in the secretion of the other cytokines tested or in CTL function using either a redirected lysis assay or in response to autologous melanoma tumor cells (Fig. S6). Thus, our data showed that, although there was no difference in CTL function between the subsets, stimulation of the TCR signaling pathway induced higher CD25 levels and triggered more autocrine IL-2 production in the CD8+BTLA+ TIL subset, which might have contributed to the increased TCR-induced proliferative response.
Figure 4. CD8+BTLA+ TIL are more responsive to TCR stimulation and produce more IL-2. (A) Sorted CD8+BTLA+ and CD8+BTLA− cells were stimulated with plate-bound anti-CD3 (OKT3) and anti-CD28 (OKT3+CD28) or not stimulated (NIL) in the absence of IL-2 for 3 d Cells were pulsed with 1 μCi of [3H]-thymidine for the last 18 h of culture. Results were shown as counts per minute (cpm) from triplicate wells (mean ± SD). * indicates significance (P < 0.05) as determined by Student t test. (B) Sorted CD8+ BTLA+ and CD8+BTLA− subsets were stimulated with 200 IU/mL IL-2, OKT3, or OKT3 plus CD28 for 72 h. Cells were analyzed for expression of CD25, LIGHT, and PD-1 by flow cytometry. The percent expression of each marker is shown. (C) Sorted CD8+ BTLA+ and CD8+BTLA− subsets were stimulated with PMA and ionomycin for 4 h and stained for expression of intracellular IL-2. The numbers indicate the MFI for each subset from 2 TIL lines.
![Figure 4. CD8+BTLA+ TIL are more responsive to TCR stimulation and produce more IL-2. (A) Sorted CD8+BTLA+ and CD8+BTLA− cells were stimulated with plate-bound anti-CD3 (OKT3) and anti-CD28 (OKT3+CD28) or not stimulated (NIL) in the absence of IL-2 for 3 d Cells were pulsed with 1 μCi of [3H]-thymidine for the last 18 h of culture. Results were shown as counts per minute (cpm) from triplicate wells (mean ± SD). * indicates significance (P < 0.05) as determined by Student t test. (B) Sorted CD8+ BTLA+ and CD8+BTLA− subsets were stimulated with 200 IU/mL IL-2, OKT3, or OKT3 plus CD28 for 72 h. Cells were analyzed for expression of CD25, LIGHT, and PD-1 by flow cytometry. The percent expression of each marker is shown. (C) Sorted CD8+ BTLA+ and CD8+BTLA− subsets were stimulated with PMA and ionomycin for 4 h and stained for expression of intracellular IL-2. The numbers indicate the MFI for each subset from 2 TIL lines.](/cms/asset/ac78ba86-40e9-4f5a-959e-6e3c867dc817/koni_a_1014246_f0004_b.gif)
Tracking the persistence of CD8+BTLA+ and CD8+BTLA− T-cell clones in vivo
The gene expression and functional data presented thus far suggested a less differentiated and more activated phenotype of the CD8+ BTLA+ TIL subset. Thus, we hypothesized that CD8+ BTLA+ TIL have longer persistence in vivo after adoptive transfer into treated patients, which might explain the positive association with clinical response.Citation2 We were interested in tracking how unique T-cells clones arising from the infused BTLA+ and BTLA− cells persisted after infusion into patients in vivo. To this end, we collected DNA from sorted CD8+BTLA+ and BTLA− subsets from the infusion product of 2 TIL-treated patients and from peripheral blood mononuclear cells (PBMCs) at multiple time points post-infusion and examined the persistence of individual clones using high-throughput CDR3 sequencing technology (Adaptive Biotechnologies, Seattle, WA) (). CDR3 sequencing of the TCR Vβ allows identification of T cells without knowing their specificities. BTLA subsets from the infusion product were used as a starting point to identify cells present in the infusion product rather than the endogenous T cells that were reconstituted in the patient following lymphodepletion. Both patients responded to TIL therapy according to RECISTCitation33 and immune-related response criteria (irRC).Citation34
Figure 5. In vivo tracking of infused CD8+BTLA+ and CD8+BTLA− subsets. TIL (100 × 106 cells) from the infusion product of treated patients were sorted into bulk CD8+ and CD8+ BTLA+ and CD8+BTLA− subsets and flash frozen. PBMCs collected at the labeled time points were treated in the same manner. DNA was isolated from the banked samples as described in Methods and sent for CDR3 sequencing at Adaptive Biotechnologies. (A) Vβ TCR diversity shown as pie charts for each subset with the total number of Vβ clonotypes found. (B) Overlap of each CDR3 sequence for the sorted CD8+BTLA+ and CD8+BTLA− subsets from the infusion product. Overlapping CDR3s are shown in blue, whereas CDR3 sequences found only in the CD8+BTLA+ subset are shown in red, and those found only in the CD8+BTLA− subset are shown in green. The axis shows the frequency of each CDR3 sequence for each subset within the infusion product. The percentage of the infusion product consisting of overlapping subsets is indicated. (C) Frequency of the unique CD8+BTLA+ and CD8+BTLA− subsets present in the blood over time.
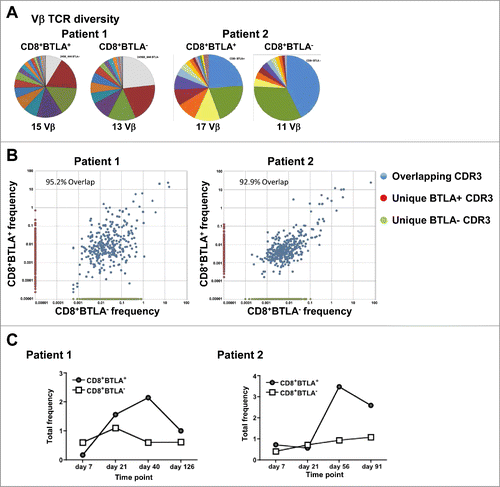
Examination of the TCR Vβ family diversity in the infused CD8+ TIL for each patient showed that the BTLA+ cells had a similar Vβ diversity to the BTLA− cells (). To determine whether the BTLA subsets were comprised of T cells with different specificities, CDR3 sequences for each subset from the infusion product were compared (). The majority of the CDR3 sequences from the infusion product overlapped between subsets for both patients (95.2% and 92.9% of CDR3 sequences overlapped between both subsets in each respective patient), supporting the concept that BTLA does not mark different clones but acts as a differentiation marker. However, as we were interested in tracking each subset in the blood of patients over time following infusion, only CDR3 sequences unique to each subset were examined (unique BTLA+, red circles; unique BTLA−, green circles). This approach of identifying unique clones was the only way of analyzing the relative persistence of each subset. The sum of all CDR3 sequences found independently within each subset was calculated as a percentage of the total number of CDR3 sequences found in the peripheral blood at each time point to separate the endogenous T cells within the blood from the infused T cells. shows that, in both patients, the frequency of total CD8+BTLA− T cells remained low (less than 1%) over all time points. However, the CD8+ BTLA+ T cells expanded and persisted at a higher frequency at the later time points. Thus, in these 2 responding patients, unique clones from the infused CD8+BTLA+ subset exhibited superior long-term persistence in the peripheral blood in vivo.
BTLA ligation provides a prosurvival signal in TIL and enhances Bcl-xL expression
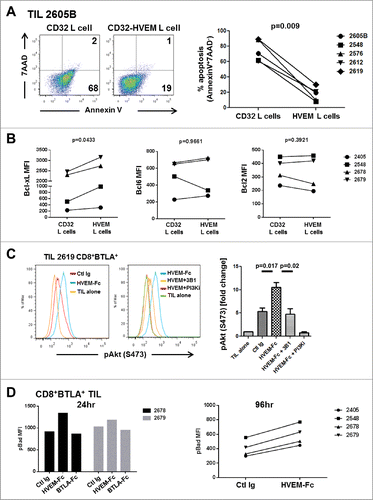
Our results so far underscore the fact that BTLA is a marker for less differentiated cells that have an enhanced responsiveness to IL-2 as well as enhanced persistence in vivo after infusion into patients. However, BTLA signaling is considered to be suppressive as its ligation has previously been shown to result in the recruitment of SHP1/2 phosphatases and decreased cell proliferation and cytokine secretion.Citation5,6 We therefore set out to determine whether BTLA signaling in our melanoma TIL was suppressive. In fact, similar to other studies using PBMC or TIL derived from tumor-draining lymph nodes, ligation of sorted CD8+ BTLA+ TIL in vitro using a plate-bound HVEM-Fc fusion protein together with anti-CD3 antibody resulted in a marked decrease in proliferation (8% versus 32%) and lower production of the proinflammatory cytokines IFN-γ and TNF-α compared to the control human IgG after 3 d of stimulation (Fig. S7). However, in an experiment examining the effects of CD3 and BTLA co-ligation using a TIL co-culture system with HVEM-expressing L cells loaded with anti-CD3 antibody we observed that the TIL were protected from cell death (, P = 0.009). As HVEM signaling has been shown to produce a prosurvival signal via the NF-κB signaling cascade, we stained the sorted CD8+BTLA+ or BTLA− subsets after co-culture with L cells for loss of IκBα as a read-out for NF-κB activation. Although a small loss was detected compared to the unstimulated control, there was no difference between the subsets cultured on either CD32-L cells or HVEM-L cells (Fig. S8A). To rule out any effect of signaling kinetics, we used another experimental system in which sorted TIL were cultured on plate-bound anti-CD3 (OKT3) with a control Ig, HVEM-Fc, or BTLA-Fc for 96 h and assessed for apoptosis. Using this model, only the CD8+BTLA+ TIL cultured with HVEM-Fc co-stimulation were protected from activation-induced cell death (AICD) (Fig. S8B). This provides further evidence that the survival phenotype was not mediated by HVEM signaling because BTLA-Fc (ligating HVEM) did not protect from apoptosis. Furthermore, examination of antiapoptotic proteins Bcl-xL, Bcl-2, and Bcl-6 after HVEM-L cell co-culture showed a significant increase in the level of Bcl-xL in the CD8+ BTLA+ TIL (, P = 0.043). Bcl-xL is an analog of Bcl-2 that has been shown to be induced by CD28 co-stimulation and mediates cell survival.Citation35 Interestingly, BTLA ligation on CD4+ T cells has been shown to result in a gene signature similar to that induced by CD28 and ICOS co-stimulation in terms of antiapoptotic gene expression.Citation9 This suggests that BTLA signaling on CD8+ T cells may, in fact, provide both a positive and negative signal.
Figure 6. For figure legend, see page 10.Figure 6 (see previous page). BTLA ligation provides a prosurvival signal to TIL. (A) CD32+ L cells or CD32+HVEM+ L cells (HVEM L cells) were pulsed with 200 ng/mL OKT3 and cultured with TIL (1×106/mL) in the presence of 100 IU/mL IL-2. After 5 days, TIL were harvested and stained for CD8, Annexin V, and 7-AAD. The left dot plots show representative staining of a TIL line gated on CD8+ cells. The right graph shows the percentage of apoptosis among all lines tested. Statistical significance was determined using a Wilcoxon signed-ranked test. (B) Sorted CD8+ BTLA+ TIL were cultured with L cells as in (A) and stained for Bcl-xL, Bcl-6, and Bcl-2 after 5 d (n = 4). (C) TIL were sorted into CD8+BTLA+ and CD8+BTLA− subsets and stimulated for 2 h with 30 ng/mL OKT3 and 10 μg/mL HVEM-Fc (plate-bound) (HVEM-Fc+OKT3), 10 μg/mL anti-BTLA blocking antibody, 3B1 (HVEM-Fc+OKT3+3B1), and 10 nM PI3K inhibitor (GSK2126458) (HVEM-Fc+OKT3+PI3Ki). TIL cultured alone or with OKT3 and a control Ig were included as negative controls. TIL were immediately fixed with prewarmed Phosflow Fix buffer I for 10 min at 37°C, permeabilized with pre-chilled Phosflow Perm buffer III, and stained with pAkt (S473) for 30 min on ice. The left histograms show representative staining from TIL 2619. The right graphs show the fold change in percent pAkt expression normalized to the TIL-alone condition (set to 1; n = 3 TIL lines). Statistical significance was determined using a 2-tailed paired Student t test. (D) CD8+ BTLA+ TIL were cultured with 30 ng/mL OKT3 plus control Ig (Ctl Ig), OKT3 plus 10 μg/mL HVEM-Fc (HVEM-Fc), or OKT3 plus 10 μg/mL BTLA-Fc (BTLA-Fc) for 24 and 96 h. Cells were stained for the presence of pBad by intracellular flow cytometry as described in Methods.
BTLA ligation induces pAkt and maintains Bad in a phosphorylated state
The above prosurvival phenotype led us to ask whether BTLA may also have a prosurvival signaling function mediated by HVEM binding in human melanoma TIL. This could potentially occur through the conserved YXN motif upstream of the ITIM and ITSM motifs in BTLA.Citation5 In the platelet-derived growth factor (PDGF) receptor, this YXN motif has been found to bind to the signaling adaptor protein Grb-2 following phosphorylation of the tyrosine, leading to recruitment of phosphatidylinositol 3’ kinase (PI-3K) and activation of Akt through serine phophorylation.Citation8,9,36 This suggests that HVEM-BTLA signaling could in fact have a prosurvival effect on TIL. To directly test whether BTLA ligation results in initiation of a prosurvival signal via phosphorylated Akt (pAkt), TIL were sorted into CD8+ BTLA+ and CD8+ BTLA− subsets and stimulated in vitro with HVEM-Fc fusion protein and anti-CD3 antibody. Ligation of BTLA resulted in a significant increase in the level of detectable pAkt (expressed as fold change over that of TIL stimulated with anti-CD3 without HVEM-Fc, which was arbitrarily set to 1) compared to the control Ig (Ctl Ig) with anti-CD3 (P = 0.017) (). Furthermore, this was specific for HVEM ligation of BTLA as preblockade of BTLA using the blocking monoclonal anti-BTLA antibody 3B1 (Genentech) significantly reduced the induction of pAkt in the presence of HVEM-Fc (P = 0.02) to levels comparable to that of the control Ig (). This activation of Akt was mediated through the PI3K pathway as addition of the pan-PI3K inhibitor GSK2126458 did not result in phosphorylation of Akt, with levels remaining similar to those with TIL alone (). To further confirm that the increased pAkt was due to signaling via BTLA, sorted CD8+BTLA− cells were also stimulated with control Ig or HVEM-Fc in the presence of anti-CD3 antibody and no significant differences in the fold change of pAkt (TIL alone levels set to 1) were observed (P = 0.61) (Fig. S9). To further elucidate the antiapoptotic role of BTLA ligation, we examined the extent of Bad phosphorylation, a downstream effect of PI3 kinase/Akt signaling. Phospho-Bad (pBad) cannot bind to Bcl-xL and Bcl-2, leaving them free to sequester Bax and Bak and prevent the release of cytochrome C from the mitochondria.Citation37 Indeed, when sorted CD8+BTLA+ TIL were stimulated with anti-CD3 and HVEM-Fc, pBad levels remained high at 24 h post stimulation compared with the Ctl Ig and BTLA-Fc co-stimulatory conditions (). pBad levels were also higher in the HVEM-Fc co-stimulated cells at 96 h after stimulation ().
Collectively, these results suggest that in the presence of TCR stimulation, BTLA ligation by HVEM facilitates survival signals in CD8+ TIL mediated through PI3K/Akt, maintenance of pBad, and induction of Bcl-xL. This would be important in the context of antigen presentation by dendritic cells or melanoma tumor cells, both of which express HVEM.Citation6,38
Discussion
The data presented in this paper provide a mechanistic basis for our recent findings from a Phase II clinical trial demonstrating that BTLA as a biomarker on adoptively-transferred TIL was associated with a positive clinical response to ACT with TIL in patients with metastatic melanoma.Citation2 We made a number of striking observations supporting a new and more complex view of the role of BTLA in activated effector-memory CD8+ T cells, not only as a marker of less-differentiated and functional T cells, but also possibly as a dual co-inhibitory and cell survival signaling molecule in human T cells. In addition, in 2 melanoma patients that responded to TIL therapy, we found that T-cell clones unique to the CD8+BTLA+ subset persisted longer in vivo in the blood after infusion and showed evidence of expansion, whereas clones associated with the BTLA− subset progressively disappeared or did not expand.
A key feature of the CD8+BTLA+ subset was its higher constitutive and TCR-induced upregulation of CD25 and autocrine production of IL-2. The ability of the BTLA+ TIL to produce more of their own IL-2 was associated with higher rates of cell division than in BTLA− cells; these are critical attributes for CD8+ memory T-cells to survive long term and maintain effector function after an immune response. Our findings were concordant with a previous study on memory CD8+ T cells in which memory CD8+CD25+ T cells both produced and responded to IL-2 in response to a variety of antigens, whereas the CD8+CD25− memory T cell population responded to fewer antigens and did not proliferate in response to stimulation with IL-2.Citation32 The ability of the CD8+ BTLA+ subset to produce more IL-2 could therefore endow them with the preferential ability to expand and persist in vivo after infusion into patients.
Another observation in this study was that CD8+ BTLA+ melanoma TIL had a highly activated, yet less differentiated, effector-memory (TEM) phenotype compared with their BTLA− counterparts. This was supported by differential gene expression studies on the sorted subsets revealing that the CD8+BTLA+ TIL were less differentiated through higher expression of memory markers such as CD28, CD25, and IL-7R. In contrast, BTLA− cells had markedly higher expression of multiple NK markers, including multiple killer cell lectin-like receptor (KLR) family members, such as KLRG1 and KLRD1, and killer-inhibitory receptor (KIR) family members, as well as DAP12.
We also observed that CD8+BTLA− TIL, although hyporesponsive to proliferative signals, nevertheless maintained a similar cytotoxic capability to the CD8+BTLA+ TIL. This phenomenon may be due to the unique signaling of KIRs on human CD8+ T cells; previous studies have found that their presence on CD8+CD28− T cells, unlike NK cells, only inhibits complex cellular functions such as proliferation, while leaving certain effector functions such as cytotoxicity essentially intact, presumably because of delayed recruitment to the TCR synapse after activation.Citation39 Additionally, although there was discordance between enrichment of multiple KIR gene transcripts in the CD8+BTLA− TIL and a few KIRs being expressed at the protein level in our present study, this might be explained by recent findings in which human CD8+ T cells, unlike NK cells, were found to generally express only one activating or inhibitory KIR.Citation40
Our gene expression studies also give some insights into the issue of inhibitory receptors and CD8+ T-cell exhaustion. Interestingly, we found significant enrichment of a T-cell deletion/tolerance signature in the CD8+BTLA− subset, supporting the more differentiated and hyporesponsive nature of these BTLA− cells. Although a significant proportion of the CD8+BTLA+ TIL expanded from melanoma metastases co-expressed PD-1 and TIM3, we found no significant enrichment of genes related to CD8+ T-cell exhaustion when we used GSEA to query 2 published gene sets: one on exhausted PD-1-expressing CD8+ T cells isolated after chronic LCMV infection in mice and the other on exhausted human PD-1+ melanoma antigen-specific (MART-1) CD8+ T cells.Citation26,41 These previously published exhaustion genes were not enriched in the sorted BTLA− TIL subset either, but these cells were characterized by a T-cell deletion gene profile as mentioned above. In addition, unlike PD-1 and TIM3, BTLA itself has not been found to be significantly enriched or overexpressed in these previously published exhaustion gene sets. Similarly, BTLA was not found to be overexpressed in a gene expression profile present in exhausted CD8+ T cells from HIV-infected patients.Citation17 It should be noted that our gene expression profiling was performed after a period of expansion of isolated TIL with IL-2 in order to generate enough cells for analysis. This may have reversed an exhausted state and its associated gene expression profile in the BTLA+ cells that may have been detectable if the TIL were analyzed directly ex vivo. Nevertheless, our results do support the view that, in the context of adoptive cell therapy, expanded TIL may not conform to the current T-cell exhaustion paradigm. Rather, expansion leads to T-cell differentiation and loss of responsiveness to IL-2 and TCR stimulation, as reflected in the expression of genes associated with T-cell deletion. A BTLA− phenotype is a key marker of this transition to a more differentiated, less cytokine-responsive state. This also supports our TCR clone tracking data in 2 responding patients using TCR Vβ CDR3 region sequencing, in which where unique clones associated with CD8+BTLA+ cells had superior persistence in vivo after TIL infusion. Our finding here is concordant with other reports showing that persistence of the infused TIL clones was a key factor associated with positive clinical response after ACT.Citation42
Our TCR Vβ CDR3 region sequencing studies, which tracked unique and shared T-cell clones in TIL from the CD8+BTLA+ and CD8+BTLA− subsets, showed that the subsets shared a majority of clones, although unique clones could also be detected in each subset (which allowed us to track the relative persistence of each subset in vivo in the patients). The presence of shared clones in the subsets suggests that BTLA+ and BTLA− cells represent discrete stages of effector-memory CD8+ T-cell differentiation, with the BTLA− subset representing a more differentiated stage toward terminal differentiation (as supported by our functional studies). The detection of unique clones in each subset may reflect differential localization of BTLA+ and BTLA− cells from the same clonal population, leading to the presence of one subset or the other in the tumor-infiltrating lymphocyte compartment. This will need to be further investigated in future studies.
The improved persistence of BTLA+ cells in vivo may be modulated by increased expansion, but might also due to increased cell survival linked to BTLA signaling or an intrinsic cell death-resistant phenotype. A prosurvival role of BTLA was initially observed in a chronic GvHD model in which adoptively transferred BTLA−/− splenocytes initially expanded in recipient mice but could not persist long term, suggesting that the memory T-cell function was compromised.Citation36 Recently, another study showed a prosurvival role of BTLA in vivo in a mouse model of vaccinia virus.Citation43 In this model, however, BTLA mediated survival and memory development through its expression on dendritic cells. We also observed a prosurvival signaling effect of BTLA through ligation with HVEM, in which we consistently saw a reduction in apoptosis in the activated TIL. Although we found that LIGHT was upregulated on CD8+ TIL after TCR activation and could potentially interact with HVEM, a prosurvival effect mediated through LIGHT is unlikely because it has no obvious signaling motif,Citation44 and it has been reported that signaling through HVEM upon binding to LIGHT might induce a different T-cell survival via NF-κB activation.Citation45 It is also possible that cis interaction or a T-T interaction between BTLA and HVEM could mediate survival via HVEM, as shown by Carl Ware's group.Citation46 However, in our system using BTLA-Fc for co-stimulation, we failed to see protection from AICD and maintenance of pBad. Furthermore, we were unable to detect NF-κB activation at early time points using our L cell system, as measured indirectly via IκB downmodulation. Thus, our results indicate that the CD8+BTLA+ TIL subset, in addition to being less differentiated and more cytokine responsive, may have improved survival capabilities through BTLA ligation. The loss of BTLA in a more highly differentiated state, leading to a loss of prosurvival Akt activation, may contribute to the enrichment of the T-cell deletion gene signatureCitation13 in the CD8+BTLA− TIL.
Mechanistically, the ability of BTLA to activate Akt and cell survival and its co-inhibitory activity during TCR activation limiting cell division and cytokine secretion can be reconciled based on its unique structural properties. Although human BTLA is similar to PD-1 in possessing a consensus ITIM and ITSM site for SHP1/2 docking as well as functioning as a monomer, it has a putative Grb-2 binding domain consisting of a YXNX motif upstream of the ITIM- and ITSM-containing domains that is not found in PD-1. Early in vitro experiments found that a synthesized 11-mer phospho-tyrosine peptide containing this domain with a phosphorylated tyrosine 226 residue from BTLA could recruit Grb-2-PI3K.Citation8 Grb-2 has also been found to be able to recruit PtdIns-3K via its interaction with c-Cbl after TCR crosslinking on Jurkat T cells.Citation47
These observations and concepts suggest that BTLA signaling may also lead to a scenario in cancer where, in the context of chronic antigen stimulation within the tumor microenvironment, BTLA ligation by melanoma tumors that express HVEMCitation6 may inhibit or arrest further cell division and differentiation of CD8+ TIL while enhancing their survival, leading to their accumulation in an incompletely differentiated state in tumors. This would explain why we and others have found that the vast majority of CD8+ TIL in melanoma are in an early effector-memory state of differentiation with few fully differentiated CTL, as well as the better persistence of TIL expressing BTLA after adoptive transfer in our clinical trials.
In summary, our study provides a molecular basis for our previously observed correlation between positive clinical outcome and CD8+BTLA+ TIL after adoptive transfer in patients with metastatic melanoma. Our data show that this effect is due to a less-differentiated state of the CD8+BTLA+ TIL subset, their ability to receive prosurvival signals through BTLA, and their ability to persist better after adoptive transfer in vivo.
Materials and Methods
Expansion of tumor-infiltrating lymphocytes from human melanoma tumors
TIL were separated from tumor cells and expanded in high-dose IL-2 (6,000 IU/mL, Novartis) as described previously.Citation19 Autologous primary melanoma tumor lines were also established by collecting cells at the 75% Ficoll layer and were used as targets in cytotoxic T lymphocyte assays.
Flow cytometry
TIL were stained using the Live/Dead® Fixable Aqua Dead Cell Stain Kit (Life Technologies, CA) according to the manufacturer's instructions. Cells were stained with a combination of antibodies (from BD unless indicated otherwise) against the following proteins: CD3 FITC (SK7), CD4 PerCP-Cy5.5 (RPT-T4), CD8 PB (RPT-T8), BTLA PE (J168), TIM3 APC (F38–2E2), PD-1 PerCP-Cy5.5 (EH12.2H7), CD27 APC-H7 (M-T271), CD28 PE-Cy7 (CD28.2), CD45RA FITC 566 (HI100), CD158a FITC, CD158b FITC, CD158e APC, NKG2D APC, NKAT2 FITC, NKB1 FITC, APC-conjugated MART-1 peptide (ELAGIGILTV) HLA-A2.1 tetramer (Beckman Coulter), and APC-conjugated gp100 peptide (IMDQVPFSV) HLA-A2.1 tetramer (Beckman Coulter). For all flow cytometry assays, data were acquired using a FACScanto II cytometer (BD Biosciences), and analyzed using FlowJo software (Treestar version 7.6.5).
Cell sorting
TIL were stained for CD8, BTLA, and CD27 and sorted using an Aria II or Influx cell sorter (BD Biosciences) directly into TIL media. Only populations with ≥95% post-sort purity were used for experiments.
Phospho-flow cytometry
Sorted cells were stimulated with increasing doses of IL-2 (50 U/mL, 200 U/mL, and 3,000 U/mL) for 20 min. At the end of the stimulation, cells were fixed in BD™ Cytofix Buffer for 10 min and permeabilized by chilled BD™ Phosphoflow™ Perm Buffer III for 20 min at 4°C. Cells were washed twice with BD™ PharMingen™ Stain Buffer and stained with fluorochrome-conjugated anti–phospho-Stat5 (Tyr694) antibody for 20 min at 4°C prior to analysis.
Intracellular cytokine staining
Sorted cells were stimulated with 25 ng/mL PMA and 1 μg/mL ionomycin in the presence of BD GolgiStopTM according to the manufacturer's instructions. After 4 h, the cells were fixed and permeabilized using BD Cytofix/CytopermTM kit and subsequently stained with IL-2 PerCP-Cy5.5, IFN-γ (PE-Cy7), and TNF-α APC antibodies.
Microarray
Total RNA was isolated from sorted CD8+ TIL subsets using a RNeasy Mini Kit (Qiagen, Valencia, CA USA). Samples were hybridized to Human Gene ST 1.0 Arrays (Affymetrix, Santa Clara, CA). Raw data that passed QC assessment were generated with Affymetrix GeneChip Command Console and imported into BRB-ArrayTools v4.2.1 (NCI, Bethesda, 587 MD USA), normalized with robust multi-array average (RMA) algorithm, and log2 transformed. The microarray data have been deposited into the NCBI GEO database (Accession #GSE43260; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43260).
Costimulation of TIL with HVEM+ L cells or HVEM-Fc fusion protein
CD32+ or CD32+ HVEM+ L cells were harvested, pulsed with 200 ng/mL OKT3 and co-cultured at a 1:1 ratio with TIL. After 12 h, TIL were harvested and stained for CD8 and intracellular IκBα PerCP-Cy5.5 (BD). After 5 days, TIL were stained for CD8, 7-AAD, Annexin-V, Bcl-xL Alexa 488 (Cell Signaling), Bcl-2 FITC (BD), and Bcl-6 PerCP-Cy5.5 (BD). Additionally, sorted CD8+ BTLA+ and BTLA− TILs were stimulated with 30 ng/mL plate-bound OKT3 and 10 μg/mL control human Ig, BTLA-Fc (R&D), or HVEM-Fc (R&D) with or without 10 μg/mL soluble anti-BTLA blocking mAb (clone 3B1) or PI3K inhibitor (10 nM GSK2126458). Cells were stained as described above.
Tracking TCR clonotypes in patients infused with TIL
TIL from the infusion product of patients treated with ACT at MD Anderson were sorted into CD8+BTLA+ and CD8+BTLA− subsets. Cell pellets were flash frozen and stored at −80°C. Peripheral blood mononuclear cells were collected post-infusion for tracking of the infused cells. Total DNA was isolated using the Qiagen AllPrep KitTM and samples were shipped to Adaptive Biotechnologies (Seattle, WA USA) for sequencing of the T cell CDR3 region using the ImmunoSEQTM assay.
Statistical analysis
For quantitative comparisons between 2 paired groups, Wilcoxon signed-rank test (2-sample, 2-tailed comparisons) was performed with Graph Pad Prism v5.0 (La Jolla, CA) with column statistics reported as mean ± S.E; P < 0.05 was considered statistically significant. P values and FDRs for gene sets used in GSEA were calculated with 1,000 permutations by phenotype. Enrichment of a particular gene set in an indicated subset of CD8+ TIL was considered significant if P < 0.05 and FDR < 0.25. We also used a comprehensive software package, BRB-ArrayTools v4.2.1 (NCI, Bethesda, MD USA), to determine statistically significant genes (P < 0.05) between CD8+ BTLA+ and CD8+BTLA− TIL subsets using a paired Student t test.
Patient tumor sample acquisition
Tumor samples were obtained from patients with Stage IIIc and Stage IV melanoma undergoing surgery at The University of Texas MD Anderson Cancer Center according to an Institutional Review Board-approved protocol and with patient consent (IRB# LAB06–0755 and LAB06–0757). Table S1 shows the characteristics of the melanoma patients, including patient age, sex, tumor location, disease stage, and anatomical sites.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
CLH and RCW contributed equally to the fluorescence-activated cell sorting, phenotypic analysis of the TIL by flow cytometry, and functional studies of the TIL subsets. RCW, HL, and EW contributed to the microarray experiment and its analysis. CLH and RCW co-wrote the manuscript. LR designed experiments, supervised the work, procured the melanoma tissue, and helped write and edit the paper.
1014246_Supplementary_Materials.zip
Download Zip (742.5 KB)Acknowledgments
The authors appreciate the dedicated melanoma surgical staff at MD Anderson Cancer Center who provided all the melanoma specimens for this study, including Drs. Jeffrey Gershenwald, Jeffrey Lee, Merrick Ross, Amy Heimberger, Paul Mansfield, Janice Cormier, and Anthony Lucci. We are grateful to Renjith Ramachandran, Christopher Toth, O.J. Fulbright, Rahmatu Mansaray, Audrey Gonzalez, Seth Wardell, and Charuta Kale for processing tumor specimens. We thank M.D. Anderson's Melanoma SPORE Tissue Bank personnel (Dr. Victor Prieto and Katie McNeil) for help in obtaining the melanoma samples used in this study as well as Dr. Luis Vence, Himabindu Pappu, and Thao Ly Do from MD Anderson's Immunomonitoring Core Lab (IMCL) for help with multiplex cytokine analysis. We thank Genentech for providing the BTLA blocking antibody (clone 3B1). GlaxoSmithKline provided the PI3Ki (GSK2126458).
Funding
This work was supported by the National Cancer Institute (NCI) grants R01-CA111999 to PH and LR and 1R21CA178580–01 to LR; by the University of Texas M.D. Anderson Cancer Center Support Grant (P30-CA16672) to the Flow Cytometry Core Facility; Award No. TL1RR024147 from the National Center for Research Resources to RCW; the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF), and the Mulva Foundation. CLH was supported by the National Institutes of Health training grant 5T32CA009598–22. This work was also supported by a Melanoma SPORE Development Grant (P50-CA093459–05-DRP21) and a Team Science Award from the Melanoma Research Alliance (MRA) to LR.
References
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005; 23:2346-57; PMID:15800326; http://dx.doi.org/10.1200/JCO.2005.00.240
- Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res 2012; 18:6758-70; PMID:23032743; http://dx.doi.org/10.1158/1078-0432.CCR-12-1177
- Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, Levy D, Kubi A, Hovav E, Chermoshniuk N, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res 2010; 16:2646-55; PMID:20406835; http://dx.doi.org/10.1158/1078-0432.CCR-10-0041
- Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother 2012; 35:651-60; PMID:23090074; http://dx.doi.org/10.1097/CJI.0b013e31827806e6
- Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 2003; 4:670-9; PMID:12796776; http://dx.doi.org/10.1038/ni944
- Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O, Olive D, Speiser DE. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest 2010; 120:157-67; PMID:20038811; http://dx.doi.org/10.1172/JCI40070
- Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res 2012; 72:887-96; PMID:22205715; http://dx.doi.org/10.1158/0008-5472.CAN-11-2637
- Gavrieli M, Murphy KM. Association of Grb-2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem Biophys Res Commun 2006; 345:1440-5; PMID:16725108; http://dx.doi.org/10.1016/j.bbrc.2006.05.036
- Wakamatsu E, Mathis D, Benoist C. Convergent and divergent effects of costimulatory molecules in conventional and regulatory CD4+ T cells. Proc Natl Acad Sci 2013; 110:1023-8; PMID:23277554; http://dx.doi.org/10.1073/pnas.1220688110
- Serriari NE, Gondois-Rey F, Guillaume Y, Remmerswaal EB, Pastor S, Messal N, Truneh A, Hirsch I, van Lier RA, Olive D. B- and T- lymphocyte attenuator is highly expressed on CMV-specific T cells during infection and regulates their function. J Immunol 2010; 185:3140-8; PMID:20693422
- Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “Exhaustion” of human CD8 T cells. Front Immunol 2013; 4:455; PMID:24391639; http://dx.doi.org/10.3389/fimmu.2013.00455
- Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals JP, Baumgaertner P, Christiansen-Jucht C, Bouzourene H, Rimoldi D, Pircher H, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One 2012; 7:e30852; PMID:22347406; http://dx.doi.org/10.1371/journal.pone.0030852
- Parish IA, Rao S, Smyth GK, Juelich T, Denyer GS, Davey GM, Strasser A, Heath WR. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood 2009; 113:4575-85; PMID:19204323; http://dx.doi.org/10.1182/blood-2008-10-185223
- Parish IA, Kaech SM. Diversity in CD8(+) T cell differentiation. Curr Opin Immunol 2009; 21:291-7; PMID:19497720; http://dx.doi.org/10.1016/j.coi.2009.05.008
- Turnbull IR, Colonna M. Activating and inhibitory functions of DAP12. Nat Rev Immunol 2007; 7:155-61; PMID:17220916; http://dx.doi.org/10.1038/nri2014
- Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer 2002; 2:850-61; PMID:12415255; http://dx.doi.org/10.1038/nrc928
- Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata P, Gupta S, Zilliox MJ, Nakaya HI, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol 2011; 186:4200-12; PMID:21383243; http://dx.doi.org/10.4049/jimmunol.1001783
- Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol 2005; 175:5895-903; PMID: 16237082; http://dx.doi.org/10.4049/jimmunol.175.9.5895
- Li Y, Liu S, Hernandez J, Vence L, Hwu P, Radvanyi L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J Immunol 2010; 184:452-65; PMID:19949105; http://dx.doi.org/10.4049/jimmunol.0901101
- Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of 676 immune function. Trends Immunol 2009; 30:306-12; PMID:19540809; http://dx.doi.org/10.1016/j.it.2009.03.013
- Brumbaugh KM, Binstadt BA, Billadeau DD, Schoon RA, Dick CJ, Ten RM, Leibson PJ. Functional role for Syk tyrosine kinase in natural killer cell-mediated natural cytotoxicity. J Exp Med 1997; 186:1965-74; PMID:9396765; http://dx.doi.org/10.1084/jem.186.12.1965
- Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 2009; 126:458-65; PMID:19278419; http://dx.doi.org/10.1111/j.1365-2567.2008.03027.x
- Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol 2006; 18:1197-209; PMID:16772372; http://dx.doi.org/10.1093/intimm/dxl060
- Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol 2005; 6:472-80; PMID:15834410; http://dx.doi.org/10.1038/ni1193
- Calnan BJ, Szychowski S, Chan FK, Cado D, Winoto A. A role for the orphan steroid receptor Nur77 in apoptosis accompanying antigen-induced negative selection. Immunity 1995; 3:273-82; PMID:7552993; http://dx.doi.org/10.1016/1074-7613(95)90113-2
- Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest 2011; 121:2350-60; PMID:21555851; http://dx.doi.org/10.1172/JCI46102
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27:670-84; PMID:17950003; http://dx.doi.org/10.1016/j.immuni.2007.09.006
- Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004; 22:745-63; PMID:15032595; http://dx.doi.org/10.1146/annurev.immunol.22.012703.104702
- Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in 695 health and disease. Immunology 2011; 134:17-32; PMID:21711350; http://dx.doi.org/10.1111/j.1365-2567.2011.03470.x
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550-7; PMID:21498393; http://dx.doi.org/10.1158/1078-0432.CCR-11-0116
- Collins ND, D'Souza C, Albrecht B, Robek MD, Ratner L, Ding W, Green PL, Lairmore MD. Proliferation response to interleukin-2 and Jak/Stat activation of T cells immortalized by human T-cell lymphotropic virus type 1 is independent of open reading frame I expression. J Virol 1999; 73:9642-9; PMID:10516077
- Herndler-Brandstetter D, Schwaiger S, Veel E, Fehrer C, Cioca DP, Almanzar G, Keller M, Pfister G, Parson W, Würzner R, et al. CD25-expressing CD8+ T cells are potent memory cells in old age. J Immunol 2005; 175:1566-74; PMID:16034095; http://dx.doi.org/10.4049/jimmunol.175.3.1566
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45:228-47; PMID:19097774; http://dx.doi.org/10.1016/j.ejca.2008.10.026
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15:7412-20; PMID:19934295; http://dx.doi.org/10.1158/1078-0432.CCR-09-1624
- Radvanyi LG, Shi Y, Vaziri H, Sharma A, Dhala R, Mills GB, Miller RG. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J Immunol 1996; 156:1788-98; PMID:8596028
- Hurchla MA, Sedy JR, Murphy KM. Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J Immunol 2007; 178:6073-82; PMID:17475832; http://dx.doi.org/10.4049/jimmunol.178.10.6073
- Bergmann A. Survival signaling goes BAD. Dev Cell 2002; 3:607-8; PMID:12431365; http://dx.doi.org/10.1016/S1534-5807(02)00328-3
- Murphy KM, Nelson CA, Sedy JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol 2006; 6:671-81; PMID:16932752; http://dx.doi.org/10.1038/nri1917
- Henel G, Singh K, Cui D, Pryshchep S, Lee WW, Weyand CM, Goronzy JJ. Uncoupling of T-cell effector functions by inhibitory killer immunoglobulin-like receptors. Blood 2006; 107:4449-57; PMID:16469873; http://dx.doi.org/10.1182/blood-2005-06-2519
- Bjorkstrom NK, Beziat V, Cichocki F, Liu LL, Levine J, Larsson S, Koup RA, Anderson SK, Ljunggren HG, Malmberg KJ, et al. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood 2012; 120:3455-65; PMID:22968455; http://dx.doi.org/10.1182/blood-2012-03-416867
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009; 10:29-37; PMID:19043418; http://dx.doi.org/10.1038/ni.1679
- Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009; 114:4099-107; PMID:19706884; http://dx.doi.org/10.1182/blood-2009-04-217604
- Flynn R, Hutchinson T, Murphy KM, Ware CF, Croft M, Salek-Ardakani S. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS One 2013; 8:e77991; PMID:24205056; http://dx.doi.org/10.1371/journal.pone.0077991
- Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A, Chauhan V, Arnott D, Eaton DL. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol 2009; 182:1509-17; PMID:19155498; http://dx.doi.org/10.4049/jimmunol.182.3.1509
- Soroosh P, Doherty TA, So T, Mehta AK, Khorram N, Norris PS, Scheu S, Pfeffer K, Ware C, Croft M. Herpesvirus entry mediator 731 (TNFRSF14) regulates the persistence of T helper memory cell populations. J Exp Med 2011; 208:797-809; PMID:21402741; http://dx.doi.org/10.1084/jem.20101562
- Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, D'Souza C, Norris PS, Pfeffer K, Murphy KM, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci 2009; 106:6244-9; PMID:19332782; http://dx.doi.org/10.1073/pnas.0902115106
- Meisner H, Conway BR, Hartley D, Czech MP. Interactions of Cbl with Grb2 and phosphatidylinositol 3′-kinase in activated Jurkat cells. Mol Cell Biol 1995; 15:3571-8; PMID:7791764