
Abstract
The question of which dendritic cells (DCs) cross-present peripheral tumor antigens remains unanswered. We assessed the ability of multiple skin-derived and lymphoid resident DCs to perform this function in a novel orthotopic murine melanoma model where tumor establishment and expansion is within the skin. Two migratory populations defined as CD103−XCR1+ and CD103+XCR1+ efficiently cross-presented melanoma-derived antigen, with the CD103−XCR1+ DCs surprisingly dominating this process. These results are critical for understanding how antitumor CD8+ T cell immunity is coordinated to tumor antigens present within the skin.
Introduction
Currently, limited information exists on the exact cell types responsible for generating an antitumor CD8+ T cell response. Thus, the overall aim of this study was to identify the DCs that are important for the cross-presentation of cutaneous melanoma antigen to naive tumor-specific T cells within the skin-draining lymph nodes. A novel orthotopic melanoma transplant model that predominantly confines peripheral tumor growth to the epidermal and dermal layers of the skin is utilized in this study to answer the important question of which DCs cross-present peripheral tumor antigen.
DCs are the master regulators of T cell immunity.Citation1 Peripheral sites, such as the skin, contain a rich network of heterogeneous DCs.Citation2 Such diversity allows individual subsets to perform a particular function, enabling a division of labor. This concept is supported by studies highlighting that some subsets of DCs are superior at presenting endocytosed antigen to CD4+ T cells on major histocompatibility complex (MHC) class II molecules, while others are better at introducing exogenous antigens into the MHC class I pathway in a process referred to as cross-presentation.Citation3-6 These differences reflect inherent variations between DC subsets that include possession of specialized intracellular processing machinery enabling external antigens to access the MHC class I peptide-loading pathwayCitation5 or the expression of defined endocytic receptors to facilitate the specific uptake of antigen from their surroundings.Citation3,4
Specific categorization of the different DC lineages remains challenging, although subclassification into independent subsets is possible.Citation7 An immediate segregation can be made of plasmacytoid DCs (pDCs) and classical DCs (cDCs). Both of these subgroups arise from a common DC precursor before terminally differentiating into their respective subgroups.Citation8,9 Traditionally, demarcation of these subgroups relies on expression of selective surface antigens and their specific immune function. For example, expression of CD317 within the DC compartment is restricted to pDCs and these cells are the primary source of type I interferon following viral infection.Citation10,11 An improved understanding of the transcription factors controlling DC development provides an alternate means to classify distinct DC subsets. Using this approach, pDCs can be separated from cDCs by regulation of their differentiation program dependent on specific transcription factors such as E2-2.Citation12,13 Furthermore, cDCs can be further divided into several subgroups displaying common functional features and properties. For example, certain mouse strains lacking the gene encoding the transcription factor Batf3 do not have the capacity to cross-present antigens.Citation14,15 However, identification of specific lineage markers exclusively expressed on cross-presenting DCs remains elusive.
Numerous studies have established that the CD8+ DC subset excels at cross-presenting antigens.Citation16 This particular subset predominantly resides in the spleen and lymphoid organs. Recent reports demonstrate that skin-derived CD103+ DCs also efficiently cross-present antigens.Citation2,17 In certain mouse strains, these subsets share a dependency on key transcription factors crucial for their development.Citation18 Thus, based on their similar functional specialization and unified gene expression signature, the CD8+ and CD103+ DCs appear to represent a distinct lineage of DCs. More recently, the expression of the XC-chemokine receptor 1 (XCR1) has been used to identify cross-presenting DCs.Citation19-23 This marker is not found on all CD8+ DCs and the existence of XCR1+ DCs that do not express either CD8 or CD103 have been reported.Citation22
In summary, a strong link exists between DC subtypes that express a common set of transcription factors and the ability to cross-present antigens to CD8+ T cells,Citation14,24 but linking this with the expression of particular surface markers remains challenging. Here, surface expression of CD8, CD103 and XCR1 is used to describe various DC populations within the skin-draining lymph nodes of mice. We report that four XCR1+ DC populations with heterogeneous surface marker expression can be identified. Using a novel transplant model of cutaneous melanoma where tumor growth is within the skin, we demonstrate that two migratory populations defined as CD103−XCR1+ and CD103+XCR1+ are superior at cross-presenting melanoma-derived antigen, with the CD103−XCR1+ population dominating this process. This report provides new insight into the functional specialization within the broad network of DCs that are responsible for skin immunosurveillance and possess the capacity to initiate antitumor CD8+ T cell immunity.
Results
Cross-presentation of cutaneous-derived melanoma antigen
Immunity to cancer is a classic biological scenario that relies heavily on cross-presentation to elicit a CD8+ T cell response. To date, the exact DC populations responsible for cross-presenting cutaneous tumor antigen remain ill-defined. In this study, we focused our attention on melanoma. Traditionally, the standard preclinical model of melanoma involves subcutaneous delivery of B16 melanoma cells.Citation25 This robust model results in tumor growth beneath the skin. However, the etiology of malignant melanoma in humans is within the epidermis. The early stages of this disease display aberrant intraepidermal growth that progresses to vertical growth and dermal invasion.Citation26 This certainly allows local epidermal- and dermal-derived DCs to sample the malignant tissue before their migration to the tumor-draining lymph nodes. Thus, to conduct studies on skin-derived DC immunosurveillance of melanoma, a more realistic model that more closely mimicks human disease is required. Therefore, we developed an orthotopic murine melanoma model where tumor establishment and expansion takes place within the skin.
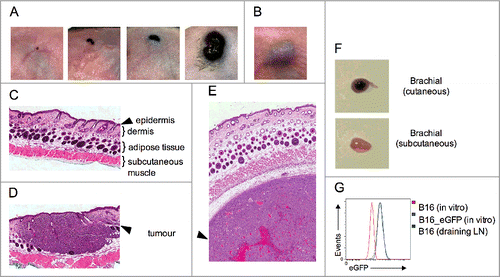
Such a model is achieved by grafting B16 cells directly onto the skin of mice in a procedure referred to as cutaneous inoculation. shows the growth of cutaneous tumors at various timepoints. It clearly shows that cutaneous tumors grow superficially whereas subcutaneous tumors grow beneath the skin (). Histological examination confirmed the macroscopic observations (), thus our novel grafting procedure provides a robust model of disease progression in the correct anatomical microenvironment. Interestingly, cutaneous-derived wild-type B16 tumors reliably form metastases in the draining lymph node (). This is not observed in the classical subcutaneous model and it is a bona-fide metastasis, as B16 cells transduced to express eGFP can be grown in vitro from tumor-draining lymph node explants (). Of critical relevance here, cutaneous inoculation results in melanoma establishment within the epidermis and dermis, mimicking the human condition and allowing skin resident DCs to sample local tumor antigens.
Figure 1. Orthotopic model of cutaneous melanoma. Wild-type B16 melanoma cells were grafted via the (A) cutaneous or the (B) conventional subcutaneous route. Disease progression over time is depicted for cutaneous lesions. (C–E) H & E staining of skin identifying the epidermal, dermal, adipose tissue and subcutaneous muscle layers on (C) uninoculated skin, (D) cutaneous, and (E) subcutaneous tumor grafts. (F) Metastatic disease in the tumor draining brachial lymph node. (G) eGFP expression on cells grown from lymph node explants (green) were compared to B16_eGFP (blue) and parental B16 (red) cells grown in vitro. Data are representative of at least two independent experiments.
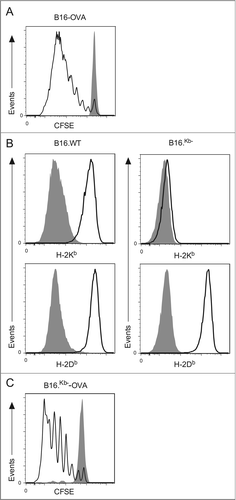
To assess tumor antigen presentation in vivo in the cutaneous model, B6 mice were inoculated with B16 cells expressing the model antigen ovalbumin (OVA). To monitor antigen presentation, we tracked the proliferation of CFSE labeled CD8+ OT.I T cells specific for an H-2Kb-restricted epitope of OVACitation27 60 h after intravenous injection into B6 mice harboring palpable cutaneous B16-OVA tumors. This approach revealed that OVA-specific T cell proliferation occurred in the draining lymph nodes (). Within this experimental model B16-OVA melanoma cells that have metastasized to the regional nodes have the capacity to present antigen directly to T cells. To circumvent this possibility, we acquired a variant of B16 that selectively lacks H-2Kb molecules and transduced it with OVA to generate B16Kb−-OVA. Treatment of wild-type B16-OVA and B16Kb−-OVA cells with IFNγ results in the expression of H-2Db molecules in both lines. However, upregulation of H-2Kb molecules after IFNγ treatment was not observed in the B16Kb−OVA cell line (), confirming previously published results that this variant has lost this particular MHC restriction element.Citation28 Considering OT.I T cells recognize a H-2Kb-restricted epitope, potential direct presentation by B16Kb−-OVA melanoma cells is abrogated and only host-derived cross-presenting DCs can drive melanoma-specific CD8 T cell proliferation. Interestingly, metastases to the skin-draining lymph nodes are either reduced or absent when this particular B16Kb− variant is grafted onto the skin. Thus, in this particular experimental setup melanoma growth is predominantly confined to the skin. To confirm cross-presentation occurs in this setting, the proliferation of OVA-specific CD8 T cells was investigated in B6 mice harboring cutaneous B16Kb−-OVA cells. Robust T cell activation was observed (), confirming that efficient cross-presentation of melanoma-derived antigen by host cells occurs in this setting.
Figure 2. Tumor-specific CD8+ T cells proliferate in mice bearing cutaneous melanomas. (A) A total of 106 CFSE-labeled CD8+ OT.I T cells were adoptively transferred into mice bearing cutaneous B16 or B16-OVA tumors. After 60 h, the tumor-draining lymph nodes were analyzed by flow cytometry for the proliferating CD45.1+CD8+Vα2+CFSE+PI− cells. Shaded and open histograms are from mice with either B16 or B16-OVA tumors respectively. (B) Wild-type B16 and B16Kb− melanoma cells were cultured in vitro with or without addition of IFNγ and analyzed for up-regulation of H2Kb and H2Db. Shaded histograms represent untreated controls. (C) A total of 106 CFSE-labeled CD8+ OT.I T cells were adoptively transferred into mice bearing cutaneous B16Kb− or B16Kb−-OVA tumors. After 60 h, the tumor-draining lymph nodes were analyzed by flow cytometry for the proliferating CD45.1+CD8+Vα2+CFSE+PI− cells. Shaded and open histograms are from mice with either B16Kb− or B16Kb−-OVA tumors respectively. Representative histograms from at least three independent experiments.
Expression of XCR1 defines DCs with a similar phenotype and ontogeny
The ability to segregate the complex skin DC network into defined populations is necessary before assessing their capacity to cross-present cutaneous melanoma antigen. Expression of either CD8 or CD103 is traditionally used to describe independent cross-presenting DC subsets. Another approach to identify these DCs is by the differential expression of CD172a (Sirpα) and CD24 (heat stable antigen), with a CD172aloCD24hi phenotype restricted to CD8+ and CD103+ DCs.Citation22,29 More recently, the expression of the chemokine receptor XCR1 has been used to identify cross-presenting DCs.Citation19-22 While many surface markers on DCs have been identified, to date they remain insufficient to completely describe unified DC populations.Citation22,30
The expression of these various surface markers on DCs isolated from skin-draining lymph nodes was investigated. DCs were isolated from lymph node suspensions as reported previouslyCitation31 and stained with the pan-DC marker CD11c as well as MHC II, CD8, CD103, XCR1, CD172a and CD24. shows our gating strategy where cDCs, identified as CD11c+MHC II+, were segregated into specific populations. Initially, cDCs were divided by expression of CD8, which is expressed on lymph node-resident DCs.Citation16 CD8+ DCs were divided further based on XCR1 expression into CD8+XCR1+ and CD8+XCR1− populations. The heterogeneous CD8− DC population was divided into two further groups: previously described CD8−CD103+XCR1+ dermal-derived DCs,Citation17 which we called CD103+XCR1+; and relatively uncharacterized CD8−CD103−XCR1+ DCs, which we called CD103−XCR1+. Our analysis of these DCs for the expression of CD172a and CD24 showed XCR1+ DCs have a unified phenotype of CD172aloCD24hi, whereas the CD8+XCR1− DCs expressed high levels of both markers (). This result suggests CD8+XCR1− DCs may be either a precursor population to a XCR1+ DC subset or they represent a distinct differentiated population.
Figure 3. Expression of XCR1 defines DCs with a similar phenotype and ontogeny. (A) Gating strategy for identifying enriched DC populations from the skin-draining lymph nodes. (B) Surface expression of CD172a and CD24 on CD8+XCR1+, CD8+XCR1−, CD103+XCR1+, and CD103−XCR1+ DCs. Shaded histograms represent fluorescence minus one control staining. Representative plots from three independent experiments are shown. (C) Quantitative RT-PCR of the transcription factors ID2, IRF8, BATF3 and IRF4 in purified DC populations. Data show the expression normalized to HPRT of two pooled biological replicates each assayed in triplicate for each DC population (mean ± SEM).
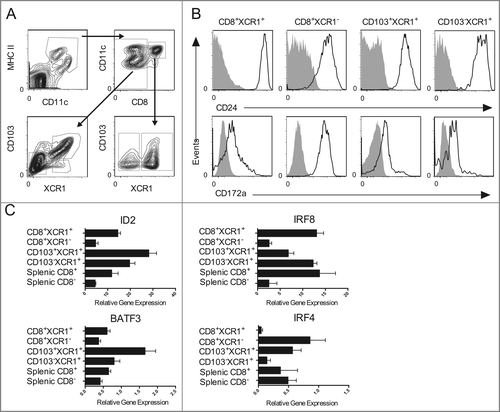
To investigate this further, the transcription factors involved in guiding the differentiation of distinct lineages of DCs was analyzed in multiple isolated populations including CD8+XCR1− DCs. Those DCs that are highly efficient at cross-presenting exogenous antigens on MHC class I molecules to CD8+ T cells are reported to share a dependence on the transcription factors ID2, IRF8, and Batf3.Citation14 DCs that are more efficient in presenting MHC class II-restricted antigens to CD4+ T cells express the lineage-specific transcription factor IRF4.Citation24,32 Splenic CD8+ and CD8− DCs were used as controls during these analyses as the correlation between discrete functions and the expression of corresponding transcription factors has been articulated clearly for these populations. The results revealed that all XCR1+ DCs expressed higher levels of ID2, IRF8, and Batf3 than the CD8+XCR1− DC subpopulation, which expressed abundant IRF4 transcripts (). These data suggest that expression of XCR1, but not CD8 or CD103, defines DCs with a similar ontogeny.
CD103−XCR1+ DCs are heterogeneous, comprised of lymphoid resident and migratory subpopulations
Examination of MHC class II expression on the various DC populations defined in revealed that CD8+XCR1+ DCs have low/intermediate expression and the CD103+XCR1+ DCs have high expression (). This phenotype is consistent with the CD8+XCR1+ DCs being immature and lymphoid resident in nature, in contrast to the CD103+XCR1+ DCs having matured following migration from the periphery. Interestingly, the CD103−XCR1+ DC subpopulation displays bimodal expression of MHC class II, indicative of a heterogeneous population of resident and migratory DCs.
Figure 4. CD103−XCR1+ DCs from the skin-draining lymph nodes are comprised of a migratory MHC IIhi population and a lymphoid resident MHC IIlo population. Analysis of MHC II expression on enriched CD8+XCR1+, CD103+XCR1+, and CD103−XCR1+ DCs isolated from the skin-draining lymph nodes of (A) wild-type B6 and (B) CCR7o/o mice. The numbers indicate the percentage of CD8+ DCs (left) and CD103+/− DCs that co-express XCR1. Histograms represent MHC II expression on each of the gated subpopulations. Data are representative plots from three independent experiments.
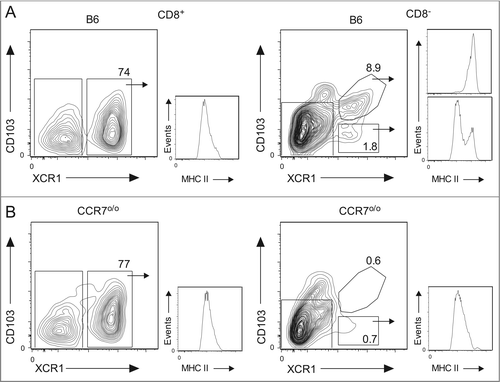
DC migration from the periphery to the draining lymph nodes depends on expression of CCR7.Citation33 To validate further that CD103−XCR1+ cells are heterogeneous and comprised partially of a trafficking population, the DC compartment within CCR7o/o mice was examined. Similar proportions of CD8+XCR1+ DCs were present within skin-draining lymph nodes of control C57BL/6 (B6) wild-type mice and CCR7o/o mice. This population has similar levels of MHC II expression in both strains, indicating that this lymph node resident population remains unaltered in the absence of CCR7 expression (). In contrast, the skin-draining lymph nodes of CCR7o/o mice completely lack the dermal-derived CD103+XCR1+MHC IIhi DCs as compared to an abundant population expressing high levels of MHC II within B6 control mice. In addition, CD103−XCR1+MHC IIhi DCs are also absent within CCR7o/o mice whereas the population of CD103−XCR1+MHC IIlo DCs remains unaffected. This provides further evidence that the CD103−XCR1+ DCs are comprised of both a lymphoid resident population and a peripheral migratory population in the skin-draining lymph nodes.
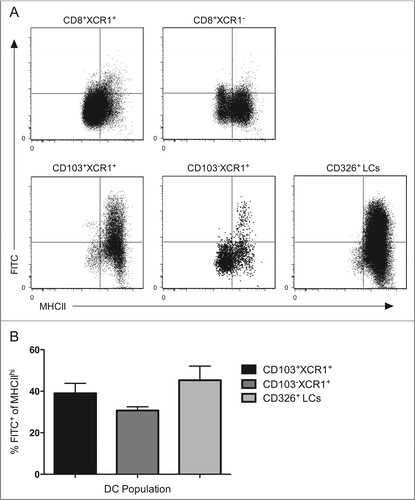
To validate further that a CD103−XCR1+ skin-emigrant DC transports antigen from the periphery to the draining lymph nodes, we painted the skin of B6 mice with FITC dissolved in the strong irritant dibutyl phthalate, known to rapidly promote DC migration.Citation34 We observed relatively few CD8+XCR1+ and CD8+XCR1− cells co-staining for FITC 4 d post skin painting, consistent with the notion that both populations are lymphoid resident within the skin-draining lymph nodes. As expected, a large proportion of the dermal-derived CD103+XCR1+ and epidermal-derived CD326+ Langerhans cells (LCs) were FITC+, consistent with published findings that many of these cellular populations efficiently migrate within this timeframe ().Citation35 Again, MHC class II expression was low/intermediate on FITC− residents and high on FITC+ trafficking DCs. Analysis of the CD103−XCR1+ DCs showed the existence of a FITC+ population restricted to only those cells expressing high levels of MHC class II. Similar proportions of FITC+ cells are observed within all MHC IIhi trafficking populations (), indicating the migratory capacity of peripheral CD103−XCR1+ is similar to well-defined trafficking DC populations following application of an irritant to the skin. Collectively, these results confirm that CD103−XCR1+ DCs are heterogeneous, comprised of a skin-resident migratory population and a blood-derived lymphoid resident population.
Figure 5. CD103−XCR1+MHC IIhi DCs migrate from the skin to the skin-draining lymph nodes with similar kinetics as CD103+XCR1+ DCs and CD326+ LCs. (A) Wild type mice were shaved, depilated and painted with a 0.5% FITC solution prepared in an inflammatory stimulating mixture of acetone and dibutyl phthalate (1:1, vol:vol) on the flank skin. After 96 h, single cell suspensions from pooled draining lymph nodes were enriched for DCs and CD8+XCR1+, CD8+XCR1−, CD103+XCR1+, CD103−XCR1+ DCs and CD326+ LCs were examined for FITC and MHC II expression by flow cytometry. (B) The percentage MHC IIhi migratory DCs positive for FITC in the draining lymph nodes 96 h after FITC painting. Data are pooled from 8 to 9 mice from three independent experiments presented as mean ± SEM. Representative plots from three independent experiments are shown.
Differential expression of CD207 distinguishes lymph node resident from migratory CD103−XCR1+ DCs
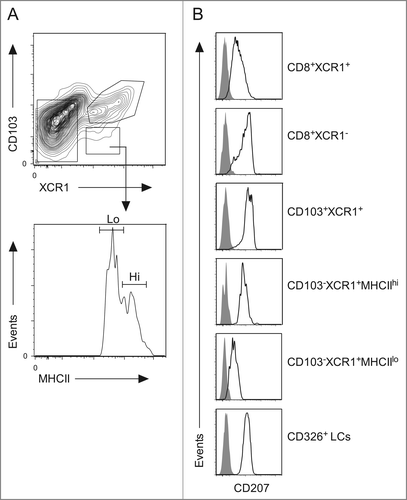
The heterogeneous CD103−XCR1+ DC population was examined for surface expression of a number of markers characteristic of the DC lineage in order to identify a potential phenotypic marker differentially expressed. As demonstrated previously, resident and migratory populations can be discriminated based on their expression of MHC class II (). Our results show that the skin-derived CD103−XCR1+MHC IIhi DCs express abundant levels of the C-type lectin CD207 similar to the levels observed on previously reported CD103+XCR1+ DCs and CD326+ LCs;Citation2 whereas expression of CD207 was considerably less abundant within the lymph node resident CD103−XCR1+MHC IIlo DCs (). Thus, a CD103−XCR1+CD207+ DC population is present within skin and migrates to the lymph nodes where a distinct resident CD103−XCR1+CD207lo DC population co-exists.
Figure 6. The resident CD103−XCR1+MHC IIlo and migratory CD103−XCR1+MHC IIhi DCs differ in their expression of CD207. (A) DC populations from the skin-draining lymph nodes were identified as per . The CD103−XCR1+ DCs were divided into MHC IIlo and MHC IIhi populations. (B) Surface expression of CD207 on CD8+XCR1+, CD8+XCR1−, CD103+XCR1+, CD103− XCR1+MHC IIlo, CD103− XCR1+MHC IIhi DCs and CD326+ LCs. Shaded histograms represent fluorescence minus one control staining. Representative plots from three independent experiments are shown.
Skin-derived CD103−XCR1+ DCs and CD103+XCR1+ DCs are the dominant migratory populations capable of cross-presenting cutaneous melanoma antigens
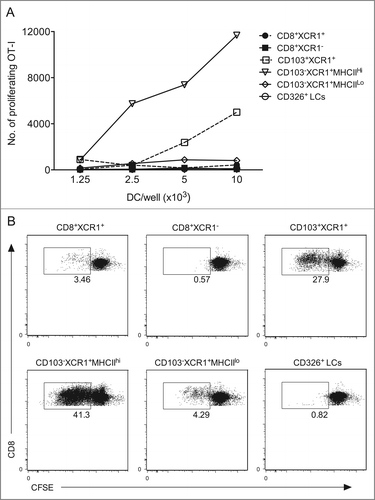
Many studies, including our own, have used ex vivo isolation of DCs to demonstrate which DCs are involved in cross-presenting antigens. DCs were isolated from tumor-draining lymph node suspensions as described previously.Citation31 Enriched DCs were stained with the pan-DC marker CD11c as well as MHC II, CD8, CD103, XCR1 and CD326 (). Next, we sorted CD11c+MHC II+ DCs by flow cytometry on the basis of differences in expression of these markers to delineate various populations. In this sorting scheme, lymphoid resident CD8+ DCs are isolated first with two populations collected based on their differential expression of XCR1 (CD8+XCR1+ and CD8+XCR1−), then from the CD8− fraction we took the CD103+XCR1+ dermal DCs followed by collection of the migratory CD103−XCR1+MHC IIhi DCs and resident CD103−XCR1+MHC IIlo DCs before isolating the LCs from the remaining DCs on the basis of CD326 expression. Following sorting, we examined these populations for their presentation of melanoma-derived OVA by co-culturing each purified DC population with CFSE-labeled OT.I cells (). The results show that CD103+XCR1+ DCs and the migratory CD103−XCR1+MHC IIhi DCs were highly efficient at cross-presenting cutaneous melanoma antigen. We consistently observe that the CD103−XCR1+MHC IIhi DCs drive the most abundant proliferation within the tumor-specific T cells, suggestive that on a per cell basis they are the most efficient DC at cross-presenting in this setting. Antigen cross-presentation was observed by all XCR1+ DC populations, with the lymph node resident CD8+XCR1+ and CD103−XCR1+MHC IIlo DCs driving modest T cell proliferation at higher DC:T cell ratios. This is in contrast to the CD8+XCR1− DC population, where no proliferation of melanoma-specific T cells is observed (), even in co-cultures containing as many as 4 × 104 of these DCs (data not shown).
Figure 7. CD103+XCR1+ and CD103−XCR1+ migratory DCs cross-present cutaneous melanoma antigen to CD8+ T cells. (A) Proliferation of 5 × 104 CFSE-labeled OVA-specific CD8+ T cells (OT.I) after 60 h of culture together with serial dilutions of DC populations isolated from the tumor draining lymph nodes of mice bearing B16Kb−-OVA melanoma. (B) Representative dot plots of CFSE dilution on CD8+ OT.I cells co-cultured with 104 of each DC population. Numbers indicate the percentage of divided cells. Data are representative of two independent experiments, which showed a similar trend.
Discussion
This study describes a novel cutaneous melanoma transplant model that was used to determine the DCs responsible for cross-presenting peripheral skin-derived tumor antigen. This new mouse model more closely reflects human melanoma than the standard subcutaneous model routinely employed in most laboratories worldwide. This is due to melanoma establishment and progression within the dermal and epidermal layers, a process not observed in the standard subcutaneous model. This model better allows for an understanding of how the rich and complex skin DC network interacts with melanoma at this site.
Using this model, we identified that the skin contains at least two populations of migratory CD103−XCR1+ and CD103+XCR1+ DCs that excel at cross-presenting solid peripheral tumor antigen to CD8+ T cells. To a much lesser extent, two lymphoid resident DCs, defined as CD8+XCR1+ and CD8−XCR1+, cross-presented tumor antigen. It is important to note that not all of these cross-presenting DCs display CD8 or CD103 on the cell surface, even though current nomenclature commonly links expression of these molecules to the unique capacity of cross-presenting antigens. An alternative means of unifying DCs with common attributes is via their transcriptome, where the differentiation of distinct DC subtypes is specifically regulated by particular sets of transcription factors.Citation36 This is demonstrated in mice lacking key transcription factors responsible for the development of cross-presenting DCs, such as ID2o/o, IRF8o/o, and Batf3o/o animals, which exhibit a severe deficit in the ability to elicit immune responses dependent on cross-presentation.Citation14,24
Recently, it has been suggested that a more appropriate nomenclature to classify ID2-IRF8-Batf3 dependent DCs as cDC1, whereas DCs that rely on the transcription factor IRF4 are classified as cDC2, with expression of these transcription factors inversely correlated.Citation37 We identified that CD8+XCR1− DCs express high levels of IRF4 mRNA, consistent with a cDC2 phenotype. This argument is supported with data showing that this population has abundant CD172a expression and is unable to cross-present melanoma antigen. Thus, CD8 expression is not a reliable marker for identifying a uniform cross-presenting DC subset. Several studies have identified a similar DC population in the spleenCitation22 and mesenteric lymph nodes,Citation30 with a functional role for this particular subset remaining to be documented.
The elevated expression of ID2, IRF8, and Batf3 mRNA observed in the XCR1+ DCs analyzed in this report is consistent with a cDC1 phenotype.Citation37 This is supported by the ability of all isolated XCR1+ DCs to cross-present tumor antigen to CD8+ T cells. Among the XCR1+ DCs, only the migratory DCs express high levels of CD207. These XCR1+CD207+ DCs can be further subdivided based on differential expression of CD103, with both migratory CD103+XCR1+ and CD103−XCR1+ DC populations exceling at cross-presenting antigen from cutaneous melanoma lesions that are present within the dermis. While both these DCs are capable of cross-presenting cutaneous melanoma antigen, others have demonstrated that a skin-derived CD103−CD207+ DC population, analogous to the CD103−XCR1+CD207+ population described in this study, fail to cross-present epidermal antigen from keratinocytes.Citation2 The inability of CD103−XCR1+ DCs to cross-present epidermal-derived antigen may be explained by inadequate access to antigen at that particular site. It is possible that CD103−XCR1+ DCs reside deeper within the dermis mitigating their ability to access epidermal-restricted antigens. In contrast, the CD103+XCR1+ DCs are known to congregate close to the epidermal-dermal junction, especially around the hair follicles.Citation38 How this subset acquires epidermal antigen that is restricted to the other side of the basement membrane remains undetermined, but this process may occur around hair follicles, which constitute a common portal of entry for invading microbesCitation39 and topical DNA application.Citation40
It is unclear why so many different DCs with the ability to cross-present are observed in this study. It is possible that the XCR1+ DCs that do not express either CD8 or CD103 are precursors in a late stage of differentiation, retaining the potential to commit to either CD8+ lymphoid resident or CD103+ skin-resident DCs. A requirement to reside in close proximity to the epidermis may exist in order for such a precursor to upregulate CD103. For example, transforming growth factor-β 1 is expressed by the skin epithelium,Citation41 where it is known to drive the upregulation of CD103 in other immune cells.Citation42 Future experiments are required to address the potential of such precursors to actively differentiate into either CD8+ or CD103+ DCs.
Alternatively, the presence of a deeper level of functional specialization may exist within an already specialized group of cells, providing an increased capacity to coordinate more effective immune regulation. It is plausible that one faction consists of the CD8+XCR1+ and CD103+XCR1+ DCs, whereas the CD8−XCR1+ and CD103− XCR1+ DCs represent another. These two groups share the capacity to cross-present, but may be strategically placed within an appropriate niche to sample antigens by unique methods within both the periphery and secondary lymphoid compartment. This view is substantiated by reports of a unique DC subset that obtains antigens by merocytosis, defined as the uptake of small particles.Citation43 This subset is capable of storing acquired antigens within compartments with reduced lysosomal degradation, resulting in prolonged antigen retention and presentation.Citation44 This “merocytic” DC subset has been identified so far only in the spleen and the reported surface antigen phenotype is in line with the CD8−XCR1+ and CD103−XCR1+ DCs described in this study. An investigation of the anatomical location of the four XCR1+ DCs analyzed in this study may reveal that the spatial relationship within the skin or lymph node is distinct, enabling each subset to have unique interactions with neighboring cellular networks. Further characterization of these DCs is required to support this concept.
In summary, a clear indication of which cells present tumor antigen to lymph node T cells has not been defined previously. However, a role for CD8+ DCs and undefined migratory populations has been reported.Citation45 This study identifies four populations of XCR1+ DCs with unique surface antigen expression that possess the ability to cross-present cutaneous tumor antigen, raising the possibility that a complex division of labor exists among DCs responsible for immunosurveillance of the skin. This concept requires further examination before it can be exploited for optimal delivery of vaccine antigen into the skin to promote enhanced anti-tumor CD8+ T cell immunity.
Materials and Methods
Mice
C57BL/6 (B6) female mice that express the CD45.2 allele were purchased from the Animal Resource Center, Murdoch, Western Australia. CCR7o/o33 and OT.ICitation27 mice on a C57BL/6.SJL-PtprcaPep3b/BoyJ background (CD45.1) were bred and maintained at the Telethon Kids Institute. All animal experiments were performed in accordance with protocols approved by the Telethon Kids Institute Animal Ethics Committee (Ethics Application ID: 243) and conformed to the National Health and Medical Research Council Australia code of practice for the care and use of animals for scientific purposes.
Cell lines
The B16.F10 melanoma cells were purchased from the ATCC. The B16Kb− cell line was a kind gift from Esteban Celis, University of Southern Florida, USA. B16 lines were transduced with retroviruses constructed using Fugene® transfection reagent (Promega) to transfect 293T cells with MSCV vectors containing a full-length membrane-bound form of OVA and eGFP, pMD.OLD.GAG.POL and pCAG-eco. The supernatant containing ecotrophic retroviruses was collected after 72 h and filtered through a 0.45 μM filter before being incubated with target cells in the presence of 8 μg/mL polybrene (Sigma). Transduction was confirmed by eGFP expression and eGFP+ cells were sorted by flow cytometry to establish purified lines. B16 cells were passaged routinely at 70–80% confluency and cultured in RPMI media (Life Technologies) supplemented with 10% FCS, 2-mercaptoethanol (50 μM), L-glutamine (2 mM), penicillin (100 U/mL) and streptomycin (100 μg/mL) in a 5% CO2 incubator at 37°C.
Cutaneous melanoma engraftment
Mice were anaesthetized with a mixture of Ketamine (100 mg/kg) and Xylazil (10 mg/kg) (Troy Laboratories) administered intraperitoneally (10 μL/g body weight). The eyes of the mice were covered with a Refresh® Lacri-Lube® (Allergan) from the time of anesthesia to when they regained consciousness to prevent them from drying out. The flank of the mouse was shaved and depilated with Veet® (Reckitt Benckiser). A small (∼2mmCitation2) area of skin on the flank was abraded using a MultiPro Dremel with a grindstone attachment. 105 B16 melanoma cells were washed, resuspended in 10 μL of Matrigel™ (BD) and applied to the abraded area. To contain the cells at the abraded site, the matrigel was allowed to set before covering it with a piece of Op-site Flexigrid™ (Smith and Nephew). The torsos of the mice were wrapped, first with a soft hypoallergenic Micropore™ tape (3M Health Care) and then by a stronger porous polyethene Transpore™ tape (3M Health Care) to protect the site. Mice were allowed to recover from anesthesia on a heating pad and monitored daily until bandages were removed 6 d post grafting.
Flow cytometry
Monoclonal antibodies specific to mouse CD8α, CD11c, MHC II, CD103, CD326, CD207, CD24 and CD172a were purchased from BD or eBioscience. The monoclonal antibody specific to XCR1 was kindly provided by R.A. Kroczek. Multi-parameter analysis was performed on a LSRFortessa (BD) and cells were sorted using a FACSAriaIII (BD). All data were analyzed with FlowJo (Tree Star). Prior to acquisition cells were stained with propidium iodide (PI; Sigma) to exclude dead cells.
DC isolation and sorting
DCs were isolated from lymphoid organs as described previously.Citation31 Briefly, single cell suspensions were prepared from pooled skin- or tumor-draining lymph nodes and enriched for conventional DCs using antibody depletion and magnetic bead enrichment. These cells were stained with anti-CD11c, -MHC II, -CD8α, -XCR1, -CD103 and -CD326 antibodies. DC subsets were sorted from PI negative events. During sorting DCs were collected in buffer containing FCS (Serana) and EDTA (Sigma) and kept on ice at all times.
Preparation of T cells
OT.I T cells were purified as described previously.Citation46 Briefly, single cell suspensions were prepared from pooled lymph nodes and spleens from OT.I female mice and enriched using antibody depletion and magnetic bead enrichment. Purity of enriched cell suspensions usually yielded >80% CD8+Vα2+ cells.
Ex vivo culture assays
Flow cytometrically sorted DCs were washed and resuspended in RPMI media (Life Technologies) supplemented with 10% FCS, 2-mercaptoethanol (50 μM), L-glutamine (2 mM), penicillin (100 U/mL) and streptomycin (100 μg/mL) and cultured in a 5% CO2 incubator at 37°C for the duration of the experiment. For ex vivo presentation assays, serial dilutions of sorted DCs were co-cultured for 60 h with 5 × 104 OT.I CD8+ T cells labeled with 2.5 μM CFSE (Sigma). Proliferation of OT.I cells was measured by flow cytometry as CFSE dilution in the CD45.1+CD8+Vα2+ cell population.
Fluorescein Isothiocyanate (FITC) painting
Mice were anaesthetized, shaved and depilated using Veet® (Reckitt Benckiser). The back and flanks of mice were painted with 100 μL of 0.5% (w/v) FITC solution (Sigma) prepared in a 1:1 mixture of acetone and dibutyl phthalate. DCs were isolated from the skin-draining lymph nodes 96 h after painting. Migratory DC were defined as FITC+ CD11c+MHCII+ cells by flow cytometry.
Preparation of RNA
Isolated DC populations were resuspended in 1 mL of Trizol solution (Life Technologies) immediately after sorting and stored at −80°C. RNA was extracted using phenol chloroform extraction and the RNEasy Mini Kit (Qiagen) according to the manufacturers' instructions. Reverse transcription of mRNA was performed using the SuperscriptIII First Strand Synthesis System (Invitrogen), with 2 μg of RNA per reaction, according to manufactures instructions.
Quantitative polymerase chain reaction
Following cDNA synthesis, quantitative polymerase chain reaction (qPCR) assays were performed using SYBR® green chemistry according to the manufacturers' instructions (Applied Biosystems). Briefly, wells contained 10 μL (1x) SYBR® green master mix, 4 μL primer mix (forward and reverse) (200 μM final concentration), 2 μL of DNase/RNase free water and 4 μL of cDNA (from first strand synthesis reaction). Cycling conditions were as follows; hold for 20 s at 95°C, cycle 40× through 95°C for 1 s, 60°C for 20 s followed by a hold for 2 min at 25°C. Amplification and detection was performed using an ABI 7900HT Fast Real-time PCR system with SDS 2.3 software (Applied Biosystems). For analysis of transcripts associated with DC ontogeny, primer sequences were: IRF4: forward 5′-TGCAAGCTCTTTGACACACA-3′; reverse 5′-CAAAGCACAGA GTCACCTGG; IRF8: forward 5′-CAGGAGGTGGATGCTTCCATC-3′; reverse 5′- GCACAGCGTAACCTCGTCTTC-3′; ID2: forward 5′-ATGAAAGCCTTCAGTC CGGTG-3′; reverse 5′-AGCAGACTCATCGGGTCGT-3′; BATF3: forward 5′- GCGCC-CGGGAACCA-3′; reverse 5′-AACCCGGTTTTTCTCT-CTCCTT-3′. For quantification of relative gene expression, the comparative ΔΔCT method was employed using HPRT expression as the reference.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Consumer and Community Participation
Telethon Kids Institute and The University of Western Australia School of Population Health facilitate a joint Consumer and Community Participation Program to support researchers, consumers and community members to work together to make decisions about research priorities, policies and practice. The Participation program supported active involvement of consumers and community members in the project. Two consumers and the Participation program staff provided input into plain language summaries, funding applications and dissemination of results to the consumer support group Melanoma WA.
Acknowledgements
We thank WR Heath for helpful discussions and are indebted to the facilities of the Telethon Kids Institute, particularly those responsible for animal husbandry and flow cytometry.
References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392:245-52; PMID:9521319; http://dx.doi.org/10.1038/32588
- Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, Devilard E, Viret C, Azukizawa H, Kissenpfennig A et al. CD207+CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med 2010; 207:189-206; PMID:20038600; http://dx.doi.org/10.1084/jem.20091964
- Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007; 316:612-6; PMID:17463291; http://dx.doi.org/10.1126/science.1137971
- Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007; 315:107-11; PMID:17204652; http://dx.doi.org/10.1126/science.1136080
- Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol 2008; 9:558-66; PMID:18376402; http://dx.doi.org/10.1038/ni.1601
- Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, Carlyle JR, Reis e Sousa C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest 2008; 118:2098-110; PMID:18497879; http://dx.doi.org/10.1172/JCI34584
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol 2002; 2:151-61; PMID:11913066; http://dx.doi.org/10.1038/nri746
- Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 2007; 8:1207-16; PMID:17922016; http://dx.doi.org/10.1038/ni1518
- Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O'Keeffe M, Bahlo M, Papenfuss A et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 2007; 8:1217-26; PMID:17922015; http://dx.doi.org/10.1038/ni1522
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 1999; 5:919-23; PMID:10426316; http://dx.doi.org/10.1038/11360
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003; 424:324-8; PMID:12819664; http://dx.doi.org/10.1038/nature01783
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008; 135:37-48; PMID:18854153; http://dx.doi.org/10.1016/j.cell.2008.09.016
- Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 2010; 33:905-16; PMID:21145760; http://dx.doi.org/10.1016/j.immuni.2010.11.023
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS et al. Batf3 deficiency reveals a critical role for CD8a+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322:1097-100; PMID:19008445; http://dx.doi.org/10.1126/science.1164206
- Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Kc W, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 2012; 490:502-7; PMID:22992524; http://dx.doi.org/10.1038/nature11531
- Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev 2010; 234:18-31; PMID:20193009; http://dx.doi.org/10.1111/j.0105-2896.2009.00870.x
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 2009; 10:488-95; PMID:19349986; http://dx.doi.org/10.1038/ni.1724
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8a+ conventional dendritic cells. J Exp Med 2010; 207:823-36; PMID:20351058; http://dx.doi.org/10.1084/jem.20091627
- Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, Hutloff A, Mages HW, Ranke K, Schaefer M et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 2009; 31:823-33; PMID:19913446; http://dx.doi.org/10.1016/j.immuni.2009.08.027
- Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8a+ dendritic cells. J Exp Med 2010; 207:1283-92; PMID:20479118; http://dx.doi.org/10.1084/jem.20100223
- Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, Guilliams M, Azukizawa H, Bogen B, Malissen B et al. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8a+ type. J Immunol 2011; 187:4411-5; PMID:21948982; http://dx.doi.org/10.4049/jimmunol.1101717
- Bachem A, Hartung E, Guttler S, Mora A, Zhou X, Hegemann A, Plantinga M, Mazzini E, Stoitzner P, Gurka S et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front Immunol 2012; 3:214; PMID:22826713; http://dx.doi.org/10.3389/fimmu.2012.00214
- Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, Fukuda Y, Yano T, Nobuoka M, Hirashima T et al. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J Immunol 2013; 190:6071-82; http://dx.doi.org/10.4049/jimmunol.1202798
- Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med 2009; 206:3115-30; PMID:20008528; http://dx.doi.org/10.1084/jem.20091756
- Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol 2001; 20:20.1; PMID: 18432774; DOI: 10.1002/0471142735.im2001s39
- Miller AJ, Mihm MC Jr. Melanoma. New Engl J Med 2006; 355:51-65; PMID:16822996; http://dx.doi.org/10.1056/NEJMra052166
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76:17-27; PMID:8287475; http://dx.doi.org/10.1016/0092-8674(94)90169-4
- Cho HI, Lee YR, Celis E. Interferon g limits the effectiveness of melanoma peptide vaccines. Blood 2011; 117:135-44; PMID:20889921; http://dx.doi.org/10.1182/blood-2010-08-298117
- Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O'Keeffe M, Shao QX, Chen WF et al. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol 2005; 174:6592-7; PMID:15905497; http://dx.doi.org/10.4049/jimmunol.174.11.6592
- Becker M, Guttler S, Bachem A, Hartung E, Mora A, Jakel A, Hutloff A, Henn V, Mages HW, Gurka S et al. Ontogenic, Phenotypic, and Functional Characterization of XCR1+ Dendritic Cells Leads to a Consistent Classification of Intestinal Dendritic Cells Based on the Expression of XCR1 and SIRPa. Front Immunol 2014; 5:326; PMID:25120540; http://dx.doi.org/10.3389/fimmu.2014.00326
- Waithman J, Zanker D, Xiao K, Oveissi S, Wylie B, Ng R, Togel L, Chen W. Resident CD8+ and migratory CD103+ dendritic cells control CD8 T cell immunity during acute influenza infection. PloS One 2013; 8:e66136; PMID:23750278; http://dx.doi.org/10.1371/journal.pone.0066136
- Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, Yamamoto K, Suematsu T, Nakamura M, Yui K et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8a− dendritic cell development. Proc Natl Acad Sci U S A 2004; 101:8981-6; PMID:15184678; http://dx.doi.org/10.1073/pnas.0402139101
- Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999; 99:23-33; PMID:10520991; http://dx.doi.org/10.1016/S0092-8674(00)80059-8
- Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med 1987; 166:1654-67; PMID:3119761; http://dx.doi.org/10.1084/jem.166.6.1654
- Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006; 25:153-62; PMID:16860764; http://dx.doi.org/10.1016/j.immuni.2006.04.017
- Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol 2012; 12:101-13; PMID:22273772; http://dx.doi.org/10.1038/nri3149
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 2014; 14:571-8; PMID:25033907; http://dx.doi.org/10.1038/nri3712
- Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA. Identification of a novel population of Langerin+ dendritic cells. J Exp Med 2007; 204:3147-56; PMID:18086865; http://dx.doi.org/10.1084/jem.20071966
- Janda JM, Abbott SL, Brenden RA. Overview of the etiology of wound infections with particular emphasis on community-acquired illnesses. Eur J Clin Microbiol Infect Dis 1997; 16:189-201; PMID:9131321; http://dx.doi.org/10.1007/BF01709581
- Fan H, Lin Q, Morrissey GR, Khavari PA. Immunization via hair follicles by topical application of naked DNA to normal skin. Nat Biotechnol 1999; 17:870-2; PMID:10471927; http://dx.doi.org/10.1038/12856
- Kane CJ, Knapp AM, Mansbridge JN, Hanawalt PC. Transforming growth factor-b 1 localization in normal and psoriatic epidermal keratinocytes in situ. J Cell Physiol 1990; 144:144-50; PMID:1694857; http://dx.doi.org/10.1002/jcp.1041440119
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol 2013; 14:1294-301; PMID:24162776; http://dx.doi.org/10.1038/ni.2744
- Reboulet RA, Hennies CM, Garcia Z, Nierkens S, Janssen EM. Prolonged antigen storage endows merocytic dendritic cells with enhanced capacity to prime anti-tumor responses in tumor-bearing mice. J Immunol 2010; 185:3337-47; PMID:20720209; http://dx.doi.org/10.4049/jimmunol.1001619
- Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to b cell antigens in nonobese diabetic mouse diabetes. J Immunol 2010; 185:1999-2003; PMID:20644171; http://dx.doi.org/10.4049/jimmunol.1001398
- McDonnell AM, Prosser AC, van Bruggen I, Robinson BW, Currie AJ. CD8a+ DC are not the sole subset cross-presenting cell-associated tumor antigens from a solid tumor. Eur J Immunol 2010; 40:1617-27; PMID:20373290; http://dx.doi.org/10.1002/eji.200940153
- Waithman J, Allan RS, Kosaka H, Azukizawa H, Shortman K, Lutz MB, Heath WR, Carbone FR, Belz GT. Skin-derived dendritic cells can mediate deletional tolerance of class I-restricted self-reactive T cells. J Immunol 2007; 179:4535-41; PMID:17878350; http://dx.doi.org/10.4049/jimmunol.179.7.4535