
Abstract
We have reported that TLR5-mediated recognition of commensal microbiota modulates systemic tumor-promoting inflammation and malignant progression of tumors at distal locations. Approximately 7–10% of the general population harbors a deleterious single nucleotide polymorphism in TLR5, implicating a novel role for genetic variation during the initiation and progression of cancer.
Commensal microbes are critical for maintaining immune homeostasis at barrier surfaces and for orchestrating innate immune responses toward invading pathogens within the periphery. Two seminal studies have recently demonstrated that the commensal microbiota was also required for the generation of a protective antitumor immune response during immunogenic chemotherapy or immunotherapy for tumors occurring outside of the intestinal tract. These studies demonstrated that immune recognition of commensal microbiota was critical for the immune system to control tumor progression of extraintestinal tumors.Citation1,2 A question that remained was how commensal microbes modulate the systemic macroenvironment of untreated cancer-bearing hosts, and what is the role of polymorphic pattern recognition receptors in these interactions.
Approximately 23% of individuals within the general population harbor polymorphisms in toll-like receptor (TLR) genes,Citation3 the most frequent variant is a dominant negative single nucleotide polymorphism in TLR5 (TLR5R392X), occurring in ∼7.5% of individuals.Citation4 Unlike other TLRs that recognize bacterial-associated molecular patterns and damage-associated molecular patterns, the only known natural ligand for TLR5, flagellin, is exclusively of bacterial origin. We found that in the absence of TLR5 signaling, malignant progression of sarcomas and ovarian tumors was significantly delayed.Citation5 Characterization of the systemic differences between TLR5-deficient and wild-type tumor-bearing mice revealed that wild-type tumor bearing mice had significantly elevated serum levels of the tumor-promoting cytokine IL-6. Corresponding to the increased levels of IL-6, compared to TLR5-deficient mice with similarly sized tumors, the number of splenic or tumor-associated myeloid-derived suppressor cells was also significantly increased in wild-type mice. Effector CD8+ T cell responses were significantly impaired in the wild-type tumor-bearing mice, suggesting that TLR5-dependent IL-6-mediated inflammation was driving global immune suppression and accelerated malignant progression.
Previous reports have shown that TLR5-deficient mice have altered commensal microbiota, resulting in increased colitis and metabolic syndrome.Citation6 However, we and others have not observed colitis or metabolic syndrome in TLR5-deficient mice,Citation5,7 underscoring both the importance of the environment the mice are housed in and mechanistic differences in the role of the microbiome in tumor progression. Quantification of ribosomal 16S bacterial genomic sequences from tumors, draining lymph nodes, and ascites fluid did not reveal the presence of bacteria at higher levels than in tumor-free mice, indicating that differences in tumor progression were not due to significant bacterial translocation. Co-housing of TLR5-deficient mice with wild-type littermates for at least 4 weeks did not completely normalize the content of the microbiota or eliminate differences in tumor progression. Consistently, the elimination of commensal intestinal bacteria with a cocktail of antibiotics was sufficient to delay the progression of distal tumors in TLR5-competent hosts, abrogating the differences observed in TLR5-deficient mice. Correspondingly, we also observed significantly reduced levels of IL-6, MDSCs infiltrating into the tumor microenvironment, and increased antitumor immune responses.
Interestingly, we observed a significant reduction in tumor-associated γδ T cells in wild-type antibiotic treated mice. γδ T cells from the tumor-microenvironment of wild-type mice were able to suppress the proliferation of tumor-specific T cells through the production of galectin-1. Galectin-1 is a lectin associated with tumor transformation, angiogenesis, and escape from immune pressure by inducing apoptosis and unresponsiveness of effector cells within the tumor environment.Citation8 Through a series of adoptive transfer and bone-marrow reconstitutions, we found that galectin-1 producing tumor-associated γδ T cells were sufficient and necessary for driving malignant progression in our pre-clinical tumor models. Additionally, granulocytic myeloid-derived suppressor cells induced galectin-1 expression in γδ T cells through the production of adenosine. Systemic, immune-mediated, and tumor-mediated elimination of IL-6 through the delivery of neutralizing antibodies, bone-marrow reconstitution, and shRNA knock-down of IL-6 in tumor cell lines, respectively, abrogated the differences in TLR5-driven malignant progression, resulting in reduced accumulation of MDSCs and galectin-1 producing γδ T cells into the tumor microenvironment and enhanced CD8+ T cell antitumor IFNγ production.
In contrast to ovarian cancer and sarcoma, breast tumors progressed significantly faster in TLR5-deficient mice. Systemic levels of IL-6 in wild-type and TLR5-deficient mice bearing advanced breast tumors were significantly reduced, indicating these differences were independent of IL-6 production. More importantly, TLR5-deficient tumor-bearing mice had significantly elevated systemic levels of IL-17, and in the absence of IL-6 tumor-promoting inflammation, IL-6 non-responsive breast tumors seemed to be driven by IL-17 tumor-promoting inflammation.
IL-17 has a controversial role in tumor progression. In ovarian cancer progression, the infiltration of IL-17 cells is associated with increased survivalCitation9 whereas in breast cancer,Citation9 IL-17 is associated with a poor survival outcome.Citation10 Our data help to clarify the role of IL-17, and suggest that in the absence of IL-6, IL-17 is the dominant tumor-promoting cytokine whereas in the presence of IL-6 responsive tumors, such as ovarian carcinoma, IL-6 drives tumor progression (Fig. 1).
Similar to what we observed in our animal models, in both human ER+ breast and ovarian cancer, TLR5R392X carriers – in whom TLR5 signal is abrogated – had significantly elevated mRNA levels of IL-17, whereas we only observed a significant increase in IL-6 in ovarian tumor samples homozygous for the ancestral allele of TLR5. Analysis of luminal ER+ breast and ovarian cancer datasets from The Cancer Genome Atlas as well as our tumor banks suggested that variances in tumor-promoting inflammation and outcome in TLR5R392X patients recapitulate the differences identified in TLR5-deficient mice. Although more samples and independent cohorts are needed to support the clinical relevance of these observations, our results pinpoint a novel role for genetic variation in orchestrating tumor-promoting inflammation through distal interactions with the microbiota. Further research is warranted on how the microbiome influences spontaneous malignant progression and responses to clinical interventions in cancer patients.
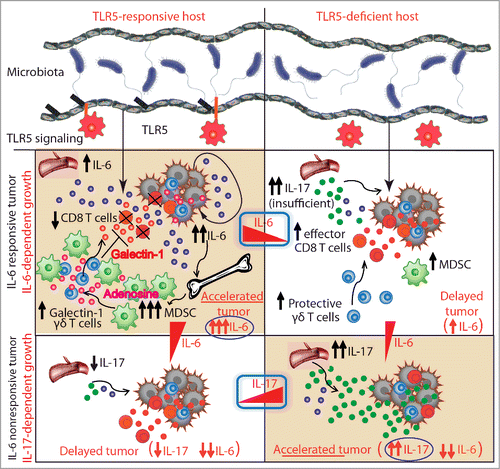
Figure 1. Commensal microbiota influence malignant progression of extraintestinal tumors by TLR5-driven modulation of the systemic tumor macroenvironment. In TLR5-responsive hosts bearing IL-6 responsive tumors, elevated systemic levels of IL-6 drive an accumulation of myeloid-derived suppressor cells (MDSCs) producing adenosine and the subsequent induction of galectin-1 expression in tumor-associated γδ T cells. This leads to significant suppression of antitumor immunity and accelerated malignant progression. TLR5-deficient tumor-bearing hosts have elevated systemic levels of IL-17, which is the dominant tumor-promoting cytokine in the absence of IL-6, when hosts are bearing IL-6 nonresponsive tumors. Differences in malignant progression are dependent on TLR5-mediated signaling and the commensal microbiota at barrier surfaces.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Viaud S, Saccheri F, Mignot Gg, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013; 342:971-6; PMID:24264990; http://dx.doi.org/10.1126/science.1240537
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013; 342:967-70; PMID:24264989; http://dx.doi.org/10.1126/science.1240527
- Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Ann Rev Immunol 2010; 29:447-91; PMID: 21219179; http://dx.doi.org/10.1146/annurev-immunol-030409-101335
- Hawn T, Verbon A, Lettinga K, Zhao L, Li S, Laws RJ, Skerrett SJ, Beutler B, Schroeder L, Nachman A et al. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires' disease. J Exp Med 2003; 198:1563-72; PMID:14623910; http://dx.doi.org/10.1084/jem.20031220
- Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, Perales-Puchalt A, Brencicova E, Escovar-Fadul X, Nguyen JM et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 2015; 27:27-40; PMID:25533336; http://dx.doi.org/10.1016/j.ccell.2014.11.009
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010; 328:228-31; PMID:20203013; http://dx.doi.org/10.1126/science.1179721
- Letran SE, Lee S-JJ, Atif SM, Flores-Langarica A, Uematsu S, Akira S, Cunningham AF, McSorley SJ. TLR5-deficient mice lack basal inflammatory and metabolic defects but exhibit impaired CD4 T cell responses to a flagellated pathogen. J Immunol 2011; 186:5406-12; PMID:21451112; http://dx.doi.org/10.4049/jimmunol.1003576
- Rabinovich GA. Galectin-1 as a potential cancer target. Br J Cancer 2005; 92:1188-92; PMID:15785741; http://dx.doi.org/10.1038/sj.bjc.6602493
- Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009; 114:1141-9; PMID:19470694; http://dx.doi.org/10.1182/blood-2009-03-208249
- Chen W-C, Lai Y-H, Chen H-Y, Guo H-R, Su I-J, Chen H. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathol 2013; 63:225-33; PMID:23738752; http://dx.doi.org/10.1111/his.12156