
Abstract
The combination of targeted therapy with BRAF and MEK inhibitors has become the standard of care in patients with BRAFV600E mutant melanoma, but responses are not durable. In addition, the impressive clinical benefits with anti-PD-1 and anti-PD-L1 antibodies (Ab) in patients with heavily pretreated metastatic melanoma and the synergistic effect of dabrafenib, trametinib and anti-PD-1 compared with single therapy alone groups support the idea that combining dabrafenib, trametinib and immunotherapy based on PD-1 blockade could be an interesting approach in the treatment of metastatic melanoma. With our mouse model of syngeneic BRAFV600E driven melanoma (SM1), we tested whether the addition of an immunostimulatory Ab targeting CD137 (4-1BB) and/or CD134 (OX40) would enhance the antitumor effect of dabrafenib, trametinib and anti-PD-1 or anti-PD-L1 therapy. In vitro studies showed that the combination group of dabrafenib, trametinib and anti-PD-1 increases CD8+ tumor infiltrating lymphocytes (TILs), as well as CD4+ T cells and tumor-associated macrophages (TAMs). An upregulation of PD-L1 was observed in the combination of dabrafenib, trametinib and anti-PD-1 therapy. Combination of dabrafenib, trametinib and anti-PD-1, with either anti-CD137 or anti-CD134, showed a superior antitumor effect, but the five-agent combination was not superior to the four-agent combinations. In conclusion, the combination of dabrafenib, trametinib, anti-PD1 or anti-PD-L1 therapy results in robust antitumor activity, which is further improved by adding the immune-stimulating Ab anti-CD137 or anti-CD134. Our findings support the testing of these combinations in patients with BRAFV600E mutant metastatic melanoma.
Introduction
The success of immune checkpoint inhibitors in advanced melanomaCitation1-5 has positioned cancer immunotherapy as one of the most exciting approaches to achieve long-term disease control of a small subset of patients with metastatic cancers. Combined BRAF and MEK inhibition prevents mitogen-activated protein kinase (MAPK) reactivation Citation6 as a mechanism of resistance to monotherapy with a BRAF inhibitor, increasing progression-free survival (PFS) and objective response rate (ORR) compared to BRAF inhibition alone,Citation7 and is now considered the standard of care treatment for patients with BRAFV600 mutant metastatic melanoma. In BRAF mutant melanoma, there is interest to combine MAPK targeted therapy and cancer immunotherapy with the goal of achieving higher response rates with prolonged duration. The rationale behind this combination is based on the potential sensitization of the immune system to target tumors by increasing antigen presentation,Citation8-10 antigen specific T-cell recognition,Citation8,11 reversing intratumoral immune suppression,Citation12 and homing of immune effector cells to the tumors,Citation9,13,14 thus improving effector functions.Citation15
PD-1 is an inhibitory T-cell receptor (TCR) with high selectivity for immune suppressive signals induced by PD-L1 expressed by cells within the tumor. An accepted mechanism of PD-L1 regulation is termed “adaptive immune resistance,” which occurs when tumor-resident cells expresses PD-L1 to protect themselves from the antitumor effector functions of cytotoxic T cells, mostly in response to interferons (IFNs).Citation16,17 This immune resistance mechanism has been characterized in tumor samples from patients treated with BRAF inhibitors, where an increase in the expression of T-cell exhaustion markers in post-dosing biopsies, including TIM3, PD-1 and PD-L1, has been described.Citation9 The increased PD-L1 expression could be suppressed with the addition of a MEK inhibitor,Citation18 providing a rational for combining target therapy and immunotherapy.
Preclinical evidence has recently shown that combined therapy of dabrafenib, trametinib and anti-PD-1 provided superior antitumor activity against the established BRAFV600E mutant murine melanoma SM1 tumor compared with anti-PD-1 plus either therapy alone, or isotype control with both dabrafenib and trametinib.Citation19 Additionally, there is growing evidence of synergistic combinations with immunostimulatory agents in cancer preclinical models.Citation20-26 Ideal candidates to enhance antitumor immunity include agents that potentiate CD8+ T-cell activation, such as the agonistic anti-CD137 (4-1BB) or anti-CD134 (OX40) Abs.
CD137 belongs to the tumor necrosis factor receptor (TNFR) superfamily and is a T cell co-stimulatory receptor.Citation27,28 Its expression has been observed to be responsible for a robust activation of CD8+ T-cells, eradication of established tumors, prevention of autoimmune diseases, and increased graft survival.Citation29-31 CD134, also a member of the TNFR superfamily, has been shown to be upregulated upon TCR engagement Citation32 and can promote co-stimulatory signals to T-cells leading to enhanced cell proliferation, survival, effector function and migration.Citation33,34 Treatment of transplantable mouse models with agonist Abs as monotherapies has shown clear signs of efficacy in the case of anti-CD137 Citation35 and anti-CD134Citation26 Abs. Beyond monotherapies, these and other immunostimulatory agents can be used in combinatorial approaches, in which synergy is often observed against transplantable tumors.Citation21,36 Moreover, synergy has also been observed on carcinogen-induced sarcomas using a combination that included anti-CD40 and anti-CD137 Abs.Citation37
Using a syngeneic mouse model of BRAFV600E mutant melanoma mouse,Citation15 we tested the hypothesis that addition of immune activating Ab to CD134 or CD137 to the combination of dabrafenib, trametinib and PD-1 blockade would increase antitumor activity.
Results
Enhanced in vivo antitumor activity with dabrafenib (D) + trametinib (T) combined with immunotherapy against SM1 tumors
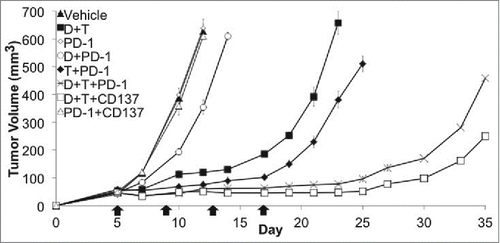
Our tumor model was the previously described SM1 BRAFV600E mutant murine melanoma,Citation15 syngeneic to fully immune-competent C57BL/6 mice, derived from a spontaneously arising melanoma in a BRAFV600E transgenic mouse. Our group has recently reported the superior antitumor activity of dabrafenib and trametinib in SM1 tumors established subcutaneously in C57BL/6 mice, when compared with tumors treated with dabrafenib or trametinib alone, or vehicle control.Citation19 We also observed a higher antitumor activity with the combination of dabrafenib, trametinib and anti-PD-1 when compared to dabrafenib and trametinib combination alone.Citation19 Here we explored combinations of dabrafenib and trametinib with the immune checkpoint inhibitor anti-PD-1, compared to the immune activating Ab to anti-CD137. In both triple combinations there were superior antitumor effects compared to dabrafenib and trametinib alone (). Consistent with a previous report,Citation38 SM1 is innately resistant to PD-1 Ab alone. However, we observed that combined therapy with dabrafenib or trametinib plus anti-PD-1 increased antitumor response compared to anti-PD-1 therapy alone, suggesting a synergistic effect of both dabrafenib and trametinib in combination with anti-PD-1. This experiment was performed in triplicate.
Figure 1. Enhanced in vivo antitumor activity with dabrafenib (D) + trametinib (T) combined with PD-1 checkpoint blockade against SM1 tumors. In vivo tumor growth curves. SM1 bearing C57BL/6 mice were treated when tumors were 3–5 mm with D 30 mg/kg and T 0.15 mg/kg combination via oral gavage daily, 4 doses of 200 μg of anti-PD-1 (PD-1), D + PD-1, T + PD-1, D + T + PD-1, D + T + anti-CD137 (CD137), PD-1 + CD137 or vehicle + isotype control Ab (4 mice in each group). This is representative graph of a three times repetition of this experiment.
Increased effector T-cell homing to the tumors associated with dabrafenib and trametinib in combination with anti-PD-1
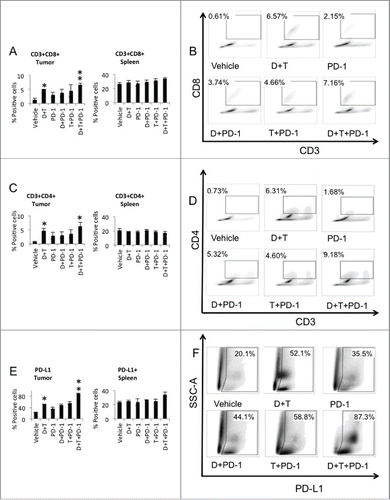
To analyze the mechanism of improved antitumor activity with the triple combination therapy, we harvested tumors and spleens on day 5 after starting treatment and stained for CD3 and CD8+. A significant increase in CD3+CD8+ cells could be observed in the tumors treated with dabrafenib and trametinib plus anti-PD-1 compared to anti-PD-1 monotherapy (p = 0.004) (). Of note, dabrafenib and trametinib without anti-PD-1 also significantly increased CD3+CD8+ cells in the tumors compared to vehicle, whereas a non-significant trend toward an increase in CD3+CD8+ cells was observed with anti-PD-1, dabrafenib and anti-PD-1 or trametinib and anti-PD-1. Effector cells harvested from the spleen did not show statistically significant difference in distribution between the treatment groups ().
Figure 2. Increased tumor infiltrating effector and helper T-cells with dabrafenib and trametinib in combination with anti-PD-1 in SM1 tumors. (A) Quantification of tumor infiltrating effector cells (TILs). TILs and splenocytes harvested at day 5 after starting treatment were counted and analyzed by flow cytometry for CD3/CD8+ staining (six mice in each group). (B) Representative flow data of percentage of CD3+CD8+ TILs in tumors is shown. (C) Quantification of tumor infiltrating helper T-cells. TILs and splenocytes harvested at day 5 after starting treatment were counted and analyzed by flow cytometry for CD3/CD4+ staining (six mice in each group). (D) Representative flow data of percentage of CD3+CD4+ TILs in tumors is shown. (E) Quantification of PD-L1 expression. TILs and splenocytes harvested at day 5 after starting treatment were counted and analyzed by flow cytometry for SSC/PD-L1 staining (six mice in each group). (F) Representative flow data of percentage for SSC/PD-L1 in tumors is shown.
Increased helper T-cells to the tumors associated with dabrafenib and trametinib in combination with anti-PD-1
Other important cell types potentially implicated in the mechanism of action of the triple combination included the analysis of CD3+CD4+ T-cells. Five days after starting treatment, tumors and spleens were harvested and stained for CD3 and CD4+. A statistically significant increase of CD3+CD4+ cells in the tumors treated with dabrafenib and trametinib or dabrafenib plus trametinib in combination with anti-PD-1 when compared to vehicle control treated tumors could be observed (). No change was observed in the distribution of CD3+CD4+ T-cells harvested from the spleen between the treatment groups ().
Upregulation of PD-L1 was seen with dabrafenib and trametinib with anti-PD-1
Upregulating the expression of ligands such as PD-L1 for inhibitory receptors on tumor-specific lymphocytes that consequently inhibit antitumor immune responses in the tumor microenvironment suggested an immune resistance mechanism induced by the activation of effector T-cells. Measurement of PD-L1 expression in the tumor cells mostly includes PD-L1 on the surface of the tumor cells but also on cells of the immune system. PD-L1 was significantly increased in the dabrafenib and trametinib treated tumors and when adding anti-PD-1 to both agents, compared to vehicle control (). Flow cytometry analysis of PD-L1 expression of SM1 spleens after 5 d of treatments did not show significant changes (). Analysis of the of PD-L1 positive macrophages in the tumors demonstrated a decrease in this subset of cells in all treatment conditions compared to vehicle except for the PD-1 treated group (Fig. S1F).
Increased MDSC number without any change in the ratio of PMN/MO-MDSC in combined treatment of dabrafenib and trametinib with anti-PD-1
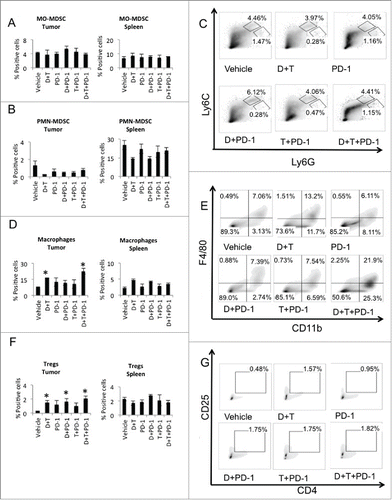
To evaluate the effect of dabrafenib, trametinib and anti-PD-1 combination on other cellular components of the tumor microenvironment, we harvested tumors and spleens 5 d after treatment started, and studied the cell populations by multiplex FACS. We looked at the myeloid-derived suppressor cells (MDSC) and its two major subsets: cells with granulocytic phenotype that express Ly6G marker (PMN-MDSC, Ly6ClowLy6GhighCD11b+) and cells with monocytic phenotype expressing Ly6C marker (MO-MDSC, Ly6ChighLy6GlowCD11b+). Dabrafenib plus trametinib significantly increased MDSCs in the tumors and spleens when compared to vehicle control group (p = 0.02 and p = 0.01 respectively) (Fig. S1B) and in combination with anti-PD-1, also in both the tumors and spleens (p = 0.01 and p = 0.03 respectively). Dabrafenib plus anti-PD-1, or trametinib plus anti-PD-1, did not change MDSCs in the tumors or spleens when compared to anti-PD-1. PMN-MDSC and MO-MDSC, the two major MDSCs subsets, were characterized, as percentage of CD11b+ cell (Fig. S1B). As shown in , there was no significant shift of the MO-MDSCs subset in the tumors among the different treatment conditions, while a non-significant decreased PMN-MDSCs () associated with both dabrafenib plus trametinib and combination treatments with anti-PD-1 could be observed. There was no significant change observed in the spleen among the different treatment groups ().
Figure 3. Dabrafenib and trametinib changed the cellular components of the tumor microenvironment. On day 5 post-treatment, tumors and spleens were isolated and stained with fluorescent labeled antibodies, analyzed by FACS. (A) MO-MDSC (CD11b+Ly6CHi Ly6GLo) presented as percentage of CD11b+ cells in tumors and spleens. (B) PMN-MDSC (CD11b+Ly6CLowLy6GHi) presented as percentage of CD11b+ cells in tumors and spleens. (C) Representative flow data of percentage of CD11b+Ly6CHi Ly6GLo and CD11b+Ly6CLowLy6GHi in tumors is shown. (D) Analysis of macrophages (F4/80+CD11b+). *p = 0.03 Vehicle vs. D + T, p = 0.03 Vehicle vs. D + T + PD-1, both in tumors. (E) Representative flow data of percentage of F4/80+CD11b+ cells in tumors is shown. (F) Analysis of T regulatory cells (Tregs, CD4+CD25+FOXp3+). *p = 0.02 Vehicle vs. D + T, p = 0.03 Vehicle vs. D + PD-1, p = 0.03 Vehicle vs. D + T + PD-1. (G) Representative flow data of percentage of CD4+CD25+ cells in tumors is shown.
Increased TAMs and Tregs in combined treatment of dabrafenib and trametinib with anti-PD-1
We then looked at mature TAMs (F4/80+CD11b+). Both dabrafenib + trametinib with or without anti-PD-1 significantly increased TAMs in the tumors when compared to vehicle control (p = 0.03 and p = 0.03 respectively) (). No significant change was seen with either dabrafenib + anti-PD-1 or trametinib + anti-PD-1. There was no significant change in macrophages in the spleen among the different treatment groups. Analysis of another immune suppressive cell population, the T regulatory cells (Fig. S1D, Tregs, CD4+CD25+FOXp3+) showed significantly increased percentage in the tumors with dabrafenib + trametinib treatment (p = 0.05), dabrafenib plus anti-PD-1 (p = 0.03) and dabrafenib + trametinib + anti-PD-1 (p = 0.03), but no other significant change observed (). These results indicated that dabrafenib and trametinib alone and in combination with anti-PD1 increase macrophage infiltration and Tregs in the tumor microenvironment. Moreover, there is a trend for both dabrafenib and trametinib +/− anti-PD-1 to decrease PMN-MDSCs, although non-significant.
Combined therapy with dabrafenib, trametinib, anti-PD-L1 and anti-CD137 improves antitumor responses against SM1 tumors
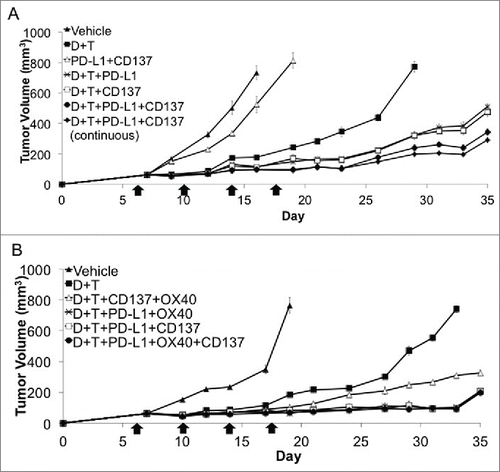
The in vitro studies suggested to us that there was margin for improving the antitumor activity if we added further immune activating Ab, with anti-CD137 being a first candidate based on prior data.Citation38 C57B/L6 mice with established subcutaneous SM1 tumors received dabrafenib, trametinib, anti-PD-L1 and anti-CD137 so that the effect of quadruple combination could be revealed. Anti-PD-L1 Ab was used in this experiment instead of anti-PD-1 Ab, since previous experiments have demonstrated similar antitumor effects in the SM1 tumor model (Fig. S1E). In replicate studies, the quadruple combination demonstrated superior antitumor response compared with either of the double or triple combination groups. In order to see if the antitumor effect mediated by quadruple combination could further be improved, continuous treatment of anti-PD-L1, instead of just four doses, was given. However, there was no improvement in antitumor activity in the quadruple combination group with continuous anti-PD-L1 treatment (). These studies suggest that antitumor effect medicated by triple combination of dabrafenib, trametinib, anti-PD-L1 could further be improved by anti-CD137.
Figure 4. Addition of immune activating antibodies to CD137 or CD134 to the combination of dabrafenib, trametinib and anti-PD-1. (A) Effects of anti-PD-L1 and anti-CD137 in combination with dabrafenib and trametinib. Tumor growth curves of established SM1 tumors in C57BL/6 mice that received dabrafenib, trametinib, anti-PD-L1 and anti-CD137 antibody. Treatment of anti-PD-L1, anti-CD137 or isotype antibody control was started at the same time with dabrafenib and trametinib when the tumor diameter reached 5 mm (4 mice in each group). This is representative graph of a three times repetition of this experiment. (B) Effects of quintuple combination of dabrafenib, trametinib, anti-PD-L1, anti-CD137 and anti-CD134. Tumor growth curves of established SM1 tumors in C57BL/6 mice that received dabrafenib, trametinib, anti-PD-L1, anti-CD137 and anti-CD134. Treatment of anti-PD-L1, anti-CD137, anti-CD134 or isotype antibody control was started at the same time with dabrafenib and trametinib when the tumor diameter reached 5 mm (4 mice in each group). This is representative graph of a three times repetition of this experiment.
Effect of quintuple combination overlapped with the effects of quadruple combination
In order to test if the antitumor effect could be further improved if an additional immunomodulating Ab was added to the combination, anti-CD134 was combined with dabrafenib, trametinib, anti-PD-L1 and anti-CD137. Anti-CD134 is another immune activating approach based on positive induction of the OX40 co-stimulatory receptor. However, there was no improvement in the antitumor activity in the quintuple combination group, compared with the quadruple combination with anti-CD134 or anti-CD137 ().
Discussion
There is a growing body of evidence to support combinatorial approaches that merge the significant response rate of BRAF inhibitor-based targeted therapy with long-term durable responses of immunotherapy in patients with advanced melanoma.Citation39 These combinations should be explored in preclinical models first to test for potential synergy between therapeutic strategies. It was previously shown that combined therapy of dabrafenib, trametinib and anti-PD-1 provided superior activity against established BRAFV600E mutant murine melanoma SM1 tumors compared with the combination of dabrafenib and trametinib.Citation19 PLX4720 is a widely used preclinical compound analogous to vemurafenib (PLX4032) that was used in combination with trametinib. By using an immune competent mouse model of BRAFV600E mutant melanoma, we demonstrated that the addition of either the agonist Ab targeting CD137 or the checkpoint inhibitor anti-PD1/L1 significantly improved the antitumor effect of targeted therapy. The quadruple therapy with dabrafenib, trametinib, anti-PD-L1 and anti-CD137 (or anti-CD134) was even more active than dabrafenib, trametinib and either anti-CD137 or anti-PD-L1.
The combination of anti-CD137 given intratumorally and anti-PD-1 potentiated cancer therapeutic immunity in the CT26 mice tumor model in a previously described report,Citation40 and this effect was explained by the co-expression of PD-1 and CD137 in the TILs. The fact that PD-L1 expression is associated with tumor resistance to CD137 co-stimulatory therapy Citation41 supports the rational for combining both agents. A previous report Citation38 assessed different monoclonal Ab-based immunotherapies against SM1 alone and in combination with PLX4720 and illustrated the significant antitumor activity of anti-CD137 alone and its enhanced antitumor activity with prior PLX4720 treatment. No antitumor activity was observed for anti-PD-1 Ab, either alone or enhanced by prior PLX4720 therapy. The different scheduling in generating combination effects with anti-PD-1 therapy might have been partially responsible for the different outcome when combining BRAF inhibition and PD-1 blockade in the SM-1 tumor model.
Other combinations of targeted therapy and immunotherapy have been explored in preclinical models, such as PLX4720 and anti-CTLA-4 Ab treatment, with no further increase in the antitumor effect of PLX4720 with CTLA-4 blockade.Citation42 To our knowledge, no previous reports have described the activity of dabrafenib and trametinib combined with anti-PD-L1 and/or anti-CD137 Ab and there is no clinical data available regarding the combination of BRAF and MEK inhibitors with immunotherapies such as anti-CTLA4 or anti-PD-1/L1, although several phase I clinical trials are ongoing.
On step further in determining the best combination of targeted therapy and immunotherapy, we combined dabrafenib and trametinib with anti-PD-L1 plus anti-CD137, anti-CD137 plus anti-CD134, anti-PD-L1 plus anti-CD134 or all five agents together. Pre-clinical studies have shown that ligation of CD134 via agonist anti-CD134 Ab can drive robust T cell-mediated antitumor immunityCitation28,43 and treatment with an agonist anti-CD134 Ab in conjunction with IL-2 augmented tumor immunotherapy.Citation44 However, there are no reports that describe the combination of BRAF and MEK inhibitors with anti-CD134 Ab. We concluded that the most active therapeutic combinations were the ones that included anti-PD-L1 Ab and that the addition of an additional immune activating Ab to a regimen containing dabrafenib, trametinib, anti-PD-L1 and either anti-CD37 or anti-CD134 did not provide any additional benefit. Potential drawbacks for combining multiple agents include the potential for an increase in the development of immune related adverse events (irAE) or overlapping toxicities (i.e. liver) that might limit the number of patients who can fully benefit from combination cancer therapies. Utilizing a spontaneous mouse tumor model such as the SM1 tumor model more faithfully recapitulates human tumor development and may potentially allow for the development of any therapy-induced toxicity, modeling tumor immunity and irAE development.
We decided to focus on characterizing the cellular components of the tumor microenvironment for the combination of dabrafenib, trametinib and anti-PD-1 because of the more advanced clinical development of this drug combination. We evaluated the frequency of effector T-cells in vivo. Previous reports documented increased numbers of TILs in biopsies of patients with melanoma treated with BRAF inhibitors Citation9,13,14 and PD-1Citation45 therapy, with a better response in those patients who had a more clonal TCR repertoire.Citation45,46 The association established between CD8+, PD-1 and PD-L1-cell densities in baseline biopsies of patients treated with anti-PD-1 therapy and the evidence of a physical interaction between PD-1 and PD-L1 cells Citation45 suggested that the increase in the proliferating CD8+ T-cells in regressing tumors could be accompanied by an increase in PD-L1 expression. In our SM1 model, we observed an increase in both CD8+ T-cells and PD-L1 expression with the combination of dabrafenib, trametinib and anti-PD-1 compared to anti-PD-1 therapy alone, correlating PD-L1 expression on tumor-resident cells and T-cell activation with treatment outcome, which has been shown by others upon release of the PD-1 immune checkpointCitation45,47,48 or upon treatment with adoptive cell transfer (ACT) of gp100 TCR transgenic activated splenocytes obtained from pmel-1 mice with both dabrafenib and trametinib.Citation19 Anti-PD-/1L1 blockade together with a BRAF inhibitor led to a significant increase in the number and activity of TILs in a syngeneic BRAF mutant melanoma model in the PTEN−/− background,Citation49 which further demonstrated synergy between combined BRAF-targeted therapy and immune checkpoint blockade. Regarding the activity of TILs in the SM1 tumor model, published data did not show any difference in the interferon-γ (IFNγ) secretion for pmel-1 ACT alone or the combination with dabrafenib, trametinib or both drugs, despite the different outcomes.Citation19
Significant changes were observed in the percent of total CD4+ T-cells with dabrafenib and trametinib with or without the addition of anti-PD-1 in the tumors compared to vehicle control. CD4+ T-cell number and function was extensively studied in a BRAFV600E mutant murine melanoma model after PLX4720 treatment, where both an increase in tumor-infiltrating CD4+ population and an increased IFNγ production and CD40L expression by these cells could be observed.Citation50 The authors attributed a regulation of the tumor microenvironment to the effector (IFNγ) and helper (CD40L) functions of CD4+ T-cells and considered the role of CD4+ T-cells to be underappreciated. This differs from other reports where no significant change in the frequency of total CD4+ T-cells between vehicle- and PLX4720-treated tumors was observed.Citation38 No difference in CD4+ T-cell infiltrate was observed in tumor biopsies from patients with metastatic melanoma undergoing treatment with a BRAF inhibitor.Citation9
Among the total CD4+ T-cell population, an increased frequency of Tregs (CD4+CD25+FoxP3+) was observed in all dabrafenib-containing treatment combinations compared to vehicle control, similarly to the results reported in the SM1 tumor model with ACT in combination with dabrafenib.Citation19 Trametinib did not increase the percent of Tregs when combined with anti-PD1 by itself, but did not attenuate the effect by dabrafenib when combined with both dabrafenib and anti-PD-1. MDSCs are used by solid tumors to escape T-cell immunityCitation51 and have been shown to correlate negatively with prognosis and overall survival.Citation52 Different subtypes of tumor-associated MDSC with distinct phenotype, morphology and immunosuppressive mechanisms have been characterized.Citation53,54 We observed an increase in the percent of total MDSCs in the tumors and spleens of the dabrafenib plus trametinib +/− anti-PD-1 treated groups compared to vehicle control. No significant shift was observed in the PMN-MDSC or MO-MDSC subset associated with any of the combination treatments.
Theoretically, a potential limitation of combining BRAF targeted therapy and immunotherapy for melanoma might be related to an increase of immune suppressive cells, such as Tregs and MDSC in the tumor microenvironment, due to the paradoxical activation of cells with wild type BRAF. Nevertheless, treatment with dabrafenib, trametinib and anti-PD-1 showed an increased antitumor effect when compared to dabrafenib or trametinib plus anti-PD-1. The increased percent of Tregs and MDSC in the dabrafenib, trametinib and anti-PD-1 combination might be responsible for the later tumor relapse. Others have observed a reduction in the frequency of intratumor Tregs (CD4+ Foxp3+) following PLX4720 therapy.Citation38 In view of these findings, a logical rationale would be to study combinations that include inhibitors of Tregs in preclinical models. Other immune suppressive cells upregulated after treatment with dabrafenib and trametinib or the combination of dabrafenib, trametinib and anti-PD-1 included TAMs (F4/80+CD11b+).
Our study showed improved antitumor activity of dabrafenib and trametinib with the addition of either the agonist Ab targeting CD137 or the checkpoint inhibitor anti-PD1/L1 and demonstrated an increased synergic effect with targeted therapy based on dabrafenib plus trametinib plus anti-PD-L1 and either anti-CD137 or anti-CD134. An increase in effector and helper T cells coincident with an upregulation of PD-L1 with dabrafenib, trametinib and anti-PD-1 could be observed. This was accompanied by a change in the tumor microenvironment consistent of an increase in Tregs, MDSC and TAMs.
Materials and Methods
Mice, cell lines and reagents
C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME), were bred and kept under defined-flora pathogen-free conditions at the AALAC-approved animal facility of the Division of Experimental Radiation Oncology, UCLA, and used under the UCLA Animal Research Committee protocol #2004-159. The SM1 murine melanoma was generated from a spontaneously arising tumor in BRAFV600E mutant transgenic mice as previously described.Citation15 Dabrafenib and trametinib were obtained under a materials transfer agreement with GSK (Brentford, UK). Dabrafenib and trametinib were dissolved in dimethylsulfoxide (DMSO; Fisher Scientific, Hanover Park, IL) and used for in vitro studies. For in vivo studies, dabrafenib and trametinib were suspended in an aqueous mixture of 0.5% hydroxypropyl methylcellulose (HPMC) and 0.2% tween 80 (Sigma-Aldrich, St. Louis, MO). One hundred μL of the suspended drug was administered by daily oral gavage into mice at 30 mg/kg of dabrafenib or/and 0.6mg/kg of trametinib when tumors reached 5 mm in diameter. Tumors were followed by caliper measurements three times per week.
Antibody treatment in in vivo model
SM1 tumors were implanted into C57BL/6 mice. When tumor diameter reached 5 mm, 4 doses of 200 μg of each anti-PD-1, anti-PD-L1, anti-CD137, anti-OX40 or isotype control Ab (all purchased from BioXCell, West Lebanon, NH) was injected intraperitoneally (i.p.) every 4 d.
Flow cytometry analysis
SM1 tumors and spleens were harvested from mice. Tumors were further digested with collagenase (Sigma-Aldrich). Splenocytes and cells obtained from digested SM1 tumors, were stained with Ab to CD3 BV605 (clone 17A2), Ly6C FITC (Clone AL-21), PD-L1/CD274 PE (Clone MIH5) (Becton Dickinson Biosciences, San Jose, CA), CD8a BV421 (Clone 53-6.7) (Biolegend, San Diego, CA), Ly-6G (Gr1) PerCP 5.5 (clone RB6-8C5), CD11b APC (clone M1/70), F4/80 Pacific blue/eFluor450 (clone BM8), CD25 APC (PC61.5), CD4+ FITC (RM4-5) (eBioscience, San Diego, CA), and analyzed with LSR-II or FACSCalibur flow cytometers (Becton Dickinson Biosciences), followed by analysis using Flow-Jo software (FLOWJO, LLC, Ashland, OR). Intracellular staining of Foxp3 PE (FJK-16s) (eBioscience) was done according to manufacturer's recommendations. After applying a gating strategy for the selection of the target population and exclusion of dead cells in tumors and spleens (Fig. S1A), the different immune cell populations were analyzed. Cells were analyzed with a LSR-II or FACSCalibur flow cytometers (BD Biosciences), followed by Flow-Jo software (Tree-Star, Ashland, OR) analysis as previously described.Citation49
Statistical analysis
Data were analyzed with GraphPad Prism (version 5) software (GraphPad Software, La Jolla, CA). A Student's t-test was used to analyze experimental data.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Material.zip
Download Zip (246.3 KB)Funding
Funded by NIH grants P01 CA168585 and P50 CA086306, the Dr. Robert Vigen Memorial Fund, the Ressler Family Foundation, the Wesley Coyle Memorial Fund and the Garcia-Corsini Family Fund (to AR). BHM was supported by the Rio Hortega Scholarship from the Hospital 12 de Octubre, Madrid, Spain. SM was supported by UCLA Final Year Dissertation Fellowship (2014–2015). SH-L was supported by NIH grant T32 CA09297, ASCO YIA, and Tower Foundation Research Grant.
References
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/10.1056/NEJMoa1200694
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369:134-44; PMID:23724846; http://dx.doi.org/10.1056/NEJMoa1305133
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/10.1056/NEJMoa1003466
- Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517-26; PMID:21639810; http://dx.doi.org/10.1056/NEJMoa1104621
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012; 119:651-65; PMID:22053109; http://dx.doi.org/10.1182/blood-2011-04-325225
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367:1694-703; PMID:23020132; http://dx.doi.org/10.1056/NEJMoa1210093
- Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010; 70:5213-9; PMID:20551059; http://dx.doi.org/10.1158/0008-5472.CAN-10-0118
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013; 19:1225-31; PMID:23307859; http://dx.doi.org/10.1158/1078-0432.CCR-12-1630
- Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology 2013; 2:e22890; PMID:23483066; http://dx.doi.org/10.4161/onci.22890
- Donia M, Fagone P, Nicoletti F, Andersen RS, Hogdall E, Straten PT, Andersen MH, Svane IM. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012; 1:1476-83; PMID:23264894; http://dx.doi.org/10.4161/onci.21940
- Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res 2013; 19:393-403; PMID:23204132; http://dx.doi.org/10.1158/1078-0432.CCR-12-1626
- Long GV, Wilmott JS, Haydu LE, Tembe V, Sharma R, Rizos H, Thompson JF, Howle J, Scolyer RA, Kefford RF. Effects of BRAF inhibitors on human melanoma tissue before treatment, early during treatment, and on progression. Pigment Cell Melanoma Res 2013; 26:499-508; PMID:23557327; http://dx.doi.org/10.1111/pcmr.12098
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012; 18:1386-94; PMID:22156613; http://dx.doi.org/10.1158/1078-0432.CCR-11-2479
- Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012; 72:3928-37; PMID:22693252; http://dx.doi.org/10.1158/0008-5472.CAN-11-2837
- Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP, McDyer JF, Fortuno L, Schleimer RP. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am J Res Cell Mol Biol 2005; 33:280-9; PMID:15961727; http://dx.doi.org/10.1165/rcmb.2004-0129OC
- Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, Maeng PJ, Lim JS. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci 2005; 40:95-103; PMID:16085391; http://dx.doi.org/10.1016/j.jdermsci.2005.06.008
- Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res 2013; 19:598-609; PMID:23095323; http://dx.doi.org/10.1158/1078-0432.CCR-12-2731
- Hu-Lieskovan SSM, Homet Moreno B, Tsoi J, Robert L, Goedert L, Koya RC, Graeber T, Comin-Anduix B, Ribas A. Improved antitumor activity of immunotherapy combined with BRAF and MEK inhibitors in BRAFV600E mutant melanoma. Sci Transl Med 2015; 18: 18;7(279):279ra41; PMID:25787767; http://dx.doi.org/10.1126/scitranslmed.aaa4691
- Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA. Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin Cancer Res 2013; 19:997-1008; PMID:23460531; http://dx.doi.org/10.1158/1078-0432.CCR-12-2214
- Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer 2007; 7:95-106; PMID:17251916; http://dx.doi.org/10.1038/nrc2051
- Ju SA, Cheon SH, Park SM, Tam NQ, Kim YM, An WG, Kim BS. Eradication of established renal cell carcinoma by a combination of 5-fluorouracil and anti-4-1BB monoclonal antibody in mice. Int J Cancer 2008; 122:2784-90; PMID:18360825; http://dx.doi.org/10.1002/ijc.23457
- Kim YH, Choi BK, Kim KH, Kang SW, Kwon BS. Combination therapy with cisplatin and anti-4-1BB: synergistic anticancer effects and amelioration of cisplatin-induced nephrotoxicity. Cancer Res 2008; 68:7264-9; PMID:18794112; http://dx.doi.org/10.1158/0008-5472.CAN-08-1365
- Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res 2000; 60:5514-21; PMID:11034096
- Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, Duret H, Yagita H, Johnstone RW, Smyth MJ et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res 2012; 72:3163-74; PMID:22570253; http://dx.doi.org/10.1158/0008-5472.CAN-12-0210
- Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, Urba WJ, Alvord G, Bunce C, Shields J. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol 2000; 164:2160-9; http://dx.doi.org/10.4049/jimmunol.164.4.2160
- Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol 2003; 3:609-20; PMID:12974476; http://dx.doi.org/10.1038/nri1148
- Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 2005; 23:23-68; PMID:15771565; http://dx.doi.org/10.1146/annurev.immunol.23.021704.115839
- Foell J, Strahotin S, O'Neil SP, McCausland MM, Suwyn C, Haber M, Chander PN, Bapat AS, Yan XJ, Chiorazzi N et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Investig 2003; 111:1505-18; PMID:12750400; http://dx.doi.org/10.1172/JCI200317662
- Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, Choi BK, Vinay DS, Kwon BS. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med 2004; 10:1088-94; PMID:15448685; http://dx.doi.org/10.1038/nm1107
- Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, Fu YX. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med 2002; 8:1405-13; PMID:12426559; http://dx.doi.org/10.1038/nm1202-796
- Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J 1990; 9:1063-8; PMID:2157591
- Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol 2000; 165:3043-50; http://dx.doi.org/10.4049/jimmunol.165.6.3043
- Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol 1998; 161:6510-7
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med 1997; 3:682-5; PMID:9176498; http://dx.doi.org/10.1038/nm0697-682
- Takeda K, Kojima Y, Uno T, Hayakawa Y, Teng MW, Yoshizawa H, Yagita H, Gejyo F, Okumura K, Smyth MJ. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J Immunol 2010; 184:5493-501; http://dx.doi.org/10.4049/jimmunol.0903033
- Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, Gejyo F, Okumura K, Yagita H, Smyth MJ. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med 2006; 12:693-8; PMID:16680149; http://dx.doi.org/10.1038/nm1405
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Investig 2013; 123:1371-81; PMID:23454771; http://dx.doi.org/10.1172/JCI66236
- Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol 2014; 32:2248-54; PMID:24958825; http://dx.doi.org/10.1200/JCO.2013.52.1377
- Palazon A, Martinez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Penuelas I, Hervas-Stubbs S, Rouzaut A et al. The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov 2012; 2:608-23; PMID:22719018; http://dx.doi.org/10.1158/2159-8290.CD-11-0314
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005; 65:1089-96; PMID:15705911
- Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012; 1:609-17; PMID:22934253; http://dx.doi.org/10.4161/onci.20226
- Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol 2010; 28:57-78; PMID:20307208; http://dx.doi.org/10.1146/annurev-immunol-030409-101243
- Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PLoS One 2012; 7:e34467; PMID:22496812; http://dx.doi.org/10.1371/journal.pone.0034467
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/10.1038/nature13954
- Cooper ZA, Frederick DT, Ahmed Z, Wargo JA. Combining checkpoint inhibitors and BRAF-targeted agents against metastatic melanoma. Oncoimmunology 2013; 2:e24320; PMID:23762807; http://dx.doi.org/10.4161/onci.24320
- Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 2014; 4:674-87; PMID:24589924; http://dx.doi.org/10.1158/2159-8290.CD-13-0458
- Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014; 20:5064-74; PMID:24714771; http://dx.doi.org/10.1158/1078-0432.CCR-13-3271
- Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res 2014; 2:643-54; PMID:24903021; http://dx.doi.org/10.1158/2326-6066.CIR-13-0215
- Ho PC, Kaech SM. BRAF-targeted therapy alters the functions of intratumoral CD4 T cells to inhibit melanoma progression. Oncoimmunology 2014; 3:e29126; PMID:25083331; http://dx.doi.org/10.4161/onci.29126
- Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 2008; 222:162-79; PMID:18364001; http://dx.doi.org/10.1111/j.1600-065X.2008.00602.x
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008; 111:4233-44; PMID:18272812; http://dx.doi.org/10.1182/blood-2007-07-099226
- Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 2010; 22:238-44; PMID:20171075; http://dx.doi.org/10.1016/j.coi.2010.01.021