
Abstract
Triple-negative breast cancer (TNBC) is a heterogeneous group of aggressive breast cancers for which no targeted treatment is available. Robust tools for TNBC classification are required, to improve the prediction of prognosis and to develop novel therapeutic interventions. We analyzed 3,247 primary human breast cancer samples from 21 publicly available datasets, using a five-step method: (1) selection of TNBC samples by bimodal filtering on ER-HER2 and PR, (2) normalization of the selected TNBC samples, (3) selection of the most variant genes, (4) identification of gene clusters and biological gene selection within gene clusters on the basis of String© database connections and gene-expression correlations, (5) summarization of each gene cluster in a metagene. We then assessed the ability of these metagenes to predict prognosis, on an external public dataset (METABRIC). Our analysis of gene expression (GE) in 557 TNBCs from 21 public datasets identified a six-metagene signature (167 genes) in which the metagenes were enriched in different gene ontologies. The gene clusters were named as follows: Immunity1, Immunity2, Proliferation/DNA damage, AR-like, Matrix/Invasion1 and Matrix2 clusters respectively. This signature was particularly robust for the identification of TNBC subtypes across many datasets (n = 1,125 samples), despite technology differences (Affymetrix© A, Plus2 and Illumina©). Weak Immunity two metagene expression was associated with a poor prognosis (disease-specific survival; HR = 2.68 [1.59–4.52], p = 0.0002). The six-metagene signature (167 genes) was validated over 1,125 TNBC samples. The Immunity two metagene had strong prognostic value. These findings open up interesting possibilities for the development of new therapeutic interventions.
Abbreviations
| AR | = | Androgen receptor |
| BC | = | Breast cancer |
| BC_CL | = | Breast cancer cell lines |
| BCSS | = | Breast cancer-specific survival |
| CCLE | = | Cancer Cell Line Encyclopedia |
| CDF | = | Consensus distribution function |
| CGP | = | Cancer Genome Project |
| EMT | = | Epithelial-mesenchymal transition |
| ER | = | Estrogen receptor |
| GE | = | Gene expression |
| HER2 | = | Human epidermal growth factor receptor 2 |
| IHC | = | Immunohistochemistry |
| IM | = | Immunomodulatory |
| LAR | = | Luminal androgen receptor |
| M | = | Mesenchymal |
| MSL | = | Mesenchymal stem-like |
| no CT | = | No chemotherapy |
| NPI | = | Nottingham Prognostic Index |
| pCR | = | Pathological complete remission |
| PR | = | Progesterone receptor |
| RMA | = | Robust multichip average |
| TILs | = | Tumor-infiltrating lymphocytes |
| TNBC | = | Triple-negative breast cancer |
| TNBC_CL | = | Triple-negative breast cancer cell lines. |
Introduction
TNBC, defined by the absence of estrogen and progesterone receptor expression and a lack of HER2 overexpression/amplification, is an aggressive disease accounting for 15%–20% of breast cancers. It differs from other molecular subtypes Citation1–3 in displaying axillary lymph node involvement, local and regional recurrence, differences in the time lag to metastasis (distant metastatic events occurring within 5 y of diagnosis), high rates of brain, lung and distant nodal metastasis and in its response to neoadjuvant treatment.
TNBC constitutes a major clinical challenge because there has been no substantial improvement in treatment for this subgroup in the recent past. Even if adjuvant chemotherapy has significantly improved outcome, reducing the risk of death by approximately 30%,Citation4 but these cancers do not respond to endocrine or targeted therapy. TNBC is, thus, currently the breast cancer subgroup with the worst outcome.Citation5 Moreover, the shape of the survival curve for this subgroup differs from that for other BC subtypes: there is a sharp decrease in survival during the first 3–5 y after diagnosis, but distant relapses, occurring after this interval, are much less common.Citation5
TNBC is a highly heterogeneous group of tumors differing in terms of their histological features, GE profiles, clinical behavior, overall prognosis Citation6 and sensitivity to systemic treatment.Citation7-9
Robust classifiers are urgently required, to improve our understanding of the molecular basis of TNBC and to define novel therapeutic interventions. Lehmann et al. recently published a classification of six molecular subtypes of TNBCCitation10 and developed a website (http://cbc.mc.vanderbilt.edu/tnbc/) Citation11 for the classification of TNBC samples on the basis of their GE profiles. This classification has been shown to be relevant, as it identifies the main biological component and pathways of TNBC. However, the large number of genes defining this TNBC molecular classification (2,188 genes) constituted a potential source of instability.Citation12,13
We developed a two-step biological network-driven gene selection process: (1) identification of the most variant genes displaying highly-correlated patterns of expression, (2) direct connection of these genes within known biological networks. This method has been reported to be efficient for the construction of molecular signatures.Citation14,15 We defined a robust TNBC molecular subtype classification, providing considerable biological insight, with great potential for use in the development of therapeutic interventions. We also identified a stromal immune module GE profile strongly correlated with TNBC prognosis.
Results
TNBC gene expression profiles identify six main gene clusters
GE profiles were obtained from 21 publicly available datasets, containing data for 3,247 primary human breast cancer samples. These profiles were processed according to the flow chart in . The training set included samples hybridized on HGU-133A Affymetrix© arrays (12 datasets, n = 1,995), to eliminate cross-platform discrepancies and to ensure robust normalization. The validation set included samples hybridized on HGU-133Plus2 Affymetrix© arrays (9 datasets, n = 1,014). We filtered out 42 outlier samples from the training set and 17 from the validation set.
Figure 1. Methodology flow chart.
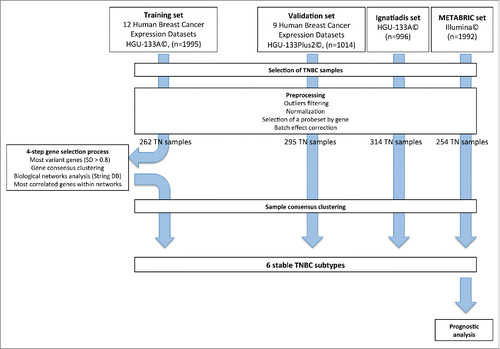
We also collected two large datasets, for the validation of our classification: the Ignatiadis set (n = 996) and the METABRIC set (n = 1,992). The processing of these two datasets has been described elsewhere.Citation16,17
Bimodal filtering on ER-PR and HER2 GE identified 262, 295, 314 and 254 TNBC samples in the training set, the validation set, the Ignatiadis set and the METABRIC set, respectively.
We developed a gene selection process based on biological networks, to decrease the intrinsic instability of molecular classification methods.
We identified the 830 most variant genes (SD > 0.8) in the training set (n = 262). A consensus clustering method and hierarchical clustering identified four main gene clusters. Further increases in cluster number yielded no significant increase in the consensus distribution function (CDF) area (Fig. S1 and Materials and Methods).
The various gene clusters were associated with different gene ontologies (Fig. S2). The clusters were thus named as follows (Fig. S3A): Immunity cluster (145 genes), Proliferation/DNA damage cluster (397 genes), AndrogenReceptor(AR)-like cluster (139 genes) and Matrix/Invasion cluster (149 genes).
The Immunity cluster was the most homogeneous, with strong correlations between the GE profiles of most of the genes within this cluster (Fig. S3B).
We used String© database software to analyze our gene selection, with the aim of decreasing the heterogeneity of each main gene cluster. We retained the genes from our initial selection that (1) had high String© database gene connection indexes (greater than 0.7, Fig. S4), (2) had similar patterns of expression to other genes within the same biological network (correlation coefficient of at least 0.5). We selected a final set of 167 genes [Immunity cluster (80), Proliferation/ DNA damage (15), AR-like(15), Matrix/Invasion (57)] (Fig. S5).
Following biological network-driven gene selection, it became clear that the original Immunity and Matrix/Invasion clusters were more accurately described by splitting them into two subclusters displaying minor differences [Immunity1 (33), Immunity2 (47), Matrix/Invasion1 (43), Matrix2 (14)] (Fig. S6A). This approach yielded an increase in the area under the CDF curve (Fig. S7).
For each of the six gene clusters identified in this way, we defined a metagene. The Immunity1 and Immunity2 metagenes displayed similar patterns of expression, with a Pearson correlation coefficient of 0.58; the Pearson correlation coefficient for the expression patterns of Matrix/Invasion1 and Matrix2 was 0.48. The Proliferation/DNA damage and Matrix metagenes displayed the strongest inverse correlation (coefficients of −0.43 and −0.60 for Matrix/Invasion1 and Matrix2, respectively) (Fig. S6B).
We validated this six-gene cluster classification, by applying hierarchical clustering based on the 167 genes selected to the validation set (n = 295). Clustering was highly consistent between the training and validation gene sets (concordance: 93–100%).
The six gene clusters identify six stable TNBC subgroups
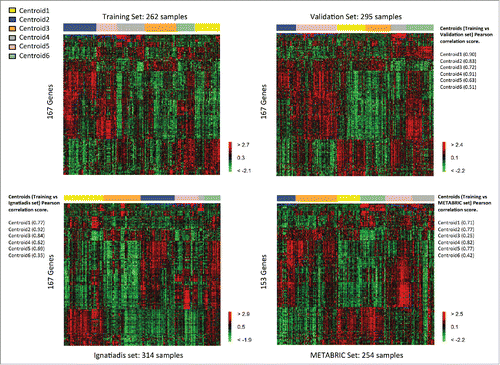
Hierarchical clustering was performed on the four TNBC datasets [training set (262), validation set (295), Ignatiadis (314) and METABRIC (254)]. For Affymetrix© arrays, we used the 167 selected genes. For the Illumina© platform, we used 153 common genes. We identified six reproducible subgroups of TNBC, for which GE patterns were similar in the training set and in the three validation sets (total of 1,125 samples). The corresponding heatmaps are shown in . The Pearson correlation coefficients for the relationships between each sample subgroup centroid in the three validation sets and the corresponding subgroup centroid in the training set are shown in .
Figure 2. Heatmaps of the selected genes in the TNBC training set (upper left) and the TNBC validation sets (upper right: validation, lower left: Ignatiadis, lower right: METABRIC).
We illustrated the dynamic links between genes within a biological network, as defined by the String© database, by showing GE levels for a “prototype sample” (Fig. S8).
We compared our sample classification with those reported by Lehmann et al. and Curtis et al. (Fig. S9). Our classification appears very different from that of Lehmann at first glance (χ2 test p value = 0.05), but the samples assigned to Centroids one and six (with high-levels of Matrix/Invasion 1 and Matrix 2 gene expression, respectively) tended to be classified as Mesenchymal (M) or Mesenchymal stem-like (MSL), the samples in Centroid 5 (strong expression of Immunity2 genes) tended to be classified as Immunomodulatory (IM), and the samples in Centroid 4 (strong expression of AR-like genes) tended to be classified as of the Luminal androgen receptor (LAR) subtype (Fig. S10A and Fig. S10B). Curtis et al. aimed at defining a new classification across all cancer subtypes, not specific to TNBC subtypes. In this classification, the TNBC samples were mostly classified as IntClust10 or IntClust4, with an even distribution.
Prognostic value of the Immunity2 metagene in TNBC
The prognostic value of the 167-gene TNBC signature was assessed with the METABRIC dataset. The 254 TNBC samples were split into two subgroups: a subgroup treated by chemotherapy (n = 139) and a subgroup not treated by chemotherapy (n = 115). The chemotherapy-naive (noCT) population and the chemotherapy-treated population were significantly different (Table S1). The patients in the noCT population were older (mean age of 61.5 y vs. 50.1 y, p < 1.2Citation10-11), more likely to be postmenopausal (77% vs. 47%, p = 5.3810-5), and their tumors were of lower grade (p = 0.01), with less lymph node involvement (81% vs. 17%, p < 2.2 Citation10-16), a lower Nottingham Prognostic Index (NPI < 3.4, 17% vs. 2%, p = 2.5710-5), and less cellularity (p = 0.03).
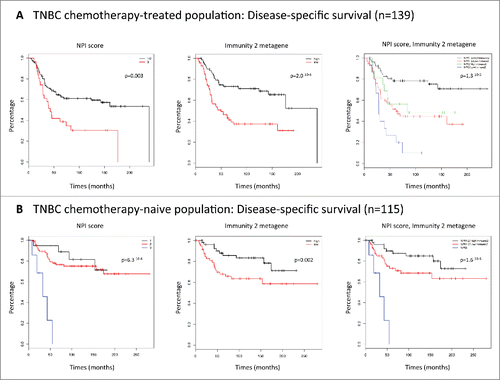
Univariate analysis identified three factors significantly correlated with a poor outcome (distant disease-free survival) in the chemotherapy-treated population: NPI > 5.4 (HR = 2.15 [1.28–3.60], p = 0.003); p53 mutation (HR = 2.42 [1.15–5.09], p = 0.02); and weak Immunity2 metagene expression (HR = 2.59 [1.54–4.34], p = 0.0002) (, ). We did not include p53 mutation status in the multivariate model, due to missing data (n = 79). A NPI > 5.4 and low-levels of Immunity2 metagene expression were retained in the multivariate model and were significantly associated with a poor outcome (HR = 2.30 [1.36–3.89], p = 0.002; HR = 2.68 [1.59–4.52], p = 0.0002, respectively) (). The combined variable, NPI score/Immunity2 metagene expression was found to be of particular interest. In a first model, a NPI score greater than 5.4 was associated with a worse prognosis: HR = 3.98 [2.00–7.92], p = 8.7210–5. For patients with NPI scores of 5.4 or below, Immunity2 metagene expression discriminated between two groups of patients with different outcomes (HR = 2.90 [1.51–5.56], p = 0.001). In a second model, NPI3 patients can also be split into two groups on the basis of Immunity2 metagene expression. The NPI3 group with high-levels of Immunity2 metagene expression had a prognosis similar to that of the NPI1/2 group with low-levels of Immunity2 metagene expression (, ).
Table 1 A. Survival analysis (disease-specific survival). Chemotherapy-treated population. Univariate and multivariate analysis.
Table 1 B. Survival analysis (disease-specific survival). Chemotherapy-treated population. Two univariate models. Combination of NPI score and Immunity2 metagene expression
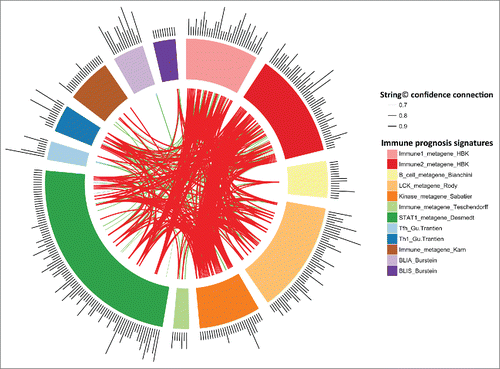
Figure 4. String Software connections between our Immunity1 and Immunity2 genes and the genes of eight previously published prognostic immune signatures. Stronger associations between genes are represented by thicker lines. Associations between genes with a coefficient < 0.9 are shown in green. Associations between genes with a coefficient ≥0.9 are shown in red. Associations between genes with a coefficient between 0.4 to 0.7 are not shown.
Univariate analysis identified four factors significantly correlated with poor outcome in the noCT population: tumor size >20 mm (HR = 2.36 [1.01–5.48], p = 0.04), lymph node-positive status (HR = 3.66 [1.65–8.11], p = 0.001), NPI score >5.4 (HR = 10.69 [2.74–41.76], p = 0.001) and low-levels of Immunity2 metagene expression (HR = 2.33 [1.09–4.95], p = 0.03) (, ). Two of these factors were retained in the multivariate model: NPI score >5.4 (HR = 12.03 [3.05–47.50], p = 0.0004) and low-levels of Immunity2 metagene expression (HR = 2.42 [1.13–5.16], p = 0.02) (). As in the chemotherapy-treated subpopulation, the combined variable, NPI score/Immunity2 metagene expression discriminated between two groups of patients with different outcomes in this noCT population (, ). The chemotherapy-naive group contained only seven patients classified as NPI3. Stratification of this subgroup defined on the basis of treatment was therefore not considered methodologically relevant.
Table 2 A. Survival analysis (disease-specific survival). Chemotherapy-naive population. Univariate and multivariate analysis.
Table 2 B. Survival analysis (disease-specific survival). Chemotherapy-naive population. Univariate analysis. Combination of NPI score and Immunity2 metagene expression.
We compared the prognostic value of the Immunity2 metagene with that of eight previously published immune signatures,Citation18-25 using the METABRIC dataset.
We generated a heatmap (Fig. S11) of the GE profiles of each of the above prognostic signatures applied to the METABRIC dataset. The samples were ordered according to our classification of low/high Immunity2 metagene expression. Expression patterns were very similar between the Immunity2 GE signature and all the other GE signatures, with the exception of the Bianchini, Karn and Burstein (BLIS) gene-expression signatures.
We first performed a univariate analysis of the prognostic value of the eight-GE signatures, as described in the corresponding original manuscripts. The Rody, Sabatier, Teschendorff, Desmedt, Gu-Trantein Tfh, Gu-Trantien Th1 and Burstein signatures were significantly correlated with the prognosis of TNBC. The Bianchini and Karn GE signatures were not correlated with the prognosis of TNBC (Fig. S12, Table S2). We then performed a multivariate analysis. We included NPI score, the Immunity2 metagene and each of the Rody, Sabatier, Teschendorff, Desmedt, Gu-Trantein Tfh, Gu-Trantien Th1, and Burstein signatures, one-by-one, in the model. In all comparisons the only significant variables remaining in the multivariate model were NPI score and the Immunity2 metagene (Table S2).
The Immunity2 metagene corresponds to B-cell and T-cell pathways
String database connections between the Immunity1 or Immunity2 genes and the genes of the eight published prognostic immune signatures Citation18-25 are provided in . The gene intersection was poor, but our immune signature nevertheless appears to be strongly correlated with other published signatures (Supplementary data), suggesting the use of similar immune pathways. The Immunity2 metagene was strongly correlated with the expression metagenes of the above signatures (coefficient greater than 0.8), except for the Bianchini, Karn and BLIS metagenes (Fig. S13).
Figure 3. (A) Kaplan–Meier plots. Disease-specific survival of the chemotherapy-treated population (n = 139). NPI score. Immunity2 metagene. NPI score/Immunity2 metagene. (B) Kaplan–Meier plots. Disease-specific survival of the noCT population (n = 115). NPI score. Immunity2 metagene. NPI score/Immunity2 metagene.
We explored the pathways relating to the immune metagenes in detail, by analyzing the correlation between the expression of the Immunity1 and Immunity2 metagenes and the metagenes defined by Gatza et al.Citation26 (IFN-α, IFNγ, STAT3, TGF-β, TNF-α) and Palmer et al.Citation27 (LB, LT, CD8+, GRANS, LYMPHS). This analysis was performed on the METABRIC dataset published by Curtis et al.Citation17
We showed that the Immunity2 metagene was highly correlated with the B-cell, T-cell and CD8+ cell metagenes (Pearson correlation scores: 0.93, 0.91, 0.87, respectively) (Fig. S14). The Immunity1 metagene was highly correlated with the interferon alpha and gamma pathways (Pearson correlation scores: 0.97, 0.94, respectively).
Furthermore, in cancer cell lines (CCLE and CGP datasets), the Immunity2 metagene displayed very low-levels of expression, similar to those of the CD8+ metagene (Fig. S15). This was true for all cell lines and BC_CLs tested.
Moreover, the IFN-y, IFN-gamma, STAT3, TGF-β, TNF-α, LB, LT, GRANS metagenes were more strongly expressed in TN BC_CLs than in HER2-positive and luminal BC_CLs (Fig. S16).
We investigated Immunity2 GE in white blood cell populations (Palmer et al.Citation27), by performing a consensus clustering of the Immunity2 genes on Palmer's dataset. This analysis identified four stable clusters of the genes of the Immunity2 signature. Some genes were more strongly expressed in B cells (GZMA, GZMB, CCR7, LY96, MS4A1, CD74 for example), others in T cells (CD3D, CCL2, CD14, CD2, LCK, IL7R), and still others in granulocytes (named Pax cells) (CD163, MNDA, NCF2, CSF2RB, FGL2) (Fig. S17). These findings suggest that, even if the “Immune2” signal is highly homogeneous within tumor samples (the entire set of genes being coordinately either over- or under-expressed), different subpopulations of cells express different subsets of these genes in the periphery.
The Immunity2 metagene is probably expressed by stromal cells
In TNBC cell lines (TNBC_CL), genes from the Immunity2 module displayed very low medians and narrow ranges of expression, suggesting that they were expressed only in the tumor stromal compartment. A similar trend was observed for all BC_CLs. The Immunity1 module genes had higher median expression levels and a broader range of expression in TNBC_CL and in all BC_CL, suggesting that Immunity1 genes were expressed by the tumor cells ().
Figure 5. (A) Boxplots of gene expression for the Immunity1 and Immunity2 metagenes, in each breast cancer cell line subtype from the CCLE. (B) Boxplots of gene expression for the Immunity1 and Immunity2 metagenes in each breast cancer cell line subtype from the CGP.
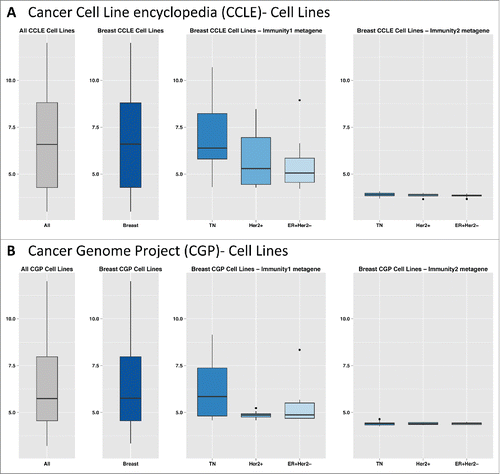
Furthermore, we explored the contributions of stromal and cancer cells to Immunity1 and Immunity2 expression in detail, by comparing our gene lists to the “stromal contribution to global GE evaluated in PDX RNAseq data”, as defined by Isella et al.Citation28 The Immunity2 metagene had a very high stromal fraction, as for the Matrix/Invasion1 and Matrix2 metagenes. The Immunity1 metagene had a very low stromal fraction, like the AR-like and Proliferation/DNA damage metagenes (Fig. S18).
The Immunity2 metagene opens up interesting new possibilities for therapeutic interventions
To highlight the new opportunities for therapeutic intervention provided by this study, we represented the existing drugs (with or without US Food and Drug Administration approval) for each metagene (Fig. S19 and Supplementary data). Some are undergoing clinical investigation in patients with TNBC.
We explored the links between PD1, PDL1, CTLA4 (and their respective metagenes) and the Immunity2 metagene. We compared the Immunity2 metagene with the TILs signature defined by Schalper et al.,Citation29 who showed that PD-L1 mRNA synthesis was associated with increases in the expression of TILs and recurrence-free survival. This analysis was performed on the METABRIC dataset. The PD1 and CTLA-4 metagenes were constructed from the genes most strongly correlated with the PD1 and CTLA-4 genes, respectively (Pearson correlation score >0.8). The PDL1 metagene was defined by Sabatier et al.Citation30
The Immunity2 metagene was highly correlated with the PD1, PDL1 and CTLA-4 metagenes (Pearson correlation: 0.90, 0.96, 0.91, respectively). The coefficient of correlation between the Immunity2 metagene and the TILs signature was up to 0.90 (Fig. S20).
In cell lines, the PD1, PDL1, CTLA-4 and TILs metagenes were very weakly expressed, like the Immunity2 metagene (Fig. S21).
Using the METABRIC dataset, we compared the prognostic value of these metagenes (PD1, PDL1, CTLA4 and TILs) with that of the Immunity2 metagene. In univariate analysis, high-levels of PD1, PDL1, CTLA-4 and TILs metagene expression were associated with a good prognosis (Fig. S22, Table S3). In multivariate analysis, we included NPI score, the Immunity2 metagene and each of the PD1, PDL1, CTLA4 and TILs metagenes, one-by-one, in the model. In all comparisons, the only significant variables remaining in the multivariate model were NPI score and the Immunity2 metagene.
Discussion
New tools for classifying TNBCs are urgently required, to improve our understanding of the molecular basis of TNBC and to identify potentially useful novel therapeutic interventions. By analyzing the GE profiles of 1,125 TNBCs, we identified a six-metagene signature (167 genes) in which the various metagenes were enriched in different gene ontologies: two clusters were enriched in immunity genes, one in proliferation/DNA damage genes, one in AR pathway genes, and two in matrix/invasion genes. This signature appeared to be particularly robust for identifying TNBC subtypes across different datasets, independently of the gene chip technology used to generate the data. Furthermore, one metagene (Immunity2) was found to be of strong prognostic value for TNBC samples.
Lehmann et al.Citation10 recently developed a classification of TNBCs in which a 2,188-gene signature was used to classify tumors. They suggested that this classification could also be used to classify xenografts or cell lines. They also developed a website (http://cbc.mc.vanderbilt.edu/tnbc/) for the classification of TNBC samples.Citation11 This study provided important biological insight into the molecular drivers of TNBC, but it also raised several key concerns. First, the normalization process involved data from different platforms. Several studies have shown that large discrepancies in signature composition and absences of concordance concerning outcome may be due to differences in the array platform and preprocessing method used.Citation12 Second, Lehmann et al. used a very large number of genes (2,188 genes) to establish their molecular signature, and this may have constituted a source of instability, due to the noise introduced.Citation12,13 As shown by Weigelt et al.,Citation31 microarray-based single-sample predictors do not allocate individual samples to a given molecular subtype reproducibly, probably because the use of large numbers of genes leads to instability of the classification when new samples are added. Third, it would be unwise to transpose this classification to various in vitro and in vivo breast cancer models (primary tumor xenografts, cell lines, cell line-derived xenografts), because the stromal environment and the original tumor are very different.Citation32,33 We found that genes from the Immunity compartment (Immunity2 module) were highly relevant for the classification of TNBC samples and that these genes were not expressed in BC_CLs. The observed lack of reproducibility between classifiers may reflect major differences in the methodology and aims of the studies concerned. Further validation will be required before these models can be used in routine clinical practice.
We developed a strategy for the definition of a GE signature based on the analysis of biological networks for the most variant genes. Within these networks, we then analyzed GE parameters, to select the genes with the most strongly correlated patterns of expression. The validation process showed that our gene matrix identified similar GE patterns across 1,125 TNBC samples. This first step in biological network analysis, which is probably less sensitive to sample fluctuations than other methods, made it possible to capture strong biological signals that might be concealed by the noise present in microarray data. Several studies have reported that the incorporation of network information improves the stability of gene selection and the biological interpretability of biomarker signatures for a given prediction accuracy.Citation14,15,34
The Immunity2 module was identified as a strong prognostic factor for disease-specific survival (strong expression of this metagene is correlated with a good outcome), regardless of the characteristics of the tumor (NPI score, tumor size, tumor grade and lymph node status). It clearly suggest the presence of an hemopoietic infiltrate, composed of activated cytotoxic T cells, B cells, myeloid cells, natural killer cells and neutrophils. This module includes adhesion molecule-associated genes (SELL, ITGB2), and genes encoding proteins involved antigen processing and presentation (CD74 or ligand, HLA-DRA), B-lymphocyte cell surface molecules (PTPRC, ITGB2, HLA-DRA), the caspase cascade (CASP1), complement pathway (C1QA, C1QB), CTL-mediated immune responses to target cells (ITGB2, CD3D, GZMB), dendritic cell regulation of Th1 and Th2 development (CD2, IL7R), granzyme-mediated apoptosis (GZMA, GZMB), IL12-mediated signaling events (CD3D, HLA-DRA, GZMA, LCK), the IL2 signaling pathway (LCK), interleukin-3, 5 and GM-CSF signaling (HCK, BLNK, CSF2RB), T-cell surface molecules (PTPRC, CD3D, CD2, ITGB2), and the T-cell receptor signaling pathway (PTPRC, CD3D, HLA-DRA, LCK).
Burstein et al.Citation25 identified four different TNBC subtypes (LAR, MES, BLIS, BLIA) with the identification of similar pathways and a prognostic value for the BLIA subgroup similar to that for the signature identified in our study. This subgroup displays an upregulation of B-cell, T-cell, and natural killer cell immune-regulating pathways and an activation of STAT transcription factor-mediated pathways. The authors showed that the prognosis was worse for basal-like immune-suppressed tumors than for basal-like immune-activated tumors, for both disease-free survival (p = 0.04) and disease-specific survival (p = 0.039).
Several recent studies have demonstrated the importance of tumor-infiltrating lymphocytes (TILs) in controlling the clinical progression of various epithelial cancers.Citation35 In breast cancer, recent advances in GE profiling have revealed an association between immune signatures and favorable outcomes.Citation29,36 A gene signature enriched in cytotoxic CD8+ T-cell genes and genes associated with natural killer cell activity has been reported.Citation37 However, the ability of CD8+ T cells to control human breast cancer is probably counteracted by the presence of immunosuppressive cells, CD4+ T-regulatory cells or macrophages: immunohistochemistry (IHC) analysis of tissue microarray data for 179 treatment-naive breast tumors revealed that high-levels of macrophages and CD4+ T cells were correlated with poor overall survival, whereas a combination of high-levels of CD8+ T cells and low-levels of macrophages and CD4+ T cells was correlated with higher overall survival.Citation38 Intratumoral B cells have also been associated with a favorable prognosis in breast cancer.Citation39 In ER-negative breast cancers, a STAT1 signaling metagene,Citation16 and a B-cell metageneCitation19 were found to be associated with better outcomes. Another group identified an immune response-based prognostic gene module (C1QA, XCL2SPP1, TNFRSF17, LY9, IGLC2, HLA-F) associated with a better prognosis than for other ER-negative breast cancers, regardless of lymph node status and lymphocytic infiltration.Citation40 According to Bertucci et al.,Citation41 the IM subtype (overlapping with medullary breast cancers, a rare form of TNBC with a prominent lymphocytic reaction) is associated with a favorable prognosis. The two immune modules identified in this study had many biological connections with other eight immune prognosis signatures published for TNBC.Citation18-25
Neoadjuvant chemotherapy is increasingly being used for TNBC, because these tumors have a poor prognosis, are assumed to be chemosensitive and no alternative specific systemic treatment is available. Patients with a complete pathologic response (pCR) after neoadjuvant chemotherapy have a better outcome than those with residual disease, and pCR is a good surrogate for long-term survival and cure in this specific subgroup.Citation9,42
The Immunity2 metagene was not found to be predictive of response to neoadjuvant chemotherapy in TNBC (272 fine needle aspirations of TNBC samples for which information about pCR or its absence was available from the eight datasets previously published by Ignatiadis et al. Citation16) (data not shown). This lack of relationship may have resulted from the use of fine needle aspiration biopsy samples. The Immunity2 genes, which are largely expressed in the stromal environment, were less strongly expressed in fine needle aspiration samples than in tumor samples (Fig. S23).
However, intratumoral immune responses are known to be correlated with clinical outcomes in TNBC. This may reflect the role of immune cells in the activity of cytotoxic chemotherapeutic agents. Some chemotherapeutic drugs, such as anthracyclines, act not only through direct cytotoxic effects, but also by activating CD8+ T-cell responses. Conflicting results have been published on the ability of other immune-based classifiers to predict outcome in TNBC. High-intratumoral levels of CD8+ T cells Citation43 or TILs Citation36,44 are associated with better clinical responses to anthracycline-based chemotherapy. West et al. Citation45 reported that high-levels of lymphocyte GE were associated with a high rate (74%) of complete pathological responses to neoadjuvant anthracycline-based chemotherapy. In 2011, Sabatier et al. Citation20 showed, by gene-expression profiling, that “Immune High” patients (59%) were more likely to present pCR than “Immune Low” patients (43%), but this difference was not significant (p = 0.29). In 2014,Citation46 they showed that “PDL1 mRNA expression high” (57%) patients presented higher rates of pCR than “PDL1 mRNA expression low” (43%) patients (p < 0.001). Wimberley et al. Citation47 showed that PDL1 protein levels in the epithelium and stroma were correlated with pCR only in hormone receptor-positive and HER2-amplified breast cancers. Denkert et al. Citation44 demonstrated the importance of TIL and immune GE signatures for predicting pCR in breast carcinoma. However no significant difference in pCR rate was detected between lymphocyte-predominant breast cancer (LPBC) and no-LPBC in the anthracycline-taxane subgroup.
However, the results of these studies suggest that clinical outcomes in ER-negative breast cancers, including TNBC in particular, are strongly influenced by tumor immune responses and are, thus, highly responsive to immunotherapies. The possible use of immunotherapy approaches to treat TNBC (tumor vaccine approaches, immune-checkpoint inhibitors, antagonists of immunosuppressive molecules and adoptive cell therapies) should be investigated in detail.Citation48
The other metagenes studied had no significant prognostic or predictive value. However, they identified sound biological networks providing opportunities for therapeutic intervention. The Immunity1 metagene included genes involved in the interferon α/β signaling pathway or cytokine signaling (STAT1, IRF7, IRF27, OAS1, OAS2, PMSB8, XAF1, IFIT1, IFITM1, ISG15, IGS20, IF6, MX1), the Toll-like receptor signaling pathway (STAT1, CXCL9, CXCL10, CXCL11, CCL5, IRF7), cell-cycle checkpoints and DNA synthesis (PSMB8, PSMB9). Patients displaying strong expression of this metagene often also had high-levels of Immunity2 and Proliferation/DNA damage metagene expression, suggesting the possible existence of common pathways. The IDO1 (indoleamine 2, 3-dioxygenase 1) gene is a particularly interesting potential target. It encodes a tryptophan-degrading enzyme known to suppress antitumor CD8+ T cells and it contributes to the inhibition of anticancer immune responses.Citation48 This immunosuppressive enzyme is actually investigated as a promising candidate target in cancer immunotherapy.
A subset of TNBC tumors strongly expresses AR-regulated genes.Citation49 AR expression has been reported to be lower in triple-negative breast tumor cells than in other types of breast cancer. The overall frequency of AR expression in carcinoma cells varies considerably between studies (0–53%).Citation50,51 We identified strong expression of AR pathway genes in 25% of our population. The biological role of androgens in TNBC remains a matter of debate. Immunohistochemical studies investigating the presence of AR in tumor cells have reported conflicting results for clinical outcome; some studies have suggested that AR expression is advantageous for survival,Citation52-54 whereas others found no significant effect.Citation55 Lehmann et al. found that the LAR subtype of TNBC displayed the lowest frequency of pCR (10%). The presence of AR in a subset of TNBC patients suggests that androgenic pathways in tumor cells could be targeted in at least some TNBC patients. The widespread availability of agents targeting the AR also makes this approach potentially appealing, as it would be straightforward to incorporate such treatment into clinical practice.
The Matrix/Invasion1 metagene included genes associated with β1 integrin cell surface interactions, ECM-receptor interaction or integrin family cell surface interactions (NID1, TGFBI, COL5A1, COL5A2, COL6A3, COL3A1, COL1A1, COL1A2, COL11A1, FN1, FBN1, THBS1, THBS2), the TGF β signaling pathway (DCN, COMP, THBS1, THBS2), the inhibition of matrix metalloproteases (MMP2, TIMP3), and the AP-1 transcription factor network (DCN, COL1A2, MMP2). Metalloproteinases (MMPs) and their tissue inhibitors are involved in several key pathways of tumor growth, invasion and metastasis.Citation56,57 The expression and activity of MMPs has been linked to advanced stages of breast cancer, greater tumor invasion and the construction of metastatic formations.Citation58,59,60 Some studies have highlighted the importance of matrix MMP expression by stromal cells as a prognostic factor in the TNBC subtype.Citation61 These molecules are thus attractive targets for drug development.Citation62
The Matrix2 metagene included genes associated with the AP-1 transcription factor network (FOS, EGR1, FABP4, DUSP1), the EGR receptor signaling pathway (FOS, DUSP1, EGR1), the Wnt or ALK signaling pathway (CAV1), the MAPK signaling pathway (FOS, DUSP1) or Trk receptor signaling mediated by the MAPK pathway (FOS, EGR1), the mTOR signaling pathway (IGF1), the PPAR signaling pathway (ADIPOQ, CD36, FABP4), and androgen-mediated signaling (FOS, EGR1). These pathways may contribute to cell motility and tumor cell invasionCitation63 and play a prominent role in epithelial-mesenchymal transition (EMT) and in stem cells. These metagenes are strongly expressed in mesenchymal cells and metaplastic breast cancers.Citation4 Metaplastic breast cancers have lineage plasticity, including spindle cell foci, and display osseous or cartilaginous differentiation.Citation64 Some drugs targeting the pathways relating to the metagenes identified here may be of particular interest for the treatment of TNBC (PI3K/mTOR inhibitor, Wnt/β catenin inhibitor).
Conclusion
In conclusion, our 167-gene TNBC molecular signature, consisting of six metagenes, appears to be particularly robust for the identification of TNBC subtypes. Furthermore, expression of the Immunity2 metagene was strongly correlated with prognosis, and many biological targets have been identified within the corresponding biological network. These findings open up interesting new possibilities for the development of new therapeutic interventions.
Patients and Methods
Data normalization and quality control
We collected 21 publicly available datasets (described in the supplementary data) containing raw GE data from microarray analyses (Affymetrix© Gene Chip Human Genome HG-U133A and HG-U133Plus2) of 3,247 primary human breast cancer samples. The data were normalized by the robust multichip average (RMA) procedure from the EMA R package.Citation65 The datasets were split into training (HGU-133A Affymetrix© arrays, 12 datasets, n = 1,995) and validation (HGU-133Plus2 Affymetrix© arrays, (9 datasets, n = 1,014) sets. We also collected two large datasets, to validate our classification: The Ignatiadis dataset (n = 996) and the METABRIC dataset (n = 1,992). Data processing for these two datasets has been described elsewhere.Citation16,17
Determination and preprocessing of triple-negative breast cancer samples
We identified the TNBC samples in each dataset, using a bimodal mixture of two Gaussian distributions for ER and HER2 gene expression, and the median value for PR expression.
The training, validation and Ignatiadis datasets
Batch effects were eliminated by the median centering of each probe-set across arrays and by a, independent quantile normalization of all arrays for each dataset. We controlled for outliers with the Array Quality Metrics R package.
The METABRIC set
We fitted a linear model (limma R package) to remove the batch effect and probes were filtered according to three criteria: probe quality,Citation66 GC content and presence in more than 5% of the samples. We centered expression values, using the R function scale().
Gene selection process
Consensus clustering was applied to the training set, to determine the optimal number of robust gene clusters for the most variant genes (standard deviation > 0.8). We investigated the enrichment of each gene cluster in particular types of genes. We then identified known biological networks, for each gene cluster separately, using String© database software version 9.1 (http://string-db.org/).Citation67
We then applied a two-step selection process: (1) we selected strong biological networks by retaining only genes for which connection scores of at least 0.7 were obtained with String© database software, (2) within each biological network, we selected groups of genes with for which expression levels were correlated, with a correlation coefficient of at least 0.5.
For each dataset (the training, validation, Ignatiadis and METABRIC sets), we applied a hierarchical clustering procedure to the TNBC GE profiles, using the selected genes to visualize the optimal number of stable TNBC subtypes.
Prognostic analysis
Prognostic analysis was performed on the METABRIC set published by Curtis et al.Citation17
Expression data were summarized by a metagene for each gene cluster (details in the supplementary material). The clinical and pathological variables available for each dataset are described in the supplementary data. Qualitative variables were compared in χ2 tests or Fisher's exact tests, as appropriate. Quantitative variables were analyzed in Student's t-tests. Survival analyses were performed separately for patients with and without chemotherapy. Survival analyses were performed, with the Kaplan–Meier estimate of the survival function. The endpoint of these analyses was breast cancer-specific survival (BCSS). Survival curves were compared in log rank tests. Hazard ratios were estimated with Cox's proportional hazard model.
Expression of the gene signature in human triple-negative breast cancer cell lines
We downloaded the GE profiles of the human cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE)Citation68 of Novartis/the Broad Institute and the Cancer Genome Project (CGP)Citation69 of the Sanger Institute. We normalized all the cell lines from different tissues together.
All statistical analyses were performed with R software (www.cran.r-project.org). P -values < 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1061176_supplemental_files.zip
Download Zip (32 MB)Acknowledgments
The authors thank Sergio Roman-Roman for the reviewing of the study and the manuscript.
Funding
This work was supported by Institut Curie, INCa (the french National Cancer Institute) Grant INCa-DGOS-4654, ANR-10-IDEX-0001-02 PSL, ANR-11-LABX-0043, CIC IGR Curie 1428 and Fundation ARC (association for Research Against Cancer).
References
- Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Ggenom 2006; 7:96; PMID:16643655; http://dx.doi.org/10.1186/1471-2164-7-96
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001; 98(19):10869-74; PMID:11553815; http://dx.doi.org/10.1073/pnas.191367098
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27(8):1160-7; PMID:19204204; http://dx.doi.org/10.1200/JCO.2008.18.1370
- Hennessy BT, Gonzalez-Angulo A-M, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 2009; 69(10):4116-24; PMID:19435916; http://dx.doi.org/10.1158/0008-5472.CAN-08-3441
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363(20):1938-8; PMID:21067385; http://dx.doi.org/10.1056/NEJMra1001389
- Turner NC, Reis-Filho JS. Tackling the diversity of triple-negative breast cancer. Clin Cancer Res 2013; 19(23):6380-8; PMID:24298068; http://dx.doi.org/10.1158/1078-0432.CCR-13-0915
- Von Minckwitz G, Blohmer JU, Costa SD, Denkert C, Eidtmann H, Eiermann W, Gerber B, Hanusch C, Hilfrich J, Huober J et al. Response-guided neoadjuvant chemotherapy for breast cancer. J Clin Oncol 2013; 31(29):3623-30; PMID:24002511; http://dx.doi.org/10.1200/JCO.2012.45.0940
- Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008; 26(8):1275-81; PMID:18250347; http://dx.doi.org/10.1200/JCO.2007.14.4147
- Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L, Valagussa P et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 2014; 384(9938):164-72; PMID:24529560; http://dx.doi.org/10.1016/S0140-6736(13)62422-8
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol J. A.. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121(7):2750-67; PMID:21633166; http://dx.doi.org/10.1172/JCI45014
- Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, Pietenpol JA. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Informat 2012; 11:147-56; PMID:22872785; http://dx.doi.org/10.4137/CIN.S9983
- Sontrop HMJ, Moerland PD, van den Ham R, Reinders MJT, Verhaegh WFJ. A comprehensive sensitivity analysis of microarray breast cancer classification under feature variability. BMC Bioinformatics 2009; 10:389; PMID:19941644; http://dx.doi.org/10.1186/1471-2105-10-389
- Abraham G, Kowalczyk A, Loi S, Haviv I, Zobel J. Prediction of breast cancer prognosis using gene set statistics provides signature stability and biological context. BMC Bioinformatics 2010; 11:277; PMID:20500821; http://dx.doi.org/10.1186/1471-2105-11-277
- Fröhlich H. Network based consensus gene signatures for biomarker discovery in breast cancer. PLoS One 2011; 6(10):e25364; PMID:22046239; http://dx.doi.org/10.1371/journal.pone.0025364
- Cun Y, Fröhlich HF. Prognostic gene signatures for patient stratification in breast cancer: accuracy, stability and interpretability of gene selection approaches using prior knowledge on protein-protein interactions. BMC bioinformatics 2012; 13:69; PMID:22548963; http://dx.doi.org/10.1186/1471-2105-13-69
- Ignatiadis M, Singhal SK, Desmedt C, Haibe-Kains B, Criscitiello C, Andre F, Loi S, Piccart M, Michiels S, Sotiriou C. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J Clin Oncol 2012; 30(16):1996-2004; PMID:22508827; http://dx.doi.org/10.1200/JCO.2011.39.5624
- Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486(7403):346-52; PMID:22522925; http://dx.doi.org/10.1038/nature10983.
- Bianchini G, Qi Y, Alvarez RH, Iwamoto T, Coutant C, Ibrahim NK, Valero V, Cristofanilli M, Green MC, Radvanyi L et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol 2010; 28(28):4316-4323; PMID:20805453; http://dx.doi.org/10.1200/JCO.2009.27.2419
- Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res 2011; 13(5):R97; PMID:21978456; http://dx.doi.org/10.1186/bcr3035
- Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra JM, Jacquemier J et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treatment 2011; 126:407-420; PMID:20490655; http://dx.doi.org/10.1007/s10549-010-0897-9
- Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol 2007; 8(8):R157; PMID:17683518; http://dx.doi.org/10.1186/gb-2007-8-8-r157
- Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, Delorenzi M, Piccart M. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 2008; 14(16):5158-65; PMID:18698033; http://dx.doi.org/10.1158/1078-0432.CCR-07-4756
- Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, Ravoet M, Le Buanec H, Sibille C, Manfouo-Foutsop G et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 2013; 123(7):1-20; PMID:23778140; http://dx.doi.org/; http://dx.doi.org/10.1172/JCI67428
- Karn T, Pusztai L, Holtrich U, Iwamoto T, Shiang CY, Schmidt M, Müller V, Solbach C, Gaetje R, Hanker L et al. Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PloS One 2011; 6(12):e28403; PMID:22220191; http://dx.doi.org/10.1371/journal.pone.0028403
- Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 2014; 21(7):1688-98; PMID:25208879; http://dx.doi.org/10.1158/1078-0432.CCR-14-0432.
- Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey-Prevot B, Potti A et al. A pathway-based classification of human breast cancer. Proc Natal Acad Sci U S A 2010; 107:6994-9; PMID:20335537; http://dx.doi.org/10.1073/pnas.0912708107
- Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics 2006; 7:115; PMID:16704732; http://dx.doi.org/10.1186/1471-2164-7-115
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C et al. Stromal contribution to the colorectal cancer transcriptome. Nature Genet 2015; 47(4):312-9; PMID:25547594; http://dx.doi.org/10.1038/ng.3224
- Schalper K A., Velcheti V, Carvajal D, Wimberly H, Brown J, Pusztai L, Rimm DL. In situ tumor PD-L1 mRNA expression is associated with increased tils and better outcome in breast carcinomas. Clin Cancer Res 2014; 20:2773-82; PMID:24647569; http://dx.doi.org/10.1158/1078-0432.CCR-13-2702
- Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D, Bertucci F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015; 6(7):5449-64; PMID:25669979
- Weigelt B, Mackay A, A'hern R, Natrajan R, Tan DS, Dowsett M, Ashworth A, Reis-Filho JS. Breast cancer molecular profiling with single sample predictors: a retrospective analysis. The Lancet Oncol 2010; 11(4):339-49; PMID:20181526; http://dx.doi.org/10.1016/S1470-2045(10)70008-5
- De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol 2003; 200(4):429-47; PMID:12845611; http://dx.doi.org/10.1002/path.1398
- Lum DH, Matsen C, Welm AL, Welm BE. Overview of human primary tumorgraft models: comparisons with traditional oncology preclinical models and the clinical relevance and utility of primary tumorgrafts in basic and translational oncology research. Curr Protoc Pharmacol 2012;Chapter 14(801):Unit 14.22; PMID:23258598; http://dx.doi.org/10.1002/0471141755.ph1422s59
- Sanavia T, Aiolli F, Da San Martino G, Bisognin A, Di Camillo B. Improving biomarker list stability by integration of biological knowledge in the learning process. BMC Bioinformatics 2012; 13 Suppl 4(Suppl 4):S22; PMID:22536969; http://dx.doi.org/10.1186/1471-2105-13-S4-S22
- Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012; 12(4):298-306; PMID:22419253; http://dx.doi.org/10.1038/nrc3245
- Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010; 28(1):105-13; PMID:19917869; http://dx.doi.org/10.1200/JCO.2009.23.7370
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008; 14(5):518-27; PMID:18438415; http://dx.doi.org/10.1038/nm1764
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011; 1(1):54-67; PMID:22039576; http://dx.doi.org/10.1158/2159-8274.CD-10-0028
- Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kölbl H, Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res 2008; 68(13):5405-13; PMID:18593943; http://dx.doi.org/10.1158/0008-5472.CAN-07-5206
- Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol 2007; 8(8):R157; PMID:17683518; http://dx.doi.org/10.1186/gb-2007-8-8-r157
- Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Mamessier E, Adélaïde J, Debono S, Houvenaeghel G, Maraninchi D, Viens P et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res 2006; 66(9):4636-44; PMID:16651414; http://dx.doi.org/10.1158/0008-5472.CAN-06-0031
- Von Minckwitz G, Untch M, Blohmer J-U, Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich J, Huober J et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol 2012; 30(15):1796-804; PMID:22508812; http://dx.doi.org/10.1200/JCO.2011.38.8595
- Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 2011; 71(14):4809-20; PMID:21646474; http://dx.doi.org/10.1158/0008-5472.CAN-11-0753
- Denkert C, von Minckwitz G, Brase JC, Bruno V. Sinn, Stephan Gade, Ralf Kronenwett, Berit M. Pfitzner, Christoph Salat, Sherene Loi, Wolfgang D. Schmitt et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015; 33(9):983-91; PMID:25534375; http://dx.doi.org/10.1200/JCO.2014.58.1967
- West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res 2011; 13(6):R126; PMID:22151962; http://dx.doi.org/10.1186/bcr3072
- Sabatier R, Finetti P, Guille A, Adelaide J, Chaffanet M, Viens P, Birnbaum D, Bertucci F. Claudin-low breast cancers: clinical, pathological, molecular and prognostic characterization. Mol Cancer 2014; 13:228; PMID:25277734; http://dx.doi.org/10.1186/1476-4598-13-228
- Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, Bossuyt V, Pusztai L, Lannin DR, Rimm DL PD-L1 Expression Correlates with Tumor-Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy in Breast Cancer. Cancer Immunol Res 2015; 3(4):326-32; PMID:25527356; http://dx.doi.org/10.1158/2326-6066.CIR-14-0133
- Stagg J, Allard B. Immunotherapeutic approaches in triple-negative breast cancer: latest research and clinical prospects. Ther Adv Med Oncol 2013; 5(3):169-81; PMID:23634195; http://dx.doi.org/10.1177/1758834012475152
- Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene. 2006; 25(28):3994-4008; PMID:16491124; http://dx.doi.org/10.1038/sj.onc.1209415
- Ogawa Y, Hai E, Matsumoto K, Ikeda K, Tokunaga S, Nagahara H, Sakurai K, Inoue T, Nishiguchi Y. Androgen receptor expression in breast cancer: relationship with clinicopathological factors and biomarkers. Inter J Clin Oncol 2008; 13(5):431-5; PMID:18946753; http://dx.doi.org/10.1007/s10147-008-0770-6
- He J, Peng R, Yuan Z, Wang S, Peng J, Lin G, Jiang X, Qin T. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol 2012; 29(2):406-10; PMID:21264529; http://dx.doi.org/10.1007/s12032-011-9832-0
- Luo X, Shi Y-X, Li Z-M, Jiang W-Q. Expression and clinical significance of androgen receptor in triple negative breast cancer. Chin J Cancer 2010; 29(6):585-90; PMID:20507730; http://dx.doi.org/10.5732/cjc.009.10673
- Rakha E A, El-Sayed ME, Green AR, Lee AHS, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007; 109(1):25-32; PMID:17146782; http://dx.doi.org/10.1002/cncr.22381
- McNamara KM, Yoda T, Miki Y, Chanplakorn N, Wongwaisayawan S, Incharoen P, Kongdan Y, Wang L, Takagi K, Mayu T et al. Androgenic pathway in triple negative invasive ductal tumors: its correlation with tumor cell proliferation. Cancer Sci 2013; 104(5):639-46; PMID:23373898; http://dx.doi.org/10.1111/cas.12121
- Peters A A, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res 2009; 69(15):6131-40; PMID:19638585; http://dx.doi.org/10.1158/0008-5472.CAN-09-0452
- Jiang Y, Goldberg ID, Shi YE. Complex roles of tissue inhibitors of metalloproteinases in cancer. Oncogene 2002; 21(14):2245-52; PMID:11948407; http://dx.doi.org/10.1038/sj/onc/1205291.
- Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 2006; 25(1):9-34; PMID:16680569; http://dx.doi.org/10.1007/s10555-006-7886-9
- Vizoso FJ, González LO, Corte MD, Rodríguez JC, Vázquez J, Lamelas ML, Junquera S, Merino AM, García-Muñiz JL. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br J Cancer2007; 96(6):903-11; PMID:17342087; http://dx.doi.org/10.1038/sj.bjc.6603666
- McGowan PM, Duffy MJ. Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Annals Oncol 2008; 19(9):1566-72; PMID:18503039; http://dx.doi.org/10.1093/annonc/mdn180
- Figueira RCS, Gomes LR, Neto JS, Silva FC, Silva IDCG, Sogayar MC. Correlation between MMPs and their inhibitors in breast cancer tumor tissue specimens and in cell lines with different metastatic potential. BMC Cancer 2009; 9:20; PMID:19144199; http://dx.doi.org/10.1186/1471-2407-9-20
- González LO, Corte MD, Junquera S, González-Fernández R, del Casar JM, García C, Andicoechea A, Vázquez J, Pérez-Fernández R, Vizoso FJ. Expression and prognostic significance of metalloproteases and their inhibitors in luminal A and basal-like phenotypes of breast carcinoma. Human Pathol 2009; 40(9):1224-33; PMID:19439346; http://dx.doi.org/10.1016/j.humpath.2008.12.022
- Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr Pharmaceut Design 2007; 13:333-46; PMID:17313364; http://dx.doi.org/10.2174/138161207779313551
- Shin S-Y, Rath O, Zebisch A, Choo S-M, Kolch W, Cho K-H. Functional roles of multiple feedback loops in extracellular signal-regulated kinase and Wnt signaling pathways that regulate epithelial-mesenchymal transition. Cancer Res 2010; 70(17):6715-24; PMID:20736375; http://dx.doi.org/10.1158/0008-5472.CAN-10-1377
- Gibson GR, Qian D, Ku JK, Lai LL. Metaplastic breast cancer: clinical features and outcomes. Am Surg 2005; 71:725-30; PMID:16468506
- Servant N, Gravier E, Gestraud P, Laurent C, Paccard C, Biton A, Brito I, Mandel J, Asselain B, Barillot E et al. EMA - A R package for Easy Microarray data analysis. BMC Res Notes 2010; 3(1):277; PMID:21047405; http://dx.doi.org/10.1186/1756-0500-3-277
- Barbosa-Morais NL, Dunning MJ, Samarajiwa S A, Darot JF, Ritchie ME, Lynch AG, Tavaré S. A re-annotation pipeline for Illumina BeadArrays: improving the interpretation of gene expression data. Nucleic Acids Res 2010; 38(3):e17; PMID:19923232; http://dx.doi.org/10.1093/nar/gkp942
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009; 37(Database issue):D412-D416; PMID:18940858; http://dx.doi.org/10.1093/nar/gkn760
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483(7391):603-607; PMID:22460905; http://dx.doi.org/10.1038/nature11003
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J et al. Europe PMC Funders Group Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012; 483(7391):570-5; PMID:22460902; http://dx.doi.org/10.1038/nature11005