
ABSTRACT
Tumor-induced, myeloid-derived suppressor cells (MDSCs)-mediated immune dysfunction is an important mechanism that leads to tumor immune escape and the inefficacy of cancer immunotherapy. Importantly, tumor-infiltrating MDSCs have much stronger ability compared to MDSCs in the periphery. However, the mechanisms that tumor microenvironment induces the accumulation and function of MDSCs are poorly understood. Here, we report that Interleukin-33 (IL-33) – a cytokine which can be abundantly released in tumor tissues both in 4T1-bearing mice and breast cancer patients, is crucial for facilitating the expansion of MDSCs. IL-33 in tumor microenvironment reduces the apoptosis and sustains the survival of MDSCs through induction of autocrine secretion of GM-CSF, which forms a positive amplifying loop for MDSC accumulation. This is in conjunction with IL-33-driven induction of arginase-1 expression and activation of NF-κB and MAPK signaling in MDSCs which augments their immunosuppressive ability, and histone modifications were involved in IL-33 signaling in MDSCs. In ST2−/− mice, the defect of IL-33 signaling in MDSCs attenuates the immunosuppressive and pro-tumoral capacity of MDSCs. Our results identify IL-33 as a critical mediator that contributes to the abnormal expansion and enhanced immunosuppressive function of MDSCs within tumor microenvironment, which can be potentially targeted to reverse MDSC-mediated tumor immune evasion.
Abbreviations:
| BrdU | = | 5-bromo-2-deoxyuridine |
| ERK | = | extracellular regulated protein kinases |
| GM-CSF | = | granulocyte-macrophage colony stimulating factor |
| IHC | = | immunohistochemistry |
| IL | = | interleukin |
| iNOS | = | inducible nitric oxide synthase |
| JNK | = | c-Jun N-terminal kinase |
| MDSC | = | myeloid-derived suppressor cells |
| NF-κB | = | nuclear factor κB |
| STAT3 | = | signal transducers and activators of transcription 3 |
| TGF-β | = | transforming growth factor-β |
| Treg | = | regulatory T cells |
Introduction
Immunosuppressive cells are the main obstacles leading to the failure of antitumor immunity and cancer immunotherapy. Among these cells, MDSCs are reported to be abnormally expanded in multiple tumor types in mice and human and have a remarkable ability to suppress T-cell response.Citation1 MDSCs in mice and human are characterized by the phenotype of CD11b+Gr-1+ and CD11b+CD33+HLA-DRlow/− respectively,Citation2 and can be further subdivided into monocytic-MDSCs (M-MDSCs) and granulocytic-MDSCs (G-MDSCs).Citation3 The inhibition of MDSC function is a promising strategy to overcome tumor-induced immune defects, which might be a key step in enhancing the efficacy of immune-based therapies.Citation4
Under steady-state conditions, immature myeloid cells take a small percentage of cells in peripheral blood and spleen with modest or no immunosuppressive ability. However, in tumor-bearing hosts they undergo abnormal expansion and acquire the ability to suppress T cell responses. Importantly, tumor-infiltrating MDSCs have much stronger suppressive ability compared to MDSCs in the periphery.Citation5 Moreover, tumor-infiltrating MDSCs have been reported to be more resistant to chemotherapy-induced apoptosis than MDSCs in the spleen.Citation6,7 Thus, tumor microenvironment is very crucial for reeducating MDSCs into potent immune suppressors, and understanding how the tumor microenvironment influences MDSC function is important to design novel strategies to reverse MDSC-mediated immune dysfunction. However, current studies have largely been restricted to peripheral MDSCs and there are few reports investigating the factors that make the functional differences between tumor-infiltrating and peripheral MDSCs.Citation5
Interleukin 33 (IL-33, accession number: NM_001164724) is a newly described member of IL-1 cytokine family and mainly expressed in structural cells such as epithelial, endothelial cells and fibroblasts.Citation8,9 The receptor of IL-33 constitutes ST2 (accession number:NM_001025602) and IL-1 receptor accessory protein (IL-1RAcP)Citation10 and ST2 is expressed on diverse immune cells.Citation9 IL-33 has been implicated in various diseases such as asthma, arthritis, viral infection and is thought to drive Th2-immune responses.Citation11,12 Unlike most interleukins, IL-33 is rarely secreted under steady-state conditions, and its secretion only occurs in necrotic cells or the cells exposed to environmental stressors such as inflammation and mechanical stress, therefore IL-33 is considered to be a danger-associated molecular pattern (DAMP) or “alarmin.”Citation13
The impact of IL-33 in tumor immunity has been reported in several recent studies but the conclusions were controversial.Citation14,15 Here we found that IL-33 was expressed at high levels in tumor microenvironment, which reduced the apoptosis of MDSCs through induction of autocrine secretion of GM-CSF thus formed an amplifying loop for MDSC accumulation. This was in conjunction with IL-33-driven induction of arginase-1 expression, which augmented the immunosuppressive function of MDSCs. The absence of IL-33 signaling in MDSCs reduced their ability to promote tumor growth. Our results identify IL-33 as a critical mediator that contributes to the enhanced function of MDSCs within tumor microenvironment, which can be potentially targeted to reverse MDSC-mediated tumor immune escape.
Results
Abundant IL-33 secretion was detected in tumor tissues
To determine the expression of IL-33 in tumor microenvironments, 4T1 mammary carcinomas were inoculated subcutaneously into female BALB/c mice. We found that IL-33 was expressed in 4T1 cells as well as in 4T1 tumors (). The mRNA level of IL-33 were dramatically upregulated in tumor tissues when compared to that in 4T1 cells (), however, we found that “ex vivo” 4T1 cells selected by 6-thioguanineCitation16 from tumor tissues had similar IL-33 levels to 4T1 cell line (data not shown), indicating that 4T1 cells might be not a main source of IL-33 within tumor tissues. To investigate what types of cells express IL-33, we prepared single-cell suspensions from 4T1 tumors, IL-33 was positively stained in epithelial cells (EpCAM+), endothelial cells (CD31+) and tumor-associated fibroblasts (PDGFRα+) (), indicating that multiple types of cells expressed IL-33 within tumor microenvironment.
Figure 1. Abundant IL-33 level is detected in tumor microenvironment. (A) 4T1 tumor cells (left) and tumor sections of 4T1-bearing mice (right) were stained with anti-IL-33 antibody and examined by confocal microscopy. Scale bar: 50 μm. (B) IL-33 mRNA in 4T1 cell line and 4T1 tumor tissue were evaluated by qPCR. (C) Single cell suspensions were prepared from tumor tissues of 4T1-bearing mice and IL-33 expression in indicated cell populations was examined. (D) IL-33 protein levels were determined in tumor supernatant and serum of 4T1-bearing mice by ELISA. (E) Immunohistochemistry staining of IL-33 in the representative human breast cancer tissues and adjacent normal tissues (scale bars: 50 μM). Staining intensity of IL-33 (F) and the percentage of IL-33-positive cells (G) was compared between tumor (n = 148) and tumor-adjacent normal tissues (n = 40). Classification of specimens according to IL-33 staining intensity (H) and the percentage of IL-33-positive cells (I) were shown. (J) IL-33 levels in tissue supernatant from nine cases of human breast cancer and serum IL-33 levels in breast cancer patients were measured by ELISA. Data are mean ± SEM. *, p < 0.05; **, p < 0.01; n.s. = not significant.
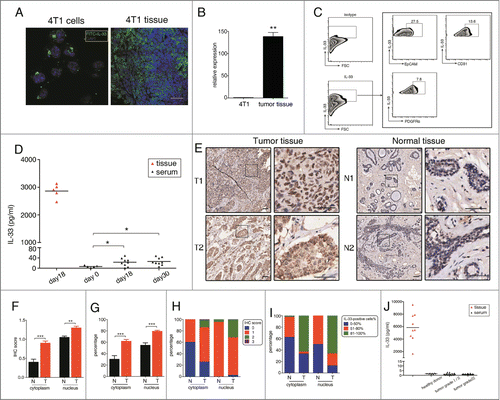
Since IL-33 is rarely secreted by living cells under steady-state conditions, we collected tumor supernatant to measure the secreted IL-33 within tumor microenvironment.Citation17 High levels of IL-33 were detected in 4T1 tumor supernatant, indicating that IL-33 was also abundantly secreted within tumor tissues. However, IL-33 levels were very low in the serum of 4T1-bearing mice and undetectable in serum of tumor-free mice (). Then we determined the expression of IL-33 receptor – ST2 on MDSCs. Both splenic and tumor MDSCs from 4T1-bearing mice expressed ST2 (Fig. S1A), interestingly, we found that approximately 45% of ST2+ cells in 4T1 tissues were also Gr-1+, indicating that MDSCs might be important target cells regulated by IL-33 within tumor microenvironment (Fig. S1B). However, 4T1 cells did not express ST2 in vitro and in vivo, and IL-33 had no influence on the proliferation and migration of 4T1 cells (data not shown).
To address IL-33 expression in human breast cancer, we examined IL-33 levels in breast cancer tissues containing 148 breast cancer specimens and 40 tumor-adjacent normal specimens by immunohistochemistry (). Elevated intensity of IL-33 staining was observed in tumor tissues compared to tumor-adjacent normal tissues, especially in cytoplasm and to a lesser extent in nucleus (). 40% (16/40) tumor-adjacent normal tissues were positively-stained for IL-33 in cytoplasm (IHC score ≥ 1), while more than 74.3% (110/148) tumor tissues have cytoplasmic expression of IL-33, with 14.2% cases (21/148) of strong staining (IHC score ≥ 2) (). Moreover, we analyzed the percentage of IL-33-positive cells in each specimen, on average, approximately 30% cells have cytoplasmic expression of IL-33 in tumor-adjacent normal tissues, this percentage increased to more than 60% in tumor tissues. The percentage of nuclear-IL-33 positive cells was also higher in tumor tissues compared to tumor-adjacent normal tissues (). Breast cancer patients were grouped based on tumor grade (grade I/II, n = 72; and grade III, n = 63), and it showed that the intensity of cytoplasmic IL-33 staining was positively associated with breast tumor grade (r = 0.17, p = 0.047), through spearman rank correlation analysis (Table S1).
Similar to the results in mice, we detected high levels of IL-33 in the supernatants of nine cases of breast cancer tissues, in contrast, IL-33 was undetectable both in serum of healthy donors and breast cancer patients, regardless of the tumor grade ().
ST2−/− mice have reduced MDSC accumulation in tumor microenvironment
To investigate whether IL-33 influences MDSC accumulation, 4T1 cells were injected subcutaneously into wild type and ST2−/− mice and 20 d later, MDSC percentages were measured in bone marrow, blood, spleen and tumor tissues. WT and ST2−/− mice had similar MDSC percentages in BM and spleen, however ST2−/− mice had slightly lower MDSC percentages in blood. Strikingly, MDSC percentages were significantly decreased in ST2−/− tumors than that in WT tumors (). Furthermore, in size-matched tumors, ST2−/− mice still had lower MDSC percentages (), demonstrating that it was not due to slower tumor growth in ST2−/− mice.Citation15 Lower G-MDSC percentages and higher M-MDSC percentages were observed in ST2−/− tumors when compared to WT tumors, whereas the percentages of mature myeloid cells (Ly6G−Ly6C− cells within CD11b+ populations) were significantly increased in ST2−/− tumors compared to WT tumors (), suggesting that MDSCs might be more prone to differentiate into mature myeloid cells in ST2−/− tumors. In contrast, tumor-free WT and ST2−/− mice had comparable MDSC percentages in BM, spleen and blood (Fig. S2A).
Figure 2. ST2−/− mice have reduced MDSC frequencies in tumor tissue but not in spleen and bone marrow. (A) and (B) WT and ST2−/− mice (n = 6) were injected subcutaneously with 3 × 105 4T1 cells. 21 d after tumor cell inoculation mice were sacrificed, the percentages of MDSCs in bone marrow, blood, spleen and tumor were analyzed by flow cytometry (gated on total live cells) (B). Representative plots were shown in (A). (C) WT and ST2−/− 4T1-bearing mice (n = 5) were sacrificed when the tumor reached 8–10 mm in diameter. MDSC percentages in tumor tissues were analyzed by flow cytometry. (D) Percentages of G-MDSCs and M-MDSCs in spleen and tumor tissues of WT and ST2−/− mice (n = 5) were analyzed by flow cytometry. (E) WT and ST2−/− mice (n = 5) were intraperitoneally injected BrdU every 12 h. 48 h later, the percentages of BrdU+ cells within MDSC population (Gr-1 gated) in tumors were analyzed. (F) Percentages of annexin V+ cells within MDSC population in tumor tissues were analyzed. Data are mean ± SEM and are representative of three independent experiments.*, p < 0.05; **, p < 0.01; ***, p < 0.001.
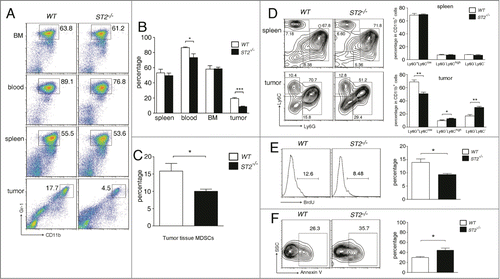
Next we compared the proliferation and apoptosis of MDSC in WT and ST2−/− 4T1-bearing mice. There were less MDSCs labeled with BrdU in ST2−/− tumors than in WT tumors (), but the percentages of BrdU+ MDSC did not significantly differ in BM and spleens between WT and ST2−/− mice (data not shown). On the other hand, more annexin V+ MDSCs were detected in ST2−/− tumor tissues when compared to WT tumors ().
Studies by us and others have shown that MDSCs are recruited to tumor tissues primarily through CXCL5/CXCR2 and CXCL12/CXCR4 axes.Citation18,19 We found comparable CXCL5 and CXCL12 mRNA levels in WT and ST2−/− tumors, and similar levels of CXCR2 and CXCR4 were observed on splenic and blood MDSCs in WT and ST2−/− tumor-bearing mice, furthermore, IL-33 had no direct influence on MDSC chemoattraction, suggesting that the reduced MDSC accumulation in ST2−/− tumors was not resulted from MDSC recruitment (Fig. S3A–C).
Taken together, the absence of IL-33/ST2 signaling reduced the survival, proliferation and accumulation of MDSCs specifically within tumor microenvironment but not in spleen and BM.
IL-33 facilitates the survival and accumulation of MDSCs through induction of autocrine GM-CSF
To investigate whether IL-33 had direct effect on MDSC expansion in vitro, MDSCs were purified from spleens of 4T1-bearing mice (>95% purity) and treated ex vivo with IL-33 (). MDSCs underwent spontaneous apoptosis without any growth factors, notably, IL-33 significantly reduced MDSC apoptosis. We also observed obvious formation of cell colonies 48 ∼72 h after IL-33 treatment (Fig. S4A).
Figure 3. IL-33 reduces the apoptosis of MDSC in GM-CSF-autocrine dependent manner. (A) Representative plots showing the MDSCs sorted from the spleen of 4T1-bearing mice 2~3 weeks after tumor cell inoculation. (B) MDSCs were treated with different doses of IL-33 for 24 h. The mRNA levels of GM-CSF and IL-6 were determined by qPCR (upper), the expression levels of untreated MDSCs were set as one. Protein levels were determined by ELISA (lower). (C) MDSCs were cultured in medium containing 40% (v/v) tumor supernatant in the presence of control IgG or anti-ST2 (40 μg/mL) for 24 h, mRNA levels of GM-CSF and IL-6 were evaluated by qPCR. (D) MDSCs were treated with IL-33 (50 ng/mL) in the presence of anti-GM-CSF (10 μg/mL) or anti-IL-6 (10 μg/mL) for 24, 48 and 72 h, then stained with annexin V and PI for analysis of apoptosis by flow cytometry. (E) Isolated MDSCs were cultured with IL-33 for 6 d, in the presence of anti-IL-6 or control IgG. Cells were stained with anti-CD11b and anti-Gr-1 antibodies, and the expression of Gr-1 on CD11b-gated cells was analyzed by flow cytometry. (F) GM-CSF mRNA levels in tumor tissues (left) and isolated MDSCs (right) of WT and ST2−/− mice were evaluated by qPCR. Data are mean ± SEM and are representative of at least three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
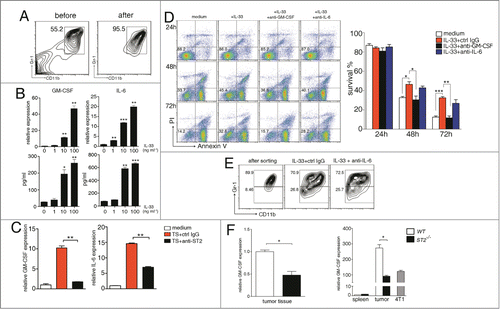
IL-33 has been reported to sustain the survival of BM cells by triggering GM-CSF expression,Citation20 we found that IL-33 dramatically upregulated the expression of GM-CSF in MDSCs both in mRNA and protein level. The expression of IL-6, another cytokine involved in MDSC accumulationCitation21 was also upregulated by IL-33 (). IL-33 did not alter the expression of G-CSF, TNF-α, VEGF, IL-1β and S100A8/A9 (data not shown), which were also reported to mediate MDSC expansion.Citation21,22 Further analysis showed that IL-33 induced GM-CSF expression primarily in G-MDSC subset, while IL-6 was equally induced in G-MDSC and M-MDSC populations (Fig. S5). Similar results were obtained in MDSCs isolated from spleens of 3LL and CT-26 bearing mice as well as in vitro induced MDSCs (Fig. S6), demonstrating that this effect of IL-33 was not 4T1 model specific.
Then we treated MDSCs with tumor supernatant and found that GM-CSF and IL-6 expression were markedly upregulated in MDSCs. Importantly, ST2 blockade dramatically reduced this effect to basal levels (), indicating IL-33 is crucial for the autocrine action of GM-CSF and IL-6 in MDSCs in tumor microenvironment.
To further address the role of GM-CSF and IL-6 on reduced MDSC apoptosis induced by IL-33, we pretreated MDSCs with GM-CSF and IL-6 blocking antibodies before IL-33 treatment. GM-CSF neutralization completely abrogated the pro-survival effect of IL-33, while neutralization of IL-6 had no obvious effect (). IL-6 has been reported to inhibit the differentiation of myeloid progenitors into mature myeloid cells,Citation23 unexpectedly, neutralization of IL-6 did not change the percentages of both Gr-1high and Gr-1low cells in cultured MDSCs in the presence of IL-33 (). These results indicated that IL-33 upregulation of IL-6 was not sufficient to block MDSC differentiation to mature myeloid cells.
GM-CSF is a crucial mediator responsible for the generation and accumulation of MDSCs.Citation23,24 We detected significantly lower GM-CSF expression in ST2−/− tumor tissues than that in WT tumors, and MDSCs isolated from ST2−/− tumors had significantly lower GM-CSF expression compared to MDSCs from WT tumors. It was noteworthy that GM-CSF level in tumor MDSCs was higher than that in 4T1 cells, indicating that MDSCs were important contributors to GM-CSF production in tumor microenvironment, while GM-CSF levels in splenic MDSCs were very low (). These results indicated that IL-33 triggered autocrine GM-CSF production by MDSCs, consequently formed an positive feedback loop facilitating their survival and accumulation in tumor microenvironment.
IL-33 significantly upregulates arginase-1 expression in MDSCs
Two arginine metabolic enzymes – arginase-1 and inducible nitric oxide synthase (iNOS) are well accepted to be a critical mediator that contributes to the immunosuppressive function of tumor-expanded MDSCs.Citation1 We found that IL-33 significantly upregulated arginase-1 and iNOS expression in splenic MDSCs. By contrast, the expression of gp91-phox and p47-phox, two subunits of the nicotinamide-adenine dinucleotide phosphate oxidase complex (NOX complex) which are responsible for ROS production in MDSCs,Citation25 were unaffected. Th2 cytokine IL-13 but not IL-4 was increased by IL-33 treatment. In contrast, Th1 cytokine IFNγ and IL-12 was downregulated in MDSCs upon IL-33 treatment (). IL-33 also upregulated arginase-1 expression in MDSCs derived from 3LL- and CT26- bearing mice as well as in vitro induced MDSCs (Fig. S6), and this upregulation was more obvious in M-MDSC subpopulation (Fig. S5).
Figure 4. IL-33 significantly upregulates arginase-1 expression by MDSCs. (A) MDSCs were treated with IL-33 (50 ng/mL) for 6 and 24 h. The mRNA levels of indicated genes were determined by qPCR, the expression levels of untreated MDSCs was set as 1. (B) 1 × 106 MDSCs were stimulated with IL-33 (50 ng/mL) for 6 and 24 h. Cell lysates were collected and arginase-1 enzyme activity was evaluated (left). Nitrites were measured in culture supernatants (right). (C) MDSCs were stimulated with IL-33 (50 ng/mL) for 24 h and stained with DCFDA in the absence or presence of PMA (50 nM). The fluorescence intensity of DCFDA was evaluated by flow cytometry. (D) MDSCs were cultured in medium containing 40% (v/v) tumor supernatant in the presence of control IgG or anti-ST2 (40 μg/mL) for 24 h. mRNA levels of arginase-1 and iNOS were evaluated by qPCR. Data are mean ± SEM and are representative of three independent experiments.*, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s. = not significant.
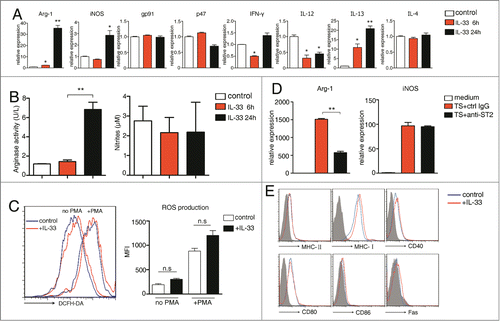
Furthermore, IL-33 treatment also enhanced arginase-1 but not iNOS enzymatic activity in MDSCs (). ROS production was not significantly altered upon IL-33 treatment (). It is noteworthy that there was no difference in cell viability between control- and IL-33 treated- group at 24-h time point. Moreover, ST2 blockade dramatically attenuated the capacity of tumor supernatant to upregulate the expression of arginase-1 but not iNOS in MDSCs ().
We also determined various surface markers reported to be associated with MDSC functions including MHC, CD80, CD86, CD40 and Fas,Citation26,27 all markers were not influenced by IL-33 (). Thus, IL-33 strongly induces the immunosuppressive activity of tumor-associated MDSC mainly through dramatic upregulation of arginase-1 level.
MDSCs in ST2−/− tumor tissues have attenuated immunosuppressive ability
To examine if IL-33 affected the immunosuppressive function of MDSCs, ST2−/− splenocytes were cultured alone or co-cultured with MDSCs derived from spleens of 4T1-bearing mice in the absence or presence of IL-33, and T cell proliferation was determined by CFSE dilution. Since IL-33 also enhanced the survival of MDSCs, exogenous GM-CSF (5 ng/mL) was added to the co-culture system to offset the pro-survival function of IL-33. Splenic MDSCs had moderate ability to suppress T cell proliferation at 1:1 ratio (MDSCs: splenocytes), however, in the presence of IL-33, these cells displayed significantly stronger ability to inhibit the proliferation of both CD4+ and CD8+ T cells. IL-33 alone had no direct influence on T cell proliferation because we used ST2−/− splenocytes as responders. The addition of arginase inhibitor, nor-NOHA, restored T cell proliferation and abrogated the effects of IL-33 on MDSCs (), supporting that enhanced activity of MDSCs upon IL-33 treatment was due to increased expression of arginase-1.
Figure 5. MDSCs in ST2−/− tumor tissues have attenuated immunosuppressive ability. (A) MDSCs isolated from spleen of 4T1-bearing WT mice were co-cultured at 1:1 ratio (MDSCs:splenocytes) with CFSE-labeled, anti-CD3/CD28 activated ST2−/− splenocytes in the absence or presence of IL-33 (50 ng/mL), arginase inhibitor (nor-NOHA, 0.5 mM) was added as indicated. The proliferation of CD4+ and CD8+ T cells were analyzed by flow cytometry 72 h later. (B) MDSCs were isolated from spleen and tumor tissues of 4T1-bearing WT or ST2−/− mice. Isolated MDSCs were co-cultured with CFSE-labeled, anti-CD3/CD28 activated splenocytes at 1:4 or 1:8 ratio (MDSCs:splenocytes). The proliferation of CD4+ and CD8+ T cells were analyzed by flow cytometry 72 h later. (C) mRNA levels of arginase-1, iNOS, gp91 and p47 in isolated MDSCs from WT and ST2−/− mice were evaluated by qPCR. (D) and (E), ST2−/− mice had higher T cell frequencies compared to WT mice in tumor microenvironments. Single cell suspensions of spleen, blood and tumors from 4T1-bearing WT and ST2−/− mice (n = 6) were stained with anti-CD4+ and anti-CD8+ antibodies, T cell frequencies were analyzed by flow cytometry. Representative plots (D) and statistical chart (E) are shown. (F) Frozen sections of tumor tissues from WT and ST2−/− mice were stained with anti-CD3 and anti-Gr-1 antibodies and examined by confocal microscopy. Representative image (left) and statistical graph (right) were shown. Scale bar: 50μm. Data are mean ± SEM and are representative of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
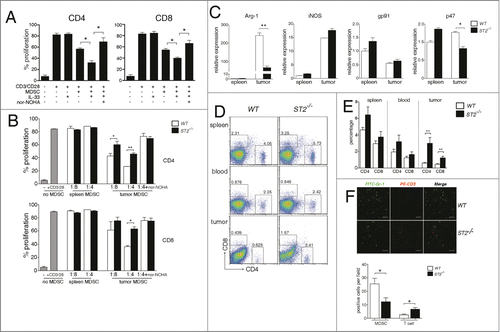
Next we found that MDSCs isolated from ST2−/− tumors manifested significantly reduced ability to suppress the proliferation of both CD4+ and CD8+ T cells, compared to MDSCs from WT tumors. The differences in suppressive ability between WT and ST2−/− tumor MDSCs were completely abrogated when arginase-1 activity was inhibited (). Splenic MDSCs from WT and ST2−/− mice both failed to suppress T cell proliferation. Moreover, it showed marked upregulation of arginase-1 and iNOS mRNA levels in tumor MDSCs compared to their spleen counterparts, whereas the upregulation of arginase-1 was less dramatic in ST2−/− tumor MDSCs, iNOS was equally upregulated in WT and ST2−/− tumor MDSCs. The expression of gp91 and p47 showed no decrease in ST2−/− tumor MDSCs compared to WT tumor MDSCs ().
Next we observed that tumor-bearing ST2−/− mice had significantly higher percentages of both CD4+ and CD8+ T cells in tumor tissues when compared to WT mice (), immunofluorescence imaging also demonstrated less MDSCs and more T cells in ST2−/− tumors than those in WT tumors (). Tumor-free ST2−/− and WT mice had same CD4+ and CD8+ T percentages in spleen and blood (Fig. S2B). These results suggested that the absence of IL-33 signaling resulted in higher T cell frequencies in tumor microenvironment rather than in periphery.
IL-33 does not influence MDSCs to induce Treg in vivo
Two reports have noted that MDSCs facilitate the generation and proliferation of regulatory T cells (Tregs) through IL-10 and TGF-β and impair immune responses indirectly.Citation28,29 IL-33 has been shown to expand CD4+CD25+Foxp3+ Tregs in cardiac allotransplatation model.Citation30 We observed that IL-33 treatment increased the expression of TGF-β but not IL-10 (). Treg percentages (in CD4+ T cells) were increased when co-cultured with MDSCs, but IL-33 failed to further induce more Tregs despite its ability to enhance TGF-β expression (). IL-33 has also been reported to induce DCs and macrophages to produce CCL17 and CCL22,Citation31 two chemokines responsible for Treg recruitment into tumors.Citation32 Both CCL17 and CCL22 were upregulated in splenic MDSCs upon IL-33 treatment (), transwell experiments also showed that supernatant of IL-33-stimulated MDSCs recruited more Tregs compared to the supernatant of unstimulated MDSCs (data not shown). Unexpectedly, ST2−/− and WT 4T1-bearing mice had comparable Treg percentages in spleen and tumor tissues (). Tumor-free WT and ST2−/− mice also had similar Treg percentages (Fig. S2C). Nevertheless, since IL-33 could upregulate CCL17 expression in isolated MDSCs derived from 3LL- and CT26- bearing mice (Fig. S6), it was possible that IL-33 may promote Treg recruitment in other tumor models, or in other disease models such as transplantation.
Figure 6. IL-33 does not influence the ability of MDSCs to induce and recruit regulatory T cells in vivo. MDSCs were treated with IL-33 (50 ng/mL) for 6 and 24 h. The mRNA levels of TGF-β, IL-10 (A) and CCL17, CCL22 (C) were determined by qPCR. (B) 4 × 105 ST2−/− splenocytes were cultured alone or co-cultured with 1 × 105 MDSCs and activated by plate-bound anti-CD3/CD28 antibodies, in the absence or presence of IL-33 (50 ng/mL) for 72 h. The percentages of Foxp3+ Tregs within CD4+ T cells were evaluated by flow cytometry. (D) The percentages of Foxp3+ Tregs (CD4+-gated) in spleen and tumor tissues from 4T1-bearing WT and ST2−/− mice were evaluated by flow cytometry. (E)–(H) ST2−/− MDSCs have attenuated ability to promote tumor growth. WT and ST2−/− mice (n = 6) were injected subcutaneously with 3 × 105 4T1 cells, tumor growth was monitored every 5 d (E). (F)–(H) MDSCs were sorted from spleens of WT or ST2−/− 4T1-bearing mice. 5 × 106 sorted MDSCs were adoptively transferred through tail vain into ST2−/− mice (n = 5) 3, 6, 9 d after 4T1 tumor cell inoculation, ST2−/− mice received PBS served as control, tumor growth was monitored (F) and compared on day 10, 20, 30 after tumor inoculation (G), mice in each group were monitored for survival (H). Data are mean ± SEM and are representative of at least three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s = not significant.
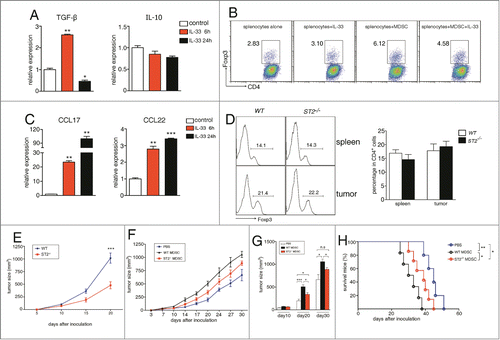
ST2−/− MDSCs have attenuated ability to promote tumor growth
Consistent with previous study,Citation15 we observed retarded 4T1 tumor growth in ST2−/− mice (). We performed adoptive transfer experiments to investigate the contribution of MDSCs to the attenuated tumor growth in ST2−/− mice. MDSCs were purified from spleen of WT and ST2−/− 4T1-bearing mice respectively and transferred intravenously into ST2−/− 4T1-bearing mice 3, 6, 9 d after 4T1 inoculation. Mice which received WT MDSCs had significantly faster tumor growth than those received PBS only (). Although ST2−/− MDSCs also promoted tumor growth, this effect was impaired compared to WT MDSCs. 30 d after tumor inoculation, mice received exogenous MDSCs still had larger tumor volumes than mice received PBS, however, the difference of tumor volume between mice received ST2−/− MDSCs and mice received PBS did not reach statistical significance (), it may be due to that endogenous ST2−/− MDSCs had been massively expanded and overwhelmed the transferred MDSCs in number.
Furthermore, mice received ST2−/− MDSCs had prolonged survival time than those received WT MDSCs (). Only the adoptively transferred MDSCs can respond to IL-33 in ST2−/− mice, we concluded that the absence of IL-33 signaling decreased the ability of MDSCs to promote tumor growth.
NF-κB and ERK are crucial transcription factors for IL-33 signaling in MDSCs
We examined which transcription factors were activated in MDSCs upon IL-33 stimulation. IL-33 strongly induced the activation of NF-κB signaling in MDSCs and to a lesser extent, p38 and ERK signaling, however the phosphorylation of JNK and STAT3 was not affected (). Inhibition of NF-κB activation dramatically suppressed the upregulation of all five genes, indicated that NF-κB activation is pivotal for IL-33 signaling in MDSCs. Inhibition of ERK activation reduced the upregulation of GM-CSF, IL-6, arginase-1, CCL17 but not CCL22, and inhibition of p38 inhibited the upregulation of arginase-1 and CCL22. Although JNK activation was not observed in our results, JNK inhibitors reduced the upregulation of CCL17. Inhibition of STAT3 activation had no influence on the upregulation of all above genes ().
Figure 7. NF-κB and ERK are crucial transcription factors involved in IL-33 signaling in MDSCs. (A) MDSCs were isolated from spleen of 4T1-bearing mice and stimulated with IL-33 (50 ng/mL) for indicated times. Levels of total and phosphorylated p65, MAPKs and STAT3 were determined by Western blot. (B) MDSCs were pre-incubated with inhibitors for indicated transcription factors for 1 h, followed by IL-33 (50 ng/mL) stimulation or 9 h. The expression of indicated genes was quantified by qPCR. Following inhibitors were used: 10 μM BAY 11-7082 (NF-κB inhibitor); 10 μM SB203580 (p38 inhibitor); 10 μM SP600125 (JNK inhibitor); 10 μM U0126 (ERK inhibitor); 25 μM AG 490 (STAT3 inhibitor). (C) MDSCs were stimulated with IL-33 for indicated times, histone modifications were examined by Western blot. Data are mean ± SEM and are representative of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
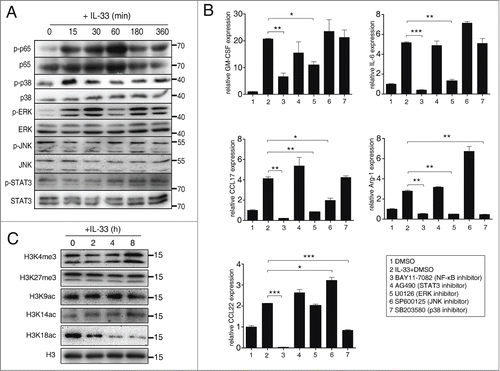
Histone modifications play important roles in regulating gene expression, we found that the level of histone H3 Lys-4 trimethylation (H3K4me3) dramatically increased in MDSCs after IL-33 treatment, the level of histone H3 Lys-14 acetylation (H3K14ac) also displayed a mild increase. The level of H3K9ac was unchanged. In contrast, the level of H3K18ac dramatically decreased in MDSCs upon IL-33 treatment. All these modifications are indicative of transcriptional activation. The level of H3K27me3, which is a marker of transcriptional silencing, was unaffected by IL-33 treatment (). This pattern change suggested that epigenetic events were involved in IL-33 signaling in MDSCs. However, the contribution of specific modifications to the expression of an individual gene needs further investigation.
Discussion
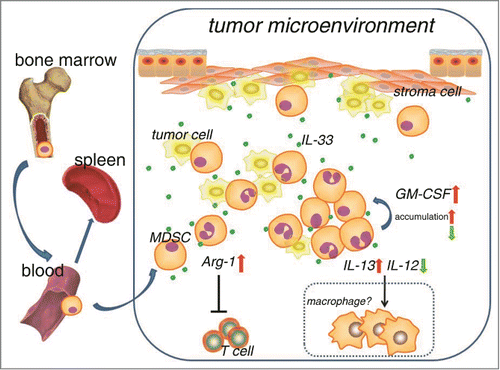
In this study, we identified alarmin IL-33 as a critical mediator by which the tumor microenvironment regulates the function of MDSCs. High level of IL-33 which can be released accompanied with tumor growth was very crucial for facilitating the intratumoral accumulation of MDSCs. IL-33 acts as an amplifier for MDSC autocrine GM-CSF signaling, and also a critical player in potentiating the immunosuppressive ability of MDSCs through induction of arginase-1 upregulation ().
Figure 8. Working model shows that IL-33 regulates the accumulation and function of MDSCs within tumor microenvironment. Dotted box represents hypothesis which has not yet been testified in our present study.
The influences of MDSCs on antigen-nonspecific T cell response are controversial, some studies have suggested that MDSCs can suppress antigen-nonspecific T cell responseCitation33, but there are also contradictory reports which indicate that MDSCs from spleen could not suppress anti-CD3/CD28 stimulated T cell proliferation.Citation5,34 Compared with peripheral MDSCs, tumor site-infiltrating MDSCs show a stronger ability to suppress adaptive immune response, facilitate tumor cell invasion and metastasis,Citation7,35 indicating the tumor microenvironment has profound influences on the function of MDSCs. However, the molecular events controlling the activation of MDSCs in tumor tissues are largely unknown, and it is also not clear what factors are mainly involved in the functional difference. A previous report has indicated that hypoxic environment can augment the function of MDSCs through increasing arginase-1 and iNOS expression.Citation5 In another study, TNFR-2 signaling has been found to promote the accumulation of MDSCs.Citation36 An important finding of our study is the positive amplifying loop for MDSC autocrine GM-CSF induced by IL-33 in tumor microenvironment. Tumor-derived GM-SCF reduces the apoptosis of tumor-associated MDSCs and impairs the therapeutic efficacy of tumor vaccines.Citation2,23,37 High-dose GM-CSF-producing tumor vaccines have low therapeutic efficacy because of MDSC recruitment.Citation38 We demonstrated here that MDSCs in tumor tissue had potent ability to produce autocrine GM-CSF, the absence of IL-33 signaling in MDSCs resulted in lowered GM-CSF levels in MDSCs as well as in tumor tissue, which impaired the proliferation and survival of MDSCs within tumor microenvironment. The induction of GM-CSF by IL-33 thus forms a positive amplifying loop for MDSC accumulation. In addition, we observed a slight decrease of MDSCs in the peripheral blood of ST2−/− mice compared to WT mice, while MDSC percentages in bone marrow were comparable between ST2−/− and WT mice. We speculated that IL-33 might stimulate the process of extramedullary hematopoiesis (EMH)Citation39 in peripheral blood during tumor growth, because we observed detectable IL-33 level in the serum of tumor-bearing mice. However, since IL-33 levels were much higher in tumor microenvironment than in blood, the decrease of MDSC percentage in ST2−/− mice was much more significant in tumor tissues than in blood and spleen.
The impact of IL-33 on antitumor immunity have controversial reports,Citation14,15,40,41 different models were used in these studies, and they mainly observed functional changes of nature killer cells and cytotoxic T cells in IL-33-administered mice or ST2−/− mice without further investigating the mechanisms of action of IL-33. IL-33 has been reported to primarily regulate innate immune systemCitation42 and we found that the major cells regulated by IL-33 within tumor tissue were myeloid cells, particularly MDSCs. Studies by us and others have demonstrated that MDSCs also facilitate tumor metastasis through production of matrix metalloproteinases (MMPs)Citation18,19 or differentiating into endothelial cells.Citation43 Our unpublished data showed that IL-33 could not alter the expression of MMP2, 9, 13, 14 in MDSCs. Whether IL-33 affects tumor metastasis remains an open question.
Our results also demonstrated that IL-33 upregulated the expression and activity of arginase-1 in MDSCs and MDSCs exposed to IL-33 had higher T cell suppression activity. The absence of IL-33 signaling reduced the suppressive abilities of MDSCs and resulted in higher T cell frequencies in tumor microenvironment. We also found that the expression of IL-13 was upregulated in MDSCs in response to IL-33, while IL-12 expression was downregulated. This polarization may induce a Th2-driven environment inside tumor tissue and favor the polarization of M2-macrophages and Th2 cells, which have been shown to be detrimental to antitumor immunity.
Human malignancies always evolve from chronic inflammation or tissue injuries, both of these pathological processes may trigger the production of IL-33. In addition, cancer patients who receive radiotherapy or chemotherapy often have massive cell necrosis which can also lead to the secretion of IL-33. A previous study has been reported that high levels of serum IL-33 are associated with poor prognostic factors and poor survival.Citation44 Therefore, reducing IL-33 concentration may be beneficial to improving the efficacy of cancer immunotherapy, especially when it is combined with radiotherapy or chemotherapy.
Taken together, our present study might open new scenarios about MDSC biology by identifying an alarmin – IL-33 as an important regulator of MDSCs within tumor microenvironment, and reveals a mechanism that how danger signal regulates antitumor immune response. MDSCs play roles in diverse pathological processes, it also reduces pathological injury in organ transplantation or autoimmune diseases.Citation45,46 Therefore, IL-33 might be a promising target for MDSC function intervention in diseases related to disorders of the immune response.
Materials and methods
Mice and cell lines
ST2−/− mice were generously provided by Dr. Andrew N. J. Mckenzie (MRC Laboratory of Molecular Biology, Cambridge). 6–8 week female ST2−/− mice and their wild-type female littermates were used for experiments. Mice on C57BL/6 background were purchased from Shanghai Slac Animal Center. All mice were housed under specific pathogen-free condition, experiments and animal care were performed according to protocols approved by the Zhejiang University Institutional Animal Care and Use Committee. The 4T1 breast cancer, 3LL Lewis lung carcinoma, CT-26 colon cancer cell line were obtained from American Type Culture Collection (ATCC) and maintained according to standard culture conditions.
Human specimens
Blood serum samples from breast cancer patients and healthy donors, 9 cases of tumor tissues from breast cancer patients were obtained from Tissue Bank in Zhejiang Cancer Hospital, Hangzhou, China. All subjects provided informed consent for obtaining the study specimens. The study protocol was approved by the Clinical Research Ethics Committee of Zhejiang Cancer Hospital. Human breast tumor tissue arrays, which contained 148 tumor and 40 tumor-adjacent normal tissues were obtained from Shanghai Biochip Company (Shanghai, China). IL-33 in tissues was evaluated by immunohistochemistry staining using anti-human-IL-33 antibody from R&D Systems (Minneapolis, MN, USA), each sample was given two immunohistochemical scores based on the cytoplasmic and nuclear staining intensity of IL-33, respectively. The percentage of cells that positively stained for IL-33 in each sample was also analyzed.
Cell sorting
M-MDSCs and G-MDSCs were sorted on a FACSAria II cell sorter (BD Biosciences, Franklin Lakes, New Jersey). To isolate MDSCs from solid tumors, tumor tissues were minced into small pieces, dissociated, prepared to single cell suspensions and sorted.
Collection of tumor tissue supernatant
Tissue supernatant was prepared as previously reported,Citation17 with some modifications. Briefly, tumor tissues from breast cancer patients or tumor-bearing mice were excised and cut into small pieces by adding 500 μL RPMI 1640 (per 400 mg tissue). Tissue fragments were centrifuged at 400 g × 10 min. The supernatant was transferred to a 5 mL syringe and filtered through a 0.22 μm filter unit.
RNA isolation and real-time quantitative-PCR
Total RNA was extracted using Trizol Reagent (Takara, Japan), real-time PCR was conducted on CFX-Touch real-time PCR machine (Bio-Rad, Hercules, CA, USA) using SYBR Green reagent (Roche, Switzerland). All primer sequences were listed in Table S2.
Western blot
Total cell lysates were subjected to SDS-PAGE and transferred to PVDF membranes, followed by immunoblotted with indicated antibodies.
T cell suppression assay
4 × 105 splenocytes from ST2−/− mice were labeled with CFSE (Invitrogen, China) and seeded into a flat-bottom 96-well plate pre-coated with 1 μg/mL anti-CD3 (eBioscience, San Diego, CA, USA) plus 5 μg/mL anti-CD28 (eBioscience) antibodies. MDSCs, IL-33 (50 ng/mL, PeproTech), nor-NOHA (0.5 mM, Merck Millipore, Germany) were added to some wells as indicated. Cells were harvested 3 d later and stained with anti-CD4+-APC and anti-CD8+-PE-Cy5.5, CFSE dilution was analyzed by flow cytometry.
BrdU labeling
16 d after tumor inoculation, mice were injected intraperitoneally with 2 mg BrdU (Sigma, China) every 12 h. Each mouse received a total of 4 injections. 48 h after the first injection, mice were sacrificed and BrdU incorporation in MDSCs was analyzed by flow cytometry.
Detection of arginase activity, NO and ROS production
Cell lysates were collected for measuring arginase activity using QuantiChrom arginase assay kit (BioAssay Systems, Hayward, CA, USA). Nitrites were measured in culture supernatants using Greiss reaction kit (Invitrogen, China). For determination of ROS production, purified MDSC were stimulated with IL-33 (50 ng/mL), resuspended and incubated in the absence or presence of 50 nM PMA, along with 2.5 DCFDA (Beyotime, China) for 25 min, fluorescence intensity of DCFDA was evaluated by flow cytometry.
Adoptive transfer experiments
MDSCs were isolated from spleen of 4T1-bearing WT and ST2−/− mice. 5 × 106 MDSCs were injected intravenously into ST2−/− mice 3, 6, 9 d after 4T1 tumor inoculation. Tumor growth was monitored using a caliper and tumor volume was calculated according to the formula: (L × W × W)/2.
Statistical analysis
Statistical analysis was performed using Student's t test, analysis of variance (anova) and log-rank test using SPSS, with a p < 0.05 considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Weihua Xiao from University of Science and Technology of China, and Dr. Wenli Mi from Fudan University for helps in breeding the ST2−/− mice.
Author contributions
Q. W., X. C. and P. X. designed the research, X. P., W. X., B. C., Y. L., C. Q., J. R., M. ZH.,Y. S., L. CH., J. H., X. W., X. S., Q. T. and Y. L. performed the experiments and analyzed the data. P. X. and Q. W. wrote the paper.
1063772_Supplemental_Material.docx
Download MS Word (15.4 KB)1063772_supplemental_files.zip
Download Zip (17.2 MB)Funding
This work was supported by grants from the National Program on Key Basic Research Project (2014CB542101), National Natural Science Foundation of China (81373115), Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents and for Innovative Research Team in Zhejiang Province (2010R50046).
References
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13:739-52; PMID:24060865; http://dx.doi.org/10.1038/nrc3581
- Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 2010; 22:238-44; PMID:20171075; http://dx.doi.org/10.1016/j.coi.2010.01.021
- Ugel S, Delpozzo F, Desantis G, Papalini F, Simonato F, Sonda N, Zilio S, Bronte V. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol 2009; 9:470-81; PMID:19616475; http://dx.doi.org/10.1016/j.coph.2009.06.014
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 2010; 207:2439-53; PMID:20876310; http://dx.doi.org/10.1084/jem.20100587
- Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res 2010; 70:3526-36; PMID:20406969; http://dx.doi.org/10.1158/0008-5472.CAN-09-3278
- Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer 2014; 134:1077-90; PMID:23983191; http://dx.doi.org/10.1002/ijc.28449
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005; 23:479-90; PMID:16286016; http://dx.doi.org/S1074-7613(05)00311-0
- Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate lymphoid cells. Trends Immunol 2012; 33:389-96; PMID:22609147; http://dx.doi.org/10.1016/j.it.2012.04.005
- Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, Gabay C, Smith DE. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine 2008; 42:358-64; PMID:18450470; http://dx.doi.org/10.1016/j.cyto.2008.03.008
- Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, Kreutzfeldt M, Hegazy AN, Schrick C, Fallon PG et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 2012; 335:984-9; PMID:22323740; http://dx.doi.org/10.1126/science.1215418
- Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol 2011; 7:321-9; PMID:21519352; http://dx.doi.org/10.1038/nrrheum.2011.53
- Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem 2012; 287:6941-8; PMID:22215666; http://dx.doi.org/10.1074/jbc.M111.298703
- Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, Lu J, Qin W, Qi Y, Xie F et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol 2015; 194:438-45; PMID:25429071; http://dx.doi.org/10.4049/jimmunol.1401344
- Jovanovic I, Radosavljevic G, Mitrovic M, Juranic VL, McKenzie AN, Arsenijevic N, Jonjic S, Lukic ML. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur J Immunol 2011; 41:1902-12; PMID:21484786; http://dx.doi.org/10.1002/eji.201141417
- Pulaski BA, Ostrand-Rosenberg S Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res 1998; 58:1486–93; PMID:9537252
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012; 21:822-35; PMID:22698406; http://dx.doi.org/10.1016/j.ccr.2012.04.025
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008; 13:23-35; PMID:18167337; http://dx.doi.org/10.1016/j.ccr.2007.12.004
- Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, Wang X, Wang J, Yu H, Cao X et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol 2012; 188:5500-10; PMID:22544933; http://dx.doi.org/10.4049/jimmunol.1103505
- Mayuzumi N, Matsushima H, Takashima A. IL-33 promotes DC development in BM culture by triggering GM-CSF production. Eur J Immunol 2009; 39:3331-42; PMID:19750479; http://dx.doi.org/10.1002/eji.200939472
- Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 2007; 67:10019-26; PMID:17942936; http://dx.doi.org/10.1158/0008-5472.CAN-07-2354
- Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One 2011; 6:e27690; PMID:22110722; http://dx.doi.org/10.1371/journal.pone.0027690
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010; 32:790-802; PMID:20605485; http://dx.doi.org/10.1016/j.immuni.2010.05.010
- Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol 2010; 185:2273-84; PMID:20644162; http://dx.doi.org/10.4049/jimmunol.1000901
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 2009; 182:5693-701; PMID:19380816; http://dx.doi.org/10.4049/jimmunol.0900092
- Yang R, Cai Z, Zhang Y, Yutzy WHT, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res 2006; 66:6807-15; PMID:16818658; http://dx.doi.org/10.1158/0008-5472.CAN-05-3755
- Nagaraj S, Nelson A, Youn JI, Cheng P, Quiceno D, Gabrilovich DI. Antigen-specific CD4(+) T cells regulate function of myeloid-derived suppressor cells in cancer via retrograde MHC class II signaling. Cancer Res 2012; 72:928-38; PMID:22237629; http://dx.doi.org/10.1158/0008-5472.CAN-11-2863
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006; 66:1123-31; PMID:16424049; http://dx.doi.org/10.1158/0008-5472.CAN-05-1299
- Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res 2010; 70:99-108; PMID:19996287; http://dx.doi.org/10.1158/0008-5472.CAN-09-1882
- Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, Wang Z, Lang M, Stolz DB, Zheng XX et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol 2011; 187:4598-610; PMID:21949025; http://dx.doi.org/10.4049/jimmunol.1100519
- Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, van Rooijen N et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 2009; 183:6469-77; PMID:19841166; http://dx.doi.org/10.4049/jimmunol.0901575
- Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer 2008; 122:2286-93; PMID:18224687; http://dx.doi.org/10.1002/ijc.23392
- Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol 2000; 165:779-85; PMID:10878351; http://dx.doi.org/ji_v165n2p779
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/10.4049/jimmunol.181.8.5791
- Zhu Y, Hu C, Li X, Shao B, Sun H, Zhao H, Li Y. Suppressive effects of aluminum trichloride on the T lymphocyte immune function of rats. Food Chem Toxicol 2012; 50:532-5; PMID:22198605; http://dx.doi.org/10.1016/j.fct.2011.12.007
- Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 2012; 122:4094-104; PMID:23064360; http://dx.doi.org/10.1172/JCI64115
- Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat 2010; 123:39-49; PMID:19898981; http://dx.doi.org/10.1007/s10549-009-0622-8
- Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 2004; 64:6337-43; PMID:15342423; http://dx.doi.org/10.1158/0008-5472.CAN-04-0757
- Van Ginderachter JA, Beschin A, De Baetselier P, Raes G. Myeloid-derived suppressor cells in parasitic infections. Eur J Immunol 2010; 40:2976-85; PMID:21061431; http://dx.doi.org/10.1002/eji.201040911
- Gao K, Li X, Zhang L, Bai L, Dong W, Shi G, Xia X, Wu L. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett 2013; 335:463-71; PMID:23499895; http://dx.doi.org/10.1016/j.canlet.2013.03.002
- Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 2014; 134:1669-82; PMID:24105680; http://dx.doi.org/10.1002/ijc.28481
- Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 2010; 107:18581-6; PMID:20937871; http://dx.doi.org/10.1073/pnas.1003059107
- Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004; 6:409-21; PMID:15488763; http://dx.doi.org/S1535610804002703
- Sun P, Ben Q, Tu S, Dong W, Qi X, Wu Y. Serum interleukin-33 levels in patients with gastric cancer. Dig Dis Sci 2011; 56:3596-601; PMID:21643739; http://dx.doi.org/10.1007/s10620-011-1760-5
- Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res 2012; 54:275-85; PMID:22535241; http://dx.doi.org/10.1007/s12026-012-8335-1
- Natarajan S, Thomson AW. Tolerogenic dendritic cells and myeloid-derived suppressor cells: potential for regulation and therapy of liver auto- and alloimmunity. Immunobiology 2010; 215:698-703; PMID:20605054; http://dx.doi.org/10.1016/j.imbio.2010.05.024