
ABSTRACT
Antitumor immunity can be enhanced by the coordinated release and delivery of antigens and immune-stimulating agents to antigen-presenting cells via biodegradable vaccine carriers. So far, encapsulation of TLR ligands and tumor-associated antigens augmented cytotoxic T cell (CTLs) responses. Here, we compared the efficacy of the invariant NKT (iNKT) cell agonist α-galactosylceramide (α-GalCer) and TLR ligands (R848 and poly I:C) as an adjuvant for the full length ovalbumin (OVA) in PLGA nanoparticles. We observed that OVA+α-GalCer nanoparticles (NP) are superior over OVA+TLR-L NP in generating and stimulating antigen-specific cytotoxic T lymphocytes without the need for CD4+ T cell help. Not only a 4-fold higher induction of antigen-specific T cells was observed, but also a more profound IFN-γ secretion was obtained by the addition α-GalCer. Surprisingly, we observed that mixtures of OVA containing NP with α-GalCer were ineffective, demonstrating that co-encapsulation of both α-GalCer and antigen within the same nanoparticle is essential for the observed T cell responses. Moreover, a single immunization with OVA+α-GalCer NP provided substantial protection from tumor formation and even delayed the growth of already established tumors, which coincided with a prominent and enhanced antigen-specific CD8+ T cell infiltration. The provided evidence on the advantage of antigen and α-GalCer coencapsulation should be considered in the design of future nanoparticle vaccines for therapeutic purposes.
Abbreviations
| NP | = | Nanoparticle |
| PLGA | = | poly lactic-co-glycolic acid |
Introduction
Current cancer immunotherapy strategies are aimed at the generation of reliable and long-lasting immune responses against tumor antigens. In this context, CTLs have received most of the attention due to their ability of specifically killing tumor cells, as well as generating a memory phenotype preventing recurrence. Various pre-requisites have to be met for a full-blown activation of CTLs such as cross-priming by an activated antigen-presenting dendritic cell (DC) and a stimulatory milieu consisting of helper T cells (Th cells), NK cells and macrophages which ensure a favorable cross-talk through secreted cytokines.Citation1,2 Therefore, when designing a new therapeutic vaccine-based strategy, it is important to include adequate stimuli in addition to antigen to activate the complete immune machinery.
Natural Killer T cells (NKT cells) are specialized immune cells that share the expression of a T cell receptor together with NK cell receptors and have the ability to bridge the innate with the adaptive immunity by the swift production of pro- and anti-inflammatory cytokines upon activation.Citation3,4 The most studied subtype of NKT cells are the invariant TCR bearing NKT cells (iNKT cells) cells that recognize glycolipid antigens such as α-GalCer presented by CD1d molecules on antigen presenting cells (APCs). This recognition by iNKT cells stimulate both iNKT cells and APCs which in turn, induce the secretion of various pro-inflammatory cytokines and activate a broad spectrum of immune cells against the tumor.Citation5 Among these IFN-γ and IL-12 are the most abundant, which can switch the immune response in favor of a Th1 driven response. Additionally, the CD40–CD154 ligation during the presentation of glycolipids to iNKT cells evokes DC maturation up-regulating other co-stimulatory ligands.Citation6,7 Based on these characteristics, iNKT cells have been called the ‘swiss army knife of the immune system’ and harnessing of iNKT cells should be considered in any antitumor vaccination modality.Citation8
α-GalCer has been regarded as a potent vaccine adjuvant.Citation4 Especially enhanced antigen-specific antitumor CD4+ and CD8+ T cell responses were documented in combination with the widely used OVA antigen.Citation9-11 Type I iNKT cell activation by systemic administration of α-GalCer was shown to protect mice from chemical or oncogene-induced primary tumor formation.Citation12 Furthermore, when loaded into tumor cells, α-GalCer inhibited the onset of transplantable tumors, and its systemic administration slowed down metastatic tumors.Citation13,14 The ability of α-GalCer to induce antigen non-specific rejection of B16.F10 lung metastases was shown to be IFN-γ dependent and to require activation of iNKT cells followed by NK cell activation.Citation15
Over the last few years, several clinical trials have been performed in cancer patients either by systemic administration of α-GalCer or adoptive transfer of α-GalCer-loaded autologous DCs.Citation16 However, systemic administration of α-GalCer and its efficacy is hampered due to its insolubility in aqueous media. Hence, some molecular modifications have been performed to increase its solubility or DCs have been ex-vivo loaded with α-GalCer improving in vivo and in vitro iNKT cell responses in mice and humans.Citation17-21
Delivery of particulate antigens or adjuvants by employing poly(lactic-co-glycolic acid) (PLGA) NP has proven advantages over soluble administration such as more efficient uptake, controlled release and improved immunogenicity.Citation22 Within the context of NKT cell stimulation, α-GalCer, MPLA and OVA coated PLGA micro and NP were injected to mice which induced a more robust and long sustained antigen-specific IgG responses than administrations in solution.Citation23 Similar α-GalCer coated silica microparticles have shown to be mostly taken up by macrophages and DCs located in the marginal zone of spleens and CD169+ macrophages in the lymph nodes of mice.Citation24,25 In addition, α-GalCer coated PLA particles were demonstrated to prevent iNKT cell anergy occurring as unresponsiveness to sequential stimulation by α-GalCer.Citation26 Nevertheless, lower in vivo and in vitro NKT cell cytokine responses were observed with both PLA and PLGA formulations of α-GalCer than administration in solution.Citation26,27 DEC205 targeted delivery of α-GalCer loaded PLGA particles to DCs were able to increase iNKT cell responses and demonstrated increased CD8+ T cell responses against OVA antigen.Citation28 In this study, we used a biological dose-response assay to quantify the actual amount of PLGA entrapped α-GalCer to obtain a more precise evaluation of nanoparticle mediated delivery. Using these well-characterized NP we challenged the co-delivery concept of protein and glycolipid antigens in detail by comparing various combinations of α-GalCer and full-length OVA. We also for the first time compared different types of adjuvants by means of in vivo and in vitro immune stimulation potentials: PLGA encapsulated α-GalCer versus more classical TLR-ligands. We demonstrate that α-GalCer and OVA loaded PLGA NP are not only superior to TLR-ligands but also induce both prophylactic and therapeutic responses in the B16.OVA mouse melanoma model.
Results
Co-encapsulation of α-GalCer and OVA elicit superior in vivo stimulation
To determine the in vivo efficacy of α-GalCer on vaccination induced antigen-specific killing, we compared responses of OVA and α-GalCer containing NP(OVA+ α-GalCer) with OVA+ TLR-ligands (R848 and poly I:C) containing NP(OVA+ TLR). Only OVA antigen encapsulating NP served as a control NP(OVA). The presence of TLR-Ligands within the OVA NP provided slightly enhanced killing of SIINFEKL peptide-pulsed syngeneic target cells over control NP (). In contrast, when co-encapsulated within the OVA nanoparticles, α-GalCer was able to generate a significant population of antigen-specific CTLs and induce the killing of SIINFEKL peptide-pulsed target cells up to 90 percent (). TLR-Ligands could not match these results with even 10 times higher doses (Fig. S1A). Interestingly, administration of the same dose of (1ng) free α-GalCer mixed with the same amount of (0.66µg) OVA loaded (NP(OVA)+ free α-GalCer) led to only 40 percent of specific killing and minimal induction of tetramer+ CTLs (). Similar results at around 40 percent killing were obtained with a mixture of NP loaded with α-GalCer alone and OVA alone (NP(OVA)+ NP(α-GalCer)) and injected at the same tail vein (). When OVA NP and free α-GalCer injected in different veins (NP(OVA)+ free α-GalCer*), the vaccine-induced responses decreased even further, suggesting that co-encapsulation of α-GalCer and the OVA antigen is crucial for the induction of a full blown CTL activation.
Figure 1. In vivo antigen-specific T cell responses induced by nanoparticles. (A) Mice were vaccinated with indicated nanoparticle combinations and transferred with SIINFEKL peptide-loaded syngeneic target cells and irrelevant peptide-loaded control cells. Primary antigen-specific in vivo killing responses (left) and SIINFEKL H-2Kb tetramer-positive cell percentages (right) obtained by OVA and α-GalCer or TLR-L injections were demonstrated. Results of three independent experiments were pooled. n> 6 for each group (B) OT-I T-cell proliferation induced by nanoparticle administration to wild type naive mice, n=5 (left). Comparison of OVA+ α-GalCer nanoparticle induced proliferation of OT-I T cells transferred to either CD1d KO or wild-type mice, n=30 (right). Parenthesis indicates encapsulation, ↔ indicates injection of NP(OVA) and free α-GalCer to different veins consequently. 0,66ug OVA, 1ng α-GalCer, 143ng R848 and 70ng poly I:C was introduced within NPs. Mean values were shown with standard error, ns: no significance, * p < .05, ** p < .01, *** p < .001
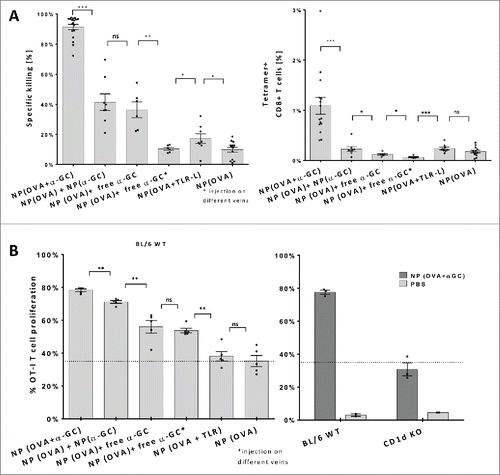
α-GalCer was also able to provide adjuvant functions to more than one epitope present within the same particle (NP (OVA+HPV+ α-GalCer)) (Fig. S2A). While the response against the HPV peptide was less than OVA in the same particle we did observed a similar killing activity as induced by another short peptide, SIINFEKL. When co-encapsulated with α-GalCer and delivered at same molar amounts SIINFEKL failed to induce comparable results as the full length OVA protein (Fig. S2A). Next, a group of mice was depleted of the CD4+ cells to question if the reduced response to short peptides was due to a possible help by CD4+ T cells to full length OVA protein which also includes MHC II associated peptides. However, no reduction of cytotoxicity was detected at these mice indicating the independence of this system from helper T cells (Fig. S2A).The enhanced stimulatory potential of co-encapsulated OVA+α-GalCer was confirmed by in vivo proliferation of CD8+ T cells derived from OT-I mice and transferred to either wild-type or CD1d KO mice, the latter incapable of presenting α-GalCer. In wild-type mice, ~35 percent of the OT-I cells proliferated after vaccination with OVA loaded nanoparticles, which could not be increased by the co-encapsulation of TLR-ligands (). In line with the observed killing capacity, co-encapsulation of OVA with α-GalCer also led to highest proliferation of OT-I T cells (). Furthermore, in CD1d KO mice, lacking the α-GalCer/CD1d presentation pathway, proliferative responses were at baseline levels as observed in WT mice vaccinated with only OVA loaded NP (). This finding demonstrates the specificity of the response and that the observed proliferation is dependent on the α-GalCer/CD1d presentation pathway. Intriguingly, the α-GalCer in separate NP was more efficient in inducing proliferation than free α-GalCer accompanying OVA NP ().
α-GalCer NP stimulate similar cytokine responses both in vitro and in vivo
Next, we isolated splenocytes from naive BL/6 or CD1d KO mice and cultured them with increasing doses of α-GalCer. Both NP (OVA + α-GalCer) and NP (α-GalCer) induced similar levels of IFN-γ secretion when compared to free α-GalCer in solution (). As expected, no IFN-γ was detected in CD1d KO mice splenocyte cultures supplemented with either NP (OVA+α-GalCer) or NP (α-GalCer) (). Furthermore, even the high-concentrations (128ng) of NP(OVA+TLR-L) were not able to stimulate IFN-γ production comparable to that of α-GalCer containing nanoparticles. Yet, they generated high IL-6 levels, demonstrating that the TLR ligand containing particles were functional since IL-6 production is entirely independent of the presence of αGalCer or CD1d (). The above mentioned in vitro responses of splenocytes were also unrelated to the presence of OVA antigen in the particles because these could neither induce IFN-γ nor IL-6 secretion.
Figure 2. Cytokine responses obtained by nanoparticle encapsulated α-GalCer and TLR-ligands. (A) IFN-g levels of 48h splenocyte cultures with indicated NPs or free α-GalCer in solution. (B) IL-6 levels of 48h splenocyte cultures with indicated NPs. Splenocytes were isolated from either naive wild-type BL/6 or CD1d KO BL/6 mice (A-B, n>4). (C) IFN-g serum levels measured 24 h after iv. injections of 1ng α-GalCer either in nanoparticles or free. Vaccinations were repeated 2 times on days 7 and 14, empty particles were used as a control, n≥3. Mean values were shown with standard error. (D) IL-2 response curves of DN32D3 NKT cell hybridoma cultured 24h with α-GalCer loaded BMDCs. α-GalCer was serially diluted in DMSO, RMPI medium or PBS 0.05% tween 20 (Vehicle) and cultured with BMDCs at a range of 25ng-0.01ng/mL. One representative graph of three experiments was shown.
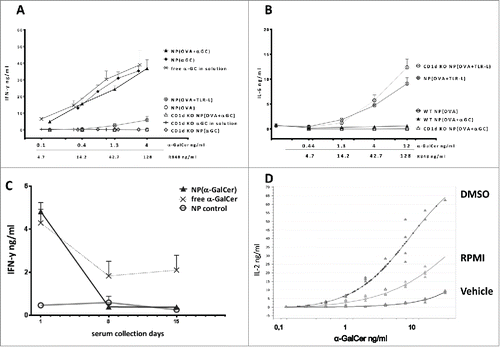
In parallel with our in vitro findings, we could demonstrate that significant IFN-γ serum levels were induced by injecting 1ng α-GalCer containing NP (). In accordance with the literature,Citation29 repeated injections of both particulate and free α-GalCer resulted in lower IFN-γ secretion as a result of iNKT cell anergy (). Together, these findings demonstrate that the activity of α-GalCer NP is entirely CD1d dependent.
We observed lower responses when the free α-GalCer was diluted with either RPMI or Vehicle (PBS %0.5 Tween) rather than DMSO (). Additionally, similar in vitro IL-2 levels were obtained when OVA+ α-GalCer NP were dissolved by DMSO to release the encapsulated α-GalCer (Fig. S1B). These findings suggest that PLGA encapsulation of α-GalCer do not interfere with its antigenic properties or presentation.
NP(OVA+ α-GalCer) induce long-term protection from B16-OVA melanoma formation
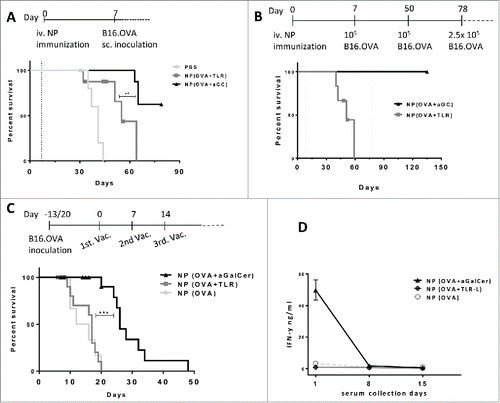
Next, we probed the prophylactic immunization potential of different nanoparticles. Mice were vaccinated with NP containing OVA antigen and 7 d later subcutaneously inoculated with 1×105 B16.OVA melanoma cells (). NP(OVA +α-GalCer) provided protection to a significant proportion of mice. Much lower protection was observed in mice that received NP(OVA+TLR-L) (). Multiple tumor inoculations were performed to test the duration of immune protection induced by NP(OVA +α-GalCer) vaccination (). Whereas all mice of the NP(OVA+TLR-L) group had to be euthanized due to tumor size limits until day 60, no tumor was observed in the NP (OVA+αGalCer) group, even if they were re-challenged with B16.OVA cells as late as day 50 and 78 (). Cumulatively, 13 out of 16 animals were tumor free at the end of 3 independent experiments (). Thus, antigen and α-GalCer co-encapsulation is capable of generating a substantial tumor resistance when administered in a protective setting.
Figure 3. Antitumor effects of (OVA+α-GalCer) nanoparticle immunization. (A) Mice were immunized on day 0 with indicated vaccines and B16.OVA cells were subcutaneously inoculated on day 7(dotted line). Survival results of two independent experiments were shown. Censored data points were shown with symbols. PBS, n= 7; NP(OVA+TLR-L), n=8 and NP(OVA+ α-GalCer), n=8. (B) Vaccinations were performed on day 0 and tumors were inoculated on day 7 as in A. NP(OVA+ α-GalCer) group was re-challenged with tumor cells on day 50 and 78 (dotted line). NP(OVA+TLR-L) n=6, NP(OVA+ α-GalCer) n=8 . (C) Mice were first subcutaneously inoculated with B16.OVA cells and vaccinations were started individually when each tumor reached ≈30mm3 and repeated three times weekly. NP(OVA), n=7, Mitv=26mm3; NP(OVA+TLR-L), n=13, Mitv=33mm3 ; NP(OVA+α-GalCer), n=19, Mitv=33mm3. Data points of mice culled due to early necrosis were censored. Censored data points were shown with symbols. Mitv: Mean initial tumor volume. *** P < .001.(D) Serum samples were collected 24 h after each vaccination of tumor-bearing mice in (C) and IFN-g levels were determined by ELISA. n=7. Mean results were shown with standard error.
Treatment with NP(OVA+α-GalCer) inhibits the growth of established B16-OVA tumors
To investigate the therapeutic efficacy of antigen + α-GalCer co-encapsulation; mice were subcutaneously inoculated with B16.OVA melanoma cells prior to immunization. The vaccinations were started when each tumor reached ~30–40mm3 and were repeated three times at weekly intervals.
While NP(OVA) or NP(OVA+TLR-L) could not provide any survival advantage over PBS injection, NP(OVA +α-GalCer) significantly inhibited tumor growth and extended the mean (overall) survival of the mice (, Fig. S3). At the same time, we observed a profound iNKT cell anergy with the mice receiving multiple treatments of NP(OVA +α-GalCer) (). Furthermore, experiments demonstrated that even a single dose of NP(OVA +α-GalCer), containing as little as 1ng of α-GalCer resulted in a significant delay of tumor growth (Fig. S3).
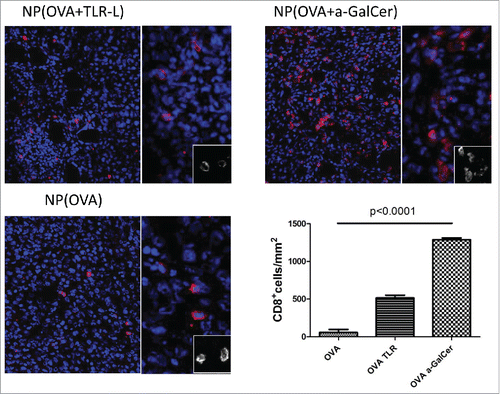
Immunofluorescence imaging of tumor specimens demonstrated increased CD8+ T cell infiltration in tumors of mice treated with NP (OVA+α-GalCer) when compared to mice treated with NP (OVA+TLR-L) or NP(OVA) (). In parallel with the microscopy results, an increased proportion of CD3+ CD8+ NK1.1- cells were detected by flow cytometry in tumor tissues (Fig. S4A and B). The intra-tumoral CD8+ T cells were also positive for SIINFEKL tetramer indicating their specificity against tumor antigen.
Figure 4. Intratumoral T cell infiltration after therapeutic vaccinations. Similar sized tumors were isolated 8 d after first vaccination. Frozen tumor tissues were screened for CTL infiltration by CD8+ (red) and DAPI (Blue) staining by Vectra automated quantitative imaging system. NP(OVA), n=2; NP(OVA+TLR-L), n=4; NP(OVA+α-GalCer), n=2.
Discussion
Induction of long-lasting anticancer T cell responses is one of the primary objectives of cancer immunotherapy. Both the limited number of tumor antigens together with their low immunogenicity form a major obstacle in generating strong antitumor responses. In an attempt to activate specific components of the immune system more directly, platforms have been developed to encapsulate antigens and adjuvants in nano-sized particles that enhance antigen-specific T cell responses.Citation30-33 Targeted delivery of these particles to DC s have provided even more efficacy.Citation28,34
In the present study, we compared the adjuvanticity of PLGA encapsulated α-GalCer with that of more classical TLR ligands in combination with OVA protein. We observed that α-GalCer served as a much better adjuvant in inducing CD8+ T cell responses when compared to TLR ligand within NPs. Even though the α-GalCer alone did not take advantage of particle formulation, the best results were surprisingly obtained when both antigen (OVA) and α-GalCer are present in the same NP. In this case, only subtle amounts of antigen and α-GalCer were enough to stimulate significant antitumor responses.
Previously, virus-like particles attached with α-GalCer failed to induce IFN-γ secretion when compared soluble α-GalCer. However, prophylactic vaccination with particulate forms of α-GalCer provided a better protection against B16.gp33 tumor formation.Citation35 In another study, α-GalCer and OVA loaded DC derived exosomes were able to control B16.OVA tumor growth better than soluble α-GalCer and OVA, while at the same time preventing development of iNKT cell anergy.Citation36 So far, only a few studies reported on the immunologic or therapeutic antitumor effect of α-GalCer in conjunction with PLGA particles.Citation26-28 In a study by Thapa et al., it was demonstrated that 500-1,000nm sized α-GalCer coated PLGA particles induced similar cytokine responses as the free glycolipid in vitro, whereas the systemic application induced 10-fold lower IFN-γ serum levels when compared to solubilized α-GalCer.Citation26 Macho Fernandez et. Al. also reported a similar discrepancy between the efficacy of soluble versus particulate α-GalCer both in vitro and in vivo, albeit less pronounced with two times lower IFN-γ serum levels.Citation27 In contrast, we find similar cytokine responses with nanoparticle or free α-GalCer both in vitro as in vivo. To carefully quantify the amounts α-GalCer in nanoparticles, we performed a biological dose-response assay. In the course of these experiments, we also observed that solvents used in diluting free α-GalCer could significantly affect its biological availability due to its insolubility in aqueous media and, therefore, might be affecting the previous results. We suggest dissolving α–GalCer in DMSO for free administration and performing a biological assay to quantify encapsulated amount in NP for a more justified comparison. Therefore, the discrepancy between our results and the previous reported studies may be due to the different solvents used in diluting free α-GalCer, different particle diameters as well as incorrect α-GalCer contents of particles due to the methods used for quantification.
Previously, Tacken et.al. demonstrated co-encapsulation of OVA and TLR-ligands in PLGA particles leading to a dramatic boost of cytotoxic T-cell mediated killing in comparison to TLR-ligands in solution when mixed with OVA NP.Citation34 Schlosser et al also made similar observations showing the importance of co-encapsulation of CpG, poly I:C and SIINFEKL in PLGA particles.Citation37 We here demonstrate that this holds not only true for TLR ligands but also for α-GalCer co-encapsulation with OVA, as it became apparent that the generation of antigen-specific T cells and killing of antigen-pulsed cells was dramatically reduced when antigen and α-GalCer were separately injected into different veins. Similar findings were reported when soluble OVA and α-GalCer were injected at different time points.Citation7 Together, all these results support the notion that co-delivery of α-GalCer and antigen to the same cell is essential. Although the underlying mechanism is not entirely clear, MyD88 dependent TLR signaling can selectively increase the cross-presentation of the phagosomes that contain both antigen and TLR-ligands, which could contribute to the enhanced responses resulting from TLR-ligand and antigen co-delivery.Citation38 Such a sub-cellular selectivity is less likely to be relevant for α-GalCer co-delivery as this is described to induce DC maturation independent of MyD88 down stream signaling.Citation9 On the other hand, α-GalCer presenting APCs gain a co-stimulatory advantage also to which CTL trafficking was promoted by an iNKT cell-mediated CCL17 driven fashion.Citation6,7, Citation9,39 This cognate licensing of DCs by iNKT cells may explain the enhanced immune responses we observed with OVA+ α-GalCer co-encapsulated NP.Citation7,39 The NP were taken up mostly by F4/80+ macrophages and to a lesser extend by DCs in the liver and spleen. An increased iNKT cell expression of CD40L demonstrate the co-stimulatory cross-talk between APCs and iNKT cells after nanoparticle vaccinations (Fig. S6A and B).The notion of cognate licensing is supported by our findings that CD40 and CD86 expressions of APCs were hampered when α-GalCer is injected in solution instead of NP (Fig. S7).
We hypothesized that the requirement of professional APCs in processing full-length proteins would ensure a co-stimulatory signal through CD1d and iNKT cell activation whereas, if they are released to circulation, short peptides might be presented by non-APCs which do not express co-stimulatory molecules and CD1d and lead to a tolerogeneic response. As expected, SIINFEKL peptide and α -GalCer loaded particles induced lower levels of specific killing in comparison to the same molar amount of full-length OVA protein and α -GalCer introduced within NP (Fig. S2A). This reduction was not due to loss of a T-cell help provided by an MHC-II epitope present on full length OVA protein. However, the same mice demonstrated similar tetramer positive CTL ratios supporting the tolerogeneic or less activated phenotype induced by non-professional APCs (Fig. S2B). Moreover, it is known that SIINFEKL peptide cargo has a very high-burst release from PLGA particles in one d when compared to OVA protein which takes d to weeks.Citation40,41 This low burst-release of full length OVA protein may also ameliorate CD8+ T cell stimulation as previously published in a setup with a long OVA peptide.Citation42
The antigen-specific in vivo killing response is an important prediction of antitumor immune responses. Concordantly, OVA+α-GalCer nanoparticles provided up to a total protection from B16.OVA melanoma formation after a single immunization, which was persistent for more than two months demonstrating a substantial memory. Finally, when tested on mice with already established tumors, OVA+α-GalCer NP exhibited a significant delay in tumor growth and extended mean survival. It is of note that these effects were obtained despite a profound iNKT cell anergy and without any OT-I T cell transfer and totally due to priming of naive T cells and generation of OVA-specific CTLs which were capable of infiltrating to the tumor sites corresponding to the extended stabilization of tumor volumes. The described CD1d-iNKT cell driven system exploited by nanoparticle delivery of the low dose α-GalCer do not require but instead replace the CD4+ T cell help. Additionally, CD154 positivity of iNKT cells was higher than basal levels until day 9 after one vaccination which may still provide co-stimulation despite the lack of IFN-y secretion. Moreover, cytotoxic immune responses may be obtained simultaneously to more than one antigen at the same time which grants a wider therapeutic potential of various stimulants and (patient-specific) antigens paving the way for more personalized cancer treatment.
Materials and methods
Reagents and Abs
PLGA (Resomer RG 502 H, lactide/glycolide molar ratio 48:52 to 52:48) was purchased from Boehringer Ingelheim (Germany). Solvents for PLGA preparation (dichloromethane) were obtained from Merck (Germany). Polyvinyl alcohol (PVA) was obtained from Sigma (The Netherlands). R848 was from Enzo Life Sciences poly I:C from Sigma-Aldrich, and endotoxin-free OVA from Hyglos. OVA (257 – 264) SIINFEKL and HPV16 E7(49–57) obtained from Anaspec. α-GalCer was purchased from Funakoshi Co. Ltd. Dimethyl sulfoxide (DMSO) ≥99.9% and Tween 20 was obtained from Sigma (The Netherlands). Atto647N was obtained from Atto-tec GmbH. RPMI 1640 medium (Life Technologies Inc..). PE-conjugated CD3, APC-Cy7 conjugated CD45.2 (BD Biosciences), PerCP conjugated CD8+α (Biolegend), and APC-conjugated H2-Kb/SIINFEKL-Tetramer (Sanquin), PE conjugated CD1d- α-GalCer dextramer (Immudex),FITC conjugated CD11b, PE/Cy7 conjugated CD11c, Alexa488 conjugated F4/80 and NK1.1 (Biolegend), PE/Cy7 conjugated CD154 (Biolegend), Celltrace CFSE, Celltrace- violet and Celltrace red (Life technologies Inc.), were used in flow cytometric cell staining. For the depletion studies, anti-mouse CD4+ antibody clone GK1.5 was obtained from BioXcell. For the detection of CD4+ populations PerCP/Cy5.5 conjugated anti-mouse CD4+ antibody clone RM4-4 was obtained from Biolegend.
Nanoparticle synthesis
PLGA NP with entrapped OVA alone or in combination with R848, poly I:C or α-GalCer were prepared using an o/w emulsion and solvent evaporation–extraction method as previously described.Citation33 In brief, 100 mg of PLGA in 3 mL of dichloromethane containing, 5 mg OVA, 1.6mg SIINFEKL or HPV16 E7, 2 mg poly I:C, 0.5 mg R848 or 0.1 mg α-GalCer and 0.5mg Atto647N was added dropwise to 25mL of aqueous 2% polyvinyl alcohol and emulsified for 120 seconds using a digital sonicator from Branson Ultrasonics (Danbury, CT). The solvent was evaporated overnight at 4°C and NP were collected by centrifugation at 14.000 rpm for 20 min, washed six times with distilled water and lyophilized.
Nanoparticle characterization
The size and polydispersity index of the NP was analyzed by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern,UK) as described before.Citation33 Encapsulation efficiency for OVA was determined by measuring the protein content of digested particles using Coomassie Plus Protein Assay Reagent (Pierce) according to the manufacturer's protocol. Encapsulation of SIINFEKL peptide was determined by measuring protein content of DMSO dissolved particles using CBQCA protein quantification kit (Life Technologies) according to the manufacturer's protocol. Concentration of R848 in NP was determined using a high-performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) equipped with a Supelcosil lC-18-T column (150 mm × 4.6 mm) with a constant flow rate of 1 mL/min and detection at a wavelength of 220nm. R848 was extracted from NP by dissolving 1 mg NP in 100μl DMSO followed by centrifugation at 14.000 rpm for 10 minutes. 25µL supernatant or standard was assayed by HPLC. The quantity of R848 and was calculated by interpolation from the standard curves. Entrapment efficiency for poly I:C was determined by measuring the absorption of digested particles or standard on a Nanodrop 2,000 (Thermo Scientific) the amount of poly I:C was calculated by interpolation from the standard curves. Nanoparticle characteristics are demonstrated at Table S1.
BMDC/DN32 cultures and quantification of α-GalCer encapsulation
For standards, stock α-GalCer was dissolved 1mg/mL in DMSO (≥99.9%) and serially diluted in DMSO(≥99.9%), RMPI 1640 medium or vehicle (PBS, 0.05% tween 20). PLGA NP were broken down with DMSO for 15 min. at 80°C. Both dissolved PLGA and standards were further diluted 1/1,000 in complete RPMI medium and cultured with 5×104 BMDCs. 24 h later supernatants were aspirated, and 105 DN32.D3 mouse iNKT hybridoma cells were added to the culture. IL-2 levels were determined by ELISA after 24-h incubation. The α-GalCer contents of dissolved PLGA NP were determined according to the standard curve of DMSO dilution obtained from the same experiment.
Mice and tissue
Wild-type C57BL/6JRccHsd (Harlan) and OT-I C57BL/6-Tg(TcraTcrb)1100Mjb/Crl (Charles River) mice were maintained under specific pathogen-free conditions at the Central Animal Laboratory (Nijmegen, the Netherlands). CD1d−/− (iNKT cell-deficient) C57BL/6 mice were bred and maintained at the animal facility of the John Radcliffe Hospital, Oxford. Drinking water and food were provided ad libitum. The experiments were performed according to guidelines for animal care of the Nijmegen Animal Experiments Committee and the Animal Ethics Committee of Cancer Research UK and the United Kingdom Home Office and in accordance with the ethical standards described in the declaration of Helsinki. Spleens were isolated under sterile conditions and stored at 4°C in RPMI 1640 medium suplemented with 100 U/mL penicilium and 100 μg/mL streptomycin until processing for maximally 2 h. Spleen cells were meshed through a 100um cell strainer by using a syringe plunger. Cell suspension was spun at 400xg for 5 min and resuspended in 3mL of 1x ammonium chloride solution for the lysis of erythrocytes. After 5 min of incubation at room temperature cells were washed with 20mL of PBS 2 times. Fresh materials were used in all experiments. Cells were counted by a hemacytometer. Fixable Viability Dye eFluor® 780 (ebioscience) or zombie violet (Biolegend) dyes were used to exclude dead cells in flow cytometry applications.
In vitro stimulation by PLGA nanoparticles
2×105 splenocytes isolated from either naïve wild-type C57BL/6 mice or CD1d knockout C57BL/6 mice were cultured with graded doses of NP or free α-GalCer in complete RPMI 1,640 medium supplemented with 10% FBS, 2.1 mM ultra-glutamine and 50 μM 2-Mercaptoethanol. The serum is routinely tested for assay performance. 48-h culture supernatants were screened for IFN-γ and IL-6 by ELISA.
In vivo cytotoxicity assay and cytokine analysis
NPs re-suspended in sterile PBS were injected into the tail veins of C57Bl/6 mice. The introduced NPs were contained 0,66µg OVA, 1ng α-GalCer, 143ng R848, and 70ng poly I:C. All groups have injected with same doses for the same components unless mentioned otherwise. Some animals were injected with free α-GalCer and NP(OVA) on opposite tail veins consequently within 1 min. Some animals were injected with PBS as a negative control. Blood samples were collected 24 h after vaccinations in anticoagulant free tubes’ kept at room temperature for 1h and serum was isolated by centrifugation. Serum samples were stored at −20°C. IFN-γ levels of serum samples were determined by ELISA. Seven to eight d after IV injections, splenocytes were prepared from wild-type C57Bl/6 mice and divided into two groups. Cells were pulsed with either 1 μg/mL relevant OVA-SIINFEKL peptide or 1 μg/mL irrelevant E1A peptide and labeled with 5 µM Celltrace-violet (relevant OVA) or 5 µM CFSE (irrelevant E1A) respectively. In order to asses killing of two epitopes by NP(OVA+HPV+ α-GalCer) vaccinations, cells were divided into three equal groups and pulsed with either 1 μg/mL relevant OVA-SIINFEKL peptide, 1 μg/mL relevant HPV16 E7 or 1 μg/mL irrelevant E1A peptide and labeled with 5 µM CFSE (relevant OVA), 5 µM Celltrace-violet (relevant HPV16 E7) or 5 µM Celltrace-red (irrelevant E1A) respectively. Stained cells were mixed and 10 × 106 total cell mixtures were injected intravenously into the immunized mice. The following day, splenocytes were isolated, stained with CD3, CD8+, CD4+, CD154 SIINFEKL tetramer and CD1d-aGalCer dextramer and cell percentages were measured by FACS Verse (BD) flow cytometer. FlowJo X software was used for analyzes of cytometry data. The PBS vaccinated mice were used to determine control ratio for each experiment.
Killing was measured by flow cytometry using the following formula:
Ratio = Irrelevant Percentage : Relevant Percentage
Percent Specific Lysis = [1-(PBS vaccinated control ratio/Experimental ratio)] × 100
For in vivo depletion of CD4+ cells, a group of mice received 500µg/mouse of anti-CD4+ (GK1.5) antibody by ip. route 2 d before vaccination with NP(OVA+aGalCer). Antibody administration was repeated on days 1, 4 and 7 with 200 µg/mouse doses. Total depletion of CD4+ T cells and iNKT cells were confirmed by FACS staining.
In vivo proliferation assay
OT-I mice splenocytes were depleted of CD1d expressing cells by B220, CD11b and CD11c magnetic beads according to the manufacturer protocol (Miltenyi Biotec). Remaining cells were stained with 5 µM CFSE and 3×106 transferred to either naive or CD1d KO mice by IV vaccination. Twenty four h later mice were vaccinated with NPs. Four d later mice were euthanized, and mononuclear cells were isolated from spleens. Proliferations of CD3+ CD8+ cells were determined by reduction of CFSE intensity.
In vivo maturation assay and fluorescent particle tracking
Naive BL/6 mice were injected with either NP(OVA+TLR-L); NP(OVA+ α-GalCer) or NP(OVA)+ free α-GalCer in the opposite vein. 0.66µg OVA and 1ng of α-GalCer doses were applied as used with in vivo cytotoxicity and proliferation assays. Sixteen h later mice were culled, and splenocytes were analyzed by flow cytometry. Red pulp macrophages were selected as CD11b+, F4/80+; B cells as B220+,CD19+; CD8α+ DCs as CD11chi, B220-, CD8α+. CD40 and CD86 expression patterns were compared.
CD154 (CD40L) expression of iNKT cells were checked 1, 5 and 9 d after vaccinations with NP(OVA+ α-GalCer) by staining liver and spleen cell suspensions with CD1d- α-GalCer dextramer and CD154 antibody. An isotype staining and non-vaccinated mice cells were used as control in all experiments.
Sixteen h after iv.injection of 2mg NP(OVA+ α-GalCer+Atto647N) particles, mice were culled and spleen and liver cells were checked for Atto647 fluorescence by flow cytometry. CD11b, CD11c, F4/80 and CD8+ antibodies were also used to discriminate major antigen presenting cells.
Tumor models
In order to assess protection from tumor onset, mice were vaccinated with PLGA NP containing 0.66µg OVA alone or together with 1ng α-GalCer or 143ng R848 and 70ng poly I:C in PBS. One-week later mice were inoculated with 1×105 B16.OVA cells on their hind flank region and tumor development was monitored daily. For one independent experiment, additional tumor cell challenges were performed to OVA+ α-GalCer immunized group on days 50 and 2.5×105 cells on day 78. For therapeutic model, mice were first inoculated with 1×105 B16.OVA cells in their hind flank region. Vaccinations were started when tumor size of a mouse reach ≈30mm3 (10–23 d after tumor cell inoculation). Allocations were performed so that all groups had similar mean tumor volumes. Vaccinations were repeated three times weekly for some groups. The animals were euthanized when tumors reach 1500mm3 volume. Mice with ulcerative tumors were culled due to ethical reasons and data points were censored.
For the detection of T cell infiltration to the tumor site, some mice with similar tumor sizes were sacrificed 8 d after first vaccination. Single cell suspensions were obtained from tumors after collagenase/DNAse treatment and consequent 1.077g/mL density gradient separation by ficoll. Leukocytes were gated by CD45.2 positivity. NK1.1, CD3, CD4+, CD8+ antibodies and SIINFEKL tetramer were used to determine T cell proportions.
Immunofluorescence staining
Tumors were isolated 24 h after second vaccination, frozen in liquid nitrogen and stored at −80 C°. Cryosections of 10µm thickness were placed on glass slides and fixed with 4% formaldehyde PBS. Blocking of slides was performed with 5% normal goat serum following staining of rat anti-mouse CD8+ antibody and consequently with A-647 conjugated goat anti-rat secondary antibody. DAPI was used as a nuclear counterstain. Stained slides were loaded onto the slide scanner of Vectra® automated quantitative multispectral imaging system. Nuance 3.0.0 software was used to build a spectral library in order to unmix the autofluorescence background and subsequently the unmixed imaged were loaded into the inForm software for cellular segmentation and quantitative analysis of CD8+ cells density in the tumor tissue.
Statistical analysis
Mann–Whitney test was used to compare two different groups in . Log-Rank(Mantel-Cox) tests were used to compare survival curves on . Graph Pad Prism v6 was used in all statistical analyzes. Raw data can be provided per request. These studies (or specific parts thereof) were performed using established protocols. These studies were conducted in a laboratory that operates under good laboratory principles. These studies were performed using standardizied assays.
Disclosure of potential conflicts of interest
No potential conflict of interest was disclosed.
1068493_Supplemental_Files.zip
Download Zip (1.5 MB)Acknowledgments
The authors present their gratitude to Melissa Wassink and Annemarie de Graaf for technical assistance.
Funding
This work was supported by ERC advanced grant PATHFINDER (269019) and KWO award KUN2009-4402 from the Dutch Cancer Society (to CF). J. Tel is recipient of NWO Veni grant 863.13.024. Carl G. Figdor is recipient of an NWO Spinoza award.
References
- Platzer B, Stout M, Fiebiger E. Antigen cross-presentation of immune complexes. Front Immunol 2014; 5:140; PMID:24744762; http://dx.doi.org/10.3389/fimmu.2014.00140
- Andersen BM, Ohlfest JR. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett 2012; 325:155-64; PMID:22809568; http://dx.doi.org/10.1016/j.canlet.2012.07.012
- Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol 2013; 13:101-17; PMID:23334244; http://dx.doi.org/10.1038/nri3369
- Carreno LJ, Kharkwal SS, Porcelli SA. Optimizing NKT cell ligands as vaccine adjuvants. Immunotherapy 2014; 6:309-20; PMID:24762075; http://dx.doi.org/10.2217/imt.13.175
- Terabe M, Berzofsky JA. The immunoregulatory role of type I and type II NKT cells in cancer and other diseases. Cancer Immunol Immunother 2014; 63:199-213; PMID:24384834; http://dx.doi.org/10.1007/s00262-013-1509-4
- Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med 2004; 199:1607-18; PMID:15197224; http://dx.doi.org/10.1084/jem.20040317
- Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol 2003; 171:5140-7; PMID:14607913; http://dx.doi.org/10.4049/jimmunol.171.10.5140
- Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the 'Swiss-Army knife' of the immune system. Curr Opin Immunol 2008; 20:358-68; PMID:18501573; http://dx.doi.org/10.1016/j.coi.2008.03.018
- Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4+ and CD8+ T cell immunity to a coadministered protein. J Exp Med 2003; 198:267-79; PMID:12874260; http://dx.doi.org/10.1084/jem.20030324
- Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M, Nakayama T, Taniguchi M, Koezuka Y, Tsuji M. Natural killer T cell ligand α-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med 2002; 195:617-24; PMID:11877484; http://dx.doi.org/10.1084/jem.20011889
- Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, Mathew B, Schmidt RR, Lunt SJ, Williams KJ et al. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest 2004; 114:1800-11; PMID:15599405; http://dx.doi.org/10.1172/JCI200422046
- Hayakawa Y, Rovero S, Forni G, Smyth MJ. Alpha-galactosylceramide (KRN7000) suppression of chemical- and oncogene-dependent carcinogenesis. Proc Natl Acad Sci U S A 2003; 100:9464-9; PMID:12867593; http://dx.doi.org/10.1073/pnas.1630663100
- Shimizu K, Goto A, Fukui M, Taniguchi M, Fujii S. Tumor cells loaded with α-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol 2007; 178:2853-61; PMID:17312129; http://dx.doi.org/10.4049/jimmunol.178.5.2853
- Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand α-Galactosylceramide. J Exp Med 2003; 198:1631-41; PMID:14657217; http://dx.doi.org/10.1084/jem.20031192
- Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, Yagita H, Godfrey DI. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of α-galactosylceramide. Blood 2002; 99:1259-66; PMID:11830474; http://dx.doi.org/10.1182/blood.V99.4.1259
- Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol 2014; 32:323-66; PMID:24499274; http://dx.doi.org/10.1146/annurev-immunol-032713-120243
- Liu Y, Goff RD, Zhou D, Mattner J, Sullivan BA, Khurana A, Cantu C 3rd, Ravkov EV, Ibegbu CC, Altman JD et al. A modified α-galactosyl ceramide for staining and stimulating natural killer T cells. J Immunol Methods 2006; 312:34-9; PMID:16647712; http://dx.doi.org/10.1016/j.jim.2006.02.009
- Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, von Blomberg BM, Scheper RJ, van der Vliet HJ et al. A phase I study of the natural killer T-cell ligand α-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res 2002; 8:3702-9; PMID:12473579
- Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, Iizasa T, Nakayama T, Taniguchi M, Fujisawa T. A phase I study of α-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res 2005; 11:1910-7; PMID:15756017; http://dx.doi.org/10.1158/1078-0432.CCR-04-1453
- Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, Hutchinson A, Geller M, Liu N, Annable R et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of α-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med 2005; 201:1503-17; PMID:15867097; http://dx.doi.org/10.1084/jem.20042592
- Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with α-galactosylceramide-loaded DCs. Nat Immunol 2002; 3:867-74; PMID:12154358; http://dx.doi.org/10.1038/ni827
- Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012; 161:505-22; PMID:22353619; http://dx.doi.org/10.1016/j.jconrel.2012.01.043
- Bershteyn A, Hanson MC, Crespo MP, Moon JJ, Li AV, Suh H, Irvine DJ. Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine. J Control Release 2012; 157:354-65; PMID:21820024; http://dx.doi.org/10.1016/j.jconrel.2011.07.029
- Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, Batista FD. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol 2010; 11:303-12; PMID:20228797; http://dx.doi.org/10.1038/ni.1853
- Barral P, Sanchez-Nino MD, van Rooijen N, Cerundolo V, Batista FD. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J 2012; 31:2378-90; PMID:22505026; http://dx.doi.org/10.1038/emboj.2012.87
- Thapa P, Zhang G, Xia C, Gelbard A, Overwijk WW, Liu C, Hwu P, Chang DZ, Courtney A, Sastry JK et al. Nanoparticle formulated α-galactosylceramide activates NKT cells without inducing anergy. Vaccine 2009; 27:3484-8; PMID:19200815; http://dx.doi.org/10.1016/j.vaccine.2009.01.047
- Macho Fernandez E, Chang J, Fontaine J, Bialecki E, Rodriguez F, Werkmeister E, Krieger V, Ehret C, Heurtault B, Fournel S et al. Activation of invariant Natural Killer T lymphocytes in response to the α-galactosylceramide analogue KRN7000 encapsulated in PLGA-based nanoparticles and microparticles. Int J Pharm 2012; 423:45-54; PMID:21575695; http://dx.doi.org/10.1016/j.ijpharm.2011.04.068
- Macho-Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B, Trottein F, Faveeuw C. Targeted Delivery of α-Galactosylceramide to CD8alpha+ dendritic cells optimizes type I NKT cell-based antitumor responses. J Immunol 2014; 193:961-9; PMID:24913977; http://dx.doi.org/10.4049/jimmunol.1303029
- Iyoda T, Ushida M, Kimura Y, Minamino K, Hayuka A, Yokohata S, Ehara H, Inaba K. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int Immunol 2010; 22:905-13; PMID:21118907; http://dx.doi.org/10.1093/intimm/dxq444
- Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. Biodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responses. J Drug Target 2003; 11:495-507; PMID:15203918; http://dx.doi.org/10.1080/10611860410001670026
- Zhang Z, Tongchusak S, Mizukami Y, Kang YJ, Ioji T, Touma M, Reinhold B, Keskin DB, Reinherz EL, Sasada T. Induction of anti-tumor cytotoxic T cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials 2011; 32:3666-78; PMID:21345488; http://dx.doi.org/10.1016/j.biomaterials.2011.01.067
- Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, Elhasi S, Samuel J, Lavasanifar A. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine 2008; 26:5046-57; PMID:18680779; http://dx.doi.org/10.1016/j.vaccine.2008.07.035
- Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, Figdor CG. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release 2010; 144:118-26; PMID:20156497; http://dx.doi.org/10.1016/j.jconrel.2010.02.013
- Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, Lambeck AJ, Figdor CG. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011; 118:6836-44; PMID:21967977; http://dx.doi.org/10.1182/blood-2011-07-367615
- McKee SJ, Young VL, Clow F, Hayman CM, Baird MA, Hermans IF, Young SL, Ward VK. Virus-like particles and α-galactosylceramide form a self-adjuvanting composite particle that elicits anti-tumor responses. J Control Release 2012; 159:338-45; PMID:22386518; http://dx.doi.org/10.1016/j.jconrel.2012.02.015
- Gehrmann U, Hiltbrunner S, Georgoudaki AM, Karlsson MC, Naslund TI, Gabrielsson S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with α-galactosylceramide on exosomes. Cancer Res 2013; 73:3865-76; PMID:23658368; http://dx.doi.org/10.1158/0008-5472.CAN-12-3918
- Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, Groettrup M. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine 2008; 26:1626-37; PMID:18295941; http://dx.doi.org/10.1016/j.vaccine.2008.01.030
- Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, Banerjee M, Overholtzer M, Roche PA, Tampé R. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014; 158:506-21; PMID:25083866; http://dx.doi.org/10.1016/j.cell.2014.04.054
- Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, Rossjohn J, Perlmutter P, Cao J, Godfrey DI et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol 2010; 11:313-20; PMID:20190758; http://dx.doi.org/10.1038/ni.1848
- Lee YR, Lee YH, Im SA, Kim K, Lee CK. Formulation and characterization of antigen-loaded PLGA Nanoparticles for efficient cross-priming of the antigen. Immune Netw 2011; 11:163-8; PMID:21860609; http://dx.doi.org/10.4110/in.2011.11.3.163
- Fischer S, Schlosser E, Mueller M, Csaba N, Merkle HP, Groettrup M, Gander B. Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J Drug Target 2009; 17:652-61; PMID:19622019; http://dx.doi.org/10.1080/10611860903119656
- Silva AL, Rosalia RA, Sazak A, Carstens MG, Ossendorp F, Oostendorp J, Jiskoot W. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: low-burst release is crucial for efficient CD8(+) T cell activation. Eur J Pharm Biopharm 2013; 83:338-45; PMID:23201055; http://dx.doi.org/10.1016/j.ejpb.2012.11.006