
ABSTRACT
Regulatory T cells (Tregs) play a crucial physiological role in the regulation of immune homeostasis, although recent data suggest Tregs can contribute to primary tumor growth by suppressing antitumor immune responses. Tregs may also influence the development of tumor metastases, although there is a paucity of information regarding the phenotype and function of Tregs in metastatic target organs. Herein, we demonstrate that orthotopically implanted metastatic mammary tumors induce significant Treg accumulation in the lungs, which is a site of mammary tumor metastasis. Tregs in the primary tumor and metastatic lungs express high levels of C–C chemokine receptor type 5 (CCR5) relative to Tregs in the mammary fat pad and lungs of tumor-free mice, and Tregs in the metastatic lungs are enriched for CCR5 expression in comparison to other immune cell populations. We also identify that C–C chemokine ligand 8 (CCL8), an endogenous ligand of CCR5, is produced by F4/80+ macrophages in the lungs of mice with metastatic primary tumors. Migration of Tregs toward CCL8 ex vivo is reduced in the presence of the CCR5 inhibitor Maraviroc. Importantly, treatment of mice with Maraviroc (MVC) reduces the level of CCR5+ Tregs and metastatic tumor burden in the lungs. This work provides evidence of a CCL8/CCR5 signaling axis driving Treg recruitment to the lungs of mice bearing metastatic primary tumors, representing a potential therapeutic target to decrease Treg accumulation and metastatic tumor growth.
Introduction
The development of distant metastases is largely responsible for the mortality associated with breast cancerCitation1 and emerging evidence supports an active role for the immune system in influencing the growth and metastasis of mammary tumors.Citation2 Tumor cell survival may be enhanced by evading immune detection through a reduction in antigen presentation,Citation3,4 secretion of suppressive proteins,Citation3 or by recruitment or generation of suppressive immune cell populations.Citation3 It has been previously shown by our group and others that myeloid cells, including CD11b+Gr1+F4/80− myeloid-derived suppressor cells (MDSCs) and CD11b+Gr1−F4/80+ macrophages, promote metastatic tumor growth through suppression of antitumor immunity using reactive oxygen species (ROS)-mediated mechanisms.Citation5,6 Under homeostatic conditions, immunosuppressive CD4+CD25+Foxp3+ Tregs function to mediate peripheral tolerance and prevent autoimmunity.Citation7 Tregs have also been associated with the growth and metastasis of several solid tumor types,Citation8 including breast cancer,Citation9 and elevated levels of tumor-infiltrating Tregs are associated with poor prognosis in many malignancies.Citation10,11
Once in the tumor, Tregs may facilitate tumor progression through a variety of immunosuppressive mechanisms including depriving T cells of co-stimulatory signals and IL-2, secretion of granzymes and perforin, or release of soluble suppressive factors such as TGF-β, IL-10, IL-35, PGE2, IDO or adenosine.Citation8,12 Expression of cell surface receptors such as CTLA-4,Citation13 CD103,Citation14 PD-1Citation15 and more recently HeliosCitation16 have aided in the identification of functionally suppressive Tregs. For a detailed review of Treg suppressive mechanisms, see refs. Citation6-7. Furthermore, Tregs may promote tumor angiogenesis by secreting VEGF-A,Citation17 and have been shown to promote tumor cell invasion through the suppression of the EMT inhibitor, maspin, in RANK+ tumor cells.Citation18 Tregs are recruited to the tumor site using chemokine receptor signaling axes such as CCL22/CCR4 in ovarianCitation19,20 and breast cancers,Citation21 CCL5/CCR5 in melanoma,Citation22 pancreaticCitation23 and colorectal cancers,Citation24 or CXCL12/CXCR4Citation25 and CCL28/CCR10+ in ovarian cancer.Citation17,25 While these chemokine/receptor pairs promote Treg accumulation in primary tumors, much less is known about whether (and how) Tregs accumulate in metastatic target organs.
As detailed further below, the data presented herein indicate that CCR5 is highly expressed by Tregs in mice implanted with metastatic mammary carcinomas, and that CCR5 may facilitate Treg recruitment to the metastatic lungs. Endogenous ligands for CCR5 include CCL3, 4, 5 and 8.Citation26 CCL8 (MCP-2) has been reported as a CCR5 agonist with a receptor affinity similar to CCL3 (MIP-1α) and stimulates GTPγS binding indicating G protein activation.Citation27,28 CCL8 is also able to stimulate receptor internalization and intracellular calcium release after binding CCR5, although the extent of receptor internalization induced by CCL8 is reduced compared to CCL3 and 4.Citation27,28 While CCL8 is a potent inhibitor of CCR5-tropic strains of HIV by blocking the CCR5-mediated interaction of HIV and the viral glycoprotein gp120,Citation26 the study of CCL8 in cancer has been limited.
CCR5 may be expressed on a range of immune cells including macrophages, dendritic cells, CD4+ and CD8+ T cells.Citation29 CCR5 expression has been shown to increase with stage of several types of solid malignancies and CCR5+ T cells have been identified in lymph nodes bearing tumor metastases in breast cancer patients.Citation30 The expression of CCR5 by Tregs is associated with a highly suppressive phenotypeCitation24 and the inhibition of CCR5 has been found to decrease tumor growth in melanoma and pancreatic cancers through a Treg mediated mechanism.Citation22,23 However, the function of CCR5+ Tregs in breast tumor metastasis and the development of metastatic foci has not been previously studied.Citation8 Several inhibitors of CCR5 are available, including Maraviroc (MVC), a small-molecule antagonist and FDA approved anti-retroviral agent used clinically for the treatment of CCR5-tropic strains of HIV.Citation31
Using transplantable mammary carcinoma models in immunocompetent Balb/c mice, we have found that metastatic tumors induce the accumulation of CCR5+ Tregs in the lungs and primary tumor. The CCR5 ligand CCL8 is elevated in the lungs of mice bearing metastatic breast tumors and our data suggest that F4/80+ macrophages are a source of CCL8 in the lungs. Tregs migrate toward CCL8 in ex vivo chemotaxis assays and this migration is inhibited by the CCR5-specific inhibitor MVC. MVC administration to tumor-bearing mice significantly reduced the proportion of Tregs in the lungs without affecting the levels of CD4+ T cells, CD8+ T cells, or CD11b+ myeloid cells. MVC treatment also significantly reduced pulmonary metastatic tumor burden without affecting primary tumor growth. Our data identify CCL8/CCR5 as a novel signaling axis that promotes Treg recruitment to the lungs of mice bearing metastatic primary mammary tumors. Importantly, our pre-clinical findings highlight the potential therapeutic utility of MVC to reduce pulmonary Treg accumulation and breast cancer metastasis to the lungs.
Results
Mice bearing metastatic tumors have increased proportions of regulatory T cells
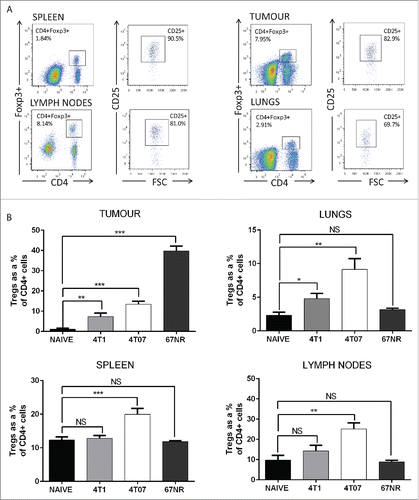
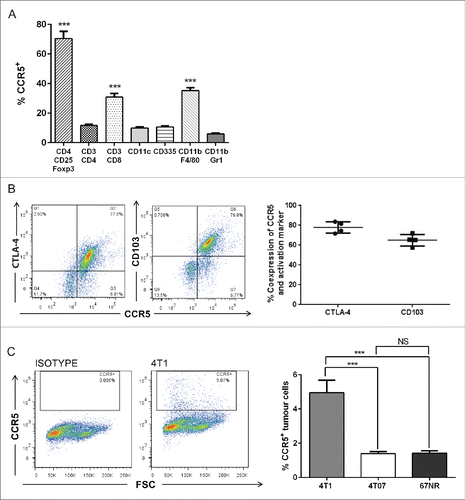
To investigate Treg levels in tissues of mice bearing metastatic and non-metastatic murine mammary carcinomas, we assessed the accumulation of CD4+CD25+FoxP3+ Tregs in the primary tumor and tissues of mice bearing 4T1, 4T07 and 67NR tumors 3 weeks after implant in comparison to the corresponding tissues of naive mice. Representative flow cytometry plots for each tissue and single stain controls for gating are shown in and Fig. S1A, respectively. The proportion of Tregs was significantly elevated in the primary tumor, spleen, lungs and lymph nodes of mice bearing 4T07 tumors compared to the corresponding tissues in naive mice (), and increased progressively from 1 week to 3 weeks post-tumor implant (Fig. S1C). In contrast, the accumulation of Tregs in lymphoid tissues was not observed in 4T1 tumor-bearing mice, as Tregs were only significantly elevated in the primary tumor and lungs. Interestingly, Treg levels were not increased in the lungs, spleen, or lymph nodes of mice bearing non-metastatic 67NR tumors, but were dramatically increased in the primary 67NR tumor compared to the naive mammary fat pad, 4T1 tumors, or 4T07 tumors (). The observed differences in Treg proportions between the different tumor types were not a result of variance in primary tumor size, as tumor weights at the time of sacrifice were not significantly different between the cell lines (Fig. S1B). These data indicate that Tregs are elevated in primary tumors of all three mammary tumor types, but are elevated in the lungs of mice bearing only metastatic 4T1 or 4T07 tumors.
Figure 1. Flow cytometric analysis and quantification of CD4+CD25+Foxp3+ Tregs in the tissues of mice bearing metastatic primary tumors. (A) Representative flow cytometry plots of Treg staining for lungs, tumor, spleen and lymph nodes of 4T1 tumor-bearing mice. (B) A greater proportion of Tregs was isolated from the tissues of mice bearing metastatic 4T1 and 4T07 tumors versus non-metastatic 67NR tumors. For the lungs, spleen and tumor, n = 10 mice per group (naive, 4T07 and 4T1) and n = 8 (67NR). For the lymph nodes, n = 6 mice per group. Significance denotes comparison of the experimental group to the equivalent organs in naive mice analyzed using a Student's unpaired two-tailed t-test *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant.
CCL8 is produced by the primary tumor and lungs of mice bearing metastatic primary tumors
We postulated that cytokines/chemokines produced by the primary tumors or by the lungs of mice bearing metastatic tumors were responsible for inducing Treg recruitment. To determine the mechanism of Treg recruitment, antibody arrays were used to profile chemokine expression by tumor cells in vitro, and by the lungs and primary tumors of mice 3 weeks after orthotopic tumor implantation (). Several chemokines were elevated in the tumor cell lysates in vitro including CCL6 and IL-16 (), and CCL2, CCL12 and CXCL1 (). Interestingly, we observed that 67NR cells produced the greatest diversity of chemokines in vitro ( and Fig. S2A), including CXCL12 (SDF-1), which was produced by 67NR, but not 4T1 or 4T07, cells in vitro (Fig. S2A). We did not observe the CCR5 ligands CCL3, 4, or 5 in 4T1 or 4T07 lysates in vitro; however, these chemokines were detected at low levels in 67NR lysates (). Lysates derived from 4T1 and 4T07 tumors expressed higher levels of CCL6, 8, 9 and decreased expression of IL-16 relative to in vitro tumor cells (). We observed a similar level of CCL8 and increased IL-16 in the 67NR primary tumor lysates in comparison to 4T1 and 4T07 tumors. Analysis of lung lysates revealed increased CCL6, 8, 9, and IL-16 in the lungs of 4T1 and 4T07 tumor-bearing mice compared to naive lungs or lungs from 67NR-bearing mice (). We also detected RARRES2, CCL2, 12, and C5/C5a in lung lysates, however, these factors were produced at similar levels by naive lungs (Fig. S2A).
Figure 2. The lungs of mice bearing metastatic breast tumors express increased levels of CCL8. (A) Chemokine arrays of lysates from in vitro tumor cells, the primary tumor and lungs from 4T1, 4T07 or 67NR tumor-bearing mice as well as the corresponding naive tissues (mammary fat pad and lungs). The lungs of tumor bearing mice produce increased CCL8 relative to naive lungs. CCL8 was also expressed by the 4T1 and 4T07 tumors but not the corresponding cell lines in vitro. (B) CCL8 levels were quantified using an ELISA for the bronchoalveolar lavage (BAL) and lung lysate from naive mice and mice bearing 4T1, 4T07 or 67NR tumors, n = 6. Significance denotes comparison of the experimental group to the equivalent organs in naive mice analyzed using a Student's unpaired two-tailed t-test *p < 0.05, **p < 0.01, NS = not significant.
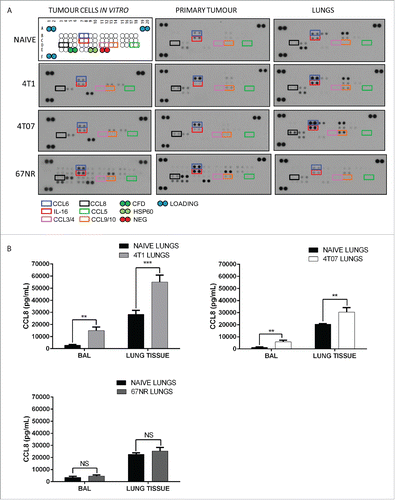
Since the expression of CCR5 by Tregs has been reported previously, and inhibitors of CCR5 are commercially available, we chose to further investigate the recruitment of CCR5+ Tregs by CCR5 ligands produced in the lungs of mice with metastatic primary tumors. The chemokines CCL8 and IL-16 share a high degree of homology between mouse and human (71% and 82% cDNA alignment, respectively),Citation32 and have both been shown to recruit cells expressing the receptor CCR5Citation33,34 although only CCL8 is a direct binding ligand of this receptor.Citation34 To further examine the expression of CCL8 in the lungs and validate the antibody array data, CCL8 levels were quantified by ELISA in the bronchoalveolar lavage (BAL) and lung tissue from naive mice or 4T1, 4T07, or 67NR tumor-bearing mice (). CCL8 was significantly elevated in the BAL and lung tissue of 4T1 and 4T07-bearing mice, but not in mice bearing non-metastatic 67NR tumors (). When taken with the lack of Treg accumulation in the lungs of naive mice and mice with 67NR tumors (), the decreased level of CCL8 in the lungs of 67NR-bearing mice suggests that CCL8 may contribute to the recruitment of Tregs to the lungs of mice bearing metastatic primary tumors.
F4/80+ macrophages are a source of pulmonary CCL8 in vivo
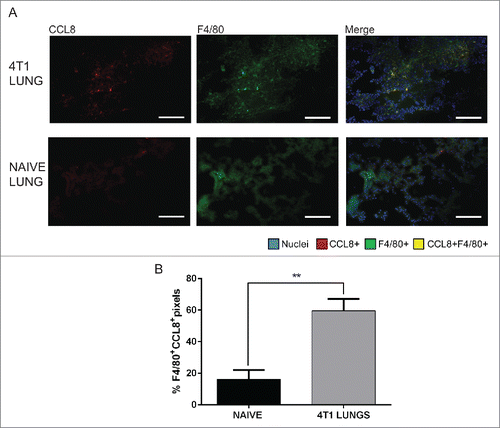
We have previously noted that CD11b+F4/80+ macrophages accumulate in the lungs of 4T1 tumor-bearing mice.Citation5 Macrophages have been shown to induce recruitment of Tregs to solid tumors through production of CCL20Citation35 and CCL22,Citation36 and have been reported to secrete CCL8 ex vivo when isolated from human BAL samples.Citation37 Based on these findings, we hypothesized that macrophages may be the source of CCL8 in the lungs of mice bearing metastatic tumors. We performed fluorescent imaging of 4T1 tumor-bearing lungs () using CCL8 and F4/80 (a pan-macrophage marker). Lower levels of F4/80+ macrophages were observed in the lungs of naive mice in comparison to the lungs of 4T1 tumor-bearing mice (data not shown), consistent with our previous findings.Citation5 Image analyses revealed that CCL8 co-localized significantly (p = 0.0035) with F4/80+ macrophages in the lungs of 4T1 tumor-bearing mice relative to the lungs from non-tumor bearing mice (). Representative images for threshold settings and secondary only control staining are shown in Fig. S3. The percentage of overlap between F4/80+pixels and CCL8+ pixels was approximately 3.7-fold higher in the lungs of 4T1 mice than naive mice (). When taken with the increased numbers of F4/80+ macrophages in the lungs of 4T1-bearing mice,Citation5 these data suggest that F4/80+ macrophages are a source of CCL8 in the lungs of tumor-bearing mice.
Figure 3. CCL8 is produced by F4/80+ macrophages in the lungs of 4T1 tumor-bearing mice, 3 weeks after orthotopic implantation. (A) Representative images of 4T1 and naïve lung samples showing CCL8+ and F4/80+ single stains and merged images. (B) Quantification of F4/80 and CCL8 overlap represented as % F4/80+ pixels that are CCL8+, n = 4. Significance denotes comparison of the experimental group to the equivalent organs in naive mice analyzed using a Student's unpaired two-tailed t-test, **p < 0.01. Images were taken with a 20X objective lens. Scale bar = 62 μm.
CCR5+ Tregs accumulate in the lungs and primary tumor of mice bearing metastatic mammary carcinomas
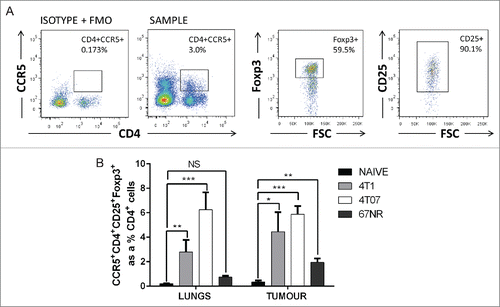
CCL8 has been previously described as a direct binding ligand of CCR5, stimulating receptor activation and intracellular Ca2+ release.Citation34 CCR5 is highly expressed by Tregs in pre-clinical models of melanoma,Citation22 pancreatic,Citation23 and colorectal cancers,Citation24 but CCR5 expression by Tregs in pre-clinical models of breast cancer has not been previously studied and the presence and potential influence of CCR5+ Tregs in metastatic sites is unknown. Therefore, we quantified CCR5+ Treg levels () in the primary tumor and lungs of mice bearing metastatic and non-metastatic tumors. We found that Tregs preferentially expressed CCR5 relative to conventional CD4+ T cells (Fig. S4A), and the proportion of CCR5+ Tregs was significantly elevated in the lungs of mice bearing 4T1 and 4T07 tumors (). CCR5+ Tregs did not significantly accumulate in the lungs of mice bearing non-metastatic 67NR tumors relative to naive mice (). Interestingly, while CCR5+ Tregs were present in the spleen, blood and lymph nodes of mice bearing 4T1 tumors, very few CCR5+ Tregs were observed in the thymus (Fig. S4B), consistent with the finding that CCR5 expression is involved in thymic egress of Tregs.Citation38 These results suggest that CCR5+ Tregs accumulate in the lungs in response to metastatic mammary tumors.
Figure 4. CCR5+CD4+CD25+Foxp3+ cells accumulate in the lungs and primary tumor of mice bearing metastatic mammary carcinomas. (A) Representative flow cytometry plots and (B) quantification of CCR5+ Tregs in the lungs and primary tumors of mice bearing 4T1, 4T07, or 67NR tumors. 4T1 and 4T07-bearing mice accumulate a higher proportion of CCR5+ Tregs compared to the lungs and mammary fat pad of naive mice (n = 8) and the lungs and primary tumor of 67NR tumor-bearing mice (n = 6). Significance denotes comparison of the experimental group to the equivalent organs in naive mice analyzed using a Student's unpaired two-tailed t-test, *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant, FMO = fluorescence minus one.
Tregs express high levels of CCR5 relative to other immune cell subsets and tumor cells
To determine whether CCR5 is expressed by tumor and other immune cells in our pre-clinical mammary carcinoma models, we assessed CCR5 levels on the tumor cell lines in vitro and on immune cells isolated from the lungs of tumor-bearing mice. CCR5 levels were significantly increased on CD4+CD25+Foxp3+ Tregs relative to Th cells (CD3+CD4+), TC cells (CD3+CD8+), dendritic cells (CD11c+), natural killer cells (CD335+), macrophages (CD11b+F4/80+) and myeloid cells (CD11b+Gr1+) (). CCR5 expression by macrophages and Tc cells were also elevated relative to other immune cell populations (). CCR5 was highly co-expressed with the receptors CTLA-4 (median 77.8%) and CD103 (median 65%) (), both of which are expressed on activated, functionally suppressive Tregs.Citation14,39 CCR5 levels were found to be low on tumor cells in vitro, with 4T1 having the highest surface expression of CCR5 (). These data reveal that Tregs in mice with metastatic mammary tumors express high levels of CCR5 relative to other immune cell populations, and suggest that inhibition of CCR5 may be a viable therapeutic strategy to target tumor-induced Treg accumulation in the lungs.
Figure 5. CCR5 is highly expressed by Tregs with a suppressive phenotype relative to other immune cell subsets. (A) Flow cytometry analysis and quantification of CCR5 expression by immune cell subsets including macrophages (CD11b+F4/80+), myeloid cells (CD11b+Gr1+), natural killer cells (CD335+ or NKp46+), Tregs (CD4+CD25+Foxp3+), dendritic cells (CD11c+) and CD4+/CD8+ T cells. Significance denotes comparison of CCR5 expression by Tregs to other immune cell subsets, n = 6, analyzed using a one-way ANOVA. (B) CCR5 is highly co-expressed on CD4+CD25+Foxp3+ Tregs with CTLA-4 (median 77.8%) and CD103 (median 65.0%), n = 4. (C) CCR5 is expressed by tumor cells (in vitro), n = 6, analyzed using a Student's unpaired two-tailed t-test. ***p < 0.001.
CCL8 induces migration of CCR5+ Tregs ex vivo
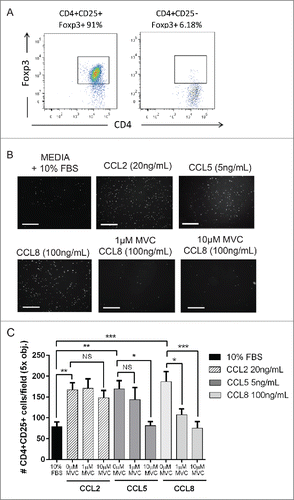
It has been previously shown that CCL8 mediates the chemotaxis of activated T cells expressing CCR5,Citation34 and we wanted to determine whether Tregs isolated from mice bearing metastatic tumors migrated toward CCL8 ex vivo in a CCR5-dependent manner. We isolated Tregs from mice bearing metastatic tumors (Fig. S5A), cultured the cells for 48 h to ensure purity of >90 % FoxP3+ Tregs (), and performed ex vivo migration assays to test the ability of Tregs to migrate toward CCL8 in the presence or absence of the CCR5 inhibitor MVC (). We found that Tregs migrated toward the chemokines CCL2, CCL5, or CCL8 ex vivo, and that Treg migration toward CCL8 was 2.8-fold greater than migration toward the negative control wells (). As expected, Treg migration toward CCL2 (a CCR5-independent ligand)Citation40 was not reduced by pre-treatment of cells with 1 µM or 10 µM MVC, whereas pre-treatment of Tregs with MVC decreased cell migration toward CCL5 (a CCR5-dependent ligand).Citation23 Similarly, pre-treatment of Tregs with MVC decreased migration toward CCL8 to negative control levels () without significantly impacting cell viability (Fig. S5B). These findings indicate that Tregs migrate in response to CCL8 and that this activity is dependent on CCR5. Moreover, these results demonstrate that CCR5-dependant Treg migration can by inhibited by MVC in vitro.
Figure 6. Tregs migrate toward CCL8 ex vivo. (A) CD4+CD25+ cells isolated from the spleens of 3 week 4T07 tumor-bearing mice using CD4+ negative selection followed by CD4+CD25+ FACS. CD4+CD25+ are enriched for Foxp3 expression (91%) following 48 h of ex vivo culture with plate bound αCD3, soluble αCD28 and rIL-2. (B–C) Tregs migrate toward CCL2, CCL5, or CCL8. The CCR5 independent ligand CCL2 was used as a positive control for migration. The CCR5-specific inhibitor, Maraviroc (MVC), inhibited the migration of cells toward CCL5 or CCL8. Data show the results from three separate experiments performed either in triplicate or duplicate analyzed using a one-way ANOVA where *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant. Scale bar = 125 μm.
Maraviroc (MVC) reduces mammary tumor lung metastasis in immunocompetent mice
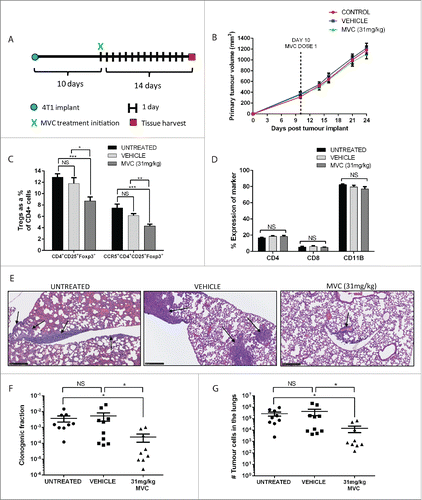
To examine the effects of MVC on Treg recruitment and metastatic growth in vivo, BALB/c mice were orthotopically implanted with 4T1 tumors. Ten days after tumor implant, mice were given 31 mg/kg MVC or vehicle (5% DMSO in acidified water) via daily oral gavage for comparison with untreated control mice (). Interestingly, while primary tumor growth was not affected by drug treatment (), mice treated with MVC exhibited reduced levels of CCR5+ Tregs and bulk Tregs in the lungs (, Fig. S6). Interestingly, MVC treatment did not reduce the proportion of CD4+ or CD8+ lymphocytes, or the proportion of CD11b+ myeloid cells, in the lungs () suggesting MVC is preferentially affecting pulmonary Treg accumulation. MVC treatment also reduced the appearance of histologically detectable macroscopic metastases in the lungs (). Analysis of metastatic tumor cells in the lungs using ex vivo clonogenic assays indicated that MVC reduces the proportion of tumor cells present in the lungs () and causes an overall 30-fold reduction in the number of metastatic tumor cells in the lungs (). These data indicate that MVC treatment decreases Treg accumulation and metastatic tumor growth in the lungs of mice bearing 4T1 murine mammary tumors.
Figure 7. Treatment of 4T1 tumor-bearing mice with MVC reduced infiltrating Treg proportions and metastatic tumor burden. (A) Mice were treated with MVC 10 d post implant of 4T1 mammary tumor cells until sacrifice at day 24. (B) 4T1 tumor volume was monitored bi-weekly using caliper measurements. (C) The percentage of CD4+CD25+Foxp3+ Tregs and CCR5+ CD4+CD25+Foxp3+ Tregs, represented as a proportion of CD4+ cells, was significantly reduced with MVC treatment. (D) The percentages of CD4+, CD8+, and CD11b+ cells were not affected by drug treatment. (E) A reduction in metastases is visible in the lungs of MVC treated mice via H&E staining. Metastatic foci in each treatment group are indicated with arrows, scale bar = 250 μm. (F) Mice treated with MVC had a reduced fraction of clonogenic tumor cells in disaggregated lung tissue and (G) a reduced number of total metastatic tumor cells in the lungs. Results from three independent experiments are shown (n = 8–9 mice), and analyzed using a Student's unpaired two-tailed t-test. Significance values are designated by *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Tregs contribute to the progression of many malignancies and are therefore attractive therapeutic targets. Although Tregs have been associated with reduced overall survival in breast, cervical, renal cancer, melanoma,Citation41 and non-small cell lung cancer (NSCLC),Citation42 many Treg-targeted treatments are hindered either due to a lack of specificity or because they disrupt maintenance of peripheral tolerance.Citation43 Emerging evidence indicates that Treg homing may be targeted using chemokine/receptor signaling axes as an alternative therapeutic strategy to the systemic elimination of Tregs.Citation22,23,43,44 In this study, we define a novel CCL8/CCR5 signaling axis for the recruitment of Tregs to the lungs of tumor-bearing mice, and demonstrate that CCR5 antagonism reduces Treg proportion and the development of metastatic tumors in the lungs. We report that CCR5+ Treg levels increase in the lungs of mice bearing metastatic mammary tumors, and that the CCR5 ligand CCL8 is expressed by macrophages that accumulate in the lungs. CCL8 induces migration of Tregs ex vivo in a CCR5-dependent manner, and treatment of tumor-bearing mice with the CCR5 antagonist MVC decreases the proportion of Tregs and metastatic tumor growth in the lungs. Our results emphasize the potential therapeutic applications of MVC for the treatment of metastatic breast cancer.
In addition to promoting primary tumor growth, intratumoural Treg accumulation has been associated with more invasive tumors.Citation10,45 In breast cancer patients, the accumulation of Tregs is associated with increased risk of relapse and death, with Treg numbers significantly elevated in patients with invasive disease.Citation46,47 Treg levels increase with progression from normal breast tissue, to ductal carcinoma in situ (DCIS), to invasive ductal carcinoma (IDC), suggesting that these cells may have an active role in promoting tumor progression and local invasion in pre-clinical breast cancer models as well as breast cancer patients.Citation46 We observed that the proportion of intratumoural Tregs increased with time post-tumor implant in mice bearing mammary carcinomas, and also that Tregs can be elevated in peripheral tissues of mice with metastatic primary tumors. Tregs were elevated in several tissues in the 4T07 model, while Treg levels were increased only in the lungs and primary tumor of 4T1-bearing mice (). While tumor cells have been recovered from the lungs of 4T07 mice,Citation48 only 4T1 tumor-bearing mice are able to form pulmonary macro-metastatic foci.Citation48 The 4T07 line is known to be more immunogenic than 4T1,Citation49 and this immunogenicity likely contributes to the inability of 4T07 tumor-bearing mice to form visible metastatic foci in the lungs and may also explain the high level of Tregs in 4T07 mice ().
Tregs migrate to the primary tumor using chemokine receptor signaling axes such as CCL5/CCR5, CCL22/CCR4, CCL17/CCR4, CCL28/CCR10, and CXCL12/CXCR4.Citation17,19,23-25 While the signaling axes regulating Treg migration to the primary tumor have been well defined, much less is known about the homing of Tregs to metastatic target organs. Using chemokine profiling of lung tissue, we identified candidate factors that may promote the recruitment of Tregs to the lungs of mice with metastatic primary tumors (). We observed increased production of CCL6 and CCL9 in the metastatic lungs and tumor (); however, these chemokines have been predominantly studied in rodents and share a low degree of homology with their human orthologues, making them less clinically relevant.Citation32 CCL3, 4 and 5 are known ligands for CCR5, although we did not observe detectable levels of these cytokines in the lungs of tumor-bearing mice. Rather, we observed CCL8 and IL-16 expression in the lungs, and CCL8 is a direct-binding ligand and agonist of CCR5.Citation26,28 IL-16, although not a direct-binding ligand of CCR5, has been shown to preferentially recruit T cells expressing CCR5,Citation33 likely due to the co-expression of the IL-16 receptor CD4 and CCR5. We found increased CCL8 in BAL and lung tissue from mice with 4T07 or 4T1 tumors; however, CCL8 was not significantly increased in the lungs of 67NR tumor-bearing mice (). Furthermore, we found that F4/80+ macrophages were a source of CCL8 in mice bearing metastatic tumors (), and we have previously shown that macrophages accumulate in the lungs of mice bearing metastatic mammary tumors.Citation5 These data suggest that CCL8 produced by macrophages that accumulate in the lungs of mice bearing metastatic tumors may induce subsequent Treg recruitment to the lungs.
Our study identified that CCR5+ Tregs were significantly increased in the lungs and primary tumor of mice bearing metastatic mammary carcinomas. We also found that CCR5+ Tregs highly co-express the markers CTLA-4 and CD103, indicating a suppressive phenotype.Citation14,50 This finding is consistent with reports that CCR5high Tregs have increased suppressive capacity compared to CCR5low Tregs,Citation24,51 and that the loss of CCR5 impairs the in vivo suppressive capacity of Tregs.Citation14 CCR5 has also been shown to promote pulmonary metastasis of tumor cells in a pre-clinical model of breast cancer using immunocompromised mice, and was associated with enhanced invasive capacity in a subpopulation of tumor cells expressing CCR5.Citation52 The expression of CCR5 by our murine tumor cell lines in vitro was low with respect to the level of CCR5 expressed by Tregs (). However, future studies are warranted to determine if the level of CCR5 expression by 4T1 and 4T07 tumor cells is consistent in vivo and represents a potential direct therapeutic target for decreasing metastatic tumor cell invasion.
While CCR5 has been previously shown to regulate Treg chemotaxis to the primary tumor in models of melanomaCitation22 and pancreatic cancer,Citation23 its role in Treg migration has not been previously examined in breast cancer. We observed that Tregs migrate toward CCL8 ex vivo, and this migration could be inhibited by administration of the CCR5 antagonist MVC without decreasing Treg viability (), indicating that the anti-migratory effect of MVC is CCR5-specific. The known CCR5 ligand CCL5 was used as a positive control for migration, as previous reports have identified this ligand as chemotactic for Tregs,Citation53 and Treg migration toward CCL5 was similarly inhibited by MVC. Importantly, treatment of Tregs with MVC did not affect migration toward the CCR5-independant ligand CCL2.Citation40,54 Our study highlights that MVC can inhibit Treg chemotaxis without disrupting the viability or function of these cells in vitro, and supports the use of MVC to therapeutically inhibit CCR5-dependent Treg migration.
Our study also demonstrates that the accumulation of CCR5+ Tregs in the lungs of mice bearing metastatic primary tumors can be reduced by MVC administration in vivo. While the accumulation of CCR5+ Tregs in the lung is reduced, their recruitment is not completely blocked. Although CCR5 expression by Tregs in our model system is high (approximately 60% in 4T1 mice, Fig. S6), this does not exclude the possibility that Tregs may migrate into the metastatic lungs using alternative chemokine signaling axes that may not be affected by MVC, and this is an area of active investigation in our laboratory. In addition, MVC is characterized as a human CCR5 inhibitor and MVC activity in mouse cells is reduced (but not absent)Citation24,52,55 due in part to the comparably lower IC50 of this drug in mouse cells.Citation56 A reduction in Treg infiltrationCitation23 has been observed in mice treated with the human CCR5 antagonistCitation57 TAK-779. Moreover, TAK-779 administration has been found to induce a dose-dependent reduction in T cell migration,Citation58-60 consistent with our observations with the administration of MVC. We therefore postulate that the MVC-mediated reduction in Treg recruitment to the lungs observed in our mouse models may be amplified in humans treated with MVC.
Our in vivo findings showing reduced Treg levels in the lungs with CCR5 antagonism contrast a previous report that showed the inhibition of CCR5 with Met-RANTES, TAK-779 or UK-484900 (an analog of MVC) did not reduce the proportion of intratumoural Tregs in a humanized immunocompetent model of colorectal cancer (hCCR5KI), despite the high expression of CCR5 and increased suppressive activity of CCR5+ Tregs relative to conventional T cells in colorectal cancer.Citation24 This discrepancy in findings may result from mechanistic differences in the function or receptor affinity of Met-RANTES and TAK-779 in vivo in comparison to MVC, or variance in the dosage of UK-484900, the time and route of administration, or micro-environmental differences in the accumulation, proliferation and function of Tregs in colorectal tumors compared to lung tissue. Additionally, the authors found that CD4+ and CD8+ cells also expressed CCR5, particularly in mice bearing CT26 tumors. Our data are consistent with these findings in that we observed CCR5 expression on CD4+ and CD8+ lymphocytes, although the percentage of CD4+ and CD8+ cells that expressed CCR5 were 2.3-fold and 6-fold lower than the proportion of Tregs expressing CCR5 in our models (). Importantly, we did not observe a reduction in CD4+ or CD8+ lymphocytes with MVC treatment () indicating that MVC was primarily affecting the recruitment of CCR5+ Tregs to the lungs.
Interestingly, treating 4T1 tumor-bearing mice with MVC did not affect primary tumor growth (), but reduced metastatic tumor growth in the lungs by 30-fold (). Further work is necessary to determine the mechanism(s) by which CCR5 antagonism contributes to reduced pulmonary metastases. A small proportion of 4T1 tumor cells express CCR5 () and therefore MVC could conceivably affect metastasis of 4T1 cells directly as observed with a subset of human breast tumor cells in immunocompromised mice.Citation61 When combined with the reduction in CCR5+ Tregs in the lungs observed with MVC treatment and the known immunosuppressive function of CCR5+ Tregs, we postulate that MVC decreases metastatic tumor growth in the lungs by reducing the Treg component of the immune suppressive microenvironment normally found in the lungs of mice with metastatic primary tumors. Importantly, our results support the inhibition of CCR5 with MVC as a potential therapeutic strategy to reduce Treg accumulation and metastatic burden without systemic elimination of the homeostatic Treg population. While the use of therapies to target Treg migration and recruitment to tissues may avoid systemic Treg depletion, the use of homing inhibitors can disrupt chemokine signaling on bystander immune cells, resulting in off-target effects. The lack of specificity of current Treg cell surface markers (CD4 and CD25) may be circumvented by targeting receptors that are highly expressed by Tregs relative to other cell populations or by exploiting therapeutic windows during which Treg recruitment is highest. In concert with previous studies showing that CCR5 is expressed by a subpopulation of breast cancer cells in vivo, our study reports the significant expression of this receptor by tumor-infiltrating Tregs, emphasizing the significance of targeting this receptor in breast cancer. Furthermore, MVC represents an attractive therapeutic due to its FDA approval and high specificity for CCR5.Citation62
Taken together, our data identify a novel CCR5/CCL8 axis that regulates Treg accumulation in the lungs of mice bearing metastatic mammary tumors, and support the development and testing of CCR5 antagonists such as MVC to decrease Treg recruitment to metastatic target organs and inhibit the growth and development of tumor metastases.
Materials and methods
Mice and tumor models
Female BALB/c mice (8–12 weeks old; Simonsen Laboratories) were housed and maintained under specific-pathogen free conditions in the Animal Resource Center at the BC Cancer Agency Research Center in micro-isolator cages with ventilated racks. Animal experiments were performed in compliance with requirements of the Canadian Council on Animal Care and the University of British Columbia Committee on Animal Ethics.
Murine mammary carcinoma cell lines 4T1, 4T07, and 67NR were a kind gift from Dr Fred Miller (Karmanos Cancer Institutes, Detroit, MI). These cell lines were isolated from a single spontaneous mammary tumor in a Balb/cfC3H mouse, but are phenotypically hetergenous and differ in their metastastic potential: 4T1 and 4T07 cells are able to metastasize from the primary tumor, while 67NR cells are unable to disseminate from the primary tumor.Citation48
Tumor cell lines were implanted orthotopically into the fourth mammary fat pad of 9–16 week old mice at a cell-line specific concentration of 105, 106 and 2 × 105 cells in a 50 μL injection volume for 4T1, 4T07 and 67NR cell lines, respectively. Cell numbers were optimized to produce consistent tumor growth rates, with tumor volumes that approach ethical restrictions 3–3.5 weeks after implantation. Cell lines were maintained for no more than 20 passages in Roswell Park Memorial Institute (RMPI) 1640 medium supplemented with D-glucose, sodium pyruvate, HEPES and 10% fetal bovine serum. Dulbecco's phosphate-buffered saline (DPBS) without calcium or magnesium (Gibco® Life Technologies) and 0.1% trypsin in citrate buffer + 0.02% EDTA were used for passaging.
Tissue processing
Spleens and lymph nodes (LNs) were mechanically disaggregated and passaged through 100 μm and 40 μm mesh filters with PBS to create single cell suspensions. Spleen samples for flow cytometry or cell sorting were treated with ammonium chloride solution (NH4Cl, 1:9 ratio, 9 min on ice according to manufacturer's recommendation, StemCell Technologies) for erythrocyte lysis. NH4Cl lysis was omitted for spleens subsequently undergoing EasySep magnetic bead isolation. Lungs and tumors were finely minced with scalpels and then agitated for 40 min at 37°C with 1 mg/mL type II collagenase (Gibco® Life Technologies) in PBS. After incubation, 0.06% DNase (Sigma Aldrich) was added and the cell suspension filtered through a 100 μm mesh filter with RPMI + 10% FBS. Lungs and tumor samples for flow cytometry were centrifuged for 20 min on Lympholyte M® (Cedarlane) to enrich for lymphocytes. Samples designated for flow cytometry or cell sorting were immediately stained with antibodies following tissue processing.
Flow cytometry
Single-cell suspensions from spleen, LN, tumor or lung were resuspended in PBS and stained for 30 min at 4°C in the dark with a fixable viability dye (eFluor® 780, eBioscience). Unless otherwise stated, all flow cytometry reagents and buffers were obtained from eBioscience. Cells were washed and resuspended in Hank's balanced salt solution with 10 mM HEPES (StemCell Technologies) + 2% FBS and 0.05% NaN3 and stained for 1 h at 4°C with surface markers to CD4 (FITC), CD25 (APC), and CCR5 (PE). To determine the % CCR5 expression by other immune cell subsets, CCR5 (APC, Biolegend) was co-stained with CD11b (PE) and Gr1 (488) (myeloid cells), CD11b (PE) and F4/80 (FITC) (macrophages), CD11c (PE) (dendritic cells), CD335 (FITC) (natural killer cells), CD3 (PE) and CD4 (FITC) (Th cells), CD3 (PE) and CD8 (FITC) (Tc cells). For the analysis of CCR5 co-expression with activation markers, a panel of antibodies including CD4 (FITC), CD25 (BV-421, Biolegend), CD103 (PerCP/Cy5.5, Biolegend), CTLA-4 (APC), and CCR5 (PE) was used. Tumor cell expression of CCR5 was analyzed using in vitro tumor cell lines. Anti-murine CD16/32 (0.5 μg/100μL, clone 2.4G2) was used to block non-specific Fc binding by antibodies. Samples from the immune panel and activation marker experiments were run on an LSR Fortessa and analyzed with FACSDiva software (BD, Mississauga, ON, Canada).
For analysis of CCR5+ Tregs, cells labeled with antibodies against CD4, CD25, and CCR5 were fixed and permeabilized for 30 min according to the manufacturer's instructions using a transcription factor buffer set (eBioscience) to allow subsequent labeling with FoxP3 antibody. For convenience, cells were incubated overnight at 4°C in 1X permeabilization buffer + 2% normal rat serum prior to staining with Foxp3 (PE-Cy7) for 40 min at 4°C in the dark and analyzed using a FACSCalibur (DxP 6-color upgrade, BD) and FlowJo software. Overnight incubation did not change the level or intensity of CD4, CD25, FoxP3, or CCR5 staining compared to samples stained and analyzed on the same day (data not shown). Treg proportions are expressed as a percentage of total CD4+ cells.
Chemokine array and ELISA
Chemokine expression by cell lines and tissues was evaluated using a Proteome Profiler™ antibody array (R&D Systems). Lung and tumor lysates were prepared through mechanical disaggregation of tissue in lysis buffer (PBS + 1% Triton X-100 + 10 μg/mL protease inhibitor cocktail (PIC; aprotinin, leupeptin, pepstatin; Sigma Aldrich) and frozen overnight at −70°C. In vitro tumor cells were resuspended at 107cells/mL in lysis buffer and frozen overnight at −70°C. The following day, tissue and in vitro tumor cell lysates were centrifuged at 10,000 × g for 10 min (4°C) to pellet cell debris, and the lysate was collected. The protein content from each sample was quantified to ensure equal loading, and the array was performed in accordance with the manufacturer's instructions. Loading accuracy was assessed by intensity of the loading control points in the corners of each blot, and heat shock 60 kDa protein 1 (HSP60) or complement factor D (CFD) were positive controls for the assay.
CCL8 concentrations in lung tissue and bronchoalveolar lavage (BAL) were assessed with a CCL8/MCP-2 DuoSet® ELISA development system (R&D Systems). BAL was collected post-mortem by flushing lungs with PBS + PIC using a 22 gauge needle. Lung tissue was homogenized in PBS + PIC and centrifuged at 330 × g to pellet cells. The pelleted cell fraction was processed in lysis buffer as above for the chemokine array. ELISA samples were assayed in triplicate according to the manufacturer's protocol and the results analyzed using ELISA Analysis software (ElisaAnalysis.com; Leading Technology Group, Australia).
Immunofluorescence
Lungs were harvested from 4T1 tumor-bearing mice 3 weeks post orthotopic implant and immediately frozen in Optimal Cutting Temperature (OCT) medium (Sakura Finetek). Serial sections of 8–10 μm were cut and stained in PBS + 4% FBS with antibodies against F4/80 (eBioscience) and CCL8 (R&D Systems) using Alexa Fluor® 488 and 594 secondary antibodies (Life Technologies). Images were captured with a Zeiss Imager Z1 using a cooled, monochrome CCD camera (Retiga 4000R, QImaging) and Northern Eclipse software. The extent of co-localization was quantified using ImageJ software after setting a threshold for each image based on serial sections stained only with the respective secondary antibodies (Fig. S3).
Regulatory T cell isolation and culture
For ex vivo chemotaxis assays, single cell suspensions of splenocytes from tumor-bearing Balb/c mice were enriched for CD4+ cells using a mouse CD4+ T cell isolation kit (EasySep; StemCell Technologies). CD4+ cells were subsequently resuspended at 107 cells/mL in PBS + 10% FBS and stained with viability dye followed by CD4 and CD25 for 1 h at 4°C in the dark (as per flow cytometry protocol). Viable, CD4+CD25+ cells (107 cells/mL) were sorted using a FACSAria II using FACSDiva software (BD) to >90% purity as validated post-sort (Fig. S5A). Gates for CD4 and CD25 were set using single stain controls for each antibody. Median yield of CD4+CD25+ cells post sorting was 2.5 × 106 total cells. Immediately following sorting, CD4+CD25+cells were resuspended to 106 cells/mL in expansion buffer (RPMI + 10% FBS, 50 μM 2-ME (Gibco® Life Technologies), 2.5 μg/mL αCD28 (eBioscience), 100 U/mL IL-2 (eBioscience), then plated into αCD3 (eBioscience) coated wells (10 μg/mL), and cultured for 40 h at 37°C with 5% CO2.
Chemotaxis
Following ex vivo culture, >90% of the CD4+CD25+ sorted cells expressed Foxp3. Median yield post culture was 1.38 × 106 total cells. Viable cells were counted using a hemocytometer and resuspended to 106cells/mL in chemotaxis buffer (RPMI + 10% FBS + 50 μM 2-ME). Using a 96-well format ChemoTx® system (Neuro Probe) with a 5 μm pore size, bottom chamber wells were prepared containing the appropriate chemokine in a final volume of 300 μL. Unless otherwise stated, all chemokines were obtained from PeproTech. RPMI + 10% FBS was used as a negative control. Immediately prior to seeding cells onto the membrane, three aliquots of 50,000 cells were removed for viability testing with 1 µM or 10 µM MVC (Selzentry®, Selleck Chemicals). The remaining Tregs (50,000 cells/well) were seeded directly onto the membrane of each well as per the manufacturer's instructions and allowed to migrate for 3 h at 37°C. Viability controls for MVC were incubated alongside the chemotaxis assay for 3 h at 37°C, and analyzed for % viability using trypan blue staining.
The entire bottom chamber fraction from each sample was collected after chemotaxis and cytospun onto slides. Cells were ethanol-fixed and DAPI stained (1 μg/mL) for imaging with a Zeiss Imager Z1 with a monochrome CCD camera (Retiga 4000R, QImaging) and Northern Eclipse software. ImageJ software was used to count the number of cells per 5X objective field. All conditions were tested in triplicate, and three photos were averaged for each slide to generate a value for the number of CD4+CD25+ cells per field of view.
Maraviroc treatment in vivo
Mice were treated with 31 mg/kg MVC (Celsentri®, ViiV Healthcare) or vehicle (5% DMSO in acidified water)Citation61 once daily by oral gavage. Drug concentration in solution was verified prior to administration using high-performance liquid chromatography (HPLC). The dose given to mice was derived using a BSA dose translation formulaCitation63 based on an equivalent human dosage of 150 mg once daily. Treatment with MVC began 10 d after orthotopic implantation of 1 × 105 4T1 cells and continued until sacrifice at 24 d post-injection. Lung tissue was enzymatically disaggregated as previously describedCitation5 and single-cell suspensions were then washed by centrifugation before NH4Cl erythrocyte lysis. Cells were washed in PBS, resuspended in medium, and aliquots of 104, 105 and 106 cells were plated in triplicate for clonogenic assays containing 60 μM 6-thioguanine (to allow for selective growth of 4T1 cells). Cells were incubated for 9–12 d (37˚C, 5% CO2) before staining colonies with malachite green for enumeration. The total number of clonogenic tumor cells in the lungs was calculated by multiplying the proportion of colony-forming tumor cells by the total number of cells recovered from the lungs. Hematoxylin and Eosin staining of formalin-fixed paraffin-embedded lung tissue was performed on 200 µm step sections, and all slides were interpreted by a Board-certified veterinary pathologist (Dr Meegan Larsen, MBed Pathology).
Statistics
One-tailed or two-tailed Student's t-tests were used for comparison of data containing less than or equal to three experimental groups. A one-way ANOVA was used for the comparison of data sets containing more than three experimental groups. GraphPad Prism was used to analyze data and perform statistical comparisons. Unless otherwise stated, all data are reported as mean + SEM.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1150398_s02_legends.zip
Download Zip (13.2 MB)Acknowledgments
The authors would like to thank Dr Meegan Larsen, DVM, DVSc from MBed Pathology for expert pathological analysis of mouse lung tissue samples. Additionally, the authors would like to thank Drs Megan Levings and Scott Patterson for protocols and helpful discussions, as well as Drs Andrew Minchinton and Alastair Kyle for invaluable assistance with HPLC analyses of MVC solutions.
Funding
This research was funded by the Canadian Institutes of Health Research (CIHR; MOP-126138) and the CIHR: Institute of Cancer Research (COP-120229). ECH and JLC were funded by Frederick Banting and Charles Best Canada Graduate Scholarships from CIHR, and ECH is funded by a Four Year Doctoral Fellowship from the University of British Columbia. MJH is funded by a Canadian Breast Cancer Foundation (CBCF; BC/Yukon Division) Post-Doctoral Fellowship. HNL was funded by a summer studentship from the CBCF (BC/Yukon Division). BJW is funded by an Alexander Graham Bell Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada. KLB is a Michael Smith Foundation for Health Research Biomedical Research Scholar.
References
- Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, Gelmon K. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010; 28:3271-7; PMID:20498394; http://dx.doi.org/10.1200/JCO.2009.25.9820 PMID:20498394
- Zindl CL, Chaplin DD. Immunology. Tumor immune evasion. Science 2010; 328:697-8; PMID:20448171; http://dx.doi.org/10.1126/science.1190310.
- Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 2006; 66:5527-36; PMID:16740684; http://dx.doi.org/10.1158/0008-5472.CAN-05-4128
- Meissner M, Reichert TE, Kunkel M, Gooding W, Whiteside TL, Ferrone S, Seliger B. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin Cancer Res 2005; 11:2552-60; PMID:15814633; http://dx.doi.org/10.1158/1078-0432.CCR-04-2146
- Hamilton MJ, Bosiljcic M, Lepard NE, Halvorsen EC, Ho VW, Banath JP, Krystal G, Bennewith KL. Macrophages are more potent immune suppressors ex vivo than immature myeloid-derived suppressor cells induced by metastatic murine mammary carcinomas. J Immunol 2014; 192:512-22; PMID:24285836; http://dx.doi.org/10.4049/jimmunol.1300096
- Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol 2010; 40:2969-75; PMID:21061430; http://dx.doi.org/10.1002/eji.201040895
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151-64; PMID:7636184.
- Halvorsen EC, Mahmoud SM, Bennewith KL. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev 2014; 33:1025-41; PMID:25359584; http://dx.doi.org/10.1007/s10555-014-9529-x
- Beyer M, Schultze JL. Regulatory T cells in cancer. Blood 2006; 108:804-11; PMID:16861339; http://dx.doi.org/10.1182/blood-2006-02-002774
- Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24:5373-80; PMID:17135638; http://dx.doi.org/10.1200/JCO.2006.05.9584
- Liu F, Lang R, Zhao J, Zhang X, Pringle GA, Fan Y, Yin D, Gu F, Yao Z et al. CD8(+) cytotoxic T cell and FOXP3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat 2011; 130:645-55; PMID:21717105; http://dx.doi.org/10.1007/s10549-011-1647-3
- Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol 2011; 23:424-30; PMID:22055883; http://dx.doi.org/10.1016/j.smim.2011.10.002
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271-5; PMID:18845758; http://dx.doi.org/10.1126/science.1160062
- Chang LY, Lin YC, Kang CW, Hsu CY, Chu YY, Huang CT, Day YJ, Chen TC, Yeh CT et al. The indispensable role of CCR5 for in vivo suppressor function of tumor-derived CD103+ effector/memory regulatory T cells. J Immunol 2012; 189:567-74; PMID:22664873; http://dx.doi.org/10.4049/jimmunol.1200266
- Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 2013; 73:3591-603; PMID:23633484; http://dx.doi.org/10.1158/0008-5472.CAN-12-4100
- Elkord E, Abd Al Samid M, Chaudhary B. Helios, and not FoxP3, is the marker of activated Tregs expressing GARP/LAP. Oncotarget 2015; 6:20026-36; PMID:26343373; http://dx.doi.org/10.18632/oncotarget.4771
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011; 475:226-30; PMID:21753853; http://dx.doi.org/10.1038/nature10169
- Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011; 470:548-53; PMID:21326202; http://dx.doi.org/10.1038/nature09707
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942-9; PMID:15322536; http://dx.doi.org/10.1038/nm1093
- Fialova A, Partlova S, Sojka L, Hromadkova H, Brtnicky T, Fucikova J, Kocian P, Rob L, Bartunkova J et al. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer 2013; 132:1070-9; PMID:22865582; http://dx.doi.org/10.1002/ijc.27759
- Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 2009; 69:2000-9; PMID:19244125; http://dx.doi.org/10.1158/0008-5472.CAN-08-2360
- Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, Cerwenka A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol 2012; 189:5602-11; PMID:23152559; http://dx.doi.org/10.4049/jimmunol.1201018
- Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 2009; 182:1746-55; PMID:19155524; http://dx.doi.org/10.4049/jimmunol.182.3.1746
- Ward ST, Li KK, Hepburn E, Weston CJ, Curbishley SM, Reynolds GM, Hejmadi RK, Bicknell R, Eksteen B et al. The effects of CCR5 inhibition on regulatory T-cell recruitment to colorectal cancer. Br J Cancer 2015; 112:319-28; PMID:25405854; http://dx.doi.org/10.1038/bjc.2014.572
- Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, Forbes B, Edelblute B, Collette B et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res 2011; 71:5522-34; PMID:21742774; http://dx.doi.org/10.1158/0008-5472.CAN-10-3143
- Blaszczyk J, Coillie EV, Proost P, Damme JV, Opdenakker G, Bujacz GD, Wang JM, Ji X. Complete crystal structure of monocyte chemotactic protein-2, a CC chemokine that interacts with multiple receptors. Biochemistry 2000; 39:14075-81; PMID:11087354; http://dx.doi.org/10.1021/bi0009340
- Haworth B, Lin H, Fidock M, Dorr P, Strange PG. Allosteric effects of antagonists on signalling by the chemokine receptor CCR5. Biochem Pharmacol 2007; 74:891-7; PMID:17669370; http://dx.doi.org/10.1016/j.bcp.2007.06.032
- Mueller A, Mahmoud NG, Goedecke MC, McKeating JA, Strange PG. Pharmacological characterization of the chemokine receptor, CCR5. Br J Pharmacol 2002; 135:1033-43; PMID:11861332; http://dx.doi.org/10.1038/sj.bjp.0704540
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ et al. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol 2001; 166:4697-704; PMID:11254730; http://dx.doi.org/10.4049/jimmunol.166.7.4697
- Zhang Y, Meng FY, Li WL, Zhou CX, Guan Z, Fan HY. Association of chemotactic factor receptor 5 gene with breast cancer. Genet Mol Res 2013; 12:5289-300; PMID:24301790; http://dx.doi.org/10.4238/2013.November.7.4
- Carter NJ, Keating GM. Maraviroc. Drugs 2007; 67:2277-88; discussion 89–90; PMID:17927288; http://dx.doi.org/10.2165/00003495-200767150-00010.
- Kersey PJ, Allen JE, Christensen M, Davis P, Falin LJ, Grabmueller C, Hughes DS, Humphrey J, Kerhornou A et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res 2014; 42:D546-52; PMID:24163254; http://dx.doi.org/10.1093/nar/gkt979
- Lynch EA, Heijens CA, Horst NF, Center DM, Cruikshank WW. Cutting edge: IL-16/CD4 preferentially induces Th1 cell migration: requirement of CCR5. J Immunol 2003; 171:4965-8; PMID:14607889; http://dx.doi.org/10.4049/jimmunol.171.10.4965
- Ruffing N, Sullivan N, Sharmeen L, Sodroski J, Wu L. CCR5 has an expanded ligand-binding repertoire and is the primary receptor used by MCP-2 on activated T cells. Cell Immunol 1998; 189:160-8; PMID:9790730; http://dx.doi.org/10.1006/cimm.1998.1379
- Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, Wang H, Chen J. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One 2011; 6:e19495; PMID:21559338; http://dx.doi.org/10.1371/journal.pone.0019495
- Wertel I, Surowka J, Polak G, Barczynski B, Bednarek W, Jakubowicz-Gil J, Bojarska-Junak A, Kotarski J. Macrophage-derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumour Biol 2015; 36:4811-7; PMID:25647263; http://dx.doi.org/10.1007/s13277-015-3133-8
- Karta MR, Gavala ML, Curran CS, Wickert LE, Keely PJ, Gern JE, Bertics PJ. LPS modulates rhinovirus-induced chemokine secretion in monocytes and macrophages. Am J Respir Cell Mol Biol 2014; 51:125-34; PMID:24498897; http://dx.doi.org/10.1165/rcmb.2013-0404OC
- Kroetz DN, Deepe GS, Jr. An aberrant thymus in CCR5−/− mice is coupled with an enhanced adaptive immune response in fungal infection. J Immunol 2011; 186:5949-55; PMID:21478401; http://dx.doi.org/10.4049/jimmunol.1003876
- Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood 2012; 119:5155-63; PMID:22403258; http://dx.doi.org/10.1182/blood-2011-11-388918
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009; 29:313-26; PMID:19441883; http://dx.doi.org/10.1089/jir.2008.0027
- Shang B, Liu Y, Jiang SJ. Prognostic value of tumor-infiltrating FoxP3(+) regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015; 5:15179; PMID:26462617; http://dx.doi.org/10.1038/srep15179
- Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, Okabe K, Matsumoto T, Sugi K et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 2012; 75:95-101; PMID:21719142; http://dx.doi.org/10.1016/j.lungcan.2011.06.002
- Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res 2011; 71:6915-20; PMID:22068034; http://dx.doi.org/10.1158/0008-5472.CAN-11-1156
- Hembruff SL, Cheng N. Chemokine signaling in cancer: Implications on the tumor microenvironment and therapeutic targeting. Cancer Ther 2009; 7:254-67; PMID:20651940
- Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 2002; 169:2756-61; PMID:12193750; http://dx.doi.org/10.4049/jimmunol.169.5.2756
- Lal A, Chan L, Devries S, Chin K, Scott GK, Benz CC, Chen YY, Waldman FM, Hwang ES. FOXP3-positive regulatory T lymphocytes and epithelial FOXP3 expression in synchronous normal, ductal carcinoma in situ, and invasive cancer of the breast. Breast Cancer Res Treat 2013; 139:381-90; PMID:23712790; http://dx.doi.org/10.1007/s10549-013-2556-4
- Takenaka M, Seki N, Toh U, Hattori S, Kawahara A, Yamaguchi T, Koura K, Takahashi R, Otsuka H et al. FOXP3 expression in tumor cells and tumor-infiltrating lymphocytes is associated with breast cancer prognosis. Mol Clin Oncol 2013; 1:625-32; PMID:24649219; http://dx.doi.org/10.3892/mco.2013.107
- Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res 2000; 2:331-4; PMID:11250725; http://dx.doi.org/10.1186/bcr77
- Parviz M, Chin CS, Graham LJ, Miller C, Lee C, George K, Bear HD. Successful adoptive immunotherapy with vaccine-sensitized T cells, despite no effect with vaccination alone in a weakly immunogenic tumor model. Cancer Immunol Immunother 2003; 52:739-50; PMID:12827306; http://dx.doi.org/10.1007/s00262-003-0405-8
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 2009; 206:1717-25; PMID:19581407; http://dx.doi.org/10.1084/jem.20082492
- Soler DC, Sugiyama H, Young AB, Massari JV, McCormick TS, Cooper KD. Psoriasis patients exhibit impairment of the high potency CCR5(+) T regulatory cell subset. Clin Immunol 2013; 149:111-8; PMID:23954573; http://dx.doi.org/10.1016/j.clim.2013.06.007
- Velasco-Velazquez M, Pestell RG. The CCL5/CCR5 axis promotes metastasis in basal breast cancer. Oncoimmunology 2013; 2:e23660; PMID:23734321; http://dx.doi.org/10.4161/onci.23660
- Chang LY, Lin YC, Mahalingam J, Huang CT, Chen TW, Kang CW, Peng HM, Chu YY, Chiang JM et al. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res 2012; 72:1092-102; PMID:22282655; http://dx.doi.org/10.1158/0008-5472.CAN-11-2493
- Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother 2008; 57:123-31; PMID:17522861; http://dx.doi.org/10.1007/s00262-007-0336-x
- Ochoa-Callejero L, Perez-Martinez L, Rubio-Mediavilla S, Oteo JA, Martinez A, Blanco JR. Maraviroc, a CCR5 antagonist, prevents development of hepatocellular carcinoma in a mouse model. PLoS One 2013; 8:e53992; PMID:23326556; http://dx.doi.org/10.1371/journal.pone.0053992
- Rosario MC, Jacqmin P, Dorr P, James I, Jenkins TM, Abel S, van der Ryst E. Population pharmacokinetic/pharmacodynamic analysis of CCR5 receptor occupancy by maraviroc in healthy subjects and HIV-positive patients. Br J Clin Pharmacol 2008; 65 Suppl 1:86-94; PMID:18333870; http://dx.doi.org/10.1111/j.1365-2125.2008.03140.x
- Saita Y, Kondo M, Shimizu Y. Species selectivity of small-molecular antagonists for the CCR5 chemokine receptor. Int Immunopharmacol 2007; 7:1528-34; PMID:17920529; http://dx.doi.org/10.1016/j.intimp.2007.07.019
- Gao P, Zhou XY, Yashiro-Ohtani Y, Yang YF, Sugimoto N, Ono S, Nakanishi T, Obika S, Imanishi T et al. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol 2003; 73:273-80; PMID:12554804; http://dx.doi.org/10.1189/jlb.0602269
- Uekusa Y, Yu WG, Mukai T, Gao P, Yamaguchi N, Murai M, Matsushima K, Obika S, Imanishi T et al. A pivotal role for CC chemokine receptor 5 in T-cell migration to tumor sites induced by interleukin 12 treatment in tumor-bearing mice. Cancer Res 2002; 62:3751-8; PMID:12097285
- Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, Obika S, Imanishi T, Tsujimura T et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol 2002; 32:2124-32; PMID:12209624; http://dx.doi.org/10.1002/1521-4141(200208)32:8%3c2124::AID-IMMU2124%3e3.0.CO;2-S
- Velasco-Velazquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, Pestell RG. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res 2012; 72:3839-50; PMID:22637726; http://dx.doi.org/10.1158/0008-5472.CAN-11-3917
- Lieberman-Blum SS, Fung HB, Bandres JC. Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin Ther 2008; 30:1228-50; PMID:18691983; http://dx.doi.org/10.1016/S0149-2918(08)80048-3
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008; 22:659-61; PMID:17942826; http://dx.doi.org/10.1096/fj.07-9574LSF