
ABSTRACT
Analysis of genomic data from patient tumors provides valuable information as to potential T-cell targets such as neoepitopes. We developed an approach to characterize, isolate and utilize neoantigens-specific T cells using MHC/peptide tetramers from fresh tumor digests and peripheral blood. This bears important implications for the implementation of T cell-based immunotherapy.
Recent developments in next generation sequencing have enabled the rapid and precise determination of mutational landscape in patient tumors. Besides common mutations (e.g., p53, Ras), all cancers contain unique somatic genetic alterations particularly abundant in carcinogen-exposed tumors (such as melanomas, colon and lung cancers). Some may be expressed as mutated proteins that could be recognized by the immune system. Approaches targeting specifically these neoepitopes may provide therapeutic benefit in the absence of off-target and normal tissue toxicities Citation1. Recent studies showed these antigens can elicit specific T-cell responses Citation2 and that treatment with cellular products enriched for such specific T cells can lead to tumor regression.Citation3
To develop an approach to rapidly isolate and expand cancer patients' T-cells specific for mutated antigens from different tissue sources (tumor digest or peripheral blood), we analyzed tumor genomic data from eight melanoma patients for non-synonymous mutations occurrence.Citation4 The latter ranged between 300 and 10,000 mutations per tumor. Next, we predicted high-affinity class I HLA-binders using available algorithms.Citation5 We used arbitrary filters to limit the number of epitopes to screen and setup a threshold of up to 48 peptides with a percentile value lower than 1%. Selected synthesized predicted epitopes were used to generate panels of fluorescent MHC-tetramersCitation6 ().
Figure 1. Predicting and using neoepitopes to identify T cells. (A) Schematic representation of the initial research steps covering next generation sequencing, identification of mutations and neoepitope/MHC tetramer generation. (B) Relative frequency of tetramer positive cells in fresh tumor digests (upper panel) and results of peripheral blood analysis of patient 3919 with mutated TRIP12/HLA-A*0101 tetramers bearing two chromophores (C1 & C2—lower panel). (C) Purity of tetramer-sorted PBLs specific for the mutated TRIP12 peptide (upper panel) and IFNγ secretion measured in co-cultures of tetramer sorted T cells with antigen presenting cells pulsed with mutated and wt TRIP12 peptides (lower panel).
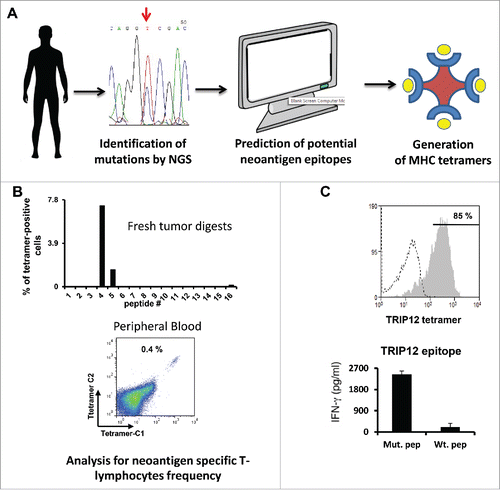
T-cells derived from fresh tumor digests were screened for neoepitope/MHC tetramer binding () and we detected neoantigen specific T-lymphocytes for at least one epitope in 4 out of 7 patient samples. Interestingly, T cells from the FTD cultures of two patients (patient 3713 and 3466) with the largest amount of non-synonymous somatic mutations (in the thousands) each bound to three neoepitope/MHC HLA-A2 tetramers, while only one or no epitope was recognized by T cells from patients harboring hundreds of non-synonymous mutations. Electronically sorted T cells were expanded over a few days 1000x or more to a high degree of purity ranging from 50%–99% tetramer-positive and displayed exquisite recognition of the mutated epitope and not of its wild-type version in co-culture ().
As surmised, all of the specificities identified in the FTDs were also detected in the expanded TIL cultures though their relative frequencies often differed dramatically between the two. Indeed, the proportion of AHNAK-specific T-cells shrank from 1% to 0.1% while TEAD-1-specific lymphocyte population was 40 times over-represented following TIL clinical expansion process. In a specific case, patient 3703, we observed an interesting phenomena as T-cells recognizing the same class I MHC mutated epitope NSDHL expressed either CD8+ or, surprisingly, CD4+. It was suggested that CD4+ T cells can have a direct role in antitumor immunity such as mediating tumor cytotoxicity, augmenting CD8+ T-cell responses and activating innate antitumor immune cells. Further characterization of unusual class I MHC-specific CD4+ T cells will shed light on their possible role in antitumor response in situ.
One novel aspect of the present work was the demonstration of the existence (and subsequent isolation) of neoepitope specific T cells directly from patient peripheral blood. Their initial frequency was, as expected, extremely low (a few in 105 cells) and usually hardly discernable but we could isolate and expand these cells to a considerably higher proportion (up to approximately 80% tetramer-positive T cells). These T cells were able to specifically recognize their cognate tumors. This underscored the potential benefit of using PBMCs as a source of neoepitope specific T cells for adoptive cell immunotherapy.
Another possible application we exemplified was the feasibility of the rapid cloning of the TCR chains expressed by neoantigen-specific T-cell populations. Beyond its therapeutic potential, this approach could provide some hints about the nature of the cellular immune response against neoantigens.Citation7
While T cells specific for mutated epitopes could be identified and isolated in 5 out of 8 patients studied, it may be possible to enhance the success rate of this approach by screening more MHC alleles and/or by improving the fidelity of the prediction algorithms. This point was well illustrated when trying to identify the optimal TEAD1 epitope from patient 3466—better predicted MHC binding was inversely correlated to in vitro measured T-cell reactivity.
To what extent the presence of neoepitope specific T cells could serve as a predictive marker for eventual clinical response? For example, patient 3919 had 0.4% of his circulating T cells specific for the mutated TRIP12 epitope but was found to be a non-responder. Alternatively, if a predicted neo epitope could not trigger T-cell reactivity, would this mean that it is not immunogenic enough, that there are no T-cell precursors generated against it, that there might be an early response to this epitope but those specific T cells have become scarce or that it is not presented at all by the tumor? Mass spectrometry analysis of eluted MHC epitopes from the tumor might shed light on this.Citation8
The clinical implementation of this approach could benefit from targeting multiple antigens simultaneously and/or combining immune checkpoint inhibitors,Citation9 bearing in mind that the number of mutations within a tumor may be useful for prospectively identifying patients who will respond favorably to checkpoint blockade.Citation10
Finally, technological development will prove crucial for the clinical implementation of this approach. One can envision a pipeline in which genetic material (DNA and RNA) from the excised tumor would be rapidly sequenced, enabling the precise identification of immunogenic mutations using improved prediction algorithm (7 d). Epitopes would be synthesized and tetramer generated within another week. Screening of patient samples (tumor digests or peripheral blood) and expansion of isolated T cells could take up to 2 weeks. This could signify that a therapeutic cellular product could be obtained within a month approximately. This is an exciting era for immune-oncology and the extension of such personalized treatments (as depicted herein) to other malignancies will undoubtedly further strengthen the benefit of immunotherapy to the masses.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/10.1126/science.aaa4971
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med 2013; 19:747-52; PMID:23644516; http://dx.doi.org/10.1038/nm.3161
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641-5; PMID:24812403; http://dx.doi.org/10.1126/science.1251102
- Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin. Invest 2015; 125:3981-91; PMID:26389673; http://dx.doi.org/doi: 10.1172/JCI82416
- Desai DV, Kulkarni-Kale U. T-cell epitope prediction methods: an overview. Methods Mol Biol 2014; 1184:333-64; PMID:25048134; http://dx.doi.org/10.1007/978-1-4939-1115-8_19
- Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 2006; 1: 1120-32; PMID:17406393; http://dx.doi.org/10.1038/nprot.2006.121
- Leisegang M, Kammertoens T, Uckert W, Blankenstein T. Targeting human melanoma neoantigens by T cell receptor gene therapy. J Clin. Invest 2016; 126:854-8; PMID:26808500; http://dx.doi.org/10.1172/JCI83465
- Kalaora S, Barnea E, Merhavi-Shoham E, Qutob N, Teer JK, Shimony N, Schachter J, Rosenberg SA, Besser MJ, Admon A et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016; PMID:26819371; http://dx.doi.org/10.18632/oncotarget.6960
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 348:124-8; PMID:25428507; http://dx.doi.org/10.1038/nature13988
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124-8; PMID:25765070; http://dx.doi.org/10.1126/science.aaa1348