
ABSTRACT
Chronic sterile inflammation has been implicated in the pathogenesis of many cancers, including skin cancer. Chronic arsenic exposure is closely associated with the development of skin cancer. However, there is a lack of understanding how arsenic induces chronic inflammation in the skin. Interleukin-1 family cytokines play a central role in regulating immune and inflammatory response. IL-1α, IL-1β and IL-18 are three pro-inflammatory cytokines in IL-1 family. Their secretion, especially the secretion of IL-1β and IL-18, is regulated by inflammasomes which are multi-protein complexes containing sensor proteins, adaptor protein and caspase-1. The data from current study show sub-chronic arsenic exposure activates AIM2 inflammasome which in turn activates caspase-1 and enhances the secretion of IL-1β and IL-18 in HaCaT cells and the skin of BALB/c mice. In addition, arsenic-promoted activation of AIM2 inflammasome and increase of IL-1β/IL-18 production are inhibited by PKR inhibitor in HaCaT cells or in the skin of PKR mutant mice, indicating a potential role of PKR in arsenic-induced sterile inflammation.
KEYWORDS:
Introduction
Chronic exposure to arsenic may promote many diseases. The primary arsenic exposure route for human is through diet and drinking water. Therefore, excessive arsenic exposure remains an important public health concern in many areas of the United States where private or unregulated wells are the resource of drinking water.Citation1
Skin cancer is the most common malignancy in the United States and some of these cases have been associated with chronic arsenic exposure. Chronic inflammation plays a critical role in skin tumor initiation and progression,Citation2 and can be induced by tumorigenic pathogens, immune deregulation, autoimmunity, environmental exposure and obesity. Chronic inflammation precedes tumor development and contributes to tumor initiation through inducing oncogenic mutations, enhancing genomic instability, and altering immune response.Citation3 Overproduction of inflammation-related cytokines and subsequently activation of their downstream transcriptional factors are two main consequences of chronic inflammation that are thought critically involved in arsenic tumorigenesis.Citation4 Cytokines can be roughly divided into two groups: pro-inflammatory cytokines and anti-inflammatory cytokines, with some cytokines have both pro- and anti-inflammatory effects under different conditions. Generally, pro-inflammatory cytokines, such as interleukin 1β (IL-1β), IL-18, IL-6 and tumor necrosis factor α (TNF-α), promote tumorigenesis; while anti-inflammatory cytokines, such as IL-4, IL-10 and IL-13, inhibit it.Citation5-7
The cytokines in interleukin-1 family play a central role in regulating immune and inflammatory response. IL-1α, IL-1β and IL-18 are translated in the cytoplasm as pro-forms. These transcripts are then proteolytically cleaved by calpain (for IL-1α) or caspase-1 (for IL-1β and Il-18) to produce the mature proteins which are released into extracellular milieu. Inflammasomes, the multiprotein complexes containing sensor proteins, adaptor protein (e.g. apoptosis-associated speck-like protein with a CARD, or ASC) and caspase-1, operates as a platform for caspase-1 cleavage and regulates maturation of IL-1β and IL-18. Without stimuli, inflammasomes remain inactivated in the keratinocytes and only the pro-forms of IL-1β is generated.Citation8 Depending on the sensor proteins, inflammasome can be categorized as NOD-like receptor protein 1 (NLRP1), NLRP3, NLRP6, NLR family CARD domain-containing protein 4 (NLRC4) and absent in melanoma 2 (AIM2) inflammasomes. It has also been suggested that calpain-regulated IL-1α maturation may also be mediated by inflammasomes.Citation9 In addition, inflammasomes are activated in human melanoma cells and in ultraviolet (UV) radiation-damaged keratinocytes.Citation10 However, whether inflammasome activation is involved in arsenic-induced inflammation is not clear.
Double-stranded RNA-activated protein kinase (PKR) is a serine/threonine PKR ubiquitously expressed in mammalian cells. It has been initially identified as an interferon-induced protein in virus-infected cells activated by dsRNA produced during the virus life cycle.Citation11 Later, it was found PKR, as a cell stress response kinase, plays important roles in the regulation of inflammatory immune responses. More recently, Lu et al discovered that PKR is required for the activation of inflammasomes which leads to secretion of pro-inflammatory cytokines, IL-1β and IL-18.Citation12 In current study, we investigated the effects of arsenic on inflammasome activity in human keratinocyte HaCaT cells as well as in the skin of PKR mutant mouse. Our data suggest that arsenic exposure activates AIM2 inflammasome and enhances the secretion of IL-1β and IL-18 through activation of PKR.
Results
Arsenic exposure increases the mRNA levels and secretion of IL-1α, IL-1β and IL-18 in HaCaT cells
We first determined the effects of arsenic on the secretion of the cytokines that may be regulated by inflammasome mechanism, including IL-1α, IL-1β and IL-18 in human keratinocyte HaCaT cells. As shown in , exposure of HaCaT cells to sodium arsenite at 0.5, 2 and 10 μM for 24, 48 and 72 h increased the mRNA levels of IL-1α, IL-1β and IL-18 in the cells and protein levels of IL-1β and IL-18 in the media. Arsenic increased the secretion of IL-1β or IL-18 in a dose and time dependent manner; but it was not for IL-1α. Notably, although the mRNA levels of IL-1β and IL-18 were readily detectable in control cells, their protein levels were barely detectable in extracellular milieu without arsenic treatment.
Figure 1. Acute arsenic treatment increases IL-1α, IL-1β and IL-18 mRNA levels as well as secretion levels in HaCaT cells. (A) HaCaT cells were treated with 0.5, 2 or 10 µM sodium arsenite for 24 h, or treated with 2 µM sodium arsenite for 24, 48 or 72 h. The mRNA levels of IL-1α, IL-1β and IL-18 in the cells were determined by real-time PCR. (B) The protein secretion levels of IL-1α, IL-1β and IL-18 in the media were determined by ELISA. (C) Cell viabilities were determined by cell counting kit-8. The experiments were repeated three times. Data are expressed as the mean ± SEM, *p < 0.05 vs. control.
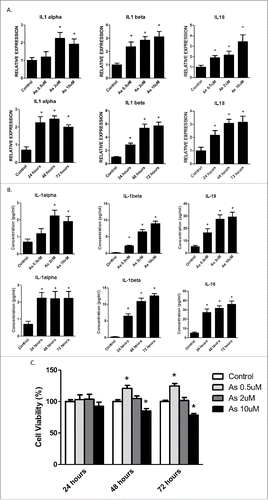
Since alterations in numbers in either cell proliferation or viability may affect cytokine secretion, the effects of arsenic at different concentrations on the growth of HaCaT cells were examined. As shown in , arsenic at 0.5 μM promoted HaCaT cell proliferation, whereas at 10 μM it caused apoptosis of HaCaT cells. At 2 μM, arsenic does not significantly affect the proliferation or viability of HaCaT cells; therefore the concentration of 2 μM was chosen for the rest of the in vitro experiments.
Arsenic promotes pro-caspase-1 cleavage in HaCaT cells via AIM2 inflammasome
To determine if arsenic-promoted secretion of IL-1α, IL-1β and IL-18 resulted from increased inflammasome activity, the level of active caspase-1 after arsenic treatment was examined. As shown in , arsenic induced pro-caspase-1 cleavage in a time-dependent manner.
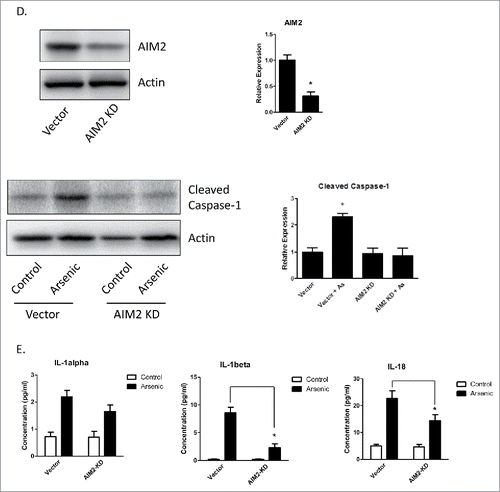
Figure 2. Arsenic activates AIM2 inflammasome in HaCaT cells. (A) HaCaT cells were treated with sodium arsenite at 2 µM for 24, 48 or 72 h. Expressions of cleaved caspase-1 were detected by Western-blot. (B) Real-time PCR showed the mRNA level of AIM2, NLRP1, NLRP3, NLRC4 and ASC in HaCaT cells after arsenic treatment. (C) The protein level of AIM2 was determined by Western-blot and quantified by densitometry. (D) HaCaT cells were transiently transfected with AIM2-shRNA plasmids. The downregulation of AIM2 was determined by Western-blot. (E) Control or AIM2-knockdown HaCaT cells were treated with arsenic for 24 h. The levels of cleaved caspase-1 were determined by Western-blot. Cytokine secretions in the media of arsenic-treated or control cells were determined by ELISA. The experiments were repeated three times. Data are represented as mean ± SEM of three experiments. *p < 0.05 vs. control.
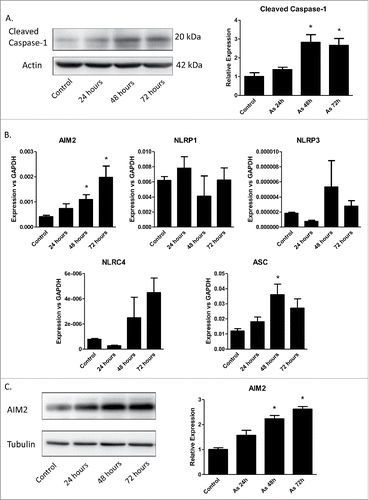
Figure 2. (Continued).
Inflammasomes can be categorized by their sensor proteins as AIM2, NLRP1, NLRP3, NLRC4 and NLRP6 inflammasomes, respectively. To find which inflammasome(s) that was involved in arsenic's action, we performed real-time PCR to analyze the changes of mRNA levels of these sensors. Among these sensors, only the mRNA level of AIM2 was increased by arsenic treatment in a time-dependent manner (). This observation was consistent with an increase of AIM2 protein level upon arsenic exposure as detected by immunoblotting ().
To confirm that AIM2 inflammasome regulated arsenic-induced pro-caspase-1 cleavage as well as the cytokines secretion, we knocked down AIM2 by transiently transfecting HaCaT cells with AIM2-shRNA plasmids. AIM2 downregulation inhibited arsenic-induced pro-caspase-1 cleavage () and secretion of IL-1β and IL-18. However, downregulation of AIM2 did no significantly affect IL-1α secretion ().
Arsenic-induced pro-caspase-1 cleavage and secretion of IL-1β and IL-18 were inhibited in the skin of AIM2 KO mice
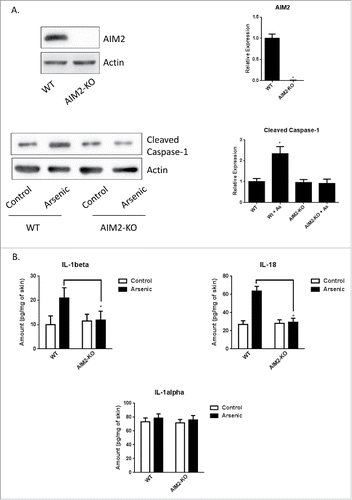
To determine the role of AIM2 in arsenic-induced pro-casepase-1 cleavage and the secretion of IL-1β and IL-18 in vivo, we treated wild type and AIM2 KO mice with arsenic in drinking water for 8 weeks. The AIM2 is deleted in the AIM2 KO mice as verified by western blot (). As expected, arsenic exposure increased the cleavage of pro-caspase-1 () and secretion of IL-1α, IL-1β and IL-18 (). AIM2 deficiency significantly decreased arsenic-induced cleavage of pro-caspase-1 and secretion of IL-1β and IL-18 observed in the wild type mice. Of note, arsenic did not affect the secretion of IL-1α by the skin of both WT and AIM2 KO mice ().
Figure 3. AIM2 deficiency inhibits arsenic-induced cleavage of pro-casepase-1 and secretion of IL-1β and IL-18 in the skin of AIM2 KO mice. (A) AIM2 deficiency was confirmed by protein gel blot (upper panel) and arsenic-induced cleavage of pro-casepase-1 was inhibited in the skin of AIM2 KO mice. (B) Arsenic-induced secretion of IL-1β and IL-18, but not IL-1α, in the skin of wildtype mice was inhibited in AIM2 KO mice. The experiments were repeated three times. Data are represented as mean ± SEM of three experiments. *p < 0.05 vs. control.
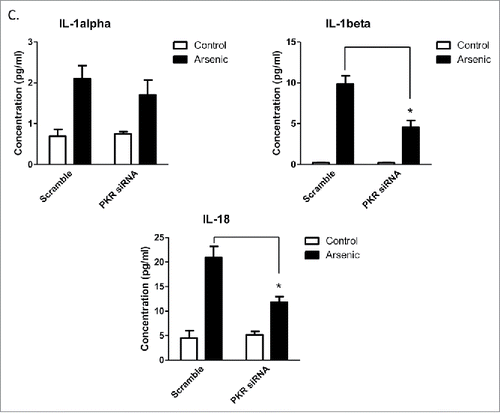
Arsenic activates AIM2 inflammasome through activation of PKR
Double-stranded RNA–dependent PKR plays a critical role in inflammation and immune response. It has been shown that PKR regulates inflammasome activation.Citation12 Therefore, we thought to determine if PKR was involved in arsenic-promoted inflammasome activation. As shown in , PKR was activated by arsenic, which was evidenced by the phosphorylation of PKR and its downstream target, eIF2α. To confirm the role of PKR in arsenic-induced AIM2 activation, we investigated the effect of inhibition of PKR, by PKR inhibitors [2-aminopurine (2-AP) or imidazolo-oxindole (C16)] or PKR siRNA, on upregulation AIM2, cleavage of pro-caspase-1 and secretion of IL-1β and IL-18. As shown in , inhibition of PKR, evidenced by decreased phosphorylation of eIF2α, abrogated arsenic-induced AIM2 upregulation, pro-caspase-1 cleavage and IL-1β/IL-18 secretion.
Figure 4. Arsenic activates AIM2 inflammasome through activation PKR. (A) HaCaT cells were treated with sodium arsenite at 2 µM for 24, 48 or 72 h. The levels of p-PKR, PKR, p-eIF2α and eIF2α were determined by Western-blot. (B) HaCaT cells were treated with 2 µM arsenic together with PKR inhibitors C16 or 2-AP for 24 h, or transiently transfected with PKR siRNA and then treated with arsenic for 24 h. The levels of p-PKR, PKR, p-eIF2α, eIF2α, AIM2 and cleaved caspase-1 were determined by Western-blot and quantified by densitometry. (D) Cytokine secretions in the media of arsenic-treated or control cells were determined by ELISA. The experiments were repeated three times. Data are represented as mean ± SEM of three experiments. *p < 0.05 vs. control.
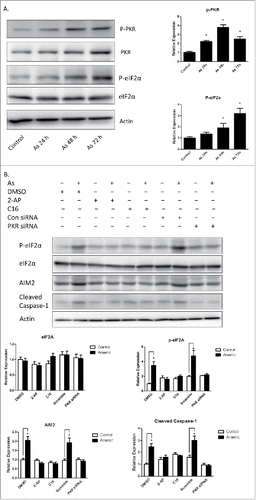
Figure 4. (Continued).
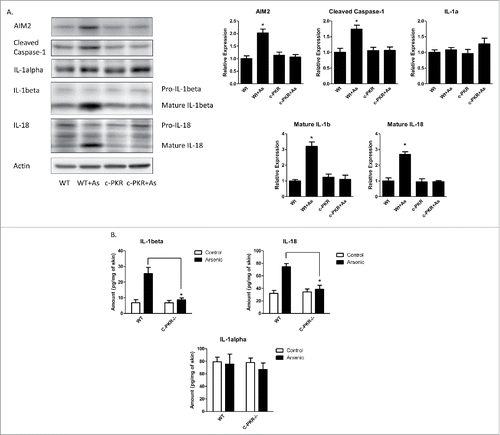
Arsenic-induced activation of AIM2 inflammasome and secretion of IL-1β and IL-18 were inhibited in the skin of C-PKR−/− mice
To investigate the role of PKR in arsenic-induced AIM2 inflammasome activation in vivo, we treated wild type and c-PKR−/− mice with arsenic in drinking water for 8 weeks. The catalytic domain in the c-terminus of PKR in the c-PKR−/− mice was disrupted and thus the function of PKR was abrogated.Citation13 As shown in , the protein levels of AIM2, cleaved caspase-1 as well as IL-1β and IL-18 were increased by arsenic treatment in the wild type mice. However, these increases were inhibited in c-PKR−/− mice. The secretion of IL-1β and 18 were also inhibited by PKR deficiency as determined by ELISA assay (). Similarly as seen in , the secretion of IL-1α was not affected by arsenic in the skin of both WT and PKR deficiency mice.
Figure 5. PKR mutation inhibits arsenic-induced activation of AIM2 inflammasome and secretion of IL-1β and IL-18, but not IL-1α, in the skin of C-PKR−/− mice. The C-PKR−/− mice and controlled C57BL/6J mice were treated with 0.25 µM sodium arsenite in drinking water for 8 weeks. The skin tissues were collected. (A) The protein levels of AIM2, cleaved caspase-1, IL-1α, IL-1β and IL-18 were determined by Western-blot and quantified by densitometry. (B) The secretion levels of IL-1α, IL-1β and IL-18 were determined by ELISA assay. The experiments were repeated three times. Data are represented as mean ± SEM of three experiments. *p < 0.05.
Discussion
Chronic inflammation contributes to tumorigenesis by enhancing oncogenic mutations, tumor promotion and angiogenesis. Tumor-promoting inflammation is driven by many factors including pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8 and CCL-5, and all of which can be regulated by IL-1β.Citation14 IL-1β has been closely associated with tumor formation, growth and angiogenesis.Citation15 IL-1β-deficient mice had decreased tumor-numbers and tumor size in a fibrosarcoma model.Citation7 They also have a reduced angiogenesis capacity compared to their wild type counterparts.Citation16
It has been reported that IL-1β is produced as a pro-form protein (31-kD form) which is biological inactive in keratinocytes without inflammatory stimuli.Citation8 Under pathological conditions, this IL-1β precursor protein is converted to a 17-kD bioactive form by a post-translational site-specific cleavage.Citation17 For keratinocytes, the chemical stimuli capable of inducing this cleavage include trinitro-chlorobenzene (TNBC), sodium dodecyl sulfate (SDS), UVCitation18 and Nickel.Citation19 The cleavage is conducted by inflammasome-controlled active caspase-1. Our data confirm this by showing that without arsenic exposure IL-1β secretion was very low, although its mRNA level was readily detectable. Arsenic exposure activated AIM2 inflammasome which in turn induced caspase-1 cleavage and converted the pro-forms of IL-1β and IL-18 into their active forms. In addition, although IL-1α is also upregulated by arsenic in HaCaT cells, their secretion is not affected by either arsenic or AIM2/PKR confirming the regulation mechanisms for IL-1α are different either between HaCaT cells and mouse skin or between IL-1α and IL-1β/IL-18.
The role of IL-18 in tumorigenesis is complicated. It has been shown IL-18 may either promote or inhibit the progress of cancer. Increase in IL-18 has been observed in various types of cancer including ovarian carcinoma, breast cancer and others. However, it has also been shown that administration of IL-18 may inhibit cancer progress in a B16 melanoma model.Citation20 Therefore, the roles of IL-18 may vary depending on the types of cancer and tissues. The implication of increased secretion of IL-18 in arsenic tumorigenesis is worth further investigating.
In this study, we discovered that arsenic induces AIM2 inflammasome activation in HaCaT cells and in the skin of the mice. Of note, the current study investigates the effects of chronic, low dose arsenic exposure on inflammatory response in the skin cells. High doses and acute exposure of arsenic to different types of cells may obtain varied results. Indeed, the data from Maier and colleagues show that treatment of mouse fibroblasts with arsenic at 50–70 μM for 2 h inhibits inflammasomes activation, pre-caspase-1 cleavage and IL-1β secretion.Citation21
Our data further support an important role of PKR in regulating inflammasome activation. Previously, it has been shown that PKR is involved in the activation of various inflammasomes in response to ATP, monosodium urate, adjuvant aluminum or live E. coli in murine macrophages.Citation12 In current study, our data confirm that PKR activates AIM2 inflammasome in response to arsenic exposure in human keratinocyte HaCaT cells since inhibition of PKR abolished arsenic-induced upregulation of AIM2 cleavage of pro-caspase-1 and IL-1β/IL-18 secretion. This PKR-mediated activation of AIM2 inflammasome was further supported by our animal data.
The detailed mechanisms for arsenic-induced AIM2 upregulation and AIM2 inflammasome activation are subject to further investigation. Since interferons activate AIM2 inflammasomeCitation22-24 and activation of PKR may increases interferons through eIF-2α,Citation25 it is possible that arsenic enhances AIM2 expression through PKR/interferons. Other studies have also shown that cytosolic DNA plays an important role in AIM2 inflammasome activation.Citation26,27 Therefore, whether cytosolic DNA is necessary for arsenic-induced AIM2 inflammasome activation is an interesting topic for further study.
Materials and methods
Materials
Sodium arsenite solution (70039), puromycin (P8833) imidazolo-oxindole (PKR inhibitor C16, I9785), 2-aminopurine (A3509) and antibody against actin (A5441) were purchased from Sigma-Aldrich. Human PKR siRNA (SR303767) and control siRNA (SR30004) was purchased from Origene. The antibodies directed against AIM2 (sc-137967, mouse), cleaved caspase-1 (sc-22166, mouse), PKR (sc-709 human; sc-6282, mouse), phospho-PKR (sc-101784), PACT (sc-377103) were purchased from Santa Cruz. The antibodies directed against cleaved caspase-1 (4199, human), AIM2 (8055, human), phospho-eIF2α (3398), eIF2α (5324) were obtained from Cell Signaling Technology. The antibodies directed against IL-1β (AB-201-NA) and IL-18 (D043-3, human; D046-3, mouse) were purchased from R&D Systems.
Animals and arsenic treatment
The AIM2−/− and wildtype C57 BL/6 J mice were purchased from the Jackson Lab. The C-PKR−/− mice in BALB/c background were generous gifts from Dr. John C. Bell at Ottawa Hospital Research Institute.Citation13 The wild type BALB/c mice were purchased from Harlan Laboratories (Indianapolis, IN). The animals were housed in a specific pathogen-free room in the Animal Facility at the University of Kentucky Medical Center. Animals were allowed to acclimatize to their new environments for 1 week prior to use. All procedures were performed in accordance with the guidelines set by the NIH and the Animal Care and Use Committee of the University of Kentucky. For arsenic treatment, mice were received 50 ppb sodium arsenite in drinking water for 8 weeks. Previously, it has been shown that exposure of mice to 10 ppb sodium arsenite alters expression of immune response genesCitation28 and sodium arsenite at 50 ppbCitation29 or 1 ppm Citation30 promote lung tumor formation in CD-1 or A/J mice, respectively. In current study, we treated the mice with 50 ppb sodium arsenite in drinking water for 8 weeks to determine the effects of arsenic on inflammasome activation.
Cell culture and chemicals treatment
Human keratinocyte HaCaT cells from Addexbio Technologies were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine and 1x antibiotic-antimycotic solution (Invitrogen, 15240–096) at 37°C in a humidified atmosphere with 5% CO2. Sodium arsenite solution was used for arsenic treatment as previously reported.Citation31 For arsenic exposure, 1×106 cells were plated into each 75 cm2 flask. On the second day, the cells were rinsed with fresh medium and cultured with new growth media containing sodium arsenite of different concentrations. For PKR inhibitors treatment, imidazolo-oxindole (C16) or 2-aminopurine (2-AP) were added into culture media for 24 h and then sodium arsenite was added. The final concentrations of C16 and 2-AP in cell culture media were 0.5 µM and 10 mM, respectively, as previously reported.Citation32
Plasmid, siRNA and transfection
Human AIM2 shRNA plasmid (TR314876), human PKR siRNA (SR303767) and control siRNA (SR30004) was purchased from Origene. For transfection, cells were transfected with plasmids or siRNA using Lipofectamine 2000 (Invitrogen, 11668–019) and selected by puromycin according to the manufacturer's protocol. The transfection was confirmed by immunoblotting analysis.
Cell viability assay
Cell viability was quantified using a Cell Counting kit-8 (CCK-8, Dojindo) according to the manufacturer's instructions. HaCaT cells were cultured in 96-well microplate at a density of 3 × 104 cells/well for 24 h. The cells were then treated with arsenic of 0.5, 2 or 10 µM for 24, 48 or 72 h. Untreated HaCaT cells were used as negative control. There were three wells in a microplate for each treatment. After treatment, CCK-8 solution (10 µL) was added to each well of the plate, and the cells were incubated at 37°C for 2 h. The optical density at a wavelength of 450 nm was measured with a SpectraMax M2e microplate reader (Molecular Devices). The results were expressed as the mean ± SEM from three independent experiments.
Cytokine assay
IL-1α, IL-1β and IL-18 productions in HaCaT cells were evaluated by using human IL-1alpha Platinum ELISA (BMS243/2, eBiosciences), human IL-1 β/IL-1F2 Quantikine HS ELISA (HSLB00C, R&D systems), human IL-18/IL-1F4 ELISA (7620, R&D systems) kit, respectively. For dose-dependent assay, cells were plated in 12-well plates at a density of 1×105 cells/well and treated with 0.5, 2 or 10 µM sodium arsenite in DMEM medium for 24 h and the media were collected and analyzed. For time dependent assay, cells were exposed to 2 µM sodium arsenite for 24, 48 or 72 h, respectively. The media were collected for ELISA assay. IL-1α, IL-1β and IL-18 productions in the mouse skin were evaluated by using mouse IL-1alpha Platinum ELISA (BMS611, eBioscience), mouse IL-1 β ELISA Kit (ELM-ILb-CL, Rabiotech), mouse IL-18 platinum ELISA kit (BMS618/3, eBioscience), respectively. Skin tissues were harvested and homogenized in extraction buffer (containing 10 mM Tris PH7.4, 150 mM NaCl, 1% Triton X-100). The homogenates were centrifuged at 10,000×g for 20 min at 4°C and the supernatants were used for the determination of cytokine levels by ELISA assay per the manufacturer's instruction. All the assays were done in triplicate. The adjusted quantities were presented as an average of the data ± standard error of mean (SEM).
Immunoblotting
Cells were washed with phosphate-buffered saline (PBS; pH 7.4) twice and lysed with RIPA buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5 % deoxycholic acid sodium, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and 10 μg/mL aprotinin] on ice for 10 min. Solubilized cells were centrifuged and the supernatant was collected. For protein extraction from skin, the skin tissues were homogenized by Kinematica Polytron 1300 D machine according to the manufacturer's instruction and then the lysis buffer was added. The immunoblotting procedure has been previously described.Citation31 Briefly, after the protein concentrations were determined, aliquots of the protein samples (40 µg) were loaded into the lanes of an SDS-polyacrylamide gel. The protein samples were separated by electrophoresis, and the separated proteins were transferred to nitrocellulose membranes. The membranes were blocked with either 5% BSA or 5% nonfat milk in 0.01 M PBS (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 h. Subsequently, the membranes were probed with primary antibodies directed against target proteins for 2 h at room temperature or overnight at 4°C. After three quick washes in TPBS, the membranes were incubated with a secondary antibody conjugated to horseradish peroxidase diluted in TPBS for 1 h. The immune complexes were detected by the enhanced chemiluminescence method (Perkin Elmer, NEL105001EA). The blots were stripped and re-probed with an anti-actin antibody. Experiments were replicated three times and results were quantified by densitometry and normalized to matched β-actin levels.
RNA extraction, reverse transcription and quantitative PCR
The mRNA expression was analyzed by quantitative real-time reverse transcription-PCR. Briefly, cells were collected for total RNA extraction by a TRIzol reagent (Invitrogen, 15596-026) according to the manufacturer's instructions. The quality of the total RNA was confirmed by the integrity of 28 S and 18 S rRNA. The first-strand cDNA was synthesized using a Reverse Transcription System (Promega, A3500). PCR was performed on a Lightcycler 480 system (Roche) using a Power SYBR Green PCR Master kit (Invitrogen, 4368706). After finishing the last cycle a melting curve analysis was performed. Standard -ΔΔCt method was used for determining changes in gene expression. The relative expression level of a given mRNA was assessed by normalizing to the housekeeping gene β-actin and comparing with control values. The primers used for analysis were as below: NLRP3 forward, 5′-aaggccgacaccttgatatg-3′; NLRP3 reverse, 5′-ccgaatgttacagccaggat-3′; NLRP1 forward, 5′-ccaattgctgagattgcaga-3′; NLRP1 reverse, 5′-agccagctacagggaagtga-3′; NLRC4 forward, 5′-atcatttgctgcgagaaggt-3′; NLRC4 reverse, 5′-ctgagccaaatcgtccaagt-3′; AIM2 forward, 5′-tctgcagccatcagaaatga-3′; AIM2 reverse, 5′-catctcctgcttgccttctt-3′; Asc forward: 5′-agctcaccgctaacgtgc-3′; Asc reverse, 5′-ctggtactgctcatccgtca-3′; IL-1α forward, 5′-aatgacgccctcaatcaaag-3′, IL-1α reverse: 5′-tgggtatctcaggcatctcc-3′; IL-1β forward: 5′-gggcctcaaggaaaagaatc-3′, IL-1β reverse: 5′-ttctgcttgagaggtgctga-3′; IL-18 forward: 5′-ggaattgtctcccagtgcat-3′, IL-18 reverse: 5′-actggttcagcagccatctt-3′; β-actin forward, 5′-agcacagagcctcgccttt-3′; β-actin reverse, 5′-agggtgaggatgcctctctt-3′.
Statistical analysis
Differences among treatment groups were tested by analysis of variance (ANOVA). Data are represented as mean ± SEM of three experiments. p < 0.05 was considered statistically significant. In cases in which significant differences were detected, specific post-hoc comparisons between treatment groups were examined by Student–Newman–Keuls tests.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr John C. Bell at Ottawa Hospital Research Institute for providing the C-PKR−/− mice.
Funding
This work was supported by the American Cancer Society [RSG-11-116-01-CNE to G.C.].
References
- Karagas MR, Stukel TA, Tosteson TD. Assessment of cancer risk and environmental levels of arsenic in New Hampshire. Int J Hyg Environ Health 2002; 205:85-94; PMID:12018020
- Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schreiber RD, Smyth MJ. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A 2008; 105:652-6; PMID:18178624; http://dx.doi.org/10.1073/pnas.0708594105
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883-99; PMID:20303878; http://dx.doi.org/10.1016/j.cell.2010.01.025
- Mantovani A, Garlanda C, Allavena P. Molecular pathways and targets in cancer-related inflammation. Ann Med 2010; 42:161-70; PMID:20384432; http://dx.doi.org/10.3109/07853890903405753
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009; 9:361-71; PMID:19343034; http://dx.doi.org/10.1038/nrc2628
- Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009; 15:79-80; PMID:19185839; http://dx.doi.org/10.1016/j.ccr.2009.01.009
- Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 2007; 67:1062-71; PMID:17283139; http://dx.doi.org/10.1158/0008-5472.CAN-06-2956
- Mizutani H, Black R, Kupper TS. Human keratinocytes produce but do not process pro-interleukin-1 (IL-1) β. Different strategies of IL-1 production and processing in monocytes and keratinocytes. J Clin Invest 1991; 87:1066-71; PMID:1999487; http://dx.doi.org/10.1172/JCI115067
- Yazdi AS, Drexler SK. Regulation of interleukin 1alpha secretion by inflammasomes. Ann Rheum Dis 2013; 72 Suppl 2:ii96-9; PMID:23253918; http://dx.doi.org/10.1136/annrheumdis-2012-202252
- Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell 2014; 5:12-20; PMID:24474192; http://dx.doi.org/10.1007/s13238-013-0001-4
- Clemens MJ. PKR–a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol 1997; 29:945-9; PMID:9375375
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012; 488:670-4; PMID:22801494; http://dx.doi.org/10.1038/nature11290
- Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW et al. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem 1999; 274:5953-62; PMID:10026221; http://dx.doi.org/10.1074/jbc.274.9.5953
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27:519-50; PMID:19302047; http://dx.doi.org/10.1146/annurev.immunol.021908.132612
- Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 2012; 13:343-51; PMID:22430787; http://dx.doi.org/10.1038/ni.2224
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, Apte RN. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A 2003; 100:2645-50; PMID:12598651; http://dx.doi.org/10.1073/pnas.0437939100
- Zepter K, Haffner A, Soohoo LF, De Luca D, Tang HP, Fisher P, Chavinson J, Elmets CA. Induction of biologically active IL-1 β-converting enzyme and mature IL-1 β in human keratinocytes by inflammatory and immunologic stimuli. J Immunol 1997; 159:6203-8.
- Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, Kummer JA, Tschopp J, French LE. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 2007; 127:1956-63; PMID:17429439; http://dx.doi.org/10.1038/sj.jid.5700819
- Gueniche A, Viac J, Lizard G, Charveron M, Schmitt D. Effect of nickel on the activation state of normal human keratinocytes through interleukin 1 and intercellular adhesion molecule 1 expression. Br J Dermatol 1994; 131:250-6; PMID:7917990; http://dx.doi.org/10.1111/j.1365-2133.1994.tb08500.x
- Cho D, Kim TG, Lee W, Hwang YI, Cho HI, Han H, Kwon O, Kim D, Park H, Houh D. Interleukin-18 and the costimulatory molecule B7-1 have a synergistic anti-tumor effect on murine melanoma; implication of combined immunotherapy for poorly immunogenic malignancy. J Invest Dermatol 2000; 114:928-34; PMID:10771473; http://dx.doi.org/10.1038/sj.jid.5600685
- Maier NK, Crown D, Liu J, Leppla SH, Moayeri M. Arsenic trioxide and other arsenical compounds inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes. J Immunol 2014; 192:763-70; PMID:24337744; http://dx.doi.org/10.4049/jimmunol.1301434.
- Fang R, Hara H, Sakai S, Hernandez-Cuellar E, Mitsuyama M, Kawamura I, Tsuchiya K. Type I interferon signaling regulates activation of the absent in melanoma 2 inflammasome during Streptococcus pneumoniae infection. Infect Immun 2014; 82:2310-7; PMID:24643540; http://dx.doi.org/10.1128/IAI.01572-14
- Sorrentino R, Terlizzi M, Di Crescenzo VG, Popolo A, Pecoraro M, Perillo G, Galderisi A, Pinto A. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am J Pathol 2015; 185:3115-24; PMID:26506473; http://dx.doi.org/10.1016/j.ajpath.2015.07.009
- Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, Fitzgerald KA, Sutterwala FS, Briken V. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-β and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol 2013; 191:3514-8; PMID:24643540; http://dx.doi.org/10.4049/jimmunol.1301331
- McAllister CS, Taghavi N, Samuel CE. Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2alpha-mediated translational control. J Biol Chem 2012; 287:36384-92; PMID:22948139; http://dx.doi.org/10.1074/jbc.M112.390039
- Ponomareva L, Liu H, Duan X, Dickerson E, Shen H, Panchanathan R, Choubey D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol Cancer Res 2013; 11:1193-202; PMID:23864729; http://dx.doi.org/10.1158/1541-7786.MCR-13-0145
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458:514-8; PMID:19158675; http://dx.doi.org/10.1038/nature07725
- Kozul CD, Hampton TH, Davey JC, Gosse JA, Nomikos AP, Eisenhauer PL, Weiss DJ, Thorpe JE, Ihnat MA, Hamilton JW. Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung. Environ Health Perspect 2009; 117:1108-15; PMID:19654921; http://dx.doi.org/10.1289/ehp.0800199
- Waalkes MP, Qu W, Tokar EJ, Kissling GE, Dixon D. Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human-relevant doses. Arch Toxicol 2014; 88:1619-29; PMID:25005685; http://dx.doi.org/10.1007/s00204-014-1305-8
- Cui X, Wakai T, Shirai Y, Hatakeyama K, Hirano S. Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol Sci 2006; 91:372-81; PMID:16543296
- Qi Y, Zhang M, Li H, Frank JA, Dai L, Liu H, Zhang Z, Wang C, Chen G. Autophagy Inhibition by Sustained Overproduction of IL6 Contributes to Arsenic Carcinogenesis. Cancer Res 2014; 74(14):3740-52; PMID:24830721
- Chen G, Ma C, Bower KA, Ke Z, Luo J. Interaction between RAX and PKR modulates the effect of ethanol on protein synthesis and survival of neurons. J Biol Chem 2006; 281:15909-15; PMID:16574643