
ABSTRACT
Despite the clinical success of anti-PD1 antibody (α-PD1) therapy, the immune mechanisms contributing to the antineoplastic response remain unclear. Here, we describe novel aspects of the immune response involved in α-PD1-induced antitumor effects using an orthotopic KrasG12D/p53R172H/Pdx1-Cre (KPC) model of pancreatic ductal adenocarcinoma (PDA). We found that positive therapeutic outcome involved both the innate and adaptive arms of the immune system. Adoptive transfer of total splenocytes after short-term (3 d) but not long-term (28 d) PD1 blockade significantly extended survival of non-treated tumor-bearing recipient mice. This protective effect appeared to be mostly mediated by T cells, as adoptive transfer of purified natural killer (NK) cells and/or granulocyte receptor 1 (Gr1)+ cells or splenocytes depleted of Gr1+ cells and NK cells did not exhibit transferrable antitumor activity following short-term PD1 blockade. Nevertheless, splenic and tumor-derived CD11b+Gr1+ cells and NK cells showed significant persistence of α-PD1 bound to these cells in the treated primary recipient mice. We observed that short-term inhibition of PD1 signaling modulated the profiles of multifunctional cytokines in the tumor immune-infiltrate, including downregulation of vascular endothelial growth factor A (VEGF-A). Altogether, the data suggest that systemic blockade of PD1 results in rapid modulation of antitumor immunity that differs in the tumor microenvironment (TME) when compared to the spleen. These results demonstrate a key role for early immune-mediated events in controlling tumor progression in response to α-PD1 treatment and warrant further investigation into the mechanisms governing responses to the therapy at the innate-adaptive immune interface.
Introduction
Dysregulated inflammatory stimuli in the pancreas create a microenvironment conducive to the stochastic accumulation of oncogenic driver mutations, such as KrasG12D/V and p53R172H, in precursor lesions that eventually lead to PDA.Citation1,2 Host–tumor interactions between stromal elements, tumor-infiltrating immune cells (e.g. myeloid derived suppressor cells (MDSCs), regulatory T cells (Treg)) and transformed cells supports tumor growth and invasion while maintaining an immune-subversive TME.Citation3 Therapies aimed at disrupting host–tumor interactions to break tumor-induced tolerance, such as inhibition of the immune checkpoint receptor PD1, have induced durable immune responses in patients with advanced malignancies.Citation4
PD1 is expressed on the surface of activated T and B lymphocytes, NK cells and monocytes. Upon binding to its ligand B7-H1 or B7-H2/B7-DC (also known as programmed cell death ligand 1 or 2 (PD-L1 or PD-L2, respectively)), PD1 dampens immune responses by inducing T cell hyporesponsiveness.Citation5,6 PD-L1 is primarily a T-cell inhibitory molecule differentially expressed on the surface of endothelial, epithelial and antigen presenting cells (APCs) with immunosuppressive properties.Citation7 The importance of the PD1/PD-L1 axis in tumorigenesis is supported by the role of PD-L1 as a biomarker whose expression levels in various malignancies have been correlated with poor prognosis.Citation8,9
Therapies that block PD1 have shown an overall response rate of 36% for PD-L1-expressing tumors;Citation4,10 however, tumor expression of PD-L1 is highly heterogeneous and not necessarily a predictor of response to α-PD1 therapy.Citation11 Although we have gained increasing knowledge regarding how PD1/PD-L1 signaling supports an immunosuppressive TME, we are still unable to identify the patient population most likely to benefit from PD1 therapy. A better understanding of how early events influences response to PD1 blockade therapy may help to gain insight into potential treatment limitations, while further advancing rational selection of drug combinations. In this study, we evaluated the contribution of innate and adaptive immunity in controlling tumor progression at the initial stages of the response triggered by α-PD1 using an orthotopic murine model of PDA. We observed the contribution of a multi-subset immune response to α-PD1 treatment that highlights a critical role for splenic T cells in mediating the antitumor response after short-term treatment in mice.
Results
Single-dose treatment with α-PD1 increases survival in an orthotopic KPC murine model of PDA
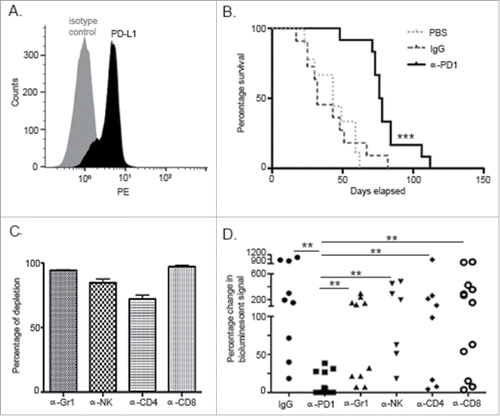
For these studies, we used an orthotopic KPC murine model of PDA that is clinically relevant to study the mechanisms of α-PD1 action. In addition to KPC cells constitutively expressing PD-L1 (), they are also highly refractory to chemotherapeutics, targeted therapies using antibodies and small molecules.Citation12,13 To uncover early events associated with treatment response, we evaluated antitumor efficacy using a single dose of α-PD1 administered 9–10 d after tumor implantation. When compared to IgG isotype or PBS vehicle controls, α-PD1 elicited robust tumor growth control in mice-bearing well established orthotopic KPC tumors as demonstrated with improved survival (32 vs. 76 d median survival time, IgG isotype control vs. α-PD1 respectively, Log-rank p = 0.0003), indicating that single-dose treatment was sufficient to modify disease outcome, thus representing a viable strategy to dissect early mechanisms of response ().
Figure 1. PD1 blockade prolonged survival in an orthotopic PDA murine model and its effect was dependent upon innate and adaptive components of the immune response. (A) Flow cytometric analysis of KPC cells growing in culture demonstrating endogenous expression of PD-L1. (B) Kaplan–Meier curve showing survival benefit of single-dose administration of α-PD1 (n = 12) compared to IgG isotype (n = 11) or PBS vehicle control (n = 9). (C) Flow cytometric analysis of PBMC demonstrating >80% depletion of each immune cell subset 24 h after antibody administration. (D) Mice received IgG isotype control (n = 9) or α-PD1 (n = 12) alone or in combination with individual depletion antibodies: α-Gr1 (n = 10), α-NK (n = 7), α-CD4+ (n = 8) or α-CD8+ (n = 12). Depletion antibodies were continuously administered every 3 d to prevent immune cell repopulation. Results are expressed as percentage of change in bioluminescence signal intensity by measuring luciferase activity using IVIS at day 0 versus day 15. Change in bioluminescent signals were compared to α-PD1 and statistical significance calculated using non-parametric Mann–Whitney test. Each symbol represents an individual mouse. Plots are showing the combined results of at least two independent experiments.**p < 0.01, ***p < 0.001.
Systemic depletion of innate and adaptive immunity abrogates efficacy of α-PD1 treatment
Since the PD1/PD-L1 signaling axis supports development and maintenance of immunosuppression within the TME, we evaluated the individual contribution of cell subsets generally involved with impaired immunity, such as Gr1+ cells (expressed on early myeloid progenitors, neutrophils, and MDSCs), NK cells, CD4+ and CD8+ T cells, in mediating the α-PD1-induced antitumor response.Citation14-17 Quantitative imaging analysis was conducted at day 15 after α-PD1 administration (24–25 d after tumor implantation) to evaluate treatment response. This time point was empirically chosen to assess α-PD1 response based on when PD1 inhibition consistently achieved its peak antineoplastic effect by using IVIS bioluminescence imaging. To account for variations in the tumor load before therapy, mice were imaged at day 0 (start of treatment) and randomized. To compare response between the treatment groups vs. α-PD1 alone, results were expressed as a difference in percentage of the total amount of bioluminescent signal obtained at day 0 vs. day 15, after normalizing day 0 readings to 100%. Assessment of tumor burden by IVIS imaging demonstrated that depletions of individual immune cell subsets tested antagonized α-PD1-mediated antitumor effects, as evidenced by significantly higher bioluminescent signal when compared to α-PD1 treatment alone ((9.07±14.03) vs. (Gr1+ cell depletion: 105.1±104.4, p = 0.0006), (NK cell depletion: 220.5±190.9, p = 0.0001), (CD4+ T cell depletion: 197.9±287.3, p = 0.0015), (CD8+ T cell depletion: 251.6±251.7, p <0.0001)), suggesting that development of α-PD1-mediated antitumor activity requires a complex engagement of the different arms of immunity ( and Fig. S1). There were no significant differences between the groups treated with α-PD1 in combination with immune subset cell depletion and IgG isotype control treatment ((380.6±391.4), (Gr1+ cell depletion: p = 0.07; NK cell depletion: p = 0.58; CD4+ T cell depletion: p = 0.27; CD8+ T cell depletion: p = 0.41)). Within-group variations in response to IgG isotype control treatment may be a function of a single static point of analysis, since Kaplan–Meier survival curve analysis of IgG isotype vs. PBS vehicle control treated mice did not show significant survival advantage (Log-rank p = 0.948, ).
α-PD1 treatment induces transient, transferable T cell-mediated antitumor responses shortly after administration
To evaluate whether PD1 inhibition is followed by persistent antitumor immunological memory, total splenocytes obtained from tumor-bearing donor mice treated with a single dose of IgG isotype control or α-PD1 for 3, 7 or 28 d (corresponding to 12–13, 16–17 or 37–38 d after tumor implantation) were adoptively transferred into untreated tumor-bearing recipient mice pre-conditioned with cyclophosphamide. Surprisingly, tumor-specific protective immunity was only observed in the group that received splenocytes from mice treated with α-PD1 3 d prior (39.5 vs. 63 d median survival time for the IgG isotype control vs. α-PD1-treated group, respectively, Log-rank p = 0.04, ). These results suggest that immunological protection elicited by α-PD1, at least in this model, is short and transient, as tumors progressed in recipient mice in spite of the transfer of splenocytes either at day 7 or 28 after treatment ().
Figure 2. Treatment with α-PD1 induced short but not long-term transferrable protective immunity. Kaplan–Meier curve showing survival benefits of adoptively transferring total splenocytes obtained from tumor-bearing donor mice at (A) day 3, (B) 7, or (C) 28 after single-dose treatment with α-PD1 (day 3: n = 9; 7: n = 8, 28: n = 8) or IgG isotype control (day 3: n = 8; 7: n = 7, 28: n = 6) into cyclophosphamide pre-conditioned tumor-bearing recipient mice. (D) Mice received α-PD1 (n = 8) alone or in combination with depletion antibodies: α-Gr1+α-NK (n = 7) or α-CD4+α-CD8+ (n = 8). Statistical significance was calculated using non-parametric Mann–Whitney test. Results are expressed as percentage of change in bioluminescence signal intensity by measuring luciferase activity using IVIS at day 0 versus day 15. Each symbol represents an individual mouse. Plots are showing the combined results of at least two independent experiments. N.S. = not significant, *p < 0.05.
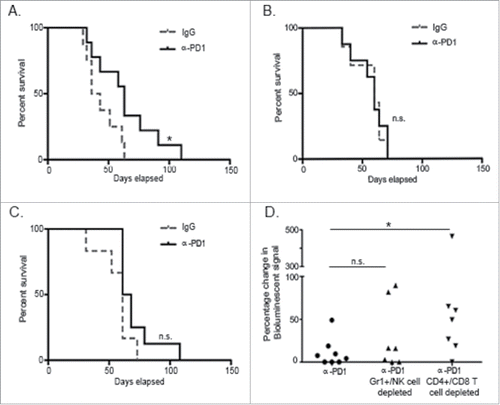
To evaluate immune populations (innate vs. adaptive) contributing to tumor-growth inhibition following adoptive transfer, we proceeded to deplete innate (Gr1+ cells and NK cells) or adaptive (CD4+ and CD8+ T cells) immune subsets in vivo in α-PD1 treated donor mice prior to the transfer. To account for differences in tumor burden at the beginning of the experiment, recipient mice were imaged at day 0 before the adoptive transfer and randomized. Splenocytes isolated from short-term (3 d) α-PD1-treated donor mice following combined depletion of Gr1+ and NK cells or CD4+ and CD8+ T cells were then transferred into untreated tumor-bearing recipient mice that were pre-conditioned with cyclophosphamide. After normalizing day 0 imaging readings to 100%, mice were re-imaged at day 15 proceeding adoptive transfer. Results were expressed as a difference in percentage of the total amount of bioluminescent signal obtained at day 0 vs. day 15. Imaging analysis of tumor progression suggests that adoptive transfer of splenocytes from α-PD1 treated donor mice depleted of Gr1+ and NK cells retained the capability to control tumor progression in recipient mice (no significant difference between α-PD1 treatment alone: 11.14 ± 16.59 vs. α-PD1treatment + Gr1+/NK cell combined depletion: 29.52 ± 39.06, p = 0.52). In contrast, transfer of splenocytes depleted of CD4+ and CD8+ T cells resulted in impaired capacity to limit tumor progression (α-PD1 treatment alone vs. α-PD1 treatment + CD4+/CD8+ T cell combined depletion: 97.91±163.6, p = 0.021, ). These results suggest that CD4+ and CD8+ T cells with antitumor activity exist in the spleen shortly after α-PD1 treatment, but most likely do not persist, at least not in sufficient numbers, to elicit an antitumor immune response at the later time points analyzed ().
α-PD1 shows persistent binding to CD11b+Gr1+ cells and NK cells in spleen and TME
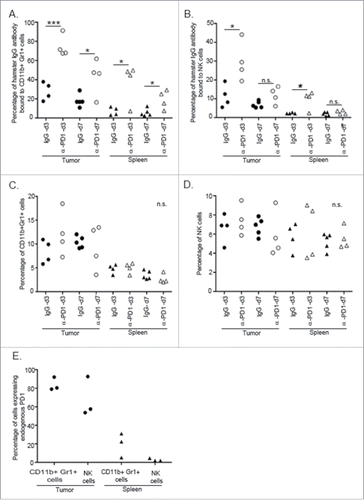
Given the transient nature of the immunological protection conferred by splenocytes transferred after short-term PD1 blockade, it was of interest to determine whether α-PD1 was still bound to splenocytes at the time of the adoptive transfer (3 vs. 7 d after treatment) and if α-PD1 reached the TME. Flow cytometric analysis of spleen and tumor at day 3 after treatment with α-PD1 showed that high amounts of the antibody was still bound to CD11b+Gr1+ cells (spleen: IgG isotype = 7.11±2.12 vs. α-PD1 = 37.49±10.16, p = 0.026; tumor: IgG isotype = 28.04±4.68 vs. α-PD1 = 74.56±5.65, p = 0.0007), and NK cells (spleen: IgG isotype = 2.30±0.14 vs. α-PD1 = 9.54±2.4, p = 0.024; tumor: IgG isotype = 11.35±3.02 vs. α-PD1 = 29.61±5.25, p = 0.023). At day 7 after treatment, α-PD1 remained bound to the cells, albeit to a lesser extent, in both the spleen ((CD11b+Gr1+ cells: IgG isotype = 5.67±1.76 vs. α-PD1 = 19.25±4.86, p = 0.023), (NK cells: IgG isotype = 1.89±0.43 vs. α-PD1 = 2.76±0.55, p = 0.25)) and tumor ((CD11b+Gr1+ cells: IgG isotype = 18.08±2.92 vs. α-PD1 = 42.97±9.56, p = 0.02), (NK cells: IgG isotype = 6.87±0.8 vs. α-PD1 = 11.82±2.26, p = 0.058), and Figs. S2–4). These differences did not result from an increase in overall percentage of infiltrating cells, as their frequencies remained unchanged (). Amounts of α-PD1 bound to CD11b+Gr1+ cells and NK cells correlated with basal levels of endogenous expression of PD1 receptor prior to treatment administration (). Even though splenic T cells have been implicated in the adoptive transfer of antitumor immunity to recipient mice after short-term PD1 blockade (), transferable antitumor activity of T cells did not appear to correlate with persistent binding of α-PD1 or shifts in the percentage of CD4+ or CD8+ T cells present in the spleen at 3 or 7 d after treatment (Fig. S5).
Figure 3. α-PD1 exhibited prolonged binding to CD11+Gr1+ cells and NK cells present in the spleen and tumor. Single-cell suspensions obtained from both spleens and tumors of mice after 3 or 7 d treatment with IgG isotype control (day 3: n = 4, 7: n = 5) or α-PD1 (day 3: n = 4, 7: n = 4) were subjected to flow cytometric analysis. Percentage of antibody that remained bound to (A) CD11b+Gr1+ cells or (B) NK cells. (C) Total percentage of CD11b+Gr1+ cells were estimated in the live CD45+ gated population and (D) of NK cells in the live CD45+CD3− gated population. (E) Baseline endogenous expression of PD1 receptor in CD11b+Gr1+ cells and NK cells at day 0 of treatment. Each symbol represents and individual mouse. Results from two independent experiments were combined. *p < 0.05, ***p < 0.001 by 2-tailed unpaired test.
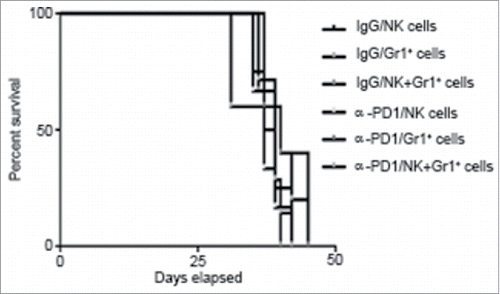
Even though effectiveness of the adoptive transfer at day 3 after treatment coincided with the presence of high amounts of α-PD1 bound to CD11b+Gr1+ cells and NK cells in both spleen and tumor, results from suggested that Gr1+/NK cells present in the spleen did not possess direct antitumor activity. These results were further confirmed by showing that adoptive transfer of spleen-purified Gr1+ cells and/or NK cells from 3 day-treated tumor-bearing donor mice into untreated tumor-bearing recipient mice were not sufficient to extend survival in tumor-bearing recipient mice when compared to IgG isotype control (). Combined results from individual immune subset depletion presented in suggest that whilst spleen-derived CD11b+Gr1+ cells and NK cells are not directly involved in tumor killing, it is likely that tumor-infiltrating CD11b+Gr1+ and NK cells participate in the α-PD1-mediated antitumor immune response. Therefore, activation of antitumor response mediated by CD11b+Gr1+ and NK cells following PD1 blockade may vary according to the location and microenvironment of these innate immune lymphocytic subsets and warrants further study.
Figure 4. Adoptive transfer of purified Gr1+ cells and/or NK cells failed to control tumor progression in untreated recipient mice. Kaplan–Meier curve showing no survival benefit of adoptively transferred NK cells and/or Gr1+ cells isolated from total splenocytes at day 3 after treatment of tumor-bearing donor mice with α-PD1 (NK: n = 6; Gr1+: 5; NK+Gr1+: n = 7) versus IgG isotype control (NK: n = 4; Gr1+: 5; NK+Gr1+: n = 6). Cells were transferred into untreated tumor-bearing recipient mice pre-conditioned with cyclophosphamide. Experiments were repeated at least four times and the results combined.
Inhibition of PD1 relieves transcriptional repression of cytokines in tumor-infiltrating leukocytes
PD1/PD-L1 signaling potentiates immunosuppression by acting as a negative regulator of costimulatory cytokines.Citation18 Because α-PD1 remained bound to immune cells subsets in the TME for as long as 7 d following administration, it was of interest to evaluate whether this phenomenon correlated with transcriptional repression relief of selected regulatory cytokines in the CD45+ tumor-infiltrating immune cells fraction (which excludes the tumor cells). Analysis of gene expression by RT-qPCR showed an increase in mRNA expression levels of interleukin (IL)−6, −8, −10, interferon gamma (IFNγ) and tumor necrosis factor α (TNF-α), as well as a decrease in VEGF-A mRNA in the tumor-associated immune-infiltrate at day 7 after treatment (). There were no differences in the expression levels of any of the cytokines studied in the spleen (data not shown). While IFNγ and TNF-α are involved in activation of cytotoxic responses in NK and T cells,Citation19,20 increased expression of pleiotropic cytokines such as IL−6, −8 and −10 are associated with improved survival of antigen-specific CD4+ T cells,Citation21 neutrophil recruitment and degranulationCitation22 and stimulation of cytotoxic CD8+ T cell responses,Citation23 respectively. In addition, VEGF-A was shown to have broad effects in inhibiting MDSC differentiation and inducing PD1 expression on CD8+ T cells.Citation24,25 These results suggest that inhibition of PD1 modulates cytokine expression in the TME within a few days after administration.
Figure 5. Inhibition of PD1 modulated the expression of pleiotropic cytokines and cell cycle-related genes in the TME. Single-cell suspensions were obtained from tumors harvested at day 3 or 7 after treatment with IgG isotype control or α-PD1. RT-qPCR showing changes in (A) in cytokine expression in the CD45+ tumor-infiltrating cells or (B) in VEGF-A and G1/S cyclins in the CD45− tumor cell enriched fraction. Results were normalized to RPL13a housekeeping gene and expressed as a fold difference relative to the IgG isotype control expression levels. Each bar represents the combined result of three independent experiments, containing pooled tumor samples from 2–3 mice in each experiment. Prior to normalizing the data, results were analyzed using the comparative ΔΔCT method and significance determined by using ANOVA followed with Bonferroni correction. (C) VEGF-A levels were determined by ELISA assay using fresh tumor tissue lysates obtained from mice treated with α-PD1 (n = 6) or IgG isotype control (n = 5) for 7 d. Each symbol represents an individual mouse. Comparison of VEGF-A protein levels between α-PD1 versus IgG isotype control treated mice was calculated using non-parametric Mann–Whitney test. N.S. = not significant. *p < 0.05, ***p < 0.001.
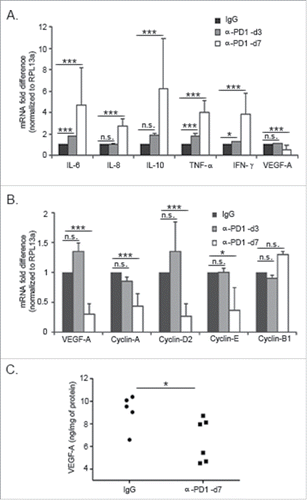
α-PD1 induces downregulation of VEGF-A and G1/S cyclins mRNA in tumor cell enriched fraction
Gene expression analysis was carried out using the tumor cell enriched fraction (CD45− cells), defined by the remaining cells collected after removal of the CD45+ tumor-infiltrating cells. Concomitant with increases in cytokine expression in the tumor infiltrate at day 7 after treatment, we observed a decrease in mRNA expression of VEGF-A and G1/S related cyclins (cyclin −A, −D2 and −E), but not in cyclin-B1, which is related to the G2/M phase of the cell cycle (). These results suggest that α-PD1 treatment could be inducing tumor cell cycle arrest. In fact, increased expression of IFNγ and TNF-α have been shown to induce senescence in a variety of mouse tumor models.Citation26 Since induction of cell cycle arrest may lead to senescence and/or apoptosis and rapid clearance in vivo, we were unable to detect the presence of cleaved caspase-3 or PARP (poly ADP ribose polymerase) by immunohistochemistry or western blot (data not shown).
Given the broad role of VEGF-A in fostering the development of immunosuppressive tumor networks while acting as an endothelial/epithelial growth factor, it was of interest to confirm downregulation of VEGF-A at the protein level.Citation25 The decrease in VEGF-A mRNA levels at day 7 after treatment was confirmed by ELISA using whole tumor extracts (α-PD1 treatment: 6.57±1.59 vs. IgG isotype control: 9.13±1.51, p = 0.030, ). Even though KPC cells secrete considerable amounts of VEGF-A in vitro (∼6 ng/mL), antibody-mediated blockade of VEGF-A did not suppress cell growth (data not shown), further suggesting that while downregulation of VEGF-A signaling may have broader implications for the immune response, it is not directly inducing senescence/apoptosis. Nevertheless, based on our findings and others, we anticipate that further studies will determine whether tumor expression of VEGF-A could be used as a biomarker for response to PD1 blockade therapy and possibly suggest that α-PD1-mediated inhibition of VEGF is an important immune-related mechanism leading to tumor remission.Citation24
Discussion
In the current study, we have uncovered early events involved in the response to α-PD1 therapy. While depletion experiments confirmed a role for T cell-mediated immunity in response to α-PD1 therapy, decrease or lack of α-PD1 antitumor activity in the absence of Gr1+ or NK cells in the primary recipient mice also suggests the importance of innate immunity mechanisms in this process. Upregulation of PD1 signaling is emerging as an important mechanism in modulating the magnitude of innate immune responses to infections agents,Citation27,28 however, its role in modulating antitumor immunity remains elusive. Although Gr1+ populations consist of different cell subsets of myeloid origin, mounting evidence points to the importance of tumor-associated granulocytes in pro-inflammatory responses and in stimulating robust cytotoxic NK cell and T cell-mediated tumor cell death.Citation29-31 Since NK cell lytic activity also supports T cell responses, it is reasonable to suggest that abrogation of Gr1+ and/or NK cells may counteract α-PD1 activity within the TME by preventing adaptive immune responses from becoming established.Citation32
Persistent antibody binding to innate immune effector cells may also facilitate the establishment of a robust immune response by preventing engagement of PD1 with its ligands for an extended period of time within the TME. In this context, the role of Fc receptors in enhancing the antitumor effects of α-PD1 cannot be excluded, as potential binding of the therapeutic antibody with Fc receptors present on neutrophils, MDSCs and NK cells could be activating alternative immunomodulatory pathways or increasing antibody half-life.Citation33 Nonetheless, this persistent antibody binding phenomenon in NK and Gr1+ cells could have potential implications for responsiveness to α-PD1 therapy and requires further validation using additional preclinical tumor models.
In terms of adaptive immunity, recent studies also question the ability of hyporesponsive PD1+ T cells to become memory cells.Citation34,35 We attempted to determine whether treatment with α-PD1 is followed by generation of functional protective immunity by transferring total splenocytes from treated tumor-bearing donor mice to an untreated recipient mice. The fact that we were only able to transfer protection during a window of time shortly after treatment suggests that response to α-PD1 involves primarily effector cells, highlighting the importance of splenic T cells as early mediators of the antitumor response. The relative absence of α-PD1 bound to splenic T cells after short-term treatment may reflect differences in antibody binding kinetics and/or be a result of clearance after single-dose administration of the antibody in C57BL6 mice at the time points analyzed (3 and 7 d, Fig. S5). The process of i.v. injection of splenocytes during transfer may have enable these spleen-derived tumor reactive T cells to enter tumors through the circulation, which was not observed in the case of Gr1+ cells and NK cells. Reasons for the apparent discrepancy between the α-PD1-mediated antitumor activity of Gr1+ cells and NK cells in the primary versus in the secondary recipient mice may be a result of complex differences in homing interactions and migratory capacities of these cells in the tumor compared to the spleen.Citation36-38 We also acknowledge that because generation of long-term immunological memory may require a few weeks or months to develop, splenocytes adoptively transferred at day 28 after PD1 blockade may have represented a population of cells with underdeveloped antitumor activity. Due to the shorter lifespan of IgG isotype control-treated mice, we were unable to conduct this experiment at later time points to better address this question.
Although more detailed studies are needed to elucidate the contribution of the systemic versus the intratumoral innate immune compartment in modulating response to α-PD1 therapy, this study provides further rationale for combination of α-PD1 with NK cell-based therapies, such as α-KIR (killer cell immunoglobulin like-receptors), to improve tumor recognition as well as with anti-angiogenic agents such as bevacizumab.Citation39,40 Novel therapeutic approaches targeting innate immunity are beginning to emerge as valuable tools for the treatment of cancers and should be explored in combination with checkpoint inhibitors to determine whether it would extend antitumor effects.
Materials and methods
Cell culture and luciferase transduction
KPC cells were established from spontaneous KPC tumors that were finely minced using scalpels and directly plated (outgrowth method), without mechanical or enzymatic dissociation as previously described.Citation41 KPC tumors were obtained from crossing the LSL-Kras-G12D with LSL-p53-R172H and PDX1-cre mice (NCI-Frederick) and further backcrossing for >10 times to attain animals with a predominantly C57BL6 background. Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 100 U/mL of penicillin/streptomycin (Gibco) at 37°C, 5% CO2 in a humidified incubator. KPC cells were transduced with the luciferase report gene using a commercially available 3-packaging plasmid lentiviral expression system from Life Technologies (catalog number K4975-00).
Mice
C57BL6 mice were bred and handled at the City of Hope animal facility, according to the protocol approved by the Institutional Care and Use Committee and to the guidelines from the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC) International. Male mice between 6–8 weeks of age were used for all experiments.
Orthotopic implantation of tumor cells and survival studies
KPC cells in the log-phase of growth (7 × 105 cells in 50 µL of PBS) were surgically implanted into the body of the pancreas under aseptic conditions as previously described.Citation42 Tumors were visualized once a week using the Xenogen IVIS imaging system (Caliper Life Sciences) 5 min after intraperitoneal (i.p) administration of 3 mg of D-luciferin/mouse (Gold Biotechnology). Bioluminescent signals were quantified using Xenogen Igor Pro imaging analysis software (Wavemetrics). To determine therapeutic efficacy of α-PD1 (Armenian hamster anti-mouse PD1, Clone J43 ),Citation43 mice received a single dose (i.p.) of 250 µg/mouse of Armenian hamster IgG isotype control (Molecular Innovations, catalog number HT-GF) or α-PD1, 9–10 d after orthotopic tumor cell implantation. Animals were humanly euthanized by CO2 inhalation after experimental end points were reached, such as tumor size, abdominal distension, reduced mobility and/or other signs of distress.
Adoptive transfer of splenocytes and purified Gr1+/NK cells
Spleens from donor mice were harvested under sterile conditions at day 3, 7 or 28 after administration of IgG isotype control or α-PD1. Spleens were gently dissociated using a 70-µm mesh filter and 1 mL syringe plunger. Cells were washed with serum-free DMEM and the erythrocytes were lysed using 10 mL of red blood cell lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0.1 mM EDTA) for 5 min, at room temperature. Cells were washed twice with PBS and counted. Alternatively, Gr1+ cells and NK cells were purified from total splenocytes (from 4 × 107 cells/donor mouse) using the Easysep mouse neutrophil or NK cell enrichment kit from StemCell Technologies. Amount of cells recovered per spleen ranged from 1–7 × 105 cells/donor mouse. Total splenocytes (3 × 107 cells/mouse) or total isolated Gr1+ cells or NK cells recovered/mouse were re-suspended in 50 µL of sterile PBS and inoculated intravenously through the retro orbital route into tumor bearing recipient mouse, 10 d after orthotopic tumor cell implantation. Recipient mice were pre-conditioned with cyclophosphamide (100 mg/kg, i.p., Sigma-Aldrich) 3 d prior to the adoptive transfer.
Selective depletion of immune cell subsets
Antibodies against CD4+ (GK1.5), CD8+ (H35) and GR1 (RB6–8C5) were purified from the supernatant of hybridoma cultures using Protein G affinity columns (Sigma Aldrich). To deplete NK cells, α-Asialo GM1 antibody (rabbit) was purchased from Wako Chemicals (code No. 986–1001). Polyclonal antibodies α-CD4+ (200 µg/mouse), α-CD8+ (200 µg/mouse), α-Gr1 (30 µg/mouse) and α-Asialo GM1 (50 µL/mouse) were administered i.p. every 3 d throughout the length of the experiment, starting 3 d prior to administration of IgG isotype control or α-PD1 (7 d after tumor implantation). Depletion was confirmed 24 h after depletion by flow cytometry using peripheral blood mononuclear cells (PBMC). For adoptive transfer of splenocytes depleted of Gr1+ cells and NK cells or CD4+ and CD8+ T cells, mice received the first dose of depleting antibodies 3 d prior to administration of α-PD1, a second dose at the day of α-PD1 administration and a third dose 48 h thereafter, to ensure maintenance of depletion of immune cell subsets. Splenocytes were processed as described above and transferred (3 × 107 cells/mouse) 3 d following treatment with α-PD1 (24 h after third depletion antibody administration).
Single cell suspensions from primary tumors
Tumors were harvested at day 3 and 7 after treatment with IgG isotype control or α-PD1. Tissue was finely minced and digested in Kreb Ringer's buffer (155 mM NaCl, 5 mM KCL, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 1% BSA) containing collagenase IV (1 mg/mL, Sigma-Aldrich) for 30 min at 37°C. After digestion, tumors were further mechanically dissociated using a 70-µm mesh filter and 1 mL syringe plunger in serum-free DMEM. Cells were centrifuged at 1200 rpm for 10 min, resuspended in 1 mL of PBS and layered on top of a 2-step percoll gradient (1030%, GE Healthcare) to clear debris. After centrifugation at 1500 rpm for 20 min, cells and debris that were trapped in the upper PBS and 10% layer were discarded. Cells collected at the 30% percoll layer were washed and resuspended in PBS prior to further manipulation.
Flow cytometry immune phenotyping
Single cell suspensions were obtained from spleens and tumors of individual mice as described above and incubated on ice with 1 µL of Fixable Viability Dye eFluor 506/107 cells in PBS for 10 min (eBioscience). Cells were washed with PBS and blocked with purified anti-mouse CD16/32 antibody (clone 93, Biolegend) containing 2% BSA for 15 min on ice. To detect the presence of hamster-originated IgG bound to immune cell subsets, anti-Armenian hamster IgG conjugated to biotin (polyclonal, catalog number 13–4113, eBioscience), which reacts with the heavy chains on the Armenian hamster IgG, were directly added to the tube after blocking step (1:100). Cells (2 × 106) were washed twice and plated in a V-bottom 96 well plate. Cells were labeled in 100 µL of PBS containing anti-mouse antibody cocktail plus Streptavidin (APC e-Fluor 780, catalog number 47–4317, eBioscience) for 45 min on ice. As an isotype control, cells were labeled with Armenian hamster IgG (APC, clone eBio299arm) instead of the biotin-streptavidin complex. Cells were analyzed using a 10-color, three laser Gallios flow cytometer (1 × 105 events/sample, Beckman Coulter). Doublets were excluded and only CD45+ (eFluor 450, clone 30-F11) live cells were used for downstream analysis using Kaluza software (Beckman Coulter). Neutrophils and MDSCs were gated as CD11b+ (FITC, clone M1-70), Gr1+ (PE, clone RB6-8C5). NK cells were gated as CD3ϵ-(PE-cyanine5.5, clone 145-2C11), CD122+ (FITC, clone TM-B1; isotype control, clone eB149-10H5) and CD49b+ (PE, clone DX5; isotype control, catalog number 12-4341-81). For detection of endogenous PD1 receptor expression, α-PD1 (APC, clone J43) was added to the cocktail. All antibodies were purchased from eBioscience. Flow cytometry gating strategies and representative contour plots showed in Figs. S1–5 were done using FlowJo.
Real time quantitative PCR (qPCR)
Separation of the immune-infiltrate from the remaining tumor was achieved by preparing single-cell suspensions from spleen and tumor as described above. To separate the immune-infiltrate from the remaining tumor, CD45+ cells were positively selected using the Mouse/Human Chimera Isolation Kit (STEMCELL Technology). The remaining CD45− fraction was considered to be the tumor enriched fraction. RNA was isolated using RNeasy extraction kit (QIAGEN) and converted to cDNA using the RevertAid First Strand cDNA Synthesis kit from Life Technologies using random hexamers. All reactions were carried out according to manufacturer's protocols. Gene expression analyses were performed in duplicates using the ABI PRISM 7300 qPCR system (Life Technologies) with SYBR Green (Kapa Biosystems). RPL13a was used as a house keeping gene for target gene normalization. Results from three independent experiments were combined and expressed as fold change compared to IgG isotype control. Primers were purchased from Integrated DNA Technologies (IDT). Primer sequences used: RPL13a: Fw-5′-GTTCGGCTGAAGCCTACCAG-3′, Rv 5′-TTCCGTAACCTCAAGATCTGCT-3′; Cyclin-A: Fw-5′AGTACCTGCCTTCACTCATTGCTG-3′ Rv-5′-TCTGGTGAAGGTCCACAAGACAAG-3′; Cyclin B1: Fw-5′-GTTAGGGTGTCTTCTCGA ATCGG-3′, Rv-5′-TTTCTGCGTTAATTTTCGTGTTCCT-3′; Cyclin-D1: Fw-5′-GGTCCATAGTGACGGTCAGGT-3′, Rv-5′- GCGTACCCTGACACCAATCTC-3′; Cyclin-D2:Fw-5′-AGACCTTCATCGCTCTGTGC-3′, Rv-5′-TAGCAGATGACGAACACGCC-3′; Cyclin-E: Fw-5′-CCTCCAAAGTTGCACCAGTT-3′, Rv-5′-CCACTTAAGGGCCTTCATCA-3′; IL-6: Fw-5′-AGTTGCCTTCTTGG GACTGA-3′, Rv-5′-GACAGGTCTGTTGGGAGTGG-3′; IL-8: Fw-5′-GCTGGGATTCACCTCAAGAA-3′, Rv-5′-GAAGTGGCAGAAGCTAACCG-3′; IL-10: Fw-5′-CATGGGTCTTGGGAAGAGAA-3′, Rv-5′-AACTGGCCACAGTTTTCAGG-3′; IFN-Y: Fw-5′-GAGGAACTGGCAAAAGGATG-3′, Rv-5′-TGAGCTCATT GAATGCTTGG-3′; TNF-α: Fw-5′-AGCCCCCAGTCTGTATCCTT-3′, Rv-5′-GAGGCACCTGACCACTCTC-3′; VEGF-A: Fw-5′-CTGTGCAGGCTGCTGTAACG-3′, Rv-5′-GTTCCCGAAACCCTGAGGAG-3′.
VEGF ELISA
Tumors were harvested at day 7 after treatment with IgG isotype control or α-PD1, minced and homogenized on ice in the presence of 5 µL/mg of protein lysis buffer (150 mM NaCl, 10 mM Tris, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 10 mM Na4P2O7, 1% Triton) containing protease inhibitor cocktail (Complete Mini, Roche). Tumor lysates were incubated on ice for 30 min, with vortexing every 5 min. Following incubation, lysates were cleared by centrifugation using a table top microcentrifuge pre-cooled to 4°C at 14000 rpm for 15 min. Protein content in the supernatant was quantified using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). VEGF concentration in tumor lysates was determined by ELISA, using 100 µg of total protein/well, according to vendor's protocol (Peprotech).
Statistical analysis
During the experimental design phase, power of analysis calculation was used to determine the number of animals (sample size) per group. Experiments were repeated at least twice and the results combined, without excluding any mice. Survival times were estimated using Kaplan–Meier curves and compared with log-rank test using Graphpad Prism 3 software. Changes in bioluminescent signal between treatment groups were compared to IgG isotype control or α-PD1 treated cohort using non-parametric Mann–Whitney test. Percentage of hamster antibody bound to immune cell subsets and overall percentage of cells were compared between treatments by tissue type and time point using unpaired 2-tailed tests and reported as mean ± standard deviation using Graphpad Prism 3 software. For RT-qPCR experiments, analysis of relative gene expression were performed using the comparative ΔΔCT method and significance determined by using ANOVA followed with Bonferroni correction on Excel.Citation44 Intratumoral expression levels of VEGF-A were compared using non-parametric Mann–Whitney test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1160184_supplementary_materials.zip
Download Zip (425.8 KB)Funding
Research reported in this publication included work performed in the Small Animal Imaging Core and Biostatistics Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Partially supported by R21CA174306 and grants from ThinkCure and the Nesvig Foundation to DJD and R01 CA72669 to BRB.
References
- Lowenfels AB, Maisonneuve P. Epidemiology and prevention of pancreatic cancer. Jpn J Clin Oncol 2004; 34:238-44; PMID:15231857; http://dx.doi.org/10.1093/jjco/hyh045
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA 2010; 107:246-51; PMID:20018721; http://dx.doi.org/10.1073/pnas.0908428107
- Whatcott C, Han H, Posner RG, Von Hoff DD. Tumor-stromal interactions in pancreatic cancer. Crit Rev Oncog 2013; 18:135-51; PMID:23237556; http://dx.doi.org/10.1615/CritRevOncog.v18.i1-2.80
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027-34; PMID:11015443; http://dx.doi.org/10.1084/jem.192.7.1027
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001; 2:261-8; PMID:11224527; http://dx.doi.org/10.1038/85330
- Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012; 1:1223-5; PMID:23243584; http://dx.doi.org/10.4161/onci.21335
- Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 2007; 13:2151-7; PMID:17404099; http://dx.doi.org/10.1158/1078-0432.CCR-06-2746
- Flies DB, Sandler BJ, Sznol M, Chen L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J Biol Med 2011; 84:409-21; PMID:22180678;
- McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol 2015; 33:2013-20; PMID:25800770; http://dx.doi.org/10.1200/JCO.2014.58.1041
- Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol 2015; 7:85-96; PMID:25755681; http://dx.doi.org/10.1177/1758834014567470
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039-49; PMID:25207767; http://dx.doi.org/10.1056/NEJMra1404198
- Manuel ER, Chen J, D'Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, Ludwig T, Chung V, Diamond DJ. Salmonella-Based Therapy Targeting Indoleamine 2,3-Dioxygenase Coupled with Enzymatic Depletion of Tumor Hyaluronan Induces Complete Regression of Aggressive Pancreatic Tumors. Cancer Immunol Res 2015; 3:1096-107; PMID:26134178; http://dx.doi.org/10.1158/2326-6066.CIR-14-0214
- Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 2006; 116:2777-90; PMID:17016559; http://dx.doi.org/10.1172/JCI28828
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol 2006; 90:51-81; PMID:16730261; http://dx.doi.org/10.1016/S0065-2776(06)90002-9
- Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 2000; 96:3838-46; PMID:11090068
- Yamaguchi T, Sakaguchi S. Regulatory T cells in immune surveillance and treatment of cancer. Semin Cancer Biol 2006; 16:115-23; PMID:16376102; http://dx.doi.org/10.1016/j.semcancer.2005.11.005
- Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 2013; 14:1212-8; PMID:24240160; http://dx.doi.org/10.1038/ni.2762
- Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, Mak TW, Ohashi PS. TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest 2007; 117:3833-45; PMID:17992258; http://dx.doi.org/10.1172/JCI32567
- Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 2007; 96:41-101; PMID:17981204; http://dx.doi.org/10.1016/S0065-2776(07)96002-2
- Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol 2009; 130:27-33; PMID:18845487; http://dx.doi.org/10.1016/j.clim.2008.08.018
- Akahoshi T, Endo H, Kondo H, Kashiwazaki S, Kasahara T, Mukaida N, Harada A, Matsushima K. Essential involvement of interleukin-8 in neutrophil recruitment in rabbits with acute experimental arthritis induced by lipopolysaccharide and interleukin-1. Lymphokine Cytokine Res 1994; 13:113-6; PMID:8061112
- Oft M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res 2014; 2:194-9; PMID:24778315; http://dx.doi.org/10.1158/2326-6066.CIR-13-0214
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015; 212:139-48; PMID:25601652; http://dx.doi.org/10.1084/jem.20140559
- Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, Terme M. Control of the immune response by pro-angiogenic factors. Front Oncol 2014; 4:70; PMID:24765614; http://dx.doi.org/10.3389/fonc.2014.00070
- Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:23376950; http://dx.doi.org/10.1038/nature11824
- Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, Garcia VE. Role Played by the Programmed Death-1-Programmed Death Ligand Pathway during Innate Immunity against Mycobacterium tuberculosis. J Infect Dis 2010; 202:524-32; PMID:20617899; http://dx.doi.org/10.1086/654932
- Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR, Jr. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A 2010; 107:13402-7; PMID:20624978; http://dx.doi.org/10.1073/pnas.1007394107
- Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood 2008; 112:4080-9; PMID:18753637; http://dx.doi.org/10.1182/blood-2008-03-143776
- Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, Akimova T, Vachani A, Litzky L, Hancock WW et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest 2014; 124:5466-80; PMID:25384214; http://dx.doi.org/10.1172/JCI77053
- Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis 2012; 33:949-55; PMID:22425643; http://dx.doi.org/10.1093/carcin/bgs123
- Andoniou CE, Coudert JD, Degli-Esposti MA. Killers and beyond: NK-cell-mediated control of immune responses. Eur J Immunol 2008; 38:2938-42; PMID:18979519; http://dx.doi.org/10.1002/eji.200838882
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/10.1038/nri2155
- Dai H, Wan N, Zhang S, Moore Y, Wan F, Dai Z. Cutting edge: programmed death-1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J Immunol 2010; 185:803-7; PMID:20548035; http://dx.doi.org/10.4049/jimmunol.1000661
- Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014; 35:51-60; PMID:24210163; http://dx.doi.org/10.1016/j.it.2013.10.001
- Sun H, Sun C, Tian Z, Xiao W. NK cells in immunotolerant organs. Cell Mol Immunol 2013; 10:202-12; PMID:23563087; http://dx.doi.org/10.1038/cmi.2013.9
- Muller WA. New mechanisms and pathways for monocyte recruitment. J Exp Med 2001; 194:F47-51; PMID:11696603; http://dx.doi.org/10.1084/jem.194.9.f47
- Jablonska J, Wu CF, Andzinski L, Leschner S, Weiss S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int J Cancer 2014; 134:1346-58; PMID:24154944; http://dx.doi.org/10.1002/ijc.28551
- Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol 2013; 10:230-52; PMID:23604045; http://dx.doi.org/10.1038/cmi.2013.10
- Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol 2011; 11:702-11; PMID:21941296; http://dx.doi.org/10.1038/nri3064
- Mack B, Eggert C, Eder K, Imrich S, Baumeister P, Harreus U, Gires O. Rapid and non-enzymatic in vitro retrieval of tumour cells from surgical specimens. PLoS One 2013; 8:e55540; PMID:23383219; http://dx.doi.org/10.1371/journal.pone.0055540
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 2009; 4:1670-80; PMID:19876027; http://dx.doi.org/10.1038/nprot.2009.171
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996; 8:765-72; PMID:8671665; http://dx.doi.org/10.1093/intimm/8.5.765
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262